

Abstract
Activation of the cystic fibrosis transmembrane conductance regulator (CFTR) channel by protein kinase A (PKA) is enhanced by protein kinase C (PKC). However, the mechanism of modulation is not known and it remains uncertain whether PKC acts directly on CFTR or through phosphorylation of an ancillary protein. Using excised patches that had been pre-treated with phosphatases, we found that PKC exposure results in much larger PKA-activated currents and shifts the PKA concentration dependence. To examine if these effects are mediated by direct PKC phosphorylation of CFTR, a mutant was constructed in which serines or threonines at nine PKC consensus sequences on CFTR were replaced by alanines (i.e. the ‘9CA’ mutant T582A/T604A/S641A/T682A/S686A/S707A/S790A/T791A/S809A). In excised patches, 9CA channels had greatly reduced responses to PKA (i.e. 5–10 % that of wild-type), which were not enhanced by PKC pre-treatment, although the mutant channels were still functional according to iodide efflux assays. Stimulation of iodide efflux by chlorophenylthio-cAMP (cpt-cAMP) was delayed in cells expressing 9CA channels, and a similar delay was observed when cells expressing wild-type CFTR were treated with the PKC inhibitor chelerythrine. This suggests that weak activation by PKA in excised patches and slow stimulation of iodide efflux from intact cells are specifically due to the loss of PKC phosphorylation. Finally, PKC caused a slight activation of wild-type channels when added to excised patches after phosphatase pre-treatment but had no effect on the mutant. We conclude that direct phosphorylation of CFTR at one or more of the nine sites mutated in 9CA is required for both the partial activation by PKC and for its modulation of CFTR responses to PKA.
The cystic fibrosis transmembrane conductance regulator (CFTR) is an ATP-dependent, phosphorylation-activated Cl− channel that mediates cAMP-stimulated Cl− secretion and may influence the activity of other membrane proteins (Picciotto et al. 1992; Gadsby & Nairn, 1999; Sheppard & Welsh, 1999). CFTR has two membrane domains (TMD1 and TMD2) each comprised of six membrane-spanning regions, two nucleotide-binding domains (NBD1 and NBD2) and a regulatory (R) domain that contains potential sites for phosphorylation by protein kinases A and C (Riordan et al. 1989). Recent crystal structures of bacterial NBDs (Hung et al. 1998; Diederichs et al. 2000; Karpowich et al. 2001) and functional studies of split CFTR channels formed by expression of half-molecules (Chan et al. 2000) suggest that NBD1 extends from aa433 to near residue 634, and the R domain from aa634 to ∼830.
Exposing wild-type CFTR channels to PKA and MgATP immediately after excision increases their open probability from approximately 0 to 0.4 (Tabcharani et al. 1991). CFTR is phosphorylated by PKA at multiple sites (Cheng et al. 1991; Picciotto et al. 1992; Rich et al. 1993; Chang et al. 1993; Gadsby & Nairn, 1999). Nine of the 10 predicted dibasic PKA consensus sequences (R, R/K, X, S/T) are situated in the R domain, which is presumed to be the main site of phosphorylation control. Mutagenesis of the dibasic and most monobasic PKA sequences strongly inhibits channel activity (Cheng et al. 1991; Chang et al. 1993; Seibert et al. 1995); however, partial activation persists when the PKA sites are removed in various combinations and no one site seems to be essential for activation (Chang et al. 1993; Ostedgaard et al. 2001).
CFTR channel responses to PKA are increased by exposing whole-cell or inside-out patches to purified PKC (Tabcharani et al. 1991; Jia et al. 1997). Stimulatory effects on iodide efflux and whole-cell CFTR current are obtained when endogenous PKC is activated by briefly treating cells with phorbol esters (Middleton & Harvey 1998; Lansdell et al. 1998; Yamazaki et al. 1999; Button et al. 2001). PKC phosphorylates S686 and S790 (Picciotto et al. 1992) but does not affect the secondary structure of the recombinant R domain (Dulhanty & Riordan, 1994). Moreover, PKC modulation is not abolished by removal of the confirmed PKC sites (Yamazaki et al. 1999; Button et al. 2001), raising the possibility that PKC may act by phosphorylating an ancillary protein. PKC weakly activates CFTR channels when added alone to freshly excised patches (Tabcharani et al. 1991; Berger et al. 1993). In the colonic cell line HT-29, phorbol 12,13-dibutyrate (PDB) does not elevate channel open probability but rather increases the incidence of open channels in patches by 16-fold, suggesting an increase in channel density (Bajnath et al. 1993).
The purpose of the present study was to investigate PKC regulation of CFTR by comparing activation of the wild-type channel with that of a mutant lacking all nine PKC consensus sequences between the Walker B motif of NBD1 and the second membrane domain (TMD2), a region that encompasses the regulatory domain. Eliminating these potential PKC sites strongly inhibited channel responses to PKA and abolished the slight activation induced by PKC alone, indicating that PKC regulates CFTR through direct phosphorylation of the channel. A preliminary account of these results has been presented (Hinkson et al. 2000).
Methods
Chemicals
Type II bovine cardiac protein kinase A catalytic subunit (PKA) and rat brain protein kinase C II (PKC) were obtained from the laboratory of Dr M. P. Walsh (University of Calgary, AB, Canada). Protein phosphatase type 2Cα (PP2Cα) was from Upstate Biotechnology (Lake Placid, NY, USA). Protein phosphatase 2A catalytic subunit (PP2A) was from Promega Corp. (Madison, WI, USA). Other chemicals were from Sigma (St Louis, MO, USA) and of the highest grade available.
Cell culture
Baby Hamster Kidney (BHK) cells stably expressing either wild-type CFTR or the mutant lacking PKC consensus sequences were plated on glass coverslips at low density 3 days before patch-clamp experiments. To assess CFTR dephosphorylation in vitro, BHK cells stably expressing histidine-tagged CFTR (i.e. 10 histidine codons added at the 3′ end of the cDNA; CFTR10His; Zhu et al. 1999) were plated in six flasks (75 cm2 each; T-75, Sarstedt no. 83.1813.502, Montréal, QC) and harvested at 80 % confluence. Cells were grown at 37 °C with 5 % CO2 in minimal essential medium (Life Technologies, Burlington, ON, Canada) containing 5 % fetal bovine serum and the selecting drug methotrexate (500 μm for cells expressing 9CA or CFTR10His, and either 0.5 or 500 μm for cells expressing wild-type CFTR, as indicated in the text).
Patch clamp
Macroscopic CFTR currents were recorded from inside-out patches with the pipette potential held at +30 mV (i.e. membrane potential Vm=−30 mV). Current traces were inverted in illustrations to avoid confusion, for example when referring to increasing chloride current, which by convention would become more negative. Pipettes were prepared on a conventional two-stage puller (PP-83, Narishige Instrument Co., Tokyo, Japan), had resistances of 4–6 MΩ when filled with 150 mm NaCl solution (154 mm total [Cl−]) and were used randomly to study wild-type or mutant channels on the same day to avoid systematic variations in tip diameter. Bath and pipette solutions initially contained (mm): 150 NaCl, 2 MgCl2 and 10Tes (pH 7.2). The bath solution was grounded through an agar bridge having the same ionic composition as the pipette. Experiments were carried out at room temperature (∼20 °C). Currents were amplified (Axopatch 1C, Axon Instruments, Inc., Foster City, CA, USA), low-pass filtered using an 8-pole Bessel-type filter (902 LPF, Frequency Devices, Haverhill, MA, USA) and recorded on microcomputer hard disk using pCLAMP version 8.2 software (Axon). Records were sampled at 500 Hz, filtered at 125 Hz and the maximum mean number of open channels (NPo) was calculated using pCLAMP or DRSCAN (Hanrahan et al. 1998) by dividing the mean plateau current (‘I’, averaged over 1 min), by the single channel amplitude (‘i’, approximately 0.3 pA) measured when the first few channels became activated (NPo=I/i). For patches containing wild-type CFTR channels, the number of functional channels N was estimated from the NPo obtained after addition of 5′-adenylylimido-diphosphate (AMP-PNP) by assuming Po≈1. Before carrying out experiments with exogenous protein kinases, excised patches were pre-treated with a cocktail of protein phosphatases for 10 min to reduce phosphorylation (see Fig. 1). Phosphatases were washed from the bath by perfusing the recording chamber for 3 min (more than eight chamber volumes) before adding kinases.
