

Abstract
The removal of N-terminal translation initiator Met by methionine aminopeptidase (MetAP) is often crucial for the function and stability of proteins. On the basis of crystal structure and sequence alignment of MetAPs, we have engineered Escherichia coli MetAP by the mutation of three residues, Y168G, M206T, Q233G, in the substrate-binding pocket. Our engineered MetAPs are able to remove the Met from bulky or acidic penultimate residues, such as Met, His, Asp, Asn, Glu, Gln, Leu, Ile, Tyr, and Trp, as well as from small residues. The penultimate residue, the second residue after Met, was further removed if the antepenultimate residue, the third residue after Met, was small. By the coexpression of engineered MetAP in E. coli through the same or a separate vector, we have successfully produced recombinant proteins possessing an innate N terminus, such as onconase, an antitumor ribonuclease from the frog Rana pipiens. The N-terminal pyroglutamate of recombinant onconase is critical for its structural integrity, catalytic activity, and cyto-toxicity. On the basis of N-terminal sequence information in the protein database, 85%–90% of recombinant proteins should be produced in authentic form by our engineered MetAPs.
Keywords: N-terminal Met, E. coli MetAP, substrate specificity, penultimate residue, ribonuclease
Removal of the translation initiator N-formyl-methionine or methionine from a recombinant protein is often critical for its function and stability, for example, in human hemoglobin, interleukin-2, growth hormones, or frog ribonucleases (Busby Jr. et al. 1987; Boix et al. 1996; Varshavsky 1996; Adachi et al. 2000; Endo et al. 2001; Liao et al. 2003). For the preparation of protein with an innate N terminus, various attempts have been made to remove the N-terminal Met. First, cyanogen bromide is used to cleave Met under extreme acidic conditions, but the method is limited to proteins without any internal Met residue (Boix et al. 1996). Second, a protease-specific oligopeptide is introduced in front of a target protein, which is then removed in vitro by the respective protease, for example, factor Xa, enterokinase, and cathepsin C (Belagaje et al. 1997). Third, the N-terminal Met of a protein is removed in vitro by the aminopeptidase of Aeromonas proteolytica (Shapiro et al. 1988; Notomista et al. 1999). Fourth, a signal peptide is introduced in front of the target protein and processed in vivo during secretion, but the yield is low (1~5 mg per liter of culture; Huang et al. 1998).
Recently, methionine aminopeptidases (MetAPs) have been used to remove the N-terminal Met in Escherichia coli and yeast. There are two types of MetAP, MetAP I and MetAP II, respectively, in eukaryotes. Only MetAP I exists in eubacteria and only MetAP II exists in archaea (Li and Chang 1995; Tahirov et al. 1998). These MetAP genes are essential for the growth of prokaryotes and eukaryotes (Chang et al. 1989; Li and Chang 1995). However, the efficiency of Met removal is limited by the side-chain radius (<3.68 Å) of the penultimate residue, the second residue after Met (Ben-Bassat et al. 1987; Hirel et al. 1989; Hwang et al. 1999; Chen et al. 2002). From the structure of E. coli MetAP and bestatin-based inhibitor complex, it has been discovered that four residues (Tyr 168, Gln 233, Met 206, and Glu 204) reside in the substrate-binding pocket (Fig. 1 ▶ from PDB 3MAT; Lowther et al. 1999; Lowther and Matthews 2000). The Met 329 and Gln 356 residues of yeast MetAP I, corresponding to Met 206 and Gln 233 of E. coli MetAP, have been replaced with Ala. With an oligopeptide as the in vitro substrate, the purified MetAP I variants (M329A, Q356A) exhibit a significant increase in catalytic activities for oligopeptides with slightly larger penultimate residues, such as Asn, His, and Met, but not for those with bulky and acidic residues (Roderick and Matthews 1993; Walker and Bradshaw 1999).
Figure 1.

Diagram of the substrate-binding pocket of E. coli MetAP. Part of E. coli MetAP (in light blue), two cobalt ions (in purple), and bestatin-based inhibitor (P1, P1′, and P2′ in orange) complex is adapted from PDB database (3MAT). The residues His 79, His 178, Glu 204, and Glu 235, involved in catalytic activity, and the residues Tyr 168, Met 206, and Gln 233, involved in substrate recognition, are colored green and yellow, respectively.
In this study, we mutated three residues, Tyr 168, M 206, and Gln 233, of E. coli MetAP by site-directed mutagenesis on the basis of the sequence alignment and structure of MetAPs (Lowther et al. 1999; Walker and Bradshaw 1999). Using the coexpression system for target protein and engineered MetAPs through the same or a separate vector, we were able to remove the N-terminal Met from bulky or acidic penultimate residues, for example, Met, His, Asp, Asn, Glu, Gln, Leu, Ile, and Trp in E. coli without further in vitro chemical or enzymatic treatment (Boix et al. 1996; Walker and Bradshaw 1999). Thus, we succeeded in producing several proteins possessing innate residues, such as onconase, an antitumor ribonuclease from frog Rana pipiens (Boix et al. 1996). The N-terminal pyroglutamate (Pyr) of the recombinant onconase is critical for its structural integrity, catalytic activity, and cytotoxicity (Liao et al. 2003). In addition to the penultimate residue, the removal of N-terminal Met was also influenced by the antepenultimate residue, the third residue after Met. On the basis of the N-terminal sequence information in the protein database, 85%–90% of recombinant proteins should be produced in authentic form by our engineered MetAP.