Figure 1. In vitro dephosphorylation of CFTR10His after metabolic labelling of unstimulated BHK cells.
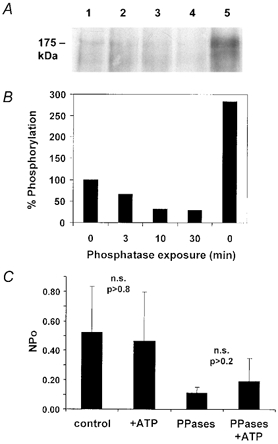
A, autoradiogram showing basal phosphorylation that remains on CFTR10His following incubation with same mixture of PP2A, PP2Cα and alkaline phosphatase used during patch-clamp experiments for 0, 3, 10 or 30 min (Lanes 1–4, see Methods). Unstimulated cells were incubated with [32P]PO4 for 4 h, then CFTR10His was affinity-purified from unstimulated cells, separated by SDS-PAGE, and exposed to film as described in the Methods. For comparison, phosphorylation on CFTR10His that had been isolated from cAMP-stimulated cells is shown in Lane 5. B, dephosphorylation as quantified by densitometry of the band at 175 kDa, normalized to the signal obtained without phosphatase exposure (Lane 1). C, mean number of channels open after excision into nominally ATP-free solution (control), bath solution containing 1 mm ATP (+ATP), after exposure to the phosphatase mixture in nominally ATP-free conditions (PPases), and treated with phosphatase mixture with 1 mm ATP present (PPases + ATP). Unpaired t tests indicate that differences in NPo in the presence or absence of ATP were not significant (n.s.) regardless of phosphatase exposure, although phosphatase treatment alone reduced NPo significantly compared with untreated controls (P < 0.02).
9CA CFTR construction and expression
PCR mutagenesis was used to construct pNUT 9CA CFTR. Mutagenic primers that replaced serines and threonines with alanines in PKC consensus sequences were synthesized by the Sheldon Biotechnology Centre at McGill University. Overlapping fragments containing the mutations were generated using wild-type CFTR cDNA or pUCF2.5 3CA (S707/790/809A) as templates. The primers substituted alanines at T582 (ACA to GCA), T604 (ACT to GCT), S641 (AGC to GCC), T682 (ACA to GCA), S686 (TCT to GCT), S790 (TCC to GCC) and T791 (ACA to GCA). The same methods were used to substitute alanines at S707 (TCT to GCT) and S809 (TCA to GCA) in pUCF2.5 3CA. A typical PCR protocol included 30 cycles of denaturation at 94 °C (30 s), annealing at 50 °C (30 s) and elongation at 72 °C (90 s using Vent DNA polymerase). Fragments were joined together in successive rounds of PCR to create a 1.5 kb cassette containing all nine mutations. E. coli strains Xl-1 blue, Top10 or DH5-α were used for plasmid propagation. The construct was confirmed by sequencing and subcloned into pNUT CFTR by replacement of the corresponding region in wild-type CFTR using flanking DraIII sites at nucleotides 1777 and 3328. pNUT 9CA CFTR was transfected into BHK cells by calcium phosphate co-precipitation, as described previously (Chang et al. 1993). Cell colonies stably expressing 9CA CFTR were selected using 500 μm methotrexate (MTX), a drug that inhibits folate metabolism and is toxic to cells unless they express a mutated (MTX-insensitive) dihydrofolate reductase gene contained in the pNUT vector (Simonsen & Levinson, 1983; Palmiter et al. 1987; Chang et al. 1993).
Iodide effluxes
Cells were incubated with iodide loading buffer (mm: 136 NaI, 3 KNO3, 2 Ca(NO3)2, 11 glucose and 20 Hepes, pH 7.4) for 1 h at room temperature. Extracellular NaI was removed by rapidly rinsing the cells six times with iodide-free efflux buffer (same as the loading buffer except NaNO3 replaced NaI). Samples were collected by completely replacing the efflux buffer (1 ml volume) with fresh solution at 1 min intervals. The first three samples were averaged to establish the baseline efflux rate, then cpt-cAMP was added and samples were collected every minute for 15 min in the continued presence of cpt-cAMP (i.e. the efflux buffer used for subsequent replacements also contained cpt-cAMP at the same concentration). The iodide concentration of each aliquot was determined using an iodide-sensitive electrode (Orion Research Inc., Boston, MA, USA) and converted to iodide content (i.e. the amount of iodide released during the 1 min interval in nanomoles). The cumulative efflux curves in Fig. 6 show the unnormalized, total amount of iodide (nmol) that was collected up to each time point indicated, i.e. the cumulative iodide effluxes calculated by adding the amount collected during each sample period to the total already collected during the preceding sample periods. The mean iodide content of samples collected before cpt-cAMP addition was subtracted from those measured after its addition to correct for background leakage (< 2 % of stimulated efflux), which accounts for the slight decrease at later time points. In some experiments (e.g. Fig. 7), cumulative results were normalized to the total iodide released during the experiment (i.e. over the entire 15 min efflux period), so that effluxes from wild-type cells ± chelerythrine could be compared on the same scale. The iodide content in each sample period was again added to the total iodide collected up to that time point, but was expressed as a percentage of the total iodide released by the cells during the experiment. Stimulated effluxes were measured in duplicate at seven different cpt-cAMP concentrations to compare the responses of 9CA and wild-type channels. The role of PKC in intact cells was tested further by inhibiting it with 10 μm chelerythrine chloride for 30 min, then measuring iodide effluxes in the continued presence of chelerythrine. Chelerythrine was added from a stock solution in dimethyl sulphoxide (DMSO). DMSO alone was added to control monolayers (final concentration 0.1 % v/v). Cell monolayers were lysed in 1 m NaOH at the end of each experiment and assayed for protein (Bradford, 1976).
Figure 6. Chlorophenylthio-cAMP (cpt-cAMP)-stimulated iodide efflux from BHK cells expressing wild-type or 9CA CFTR.
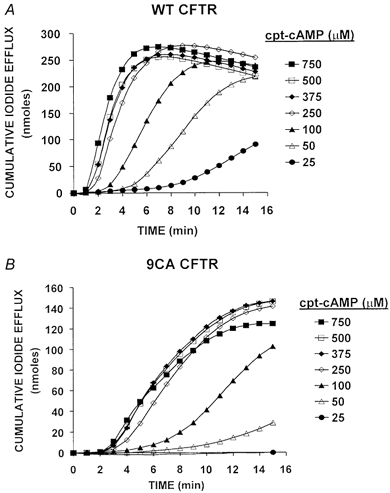
A, cumulative iodide efflux determined at 1 min intervals during exposure of cells expressing wild-type CFTR to cpt-cAMP. B, cumulative iodide effluxes determined as in A using cells that express 9CA. Values shown are the means of duplicate samples at each cpt-cAMP concentration. Variation between samples was < 15 % of the mean values.
Figure 7. Effect of chelerythrine on cAMP-stimulated iodide efflux from cells expressing wild-type (WT) or 9CA CFTR.
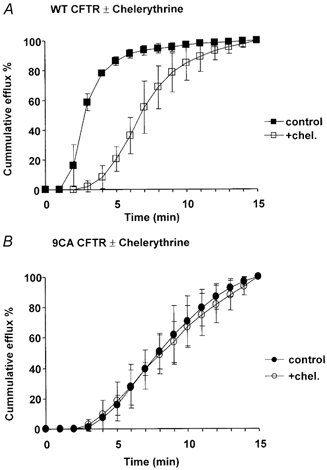
Iodide efflux expressed cumulatively as a percentage of the total released during 15 min stimulation by 100 μm cpt-cAMP. A, time course of cpt-cAMP-stimulated iodide efflux from cells expressing wild-type CFTR under control conditions (▪) or in the presence of 10 μm chelerythrine (□). B, time course of cpt-cAMP-stimulated iodide efflux from cells expressing 9CA CFTR as in A. Symbols indicate means ±s.e.m., n = 6 cell monolayers.