Results
Production of MetAP and target proteins
The gene encoding engineered MetAP was expressed itself or coexpressed with target protein in E. coli BL21(DE3). The MetAP was soluble and purified to electrophoretical homogeneity (Fig. 2A ▶, lane 2). The yields of MetAP varied with the type of coexpressed target protein (Fig. 2A ▶, lanes 5 and 7), for example, 50 mg purified protein was obtained per 1 L of culture in the absence of target protein. The yield of coexpressed target proteins varied with the property of the protein, for example, the amount of frog ribonucleases (onconase and RC-RNaseL1) is different as shown in Fig. 2B ▶, lanes 6 and 8).
Figure 2.
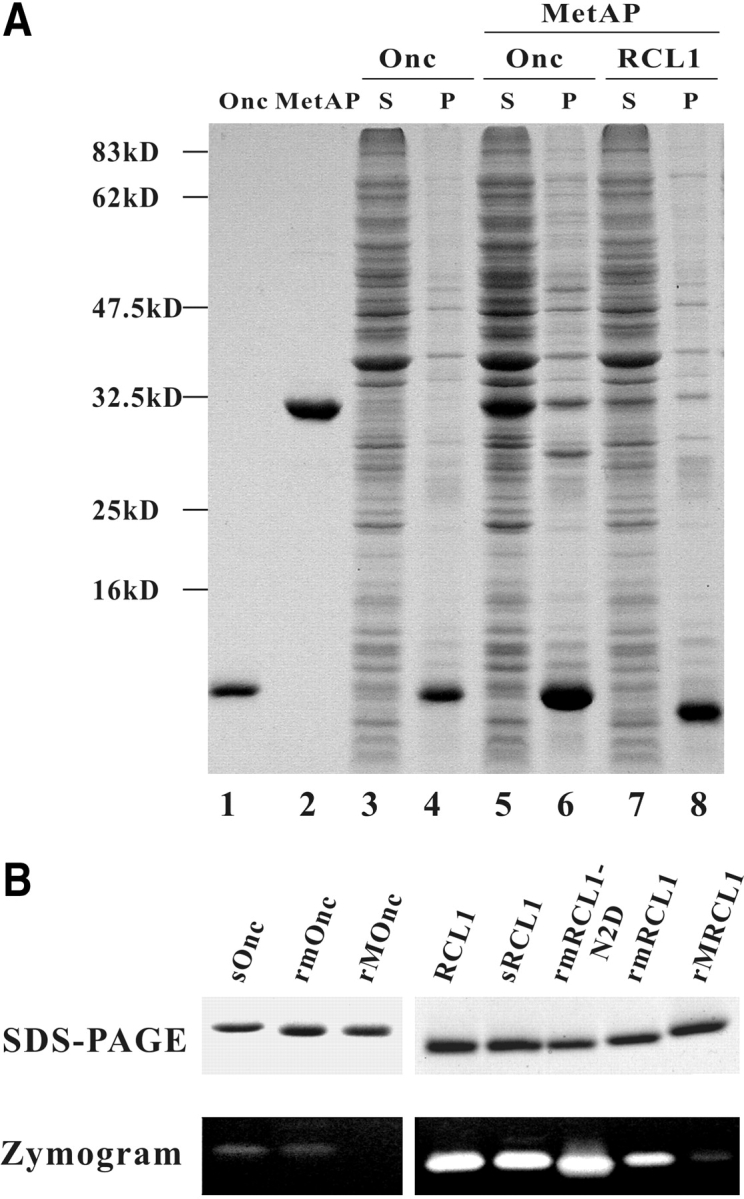
Expression and analyses of MetAP and target ribonucleases. (A) Coexpression of MetAP and target ribonucleases. The protein component of supernatant and insoluble pellet of 30-μL transformed E. coli BL21(DE3) culture was analyzed by 13.3% SDS-PAGE and Coomassie blue staining. (Lane 1) Two micrograms onconase; (lane 2) three micrograms MetAP; (lanes 3,4) supernatant and pellet of E. coli expressing onconase only; (lanes 5,6) supernatant and pellet of E. coli coexpressing engineered MetAP and onconase; (lanes 7,8) supernatant and pellet of E. coli coexpressing engineered MetAP and RC-RNaseL1. S and P represent the supernatant and insoluble pellet of E. coli lysate, respectively. (B) Analyses of recombinant ribonucleases. (Top panel) Two micrograms of purified ribonuclease was separated by 13.3% SDS-PAGE and stained by Coomassie blue. (Bottom panel) Ten nanograms onconase and 1 ng RC-RNaseL1 were separated by RNA-casting SDS-PAGE and stained by Toluidine blue O. sOnc and sRCL1 represent secretory onconase and RC-RNase L1, respectively, which were purified from culture medium; rmOnc, rmRCL1, and rmRCL1-N2D represent Met-tagged onconase, RC-RNase L1, and RC-RNase L1-N2D, respectively, which are treated with MetAP-*TG in vivo; rMOnc and rMRCL1 represent Met-tagged onconase and RC-RNase L1, respectively, without MetAP treatment; RCL1 represents native RC-RNase L1 purified from bullfrog liver.
Two residues involved in substrate specificity
Residue Gln 233, adjacent to two Co++ ions and two catalytic residues, Glu 204 and Glu 235, in the substrate-binding pocket of E. coli MetAP (Fig. 1 ▶) was replaced by a small residue, for example, Gly, Thr, Asp, or Asn, and coexpressed with the onconase that begins with MQD. The percentage of Met removal was calculated by dividing the amount of proteins initiated with the penultimate residue by the amount of total proteins (i.e., proteins initiated with the penultimate residue plus unprocessed protein initiated with Met) after Edman degradation. The Met preceding Gln was removable by MetAP-Q233G (74% of Met removal) and MetAP-Q233T (88%). Furthermore, a second residue, Met 206, in the pocket (Fig. 1 ▶) was replaced by a small residue, for example, Gly, Thr, or Val. The Met of MQD-Onc was also removable by MetAP-M206G (85% of Met removal), MetAP-M206T (86%), and MetAP-M206V (61%). To make the MetAP more competent, we made a double mutation at residues Met 206 and Gln 233. For the substrate MQD-Onc, the double mutants, MetAP-M206G-Q233G (96%), MetAP-M206T-Q233G (91%), MetAP-M206T-Q233T (93%), and MetAP-M206V-Q233T (90%) variants, designated as MetAP-*GG, MetAP-*TG, MetAP-*TT, and MetAP-*VT, respectively, possess high in vivo catalytic activities (Table 1; data not shown). For other substrates, only MetAP-*TG exerted high catalytic activity for MLD-Onc (86% Met removal) and MID-Onc (22%; Table 1), whereas the MetAP-*GG did not, regardless of the apparent large pocket for substrate binding (data not shown).
Table 1.
Effect of penultimate residue of substrate on the in vivo catalytic activity of engineered MetAPs
| Catalytic activity (%)a | |||||
| Substrate | MetAP-YMQb | MetAP-*TG | MetAP-GTG | MetAP-TTG | Max. side-chain length (Å)c |
| M↓GD-Oncd | 100c | 95 | —e | —e | 0.00 |
| M↓AD-Onc | 100 | 99 | — | — | 1.51 |
| M↓PD-Onc | 80–90 | 93 | — | — | 2.40 |
| M↓SD-Onc | 80–90 | 99 | — | — | 2.41 |
| M↓TD-Onc | 80–90 | 85 | — | — | 2.54 |
| M↓VD-Onc | 80–90 | 80 | — | — | 2.55 |
| M↓ND-Onc | ~20 | 85 | — | — | 3.68 |
| M↓DD-Onc | ~20 | 68 | 27 | 80 | 3.74 |
| M↓LD-Onc | ~20 | 86 | 90 | 3.90 | |
| M↓ID-Onc | ~20 | 22 | 94 | 68 | 3.91 |
| M↓HD-Onc | 0 | 92 | — | — | 4.64 |
| M↓QD-Onc | 0 | 91 | 97 | 4.93 | |
| M↓ED-Onc | 0 | 33 | 35 | 58 | 4.97 |
| M↓FD-Onc | 0 | 5 | 23 | — | 5.10 |
| M↓M↓D-Onc | 0 | 100 (73)f | — | — | 5.46 |
| M↓KD-Onc | 0 | 3 | 7 | — | 6.37 |
| M↓YD-Onc | 0 | 1 | 20 | — | 6.43 |
| M↓WD-Onc | 0 | 2 | 78 | — | 6.64 |
| M↓RD-Onc | 0 | 2 | 4 | — | 7.40 |
a Activity was expressed by the percentage of terminal residue removed from Met-tagged onconase with a different penultimate residue.
b MetAP-YMQ represents the wild-type E. coli Met aminopeptidase.
c The maximal side-chain length of amino acid residue and the efficiency of Met removal by wild-type MetAP were adapted from the results of Hirel et al. (1989).
d The vertical arrow represents the site cleaved by MetAP.
e Dashes indicate the result was not measured.
f The value in parentheses represents the percent of penultimate residue that is removed by MetAP.
A third residue involved in substrate specificity
A third residue, Tyr 168, in the substrate-binding pocket of MetAP (Fig. 1 ▶) was further replaced with a small residue, Gly, Ser, Thr, Val, Asp, or Glu. For the substrate MID-Onc, the efficiency of Met removal by the parental MetAP-*TG was 22%, but it increased markedly to 94% by Y168G mutation and to 68% by Y168T mutation, which were designated as MetAP-GTG and MetAP-TTG, respectively (Table 1). In contrast, for substrates with an acidic penultimate residue, for example, MDD-Onc or MED-Onc, the third Y168T mutation exhibited higher activity than that of the Y168G mutation (Table 1). The result suggests that the hydroxyl group of residue 168 of E. coli MetAP with a short side chain (Thr) may play a crucial role in the recognition of the acidic penultimate residue.
The substrate specificity of engineered MetAPs
The substrate specificities of MetAP variants were determined by using MXD-Onc as a substrate that possesses a different penultimate residue (Table 1). The Met preceding the moderate penultimate residue—that is, Asn (85%), Leu (86%), His (92%), Gln (90%), and Met (100%)—as well as a small and uncharged residue—that is, Gly (95%), Ala (99%), Pro (93%), Ser (99%), Thr (85%), and Val (80%)—were mostly removed by the MetAP-*TG. For proteins with a bulky penultimate residue, Ile and Trp, most of Met was removed by MetAP-GTG, at 94% and 78%, respectively. For protein with acidic penultimate residues, Asp and Glu, the Met was also removable by MetAP-TTG, at 80% and 58%, respectively. However, the Met preceding bulky basic penultimate residues, Lys and Arg, is still not removable by these variants.