Metabolic labelling and in vitro dephosphorylation of CFTR
BHK cells expressing CFTR10His (i.e. with 10 histidines added to the C terminus) were grown to 80 % confluence, rinsed with phosphate-free medium and incubated with 0.41 mCi ml−1[32P]PO4 solution for 4 h at 37 °C and 5 % CO2. After removing the labelling solutions and rapidly washing with PBS (phosphate buffered saline) at 4 °C, cells were lysed (mm: 150 NaCl, 20 Tris-HCl, pH 7.4, 1 EDTA with 50 μm sodium vanadate, 1 % Triton X-100) on ice for 30 min, centrifuged at 10 000 g for 5 min, and reacted with Ni2+-NTA (nitrilotriacetic acid) agarose beads in binding buffer (mm: 15 imidizole, 500 NaCl, 20 Tris-HCl, pH 7.4, 1 EDTA with 50 μm sodium vanadate and 10 % glycerol) for 30 min at 4 °C with agitation. Beads were washed twice with cold binding buffer and twice with patch-clamp solution (mm: 150 NaCl, 2 MgCl2, 10 Tes, pH 7.2), then resuspended in patch-clamp solution with 20 mm MgCl2 and 95 U ml−1 alkaline phosphatase, 2 U ml−1 PP2A and 1 U ml−1 PP2Cα for 0, 3, 10 or 30 min. Dephosphorylation was stopped by washing the beads three times with cold binding buffer. The beads were resuspended in elution buffer (mm: 500 NaCl, 300 imidazole, 50 Na3PO4, pH 7.0 with 10 % glycerol and 0.2 % Triton X-100) and loaded along with eluate and sample-loading buffer onto an SDS-PAGE gel. Gels were exposed to X-ray film and radiolabelled proteins were visualized by autoradiography.
In vitro phosphorylation of CFTR by PKA or PKC
Phosphorylation of immunoprecipitated wild-type CFTR was carried out over the range 0–400 U ml−1 PKA for comparison with channel activity elicited by the same concentrations of PKA in patch-clamp experiments. BHK cells expressing wild-type CFTR were pre-treated with 5 μm chelerythrine chloride and 500 nm H89 in culture medium for 30 min at 37 °C to reduce basal phosphorylation, and then lysed in RIPA buffer (1 % Triton X-100, 0.1 % deoxycholic acid, 0.1 % SDS, 150 mm NaCl, 20 mm Tris-HCl, pH 8) supplemented with EDTA-free protease inhibitor cocktail (Roche Diagnostics, Laval, QC, Canada). Cellular debris was removed by centrifugation (21 000 g for 15 min) and the supernatant (containing cell lysate) was collected for phosphorylation and quantification of CFTR. CFTR was immunoprecipitated from the lysates by overnight incubation with 2 μg ml−1 M3A7 ascites (monoclonal anti-CFTR antibody) on a bench-top rotator at 4 °C. Protein G that had been immobilized on Sepharose 4B fast-flow beads was added during the last hour. The lysate-M3A7-protein G bead mixture was washed twice by centrifugation (21 000 g for 30 s) in RIPA buffer to remove unbound protein and three times in phosphorylation buffer (mm: 140 NaCl, 4 KCl, 2 MgCl2, 0.5 CaCl2, 10 Tris-HCl, pH 7.4) to remove detergent. After the final wash, the beads were incubated in phosphorylation buffer that had been supplemented with 20 μm MgATP, 10 μCi γ[32P]ATP (Amersham Pharmacia Biotech Inc., Baie d'Urfé, QC, Canada) and 10 μg BSA at 30 °C. Protein kinases (PKA or PKC + the lipid activator DiC8) were added and the reaction was stopped 10 min later by the addition of ice-cold RIPA buffer. The beads were washed in RIPA and phosphorylation buffers, then radiolabelled proteins were eluted in 2 × SDS-PAGE sample buffer for 10 min at 21 °C. After brief centrifugation (21 000 g, 2 min) the supernatant was run on an SDS-PAGE gel, transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore Corp., Bedford, MA, USA), and exposed to a Storage Phosphor Screen. Phosphorylation was detected using a PhophorImager and quantified with Image QuaNT software (Amersham Biosciences, Piscataway, NJ, USA). Radiolabelling was normalized to the amount of CFTR, which was estimated by Western blotting with the monoclonal anti-R domain antibody L11E8 and comparing the signal intensity with those obtained with known amounts of glutathione-S-transferase (GST)-R domain fusion protein isolated after expression in E. coli (Luo et al. 2000).
Immunoblotting of wild-type and mutated CFTR
BHK cells stably expressing wild-type or 9CA CFTR were washed twice with ice-cold PBS, harvested by scraping, and lysed on ice for 30 min in RIPA buffer supplemented with protease inhibitor cocktail. The lysate was centrifuged (15 000 g, 10 min at 4 °C) and an aliquot of supernatant was assayed for protein using bicinchoninic acid (Pierce Chemical Co., Rockford, IL, USA). SDS-PAGE loading buffer (2 ×) was added to an equal volume of supernatant containing 20 μg protein, subjected to 7.5 % SDS-PAGE, and transferred to a PVDF membrane. The monoclonal anti-CFTR antibody M3A7 (Kartner & Riordan, 1998) was used as the primary antibody. The secondary antibody (goat anti-mouse conjugated to peroxidase; Jackson ImmunoResearch Lab. Inc., West Grove, PA, USA) was detected using the enhanced chemiluminescence kit from Amersham Pharmacia Biotech. CFTR expression level was assessed by densitometry of scanned Western blots using Image QuaNT software as described above.
Statistics
Results are reported as the means ±s.e.m.; n = number of observations. Differences were assessed using Student's t test, with P < 0.05 considered significant.
Results
Basal phosphorylation and channel activity in excised patches
To reduce basal phosphorylation and standardize experimental conditions before testing exogenous kinases, excised patches were routinely pre-treated with a mixture of phosphatases for 10 min and rinsed for 3 min. The phosphatases used (PP2A, PP2C and alkaline phosphatase) were those found to be most effective in dephosphorylating [32P]PO4-labelled CFTR and/or recombinant R domain protein in previous studies (Berger et al. 1993; Becq et al. 1994; Travis et al. 1997; Luo et al. 2000). To assess the effectiveness of the phosphatase pre-treatment under conditions approximating those present during the present patch-clamp studies, in vitro dephosphorylation of radiolabelled CFTR was measured under similar conditions. Polyhistidine-tagged CFTR was purified from cells that had been metabolically labelled by incubation with [32P]PO4 for 4 h. The phosphorylation remaining on CFTR10His after incubation with phosphatases for different time intervals was determined by autoradiography. The autoradiogram in Fig. 1 shows dephosphorylation of CFTR by the phosphatase mixture under control conditions (lanes 1–4). For comparison, CFTR from cells that had been stimulated with cAMP agonists (10 μm forskolin, 200 μm dibutyryl-cAMP and 1 mm 3-isobutyl-1-methylxanthine (IBMX)) during the last 10 min of metabolic labelling is also shown (lane 5). Optical density of the band at ∼175 kDa declined by 70 % within the first 10 min of exposure to phosphatase mixture, but the remaining 30 % was still detectable after 30 min. In these experiments, radiolabelling was about 3-fold higher on CFTR isolated from stimulated cells than on CFTR from unstimulated cells, and ∼10-fold that on CFTR that had been treated with phosphatases.
When membrane patches were excised from unstimulated cells into bath solution containing (mm): 150 NaCl, 2 MgCl2, 10 Tes, pH 7.2, brief openings were observed before and after treatment with the same phosphatases as above, although the mean number of open channels (NPo) was low compared with that during PKA stimulation (i.e. NPo < 2.0 vs. NPo > 200 with 400 U PKA). This activity was observed when the bath contained only trace ATP or with 1 mm ATP (Fig. 1C), and may reflect non-hydrolytic gating induced by stably bound nucleotide (Aleksandrov et al. 2002) that was present when the patches were first excised. Single channel conductance calculated from the unitary current and holding potential was consistent with CFTR channels (8–10 pS; e.g. Kartner et al. 1991), and openings were not observed when patches were from untransfected BHK cells lacking CFTR. Exposure to the mixture of three phosphatases reduced NPo from 0.66 ± 0.208 (n = 6 patches) to 0.11 ± 0.38 (n = 9 patches) under nominally ATP-free conditions. NPo was also low when patches were exposed to alkaline phosphatase alone (NPo = 0.127 ± 0.068, n = 13 patches), and when cells were pre-treated with H89 (10 μm) and chelerythrine (1 μm) for 30 min with the goal of inhibiting basal phosphorylation before patches were excised (NPo= 0.197 ± 0.034, n = 8 patches). Thus open probability was very low under these conditions, since a Po < 0.0005 would be expected when mean NPo is 0.11 and patches contain N = 252 ± 70.4 (mean ±s.e.m., n = 11 patches), as indicated by the macroscopic currents measured during stimulation of wild-type channels with PKC and PKA (assuming a single channel current i = 0.3 pA and Po near 1.0 with AMP-PNP, see Fig. 2). Since basal activity was negligible compared with currents measured during kinase stimulation, and since longer phosphatase exposures did not reduce phosphorylation further, we used a protocol in which excised patches were treated with a mixture of PP2A, PP2C and alkaline phosphatase for 10 min, then washed for 3 min before testing exogenous kinases
Figure 2. Effect of PKC pre-treatment on the concentration-dependent activation of CFTR channels by PKA.