Effect of antepenultimate residue on substrate specificity
Using MXD-Onc as a substrate, we demonstrated the importance of the penultimate residue on the substrate specificity of engineered MetAP in vivo (Table 1). However, we also found that the substrate specificity of MetAP was influenced by the antepenultimate residue. With MAX-Onc as a substrate, the removal of terminal Met was not significantly altered by the change of the antepenultimate residue, whereas both terminal Met and penultimate Ala were removable by the engineered MetAPs when the antepenultimate residue was small, that is, Gly or Ala (Table 2). With MLX-Onc or MQX-Onc as a substrate, the terminal Met and penultimate Leu/Gln residues were also removable by engineered MetAPs when the antepenultimate residue was small or moderate, such as Gly, Ala, Ser, Thr, Val, or Asn, but not bulky or charged, such as Asp, Glu, Phe, Trp, Lys, or Arg. Furthermore, both Met and Phe/Trp residues were removable by MetAP-GTG when the antepenultimate residue was Ala but not Asp (Tables 1, 2). With respect to wild-type MetAP, the N-terminal Met and penultimate Ala of MAA- or MAG-initiated onconase and RC-RNase 4 were also removable (Tables 2, 3).
Table 2.
Effect of antepenultimate residue of substrate on the in vivo catalytic activity of engineered MetAPs
| Catalytic activity (%)a | Catalytic activity (%) | |||||
| Substrate | MetAP-YMQb | MetAP-*TG | MetAP-GTG | Substrate | MetAP-*TG | MetAP-GTG |
| M↓A↓G-Oncc | 80 (63)d | 80 (57) | 86 (16) | M↓L↓L-Onc | 88 (24) | 93 (55) |
| M↓A↓A-Onc | 85 (77) | 100 (25) | 100 (30) | M↓L↓H-Onc | 75 (36) | 100 (51) |
| M↓A↓P-Onc | 88 (64) | 72 (38) | N.D.e | M↓LM-Onc | 100 | 100 |
| M↓AV-Onc | —f | 100 | — | |||
| M↓AN-Onc | — | 100 | — | M↓Q↓A-Onc | 100 (100) | N.D.e |
| M↓AD-Onc | — | 100 | — | M↓Q↓P-Onc | 25 | 54 (100) |
| M↓AL-Onc | — | 100 | 100 | M↓Q↓T-Onc | 76 (26) | 86 (79) |
| M↓AQ-Onc | — | 97 | — | M↓Q↓N-Onc | 100 (65) | 83 (90) |
| M↓AE-Onc | — | 96 | — | M↓QD-Onc | 91 | 97 |
| M↓AF-Onc | — | 100 | — | M↓QE-Onc | 71 | — |
| M↓AM-Onc | — | 96 | 96 | M↓QF-Onc | 80 | — |
| M↓AK-Onc | — | 100 | — | M↓Q↓M↓-Onc | 100 (59) | 100,100 (73) |
| M↓AR-Onc | — | 94 | — | M↓QK-Onc | 78 | — |
| M↓QW-Onc | 95 | — | ||||
| M↓L↓G-Onc | — | 100 (100) | 87 (46) | M↓QR-Onc | 83 | — |
| M↓L↓A-Onc | — | 100 (100) | 100 (80) | |||
| M↓L↓P-Onc | — | 5 | 55 (95) | M↓IA-Onc | 36 | 96 |
| M↓L↓S-Onc | — | 82 (59) | 87 (69) | M↓F↓A-Onc | 2 | 36 (100) |
| M↓L↓T-Onc | — | 91 | 72 (54) | M↓KA-Onc | 2 | 9 |
| M↓L↓V-Onc | — | 76 (27) | 97 (69) | M↓YA-Onc | 8 | 22 |
| M↓L↓N-Onc | — | 92 (58) | 100 (68) | M↓W↓A-Onc | 2 | 100 (89) |
| M↓LD-Onc | — | 90 | 98 | M↓RA-Onc | 2 | 8 |
a Activity was expressed by the percentage of terminal residue removed from Met-tagged onconase with different penultimate and antepenultimate residue.
b MetAP-YMQ represents the wild-type E. coli Met aminopeptidase.
c The vertical arrow represents the site cleaved by MetAP.
d The value in parentheses represents the percent of penultimate residue that is removed.
e N.D. indicates that no protein is detectable under the same culture conditions.
f Dashes indicate the result was not measured.
Table 3.
The substrate specificity of MetAP-*TG in Escherichia coli
| Substratea | Solubility | Catalytic activity (%) |
| M↓QDWL-Onc | − | 91 |
| M↓QDWL-Oncb | − | 80 |
| M↓QDWE-RC3 | − | 96 |
| M↓QDWD-RC6 | − | 82 |
| M↓QDWA-RC4 | − | 93 |
| M↓Q↓AWA-RC4 | − | 100 (100)c |
| M↓A↓AWA-RC4 | − | 100 (18) |
| M↓A↓PWA-RC4 | − | 79 (63) |
| M↓Q↓NWE-RC2 | − | 80 (56) |
| M↓Q↓NWA-RCL1 | − | 96 (62) |
| M↓QDWA-RCL1 | − | 99 |
| M↓L↓AGA-CA150 | + | 100 (71) |
| M↓LDAGA-CA150 | + | 88 |
| M↓LDAGA-CA150b | + | 77 |
| M↓QDIL-GST | +d | 87 |
| M↓QDIL-GSTb | +d | 71 |
| M↓QDIL-GST | −e | 85 |
| M↓QDIL-GSTb | −e | 70 |
a (Onc) Onconase (AF332139); (RC2) RC-RNase2 (AF242553); (RC3) RC-RNase3 (AF242554); (RC4) RC-RNase4 (AF242555); (RC6) RC-RNase6 (AF242556); (RCL1) RC-RNase1 (AF288642); (the WW domain of transcription elongation regulator 1) CA150 (nt 1295-1717 of NM_006706); (GST) glutathione S-transferase (MI4654).
b The gene of the target protein was cloned in pET22b and cotransformed with MetAP-*TG, which was cloned in pET29b. The expressions of both genes were driven by T7 RNA polymerase simultaneously.
c The value in parentheses represents the percent of penultimate residue that is removed.
d,e The majority of recombinant GST is solubled, but some is insolublee under the culture condition as described in the text.
The removal of N-terminal Met from the penultimate Ala/Leu/Gln residue by MetAP-*TG was reduced when the antepenultimate residue was Pro. In contrast, most of the newly exposed Leu/Gln residue preceding Pro was removed by MetAP-GTG, although only half of the N-terminal Met was removed (Table 2). This result suggests that the Pro residue at the antepenultimate site may exert a steric hindrance on the catalytic activity of the MetAP enzyme.
Applications in other recombinant proteins
To investigate the possible application of engineered MetAPs in the production of recombinant proteins, we used several soluble or insoluble proteins as substrates. The Met of insoluble MQD-initiated bullfrog ribonucleases (RC-RNase 3, RC-RNase 4, RC-RNase 6, and RC-RNase L1-N2D) was removed by MetAP-*TG, and both Met and Gln of insoluble MQN-initiated bullfrog ribonucleases (RC-RNase 2 and RC-RNase L1) were removed by the enzyme (Table 3). The terminal Met of MQD-initiated glutathione S-transferase (GST) of Schistosoma japonicum, either soluble or insoluble, was also removable by the MetAP-*TG. Both Met and Leu of the soluble MLA-initiated WW domain of CA150 protein, a transcription elongation regulator, were also removable (Table 3). To increase the competence of the engineered MetAP, we cloned the MetAP-*TG gene in the vector pET29b, which possesses kanamycin-resistance, and we cloned the gene of target protein, for example, onconase, the WW domain of CA150, or GST, in a separate expression vector pET22b, which possesses ampicillin resistance and is coexpressed in E. coli BL21(DE3). We found that the Met of these MQD- or MLD-initiated proteins was also removable, but with a slightly lower efficiency (10%–15% less; Table 3). This reduction may be due to lower copy number of the plasmid and the RNA transcript encoding the engineered MetAP.
From the eukaryotic curated (NP) protein sequence entries of the NCBI RefSeq database (dated January 7, 2003) using the SignalP program, we found that 37.4% of 5298 secretory proteins possesses a small residue, such as Gly, Ala, Cys, Pro, Ser, Thr, or Val, at N termini and 58.5% of 17,978 nonsecretory proteins possesses a small penultimate residue. The N-terminal Met of these recombinant proteins may be processed by wild-type MetAP. On the basis of the results we obtained by the coexpressed MetAPs, up to 84.5% of secretory proteins and 90.2% of nonsecretory proteins should be produced in authentic form.
Properties of recombinant ribonucleases
N terminus
The N terminus of MetAP-processed MQD-initiated on-conase was Gln, determined by Edman degradation, but it was a mixture of Gln/Pyr soon after refolding from inclusion bodies determined by mass spectrum analysis. One value (11,818 daltons) agreed with the mass of Pyr-initiated onconase (secretory or native onconase) and the other (11,835 daltons) agreed with that of Gln-initiated onconase calculated by the ExPASY server from the DNA sequence (Wilkins et al. 1998). The reduction of 17 daltons indicates that Pyr is derived from Gln through deamination. Similar results were also observed in the recombinant RC-RNase L1 proteins.
Catalytic activity
The catalytic activity of onconase was markedly reduced by the addition of Met and disruption of Pyr at the N terminus (100-fold less). The catalytic activity of onconase (rm-Onc) was restored after Met removal and refolding, similar to that of secretory or native onconase assayed by both zymogram (Fig. 2B ▶, left panel) and the acid-insoluble method (data not shown). The catalytic activity of demethi-oninylated and refolded rmRCL1-N2D beginning with MQD was similar to that of ribonuclease isolated from bullfrog liver (RCL1) or secretory ribonuclease purified from culture medium (sRCL1), but that of MetAP-processed wild-type ribonuclease (rmRCL1) beginning with MQN was not fully restored (Fig. 2B ▶, right panel), probably due to the partial deletion of terminal Gln. These results reveal that the removal of N-terminal Met by engineered MetAP is crucial for the catalytic activity of MQD-initiated frog ribonuclease.