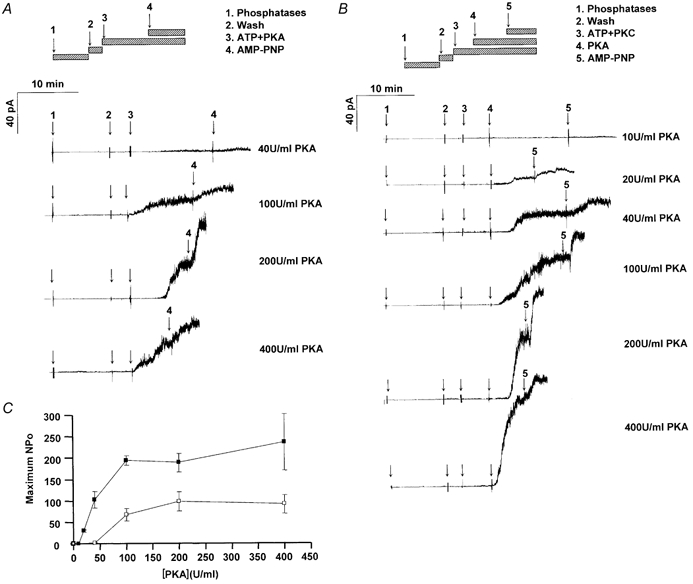
Macroscopic currents activated by different concentrations of PKA recorded without or with PKC pre-treatment. A, as indicated by the hatched bars, patches were excised from BHK cells over-expressing wild-type CFTR, incubated with a mixture of protein phosphatases for 10 min, washed for 3 min, and stimulated by the addition of 1 mm ATP and various concentrations of PKA (no PKC pre-treatment). AMP-PNP (1 mm) was added to maximally increase Po and improve the estimate of channel number. B, effect of adding ATP and 3.78 nm PKC (with 5 μm DiC8 lipid activator) 5 min before adding PKA. Note the large currents (approx. 90 pA) activated by 400 units ml−1 PKA. C, summary of [PKA] dependence (in units ml−1) on the maximum number of channels open (i.e. maximum NPo) when patches were (▪) or were not (□) pre-treated with PKC.
Effect of PKC phosphorylation on the response to PKA
To study the modulation of wild-type CFTR channels by PKC we examined their activation by various PKA concentrations without (Fig. 2A) or with (Fig. 2B) PKC pre-treatment. Figure 2A shows that activation by 40 U ml−1 was barely detectable when patches had not been exposed to PKC, but increased as PKA concentration was elevated so that a maximal response was obtained with 200 U ml−1 PKA (vs. 100 U ml−1 after PKC pre-treatment). By contrast, activation was easily observed with 20 U ml−1 PKA when patches were pre-treated with PKC (Fig. 2B), and the response to 40 U ml−1 PKA was also larger than without PKC pre-treatment (compare with Fig. 2A). The plateau response with 400 U ml−1 PKA after PKC pre-treatment was about twice that observed when PKA was the only kinase added. The delay between PKA addition and current response declined from 3.7 ± 2.5 to 0.6 ± 0.5 min as PKA activity was increased from 100 to 400 U ml−1 (P < 0.025). AMP-PNP was added to further increase Po and improve estimates of channel number (Hwang et al. 1994; Mathews et al. 1998). AMP-PNP caused similar stimulation (expressed as a percentage increase) regardless of the PKA activity present. Relationships between maximum NPo and [PKA] with and without PKC pre-treatment are summarized in Fig. 2C. Note that PKC shifted the EC50 for PKA to lower concentrations and more than doubled the plateau NPo, further evidence that PKC enhances channel responsiveness to stimulation by PKA.
The relationship between PKA-induced channel activity and protein phosphorylation was compared by measuring in vitro phosphorylation under conditions similar to those present during patch-clamp experiments. CFTR became radiolabelled more efficiently by PKA than PKC in the presence of γ[32P]ATP when measured at low kinase concentrations where CFTR phosphorylation was a linear function of the kinase activity used in the assay (Fig. 3A, activity units as defined in legend). CFTR radiolabelling did not become saturated even at the highest PKC activity used (450 U ml−1; Fig. 3B). By contrast, CFTR phosphorylation was maximal at 100 U ml−1 PKA and was about 10-fold higher with PKA than with PKC (compare axes of Fig. 3B and C). Interestingly, PKA activation of CFTR channels (i.e. their functional response to PKA) mirrored their phosphorylation by PKA in biochemical assays but only if patches were pre-treated with PKC (Fig. 3C). When patches were not pre-treated with PKC, higher PKA activities were required to stimulate channels than to phosphorylate CFTR protein. To facilitate these comparisons, Fig. 3C shows phosphorylation (▵) and channel activity (squares, replotted from Fig. 2C) on the same graph as functions of PKA activity. Note that the functional response only mirrored PKA phosphorylation when the patches used for recording had been exposed to PKC. This correspondence between radiolabelling by PKA alone (without PKC pre-treatment) and channel activation by PKA after PKC pre-treatment implies that PKC acts independently of PKA phosphorylation, for example by enabling channels to undergo a conformational change in response to PKA phosphorylation rather than by enhancing the level of PKA phosphorylation.
Figure 3. In vitro phosphorylation of wild-type CFTR by PKC and PKA.
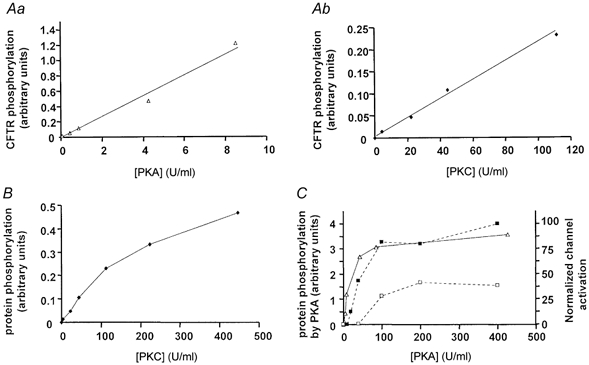
A, in vitro phosphorylation by PKA (a) and PKC (b) at low kinase activities as measured in arbitrary units using a PhosphorImager and Image QuaNT software (Molecular Dynamics). B, in vitro phosphorylation by high PKC activities. C, correlation between PKA dependence of phosphorylation (▵) and channel activation without (□), or with (▪) PKC pre-treatment. One unit is the amount of kinase catalysing the transfer of 1 pmol of phosphate from ATP to casein (for PKA) or to neurogranin (PKC) at 30 °C.
Expression of a CFTR mutant that lacks PKC consensus sequences in NBD1 and the R domain
PKC could act by directly phosphorylating CFTR or indirectly through phosphorylation of an ancillary protein (reviewed by Hanrahan et al. 2002). To distinguish these possible mechanisms we constructed a mutant (9CA) in which all PKC consensus sequences between the Walker B motif of NBD1 and second transmembrane domain (TMD2; i.e. the seventh membrane-spanning segment; T582A, T604A, S641A, T682A, S686A, S707A, S790A, T791A and S809A) were eliminated (Fig. 4A and B). pNUT 9CA was transfected into BHK cells and clonal cell lines stably expressing the mutant were selected in medium containing 500 μm methotrexate (MTX). Parallel transfections with wild-type CFTR cDNA were also performed, and cell lines expressing wild-type protein were selected under the same conditions. When levels of mutant and wild-type proteins were compared in clonal cell variants by probing Western blots with the monoclonal antibody M3A7 (20 μg aliquots of cellular protein), expression of the mutant was variable but consistently lower than wild-type (Fig. 4C). Abundance of the mature, complex-glycosylated CFTR (i.e. band ‘C’, apparent Mr∼175 000) relative to immature protein appeared similar for mutant and wild-type proteins. In an attempt to match channel expression as closely as possible, transfection with wild-type CFTR was repeated but stable cell lines were selected using 100- or 1000-fold lower concentrations of MTX (5 or 0.5 μm, respectively). The variants were compared and cell lines having the lowest expression of wild-type CFTR and highest expression of 9CA were chosen for subsequent experiments. Wild-type CFTR expression was still 3–4-fold higher than that of the mutant in these lines, but expression level was taken into account when comparing channel activities (see below) and would not explain the dramatic difference in their responsiveness to PKA activation.