In vitro catalytic activities of MetAP variants
For oligopeptides ranging from tetra- to dodecapeptides with Ala at the penultimate site, the wild-type MetAP and MetAP-*TG possessed high catalytic activity, whereas the MetAP-*GG, MetAP-*TT, and MetAP-GTG variants did not (Table 4). For the oligopeptide with Gln at the penultimate residue, wild-type and most engineered MetAPs did not exhibit significant catalytic activities, whereas the MetAP-GTG exerted 43% catalytic activity for the dodeca-peptide MQDWLTFQKKHI in comparison with that for MADWLTFQKKHI, which is identical to the N-terminal α-helix of rM-Onc. The low in vitro catalytic activity of the purified MetAP for MQD-initiated oligopeptides suggests that some factor(s) may be required for the in vivo removal of Met from a bulky penultimate residue.
Table 4.
In vitro catalytic activity of engineered MetAPs toward synthetic oligopeptides
| Specific activity (unit/mg)a | |||||
| Substrate | MetAP YMQ (wt) | MetAP-*GG | MetAP-*TT | MetAP-*TG | MetAP-GTG |
| MADY | 138 ± 8 | 0.51 ± 0.01 | 0.38 ± 0.03 | 72 ± 10 | 1.68 ± 0.51 |
| MADYLT | 138 ± 6 | 0.29 ± 0.01 | 0.12 ± 0.01 | 116 ± 4 | 2.50 ± 0.42 |
| MQDYLT | 0.43 ± 0.01 | 0.29 ± 0.02 | 0.13 ± 0.01 | 0.42 ± 0.02 | 0.23 ± 0.01 |
| MADWLTFQKKHI | 85 ± 13 | 3.28 ± 0.04 | 1.00 ± 0.08 | 73 ± 15 | 9.80 ± 0.26 |
| MQDWLTFQKKHI | 0.62 ± 0.08 | 0.50 ± 0.02 | 0.25 ± 0.01 | 0.59 ± 0.07 | 4.24 ± 0.14 |
a One unit of activity is defined as 1 μmole of amino acid produced per min under the assay condition described in the text using pure Met as a standard.
Discussion
Several attempts have been made to remove the translation initiator Met or fMet for the production of a recombinant protein with an identical N-terminal residue to that of native protein. Most of these in vitro methods, however, are limited by harsh conditions, high cost, or low yields. The method for the in vivo removal of Met by MetAP has been limited by the size, charge, and hydrophobicity of the penultimate residue of target proteins (Ben-Bassat et al. 1987; Hirel et al. 1989; Hwang et al. 1999). The residues responsible for substrate binding have been verified by the X-ray crystallography of E. coli MetAP and its inhibitor complex (Lowther et al. 1999; Lowther and Matthews 2000). The corresponding residues, Met 326 and Gln 359, of yeast MetAP I have been verified using an in vitro assay system. The M326A and/or Q359A mutants exhibit a significant increase of catalytic activity for some oligopeptides with a slightly larger penultimate residue, such as Met, Asn, or His. In this study, three residues, Y168G, M206T, and Q233G, were mutated in the substrate-binding pocket of E. coli MetAP and it was coexpressed with its target proteins through the same or a separate vector in E. coli without the need for further chemical or enzymatic treatment for Met removal. Our results showed that the Met of most recombinant proteins could be removed even if the penultimate residue is bulky, acidic, or hydrophobic. However, the efficiency of Met removal from the basic penultimate residue is still low, for example, Lys at 9% Met removal and Arg at 8%. We suspect that resistance of MK- or MR-initiated proteins to the engineered MetAP could be due to the charge repulsion and steric hindrance of the substrate by the residues of MetAP in the substrate-binding pocket. Thus, substitution of positively charged or bulky residue with a small or hydrophilic residue in the substrate-binding pocket may improve the substrate recognition and catalytic activity of MetAP.
In addition to the penultimate residue, the antepenultimate residue of a substrate may affect MetAP activity. The catalytic activity of human or yeast MetAP was reduced when the penultimate Val/Thr residue was followed by a Pro, for example, the MVP-initiated human β-globin (Prchal et al. 1986) or MVP- or MTP-initiated yeast iso-1-cytochrome c (Moerschell et al. 1990). In contrast, both terminal Met and penultimate Ala residues were removed by E. coli MetAP when the antepenultimate residue was Pro, for example, MAP-initiated human interleukin-2 (Ben-Bassat et al. 1987). Thus, other aminopeptidase(s) could be responsible for the removal of the penultimate Ala residue in vivo (Ben-Bassat et al. 1987). Our results showed that the N-terminal Met and penultimate Ala/Leu/Gln/Phe/Trp residue were removed in vivo by engineered MetAP when the antepenultimate residue was small. The terminal Met and penultimate Ala of MAA-initiated onconase or RC-RNase 4 were also removed by the wild-type E. coli MetAP (Tables 2, 3). Once the terminal Met is removed by a MetAP variant, the small antepenultimate residue may serve as a penultimate residue and enable the subsequent cleavage of a new terminal residue of the demethioninylated protein by the MetAP. Thus, instead of possible involvement of other proteases, we suggest that the E. coli MetAP may possess other aminopeptidase activities for the terminal Ala, Leu, Gln, Phe, and Trp (Ben-Bassat et al. 1987). The removal of N-terminal bulky residues, such as Leu, Gln, Phe, and Trp, and the subsequent exposure of small residues, such as Ala, Ser, and Thr, may protect the processed proteins from degradation. As predicted from the N-end rule of protein stability, proteins possessing a bulky N-terminal residue, known as destabilizing residues, have short half-lives and those possessing small terminal residues, known as stabilizing residues, have long half-lives (Tobias et al. 1991; Varshavsky 1996).
In addition to Met removal, modification to the N-terminal residue is often crucial for the function of a protein. For example, the acetylation of the N-terminal Asp/Glu residue of actin strengthens its interaction with myosin (Abe et al. 2000). Also, the N-myristoylation of the N-terminal Gly of SOS3 protein is essential for conferring salt tolerance in plants (Ishitani et al. 2000). The conversion of the N-terminal Gln to Pyr is found in all known cytotoxic frog ribonucleases, some mammalian ribonucleases, immunoglobulins, and several hormones (thyrotropin releasing hormone, luteinizing hormone releasing hormone, gonadotropin-releasing hormone, corticotropin-releasing hormone, and gastrin [Busby Jr. et al. 1987; Fischer and Spiess 1987; Boix et al. 1996; Liao et al. 2003]).
Three hydrogen bonds contributed by N-terminal Pyr of frog ribonuclease are essential for its structural integrity, catalytic activity, and cytotoxicity (Huang et al. 1998; Leu et al. 2003; Liao et al. 2003). In this study, a large amount of demethioninylated proteins (10~50 mg from 1 L of culture) was prepared in vivo by engineered MetAP, and the terminal Gln was subsequently converted to Pyr in vitro under mild conditions instead of using chemical and enzymatic treatments (Shapiro et al. 1988; Notomista et al. 1999; Liao et al. 2003). For prevention of subsequent cleavage of the penultimate residue, the antepenultimate residue of any protein of interest may be replaced by bulky or acidic residues if they do not alter the properties of the protein, such as RCL1-N2D. Thus, the engineered MetAP and the expression system developed here in E. coli BL21(DE3) are able to produce large quantities of soluble or insoluble authentic protein.
The wild-type MetAP exhibited high catalytic activities for MAD-initiated proteins in vivo and for MAD-initiated oligopeptides in vitro. However, the engineered MetAP-*TG demonstrated high catalytic activities only for MQD-initiated proteins in vivo, but not for MQD-initiated oligopeptides in vitro. Although yeast MetAP I has been found to be associated with ribosomal proteins and mammalian MetAP II is associated with eukaryotic translation initiation factor (eIF-2; Wu et al. 1993; Vetro and Chang 2002), the possible association of cellular protein(s) with E. coli MetAP is not known. The MetAP-*TG retains high catalytic activity for MAD-initiated oligopeptides in vitro, but it does not exhibit significant catalytic activity for MQD-initiated oligopeptides. This is probably due to the relatively rigid structure of the purified enzyme and the limited space for the binding of bulky penultimate residues in vitro. In contrast, it may exhibit a higher flexibility in binding bulky penultimate residues if it is assisted by cellular proteins in vivo. Therefore, it is possible that the removal of terminal Met from bulky penultimate residues by E. coli MetAP may be assisted by other cellular protein(s) in vivo during the elongation phase of protein synthesis.
The amino acid residues responsible for catalytic activity and substrate specificity of MetAP are conserved in most living organisms (Tahirov et al. 1998), for example, the Met 206 and Gln 233 of E. coli MetAP and the Met 329 and Gln 356 of yeast MetAP I (Ben-Bassat et al. 1987; Hirel et al. 1989; Hwang et al. 1999). Thus, mutations of the corresponding residues of human MetAP will likely destroy its function. Furthermore, mutations of the penultimate or antepenultimate residues of any desired target protein may also alter their stability and function. Therefore, both of these features may provide important clues in the study of human genes and associated diseases and may help investigators explore the possibility of gene therapy, such as for hemoglobin Long Island with reduced oxygen affinity, which is caused by the His-Pro substitution at the third residue of β-globin and the failure to remove the N-terminal Met (Prchal et al. 1986).