Figure 4. Mutagenesis of PKC consensus sequences on CFTR.
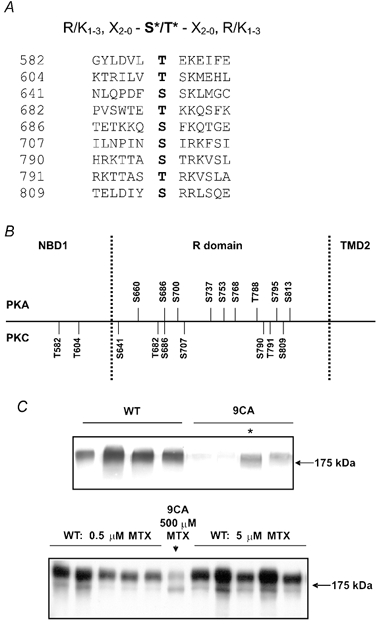
A, putative PKC sites between the Walker B motif of NBD1 and TM7. The strongest consensus is shown at the top (based on Kennelly & Krebs, 1991). Amino acids mutated in the 9CA mutant are shown with the flanking sequences and are numbered on the left. B, relative locations of putative PKA and PKC sites. C, Western blots of BHK cells stably expressing wild-type CFTR (WT) or mutated (9CA) CFTR. The upper blot shows CFTR expression in four cell lines expressing wild-type (left) or 9CA (right) that had been independently selected using 500 μm methotrexate (MTX). The asterisk indicates the line with highest 9CA expression. The lower blot shows cell variants that had been selected using 1000-fold lower drug concentration (0.5 μm; five lanes on the left) or 100-fold lower drug concentration (5 μm methotrexate; five lanes on the right). For comparison, the centre lane was loaded with the same amount of lysate containing 9CA protein (marked with an asterisk in upper blot).
Activation of 9CA channels by PKA in excised patches
To assess the role of PKC consensus sequences in NBD1 and the R domain in modulating activation by PKA, the dependence of 9CA channel activity on PKA concentration was assessed using inside-out patches and a protocol similar to that described above for wild-type channels (see Fig. 2). Patches were pre-incubated with alkaline phosphatase + PP2Cα+ PP2A for 10 min, the chamber was rinsed of phosphatases, then ATP and PKC were added and followed 5 min later by the addition of various concentrations of PKA. Unlike wild-type CFTR, 9CA channels were only slightly activated by 40 U ml−1 PKA despite this PKC pre-treatment (Fig. 5A), and were not stimulated significantly by elevating PKA activity further (200–400 U ml−1). The PKA concentration dependence of 9CA channel activation is summarized in Fig. 5B and C, along with the mean values for wild-type channels from Fig. 2C for comparison. The results are plotted on a logarithmic scale because of the large difference in NPo values. Note that NPo reached only ∼1.3 for 9CA channels, which is about 100-fold lower than for PKC pre-treated wild-type channels. Since the NPo of 9CA channels was still ∼30-fold lower than wild-type after allowing for their reduced expression, the results strongly suggest that at least one PKC consensus sequence mutated in 9CA is essential for normal responses to PKA.
Figure 5. Responsiveness of 9CA channels to PKA in excised patches.
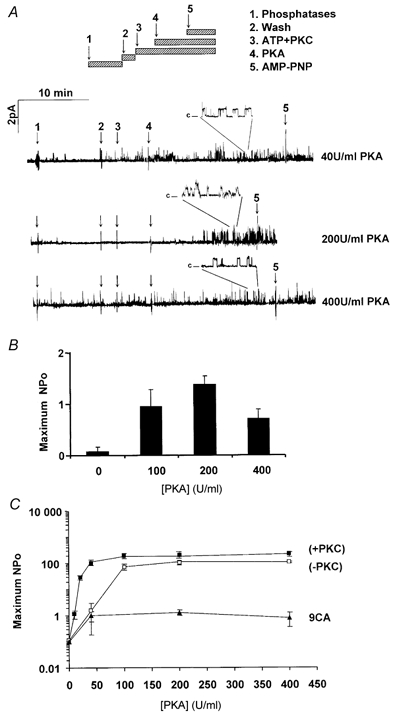
Recordings of 9CA mutant channels using the inside-out configuration (Vp = +30 mV). Following excision, patches were treated with a mixture of three phosphatases (see Fig. 1) for 10 min to remove basal CFTR phosphorylation. After a 3 min wash, they were treated with 3.78 nm PKC + 5 μm DiC8 and 1 mm ATP for 5 min, then PKA was added at the concentrations indicated. A, recording obtained from patches exposed to different concentrations of PKA following pre-treatment with PKC. The insets show the same recordings with the time base expanded 10 ×. B, maximal NPo of 9CA channels activated by PKA after pre-treatment with PKC (means ±s.e.m; n = 5–8 patches) C, comparison of the relationship between maximum NPo and PKA concentration using excised membrane patches containing wild-type (squares) or 9CA mutant channels (triangles). Dark symbols indicate patches that were pre-treated with PKC.
Activation of wild-type and 9CA channels in intact cells
To investigate PKC modulation of CFTR in intact cells we examined the effects of chelerythrine on chlorophenylthio-cAMP- (cpt-cAMP) stimulated iodide efflux from BHK cells stably expressing wild-type or 9CA channels. Efflux rate increased with [cpt-cAMP], as revealed by the slopes of the cumulative efflux curves, which became steeper over the range 25–250 μm and then remained constant up to 750 μm cpt-cAMP. Stimulated efflux from cells expressing the mutant was lower at all concentrations of cpt-cAMP and time points examined (note the different vertical scales in Fig. 6A and B) compared with cells expressing wild-type CFTR, although their dependence on cpt-cAMP concentration was similar. Concentration dependence may reflect factors such as membrane permeability to cpt-cAMP and affinity of PKA regulatory subunits for cpt-cAMP, rather than responsiveness to PKA. Regardless, since cells expressing 9CA CFTR still lost ∼140 nmol of iodide during 15 min stimulation with 750 μm cpt-cAMP (vs. 250 nmol iodide from cells expressing wild-type), 9CA channels are clearly functional in intact cells and can mediate large iodide effluxes, although they respond more slowly than wild-type channels.
To examine if iodide efflux from cells expressing 9CA is slow due to the removal of PKC sites or to a general disruption of channel function caused by mutagenesis, we compared iodide effluxes from control cells and those that had been incubated with the PKC inhibitor chelerythrine chloride (10 μm). Chelerythrine caused a delay in cpt-cAMP-stimulated iodide efflux from cells expressing wild-type CFTR that was reminiscent of cells expressing 9CA (Fig. 7A). By contrast, iodide efflux from cells expressing the mutant, which was already slow compared with that from cells expressing wild-type CFTR, was not delayed further by chelerythrine (Fig. 7B). Thus the effects of mutagenesis and chelerythrine were not additive, and the slow iodide efflux from cells expressing 9CA channels could be attributed to a reduction of PKC phosphorylation on the channels.
Partial activation of CFTR channels by PKC alone
Exogenous PKC partially activated CFTR channels when added to freshly excised patches in the absence of PKA (Tabcharani et al. 1991; Berger et al. 1993). It remains unclear if they were activated by PKC per se or if the activation was caused by residual PKA phosphorylation, with PKC exerting a permissive action (Jia et al. 1997). We re-examined the effect of PKC alone on wild-type and 9CA channels using patches that had been pre-treated with phosphatases as described above. When MgATP (1 mm) was added and channel activity recorded before and after addition of 3.8 nm PKC and its lipid activator DiC8 (5 μm), PKC increased the NPo of wild-type channels by about 8-fold, from 0.15 ± 0.06 to 1.20 ± 0.39 (means ±s.e.m.; P < 0.002, n = 8 patches, Fig. 8A). Although this response was highly significant statistically, it represents only 1 −2 % of the stimulation induced by PKA. A decrease in channel closed time was apparent from inspection of the traces but could not be quantified due to the large number of channels. Using the 9CA mutant we investigated whether the PKC sites on CFTR that modulate responses to PKA might also mediate the weak activation by PKC alone (Fig. 8B). When recording from patches with 9CA channels, no stimulation of NPo by PKC alone was detected although a low level of basal activity was clearly detected (0.15 ± 0.07 vs. 0.10 ± 0.08; means ±s.e.m.; n = 8 patches; P > 0.2). Interestingly, the activity of 9CA channels in freshly excised patches without kinases (NPo = 0.11 ± 0.07; mean ±s.e.m.; n = 8 patches) was lower than that of wild-type channels under the same conditions (0.66 ± 0.21; mean ±s.e.m.; n = 6 patches), but was similar to that of wild-type channels exposed to phosphatases (0.11 ± 0.38; mean ±s.e.m.; n = 9 patches). Thus basal phosphorylation of wild-type channels at the sites mutated in 9CA may contribute to the low activity of wild-type channels patches before they are exposed to exogenous phosphatases.
Figure 8. Effect of PKC (added alone) on the activity of wild-type and 9CA mutant CFTR channels in excised patches.
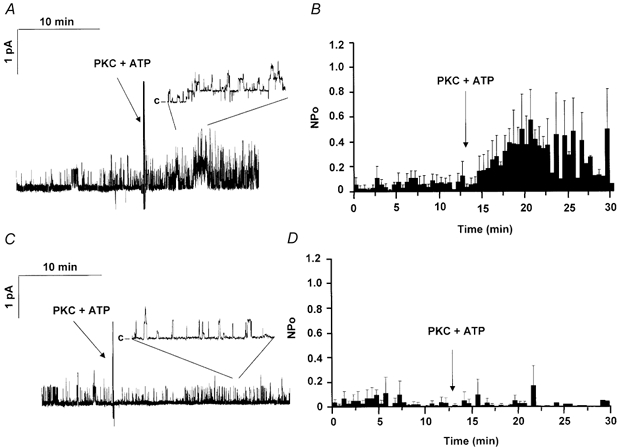
A, trace showing effect of adding PKC (3.78 nm), DiC8 (5 μm) and ATP (1 mm) to a patch containing wild-type channels (30 min recording). All patches were pre-treated with protein phosphatases (alkaline phosphatase + PP2Cα+ PP2A) before the beginning of the recording. The inset shows an expanded view of the same trace (10 × for WT and 20 × for 9CA channels). B, mean NPo calculated for 30 s intervals. C, trace obtained under the same conditions as in A except that the patch was from a cell expressing 9CA channels. D, mean NPo values calculated for 30 s intervals using 9CA channel. All data show the means ±s.e.m., n = 8–10 patches.
Discussion
The present results indicate that CFTR responses to PKA are modulated by phosphorylation of PKC sites on CFTR itself, rather than on an ancillary protein. The PKC dependence of wild-type CFTR channels was partially (∼50 %) circumvented by exposure to high PKA activity. Since activation by high PKA activity in the absence of PKC was not observed for 9CA channels (regardless of PKC), it may involve phosphorylation of at least one of the sites on wild-type CFTR that are mutated in 9CA. PKC alone weakly activated CFTR channels (< 5 %) after pre-treatment with phosphatases, and this small response to PKC alone required at least one of the nine consensus sequences since it did not occur with 9CA channels. However the issue of whether PKC phosphorylation itself activates the channel or permits activation by residual phosphorylation at PKA sites remains unresolved, since phosphatase pre-treatment did not completely remove basal phosphorylation. The results demonstrate direct control of CFTR by PKC, which may act independently or downstream of PKA phosphorylation, and suggest that convergent regulation by these kinases may be more complex than previously thought.
Effects of PKC on CFTR protein expression and channel activity
PKC activation reduces CFTR mRNA expression (Trapnell et al. 1991; Bargon et al. 1992; Dechecchi et al. 1992; Kang-Park et al. 1998) and this decrease in expression is at least partly due to a reduction in mRNA stability (Kang-Park et al. 1998). The reduction in CFTR mRNA expression may be mediated by PKCβ (Umar et al. 2000) but does not involve PKCδ or PKCɛ, which are thought to acutely regulate channel activity (Liedtke et al. 2001). In the present study, 9CA expression was consistently lower than that of wild-type CFTR, which seems paradoxical in light of previous work showing CFTR degradation in HT-29 colon cells is enhanced by PKC stimulation (Breuer et al. 1993). An effect of PKC on some protein involved in CFTR turnover might explain the discrepancy. However, low expression of 9CA is probably due to partial misprocessing, since several residues mutated in 9CA are situated in, or near, NBD1 where mutations are prone to cause misfolding (reviewed in Hanrahan et al. 2002). Nevertheless, large cAMP-stimulated iodide effluxes were measured from cells expressing 9CA channels, indicating significant expression of functional channels. Finding that mutation of potential PKC sites has a more pronounced effect on PKA activation than does mutagenesis of the dibasic PKA consensus sequences (Chang et al. 1993) reinforces the importance of PKC in channel regulation. Iodide efflux was clearly delayed from cells expressing 9CA, and a similar lag in cumulative iodide release was observed when cells expressing wild-type channels were exposed to the PKC inhibitor chelerythrine. This, and the fact that chelerythrine had no additional effect on cells expressing 9CA, suggest that the altered behaviour of the mutant is due to reduced phosphorylation at one or more PKC consensus sequences that were eliminated. AMP-PNP did not cause ‘locking open’ of the PKA-activated 9CA channels, but the significance of this abnormality is not clear since loss of locking by AMP-PNP is not always associated with abnormal gating when ATP is the only nucleotide present.
PKC pre-treatment shifted the activation of wild-type channels to lower [PKA], further evidence that it regulates CFTR by enhancing responses to PKA (Tabcharani et al. 1991; Jia et al. 1997; Liedtke & Cole 1998). In previous studies of CFTR expressed in CHO cells we found that PKC was strictly required for activation by PKA in excised patches after prolonged rundown, although the PKC dependence in BHK cells in whole-cell and excised patches was eventually overridden during prolonged PKA stimulation (Jia et al. 1997; and Fig. 2A in the present study). If this reflects PKA phosphorylation of modulatory PKC sites, it might explain the inability of high [PKA] to activate 9CA channels (Fig. 5C).
One PKC site is also a strong dibasic PKA consensus sequence (S686; KKQSFK) and would probably be phosphorylated efficiently by either kinase, therefore it is unlikely to mediate delayed activation by PKA. Two other PKC consensus sequences that could serve this role are ‘low probability’ PKA sites (S641 and S790; DFSSK and STRK, respectively), although their phosphorylation by PKA remains to be demonstrated (Picciotto et al. 1992; Seibert et al. 1995; Townsend et al. 1996). PKC sites probably mediate weak activation by PKC alone, since the response to PKC was abolished in the 9CA mutant. PKC does phosphorylate the recombinant R domain between residues 761 and 770 (Picciotto et al. 1992), which has a consensus sequence for PKA (S768; RRRQSV) but not PKC. Moreover, PKC can phosphorylate a synthetic peptide containing aa653–668, which has no PKC consensus sequence but does have a strong PKA site (S660; RRNS; Picciotto et al. 1992). Further studies of individual sites are needed to evaluate the importance of such cross-phosphorylation by the other kinase.
In vivo phosphorylation of two residues (S686 and S790) by PKC has been demonstrated, but removing them did not abolish the stimulation of Xenopus CFTR by phorbol ester (Button et al. 2001). The PKC dependence of SS686, 790AA suggested that additional PKC sites, perhaps situated on other ancillary proteins, might participate in modulation by PKC. Results with a synthetic peptide comprising amino acids 693–716 of human CFTR suggest that S707 may also be a PKC substrate, although this has not been tested by direct sequencing or mutagenesis (Picciotto et al. 1992). Regardless, the present results show that PKC control of CFTR responses to PKA requires at least one of the nine sequences altered in 9CA. Since the mutant was also unresponsive to PKC alone, the same sequences may also be required for partial activation by PKC alone.
Cyclic AMP-stimulated halide efflux from cells expressing 9CA was surprisingly robust considering its lower expression and comparison of patch-clamp data from wild-type and mutant channels. Whilst this argues against the weak kinase responses of 9CA being an artefact of mutagenesis, a caveat of efflux assays is their tendency to underestimate differences between cell lines (in this instance due to the offsetting effects of reduced anion conductance and increased iodide driving force in 9CA cells). Anion conductance and iodide efflux only change in parallel when the driving force remains constant, but since the outward driving force collapses as iodide is lost and the membrane depolarizes towards the I− equilibrium potential during efflux assays, stimulation of conductance in wild-type cells might have been larger than it appears in Fig. 6A.
Possible mechanism of regulation by PKC
PKC effects are mediated by direct phosphorylation of CFTR, but the mechanism remains obscure. The [PKA] dependence of channel activation was similar to that of radiolabelling (i.e. phosphorylation) by PKA under comparable conditions, but only if channels had been pre-treated with PKC during the patch-clamp experiments. Taken together these findings imply that PKC regulation is independent from phosphorylation by PKA, and may act, for example, by improving the coupling between PKA phosphorylation and channel activation. Our previous studies indicated a change in PKA phosphorylation following PKC stimulation (Chang et al. 1993), but the experimental conditions used in that study were very different from the ones used in the present work. Treating cells with PMA before forskolin led to more [32P]orthophosphate incorporation than when the order was reversed (Chang et al. 1993), but phosphorylation at identified PKA sites needs to be measured to evaluate whether stimulation of PKC enhances the phosphorylation of CFTR by PKA. The PKC isotype regulating CFTR may vary with cell type since PKCα is implicated in the secretory response to carbachol in colonic HT-29cl.19A cells (Van den Berghe et al. 1992), whereas α1-adrenergic stimulation of CFTR in Calu-3 cells is mediated by PKCɛ (Liedtke & Cole, 1998; Liedtke et al. 2001). Regulation by PKC is likely to be a general property of CFTR in other cells (e.g. Walsh, 1991; Collier & Hume, 1995; Middleton & Harvey, 1998) and independent of the isotype, since specificity was not observed when different PKCs were added to excised patches (Berger et al. 1993).
Physiological significance of PKC modulation
Why is CFTR regulated by both PKA and PKC? Some physiological agonists presumably activate both kinases simultaneously to elicit a concerted secretory response. Thus, physiological control of submucosal gland cells involves stimulation of PKA and PKC pathways via β- and α1-adrenergic receptors, respectively. By contrast, the prostaglandins E1 and F2α regulate CFTR activity in intestinal cells by stimulating either PKA or PKC, respectively, but not both (Weymer et al. 1985; Yurko-Mauro & Reenstra, 1998). Converging kinases presumably allow integration of signals from multiple secretagogues and stimuli. The present results indicate that the PKC sites that modulate channel responses to PKA are in the distal region of NBD1 and/or the R domain of CFTR.
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) and Canadian Cystic Fibrosis Foundation (CCFF) to J.W.H., and from the National Institutes of Health (NIDDK) to J.R.R. The author V.C. is a CCFF/CIHR postdoctoral fellow, and J.W.H. is a senior scientist of the CIHR.
References
- Aleksandrov L, Aleksandrov AA, Chang X-B, Riordan JR. The first nucleotide binding domain of CFTR is a site of stable nucleotide interaction whereas the second is a site of rapid turnover. J Biol Chem. 2002;277:15419–15425. doi: 10.1074/jbc.M111713200. [DOI] [PubMed] [Google Scholar]
- Bajnath RB, Groot JA, De Jonge HR, Kansen M, Bijman J. Synergistic activation of non-rectifying small-conductance chloride channels by forskolin and phorbol esters in cell-attached patches of the human colon carcinoma cell line HT-29cl. 19A. Pflugers Arch. 1993;425:100–108. doi: 10.1007/BF00374509. [DOI] [PubMed] [Google Scholar]
- Bargon J, Trapnell BC, Yoshimura K, Dalemans W, Pavirana A, Lecocq J-P, Crystal RG. Expression of the cystic fibrosis transmembrane conductance regulator gene can be regulated by protein kinase C. J Biol Chem. 1992;267:16056–16060. [PubMed] [Google Scholar]
- Becq F, Jensen TJ, Chang X-B, Savoia A, Rommens JM, Tsui L-C, Buchwald M, Riordan JR, Hanrahan JW. Phosphatase inhibitors activate normal and defective CFTR chloride channels. Proc Natl Acad Sci U S A. 1994;91:9160–9164. doi: 10.1073/pnas.91.19.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger HA, Travis SM, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by specific protein kinases and phosphatases. J Biol Chem. 1993;268:2037–2047. [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Breuer W, Glickstein H, Kartner N, Riordan JR, Ausiello DA, Cabantchik IZ. Protein kinase C mediates down-regulation of cystic fibrosis transmembrane conductance regulator levels in epithelial cells. J Biol Chem. 1993;268:13935–13939. [PubMed] [Google Scholar]
- Button B, Reuss L, Altenberg GA. PKC-mediated stimulation of amphibian CFTR depends on a single phosphorylation consensus site. Insertion of this site confers PKC sensitivity to human CFTR. J Gen Physiol. 2001;117:457–467. doi: 10.1085/jgp.117.5.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KM, Csanády L, Seto-Young D, Nairn AC, Gadsby DC. Severed molecules functionally define the boundaries of the cystic fibrosis transmembrane conductance regulator's NH2-terminal nucleotide binding domain. J Gen Physiol. 2000;116:163–180. doi: 10.1085/jgp.116.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X-B, Tabcharani JA, Hou Y-X, Jensen TJ, Kartner N, Alon N, Hanrahan JW, Riordan JR. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all ten PKA consensus phosphorylation sites. J Biol Chem. 1993;268:11304–11311. [PubMed] [Google Scholar]
- Cheng SH, Rich DP, Marshall J, Gregory RJ, Welsh MJ, Smith AE. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991;66:1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- Collier ML, Hume JR. Unitary chloride channels activated by protein kinase C in guinea pig ventricular myocytes. Circ Res. 1995;76:317–324. doi: 10.1161/01.res.76.2.317. [DOI] [PubMed] [Google Scholar]
- Dechecchi MC, Rolfini R, Tamanini A, Gamberi C, Berton G, Cabrini G. Effect of modulation of protein kinase C on the cAMP-dependent chloride conductance in T84 cells. FEBS Lett. 1992;311:25–28. doi: 10.1016/0014-5793(92)81358-s. [DOI] [PubMed] [Google Scholar]
- Diederichs K, Diez J, Greller G, Muller C, Breed J, Schnell C, Vonrhein C, Boos W, Welte W. Crystal structure of MalK, the ATPase subunit of the trehalose/maltose ABC transporter of the archeon Thermococcus litoralis. EMBO J. 2000;19:5951–5961. doi: 10.1093/emboj/19.22.5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulhanty AM, Riordan JR. Phosphorylation by cAMP-dependent protein kinase causes a conformational change in the R domain of the cystic fibrosis transmembrane conductance regulator. Biochemistry. 1994;33:4072–4079. doi: 10.1021/bi00179a036. [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Nairn AC. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev. 1999;79:S77–107. doi: 10.1152/physrev.1999.79.1.S77. [DOI] [PubMed] [Google Scholar]
- Hanrahan JW, Gentzsch M, Riordan JR. The cystic fibrosis transmembrane conductance regulator (ABCC7) In: Holland B, Higgins CF, Kuchler K, Cole SPC, editors. ABC Proteins: From Bacteria to Man. New York: Blackwell Science Inc; 2002. pp. 589–618. [Google Scholar]
- Hanrahan JW, Kone Z, Mathews CJ, Luo J, Jia Y, Linsdell P. Patch-clamp studies of cystic fibrosis transmembrane conductance regulator chloride channel. Methods Enzymol. 1998;293:169–194. doi: 10.1016/s0076-6879(98)93014-2. [DOI] [PubMed] [Google Scholar]
- Hinkson DAR, Riordan JR, Chang XB, Hanrahan JW. Regulation of CFTR by protein kinase C. Pediatr Pulmonol Suppl. 2000;20:176. [Google Scholar]
- Hung L-W, Wang IX, Nikaido K, Liu PQ, Ames GFL, Kim S-H. Crystal structure of the ATP-binding subunit of an ABC transporter, the istidine permease of Salmonella typhimurium. Nature. 1998;396:703–707. doi: 10.1038/25393. [DOI] [PubMed] [Google Scholar]
- Hwang T-C, Nagel G, Nairn AC, Gadsby DC. Regulation of the gating of cystic fibrosis transmembrane conductance regulator Cl channels by phosphorylation and ATP hydrolysis. Proc Natl Acad Sci U S A. 1994;91:4698–4702. doi: 10.1073/pnas.91.11.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Mathews CJ, Hanrahan JW. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J Biol Chem. 1997;272:4978–4984. doi: 10.1074/jbc.272.8.4978. [DOI] [PubMed] [Google Scholar]
- Kang-Park S, Dray-Charier N, Munier A, Brahimi-Horn C, Veissiere D, Picard J, Capeau J, Cherqui G, Lascols O. Role for PKCα and PKCɛ in down-regulation of CFTR mRNA in a human epithelial liver cell line. J Hepatol. 1998;28:250–262. doi: 10.1016/0168-8278(88)80012-6. [DOI] [PubMed] [Google Scholar]
- Karpowich N, Martsinkevich O, Millen L, Yual Y-R, Dai PL, MacVey K, Thomas PJ, Hunt JF. Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure. 2001;9:571–586. doi: 10.1016/s0969-2126(01)00617-7. [DOI] [PubMed] [Google Scholar]
- Kartner N, Hanrahan JW, Jensen TJ, Naismith AL, Sun S, Ackerley CA, Reyes EF, Tsui L-C, Rommens JM, Bear CE, Riordan JR. Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell. 1991;64:681–691. doi: 10.1016/0092-8674(91)90498-n. [DOI] [PubMed] [Google Scholar]
- Kartner N, Riordan JR. Characterization of polyclonal and monoclonal antibodies to cystic fibrosis transmembrane conductance regulator. Methods Enzymol. 1998;292:629–652. doi: 10.1016/s0076-6879(98)92049-3. [DOI] [PubMed] [Google Scholar]
- Kennelly PJ, Krebs EG. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem. 1991;266:15555–15558. [PubMed] [Google Scholar]
- Lansdell KA, Delaney SJ, Lunn DP, Thomson SA, Sheppard DN, Wainwright BJ. Comparison of the gating behaviour of human and murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in mammalian cells. J Physiol. 1998;508:379–392. doi: 10.1111/j.1469-7793.1998.379bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke CM, Cody D, Cole TS. Differential regulation of Cl− transport proteins by PKC in Calu-3 cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L739–747. doi: 10.1152/ajplung.2001.280.4.L739. [DOI] [PubMed] [Google Scholar]
- Liedtke CM, Cole TS. Antisense oligonucleotide to PKC-ɛ alters cAMP-dependent stimulation of CFTR in Calu-3 cells. Am J Physiol. 1998;275:C1357–1364. doi: 10.1152/ajpcell.1998.275.5.C1357. [DOI] [PubMed] [Google Scholar]
- Luo J, Zhu T, Evagelidis A, Pato M, Hanrahan JW. Role of protein phosphatases in the activation of CFTR (ABCC7) by genistein and bromotetramisole. Am J Physiol Cell Physiol. 2000;279:C108–119. doi: 10.1152/ajpcell.2000.279.1.C108. [DOI] [PubMed] [Google Scholar]
- Mathews CJ, Tabcharani JA, Chang X-B, Jensen TJ, Riordan JR, Hanrahan JW. Dibasic protein kinase A sites regulate bursting rate and nucleotide sensitivity of the cystic fibrosis transmembrane conductance regulator chloride channel. J Physiol. 1998;508:365–377. doi: 10.1111/j.1469-7793.1998.365bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton LM, Harvey RD. PKC regulation of cardiac CFTR Cl− channel function in guinea pig ventricular myocytes. Am J Physiol. 1998;275:C293–302. doi: 10.1152/ajpcell.1998.275.1.C293. [DOI] [PubMed] [Google Scholar]
- Ostedgaard LS, Baldursson O, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by its R domain. J Biol Chem. 2001;276:7689–7692. doi: 10.1074/jbc.R100001200. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Behringer RR, Quaife CJ, Maxwell F, Maxwell IH, Brinster RL. Cell lineage ablation in transgenic mice by cell-specific expression of a toxin gene. Cell. 1987;50:435–443. doi: 10.1016/0092-8674(87)90497-1. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Cohn JA, Bertuzzi G, Greengard P, Nairn AC. Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1992;267:12742–12752. [PubMed] [Google Scholar]
- Rich DP, Berger HA, Cheng SH, Travis SM, Saxena M, Smith AE, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by negative charge in the R domain. J Biol Chem. 1993;268:20259–20267. [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J-L, Drumm ML, Iannuzzi MC, Collins FS, Tsui L-C. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Seibert FS, Tabcharani JA, Chang X-B, Dulhanty AM, Mathews CJ, Hanrahan JW, Riordan JR. cAMP-dependent protein kinase-mediated phosphorylation of cystic fibrosis transmembrane conductance regulator residue ser-753 and its role in channel activation. J Biol Chem. 1995;270:2158–2162. doi: 10.1074/jbc.270.5.2158. [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- Simonsen CC, Levinson AD. Isolation and expression of an altered mouse dihydrofolate reductase cDNA. Proc Natl Acad Sci U S A. 1983;80:2495–2499. doi: 10.1073/pnas.80.9.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabcharani JA, Chang X-B, Riordan JR, Hanrahan JW. Phosphorylation-regulated Cl− channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991;352:628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- Townsend RR, Lipniunas PH, Tulk BM, Verkman AS. Identification of protein kinase A phosphorylation sites on NBD1 and R domains of CFTR using electrospray mass spectrometry with selective phosphate ion monitoring. Protein Sci. 1996;5:1865–1873. doi: 10.1002/pro.5560050912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell BC, Chu C-S, Paakko PK, Banks TC, Yoshimura K, Ferrans VJ, Chernick MS, Crystal RG. Expression of the cystic fibrosis transmembrane conductance regulator gene in the respiratory tract of normal individuals and individuals with cystic fibrosis. Proc Natl Acad Sci U S A. 1991;88:6565–6569. doi: 10.1073/pnas.88.15.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis SM, Berger HA, Welsh MJ. Protein phosphatase 2C dephosphorylates and inactivates cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci U S A. 1997;94:11055–11060. doi: 10.1073/pnas.94.20.11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar S, Sellin J, Morris AP. Murine colonic mucosa hyperproliferation. II. PKC-beta activation and cPKC-mediated cellular CFTR overexpression. Am J Physiol Gastrointest Liver Physiol. 2000;278:G765–778. doi: 10.1152/ajpgi.2000.278.5.G765. [DOI] [PubMed] [Google Scholar]
- Van den Berghe H, Vaandrager AB, Bot AGM, Parker PJ, De Jonge HR. Dual role for protein kinase Cα as a regulator of ion secretion in the HT29cl. 19A human colonic cell line. Biochem J. 1992;285:673–679. doi: 10.1042/bj2850673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh KB. Activation of a heart chloride current during stimulation of protein kinase C. Mol Pharmacol. 1991;40:342–346. [PubMed] [Google Scholar]
- Weymer A, Huott P, Liu W, McRoberts JA, Dharmsathaphorn K. Chloride secretory mechanism induced by prostaglandin E1 in a colonic epithelial cell line. J Clin Invest. 1985;76:1828–1836. doi: 10.1172/JCI112175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Britton F, Collier ML, Horowitz B, Hume JR. Regulation of recombinant cardiac cystic fibrosis transmembrane conductance regulator chloride channels by protein kinase C. Biophys J. 1999;76:1972–1987. doi: 10.1016/S0006-3495(99)77356-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurko-Mauro KA, Reenstra WW. Prostaglandin F2α stimulates CFTR activity by PKA- and PKC-dependent phosphorylation. Am J Physiol. 1998;275:C653–660. doi: 10.1152/ajpcell.1998.275.3.C653. [DOI] [PubMed] [Google Scholar]
- Zhu T, Dahan D, Evaglelidis A, Zheng S-X, Luo J, Hanrahan JW. Association of cystic fibrosis transmembrane conductance regulator and protein phosphatase 2C. J Biol Chem. 1999;274:29102–29107. doi: 10.1074/jbc.274.41.29102. [DOI] [PubMed] [Google Scholar]