Materials and methods
Construction of expression plasmids
The rpr (AF332139) gene encoding for the target protein, onconase, from R. pipiens was tagged with the NdeI and BamHI sites at the 5′ and 3′ ends, respectively, and inserted into expression vector pET22b (Novagen; Huang et al. 1998; Liao et al. 2000, 2003). The other target proteins were also cloned by the same method including RC-RNase L1 (AF288642.2), RC-RNase 2 (AF242553), RC-RNase 3 (AF242554), RC-RNase 4 (AF242555), RC-RNase6 (AF242556) from Rana catesbeiana, the WW domain of a transcription elongation regulator 1, CA150 (nt 1295–1717 of NM_006706), and glutathione S-transferase (M14654). The Escherichia coli methionine aminopeptidase gene (accession no. M 15106) or its mutant was fused with the T7 promoter by three steps of PCR and inserted into the same pET22b vector containing a target protein, as described earlier, through SacI and HindIII. The first PCR was performed using oligonucleotide 1 (5′-CGCG GAGCTCGATCCCGCGAAATTAATACG-3′) and oligonucleo-tide 2 (5′-CTTGATTGAGATAGCCATTATCTCCTTCTTAAAG TTAAACAAAATTATTTCTAGAGG-3′) as the 5′ and 3′ primers, respectively, and pET22b as the template. The second PCR was performed using the earlier-mentioned PCR product as the 5′ megaprimer and oligonucleotide 3 (5′-CCGGAAGCTTTTATTC GTCGTGCGAGATTATCG-3′) as the 3′ primer and E. coli MetAP gene as the template (Ben-Bassat et al. 1987; Huang et al. 1998). The third PCR was to amplify the second PCR product using oligonucleotide 1 and oligonucleotide 3 as primers. Therefore, the genes encoding target protein and E. coli MetAP have their own T7 RNA polymerase promoter, but their encoded mRNAs may be located in the same RNA molecule for cotranslation because only one transcription terminator exists downstream of the MetAP gene. Alternatively, the MetAP gene alone may be directly subcloned into vector pET29b through the SacI and HindIII sites. Residues Tyr 168, Met 206, and Gln 233 in the putative substrate-binding site of E. coli MetAP were mutated by site-directed mutagenesis on the basis of the crystal structure (Fig. 1 ▶, PDB code 3MAT) and amino acid sequence alignment of MetAPs (Ben-Bassat et al. 1987; Lowther et al. 1999; Walker and Bradshaw 1999). For the expression of secretory ribonucleases, the ribonuclease gene was cloned and expressed as described previously (Huang et al. 1998).
Expression and purification of MetAP and target proteins
The MetAP was expressed alone or coexpressed with a target protein, that is, onconase and RC-RNaseL1, in E. coli BL21 (DE3) in the presence of 0.5 mM isopropyl-β-D-thiogalactopyranoside at 37°C. After sonication, the MetAP in soluble fraction was dialyzed against buffer A (20 mM HEPES at pH 8.0, 50 mM KCl) and purified to homogeneity by phosphocellulose (Whatman P-11) and DEAE-cellulose (Whatman DE52) column chromatographies with a yield of 50 mg from 1 L of culture.
Most of the coexpressed target protein, for example, onconase or RC-RNase L1, existed in insoluble cell lysate. These proteins were denatured, renatured, and purified as described previously (Liao et al. 2003). The purified ribonucleases were stored in 20 mM sodium phosphate (pH 7.0) for Pyr formation at the N terminus. Alternatively, the ribonuclease secreted into culture medium through the pelB system was concentrated and purified to electrophoretical homogeneity with a yield of 1~5 mg per 1 L of culture (Huang et al. 1998).
Analysis of N termini of recombinant proteins
Proteins in the cell pellet or in the soluble fraction were separated by 13.3% SDS-PAGE and contact-transferred to ProBlott membrane (Applied Biosystems) in transfer buffer (10 mM Tris-HCl at pH 7.5, 50 mM NaCl, 2 mM EDTA, 0.5 mM 2-mercaptoethanol) overnight. The N-terminal five residues of the transferred protein were determined by Edman degradation. The percentage of Met removal was calculated by dividing the amount of protein initiated with the penultimate residue by the amount of total proteins (i.e., proteins initiated with the penultimate residue and unprocessed protein initiated with Met) after the first degradation cycle. The same calculation was made from five consecutive degradation cycles, for example, MQNWL for onconase. The efficiency of Met removal is reported as the mean values of at least three cycles. The mass spectrum analysis was carried out as described previously (Liao et al. 2003).
In vitro MetAP activity assay
The oligopeptide substrates used in this experiment were synthesized on the 432A Synergy peptide synthesizer (Applied Biosystems). The enzymatic activity of purified MetAP was assayed as described by Ben-Bassat et al. (1987). Briefly, the diluted MetAP enzyme in 10 μL was added to 90 μL of the substrate solution containing 4 mM oligopeptide, 0.1 M potassium phosphate buffer (pH 7.5), and 0.2 mM CoCl2, incubated for 10 min at 37°C and stopped in boiling water for 2 min. After addition of 0.9 mL of color development mixture containing 0.2 mg of L-amino acid oxidase, 0.03 mg of horseradish peroxidase, and 0.2 mg of o-dianisidine in 0.1 M Tris-HCl (pH 7.4), the tubes were incubated for 10 min at 37°C and the optical density at 440 nm was recorded. One unit of activity is defined as 1 μmole of amino acids produced per min under the assay conditions and the pure Met was used as standard. The N-terminal residues of MetAP-treated oligopeptide were further verified by Edman degradation.
Ribonuclease activity assay
Ribonuclease activity was analyzed by zymogram assay on RNA-casting PAGE (Liao and Wang 1994). In addition, the ribonuclease activity of each purified ribonuclease was determined by the release of acid-soluble nucleotides from bakers’ yeast total RNA after ribonuclease digestion. One unit of enzyme activity was defined as the amount of enzyme producing one A260 acid-soluble material for 15 min at 37°C (Huang et al. 1998).
Acknowledgments
We thank Mr. T.T. Lee for the computation and statistical analyses of the N-terminal residues of proteins; Ms. C.C. Lin for oligopeptide synthesis; and the Core Facilities for Proteomic Research, Institute of Biological Chemistry, Academia Sinica, for the analyses of amino acid sequence and mass spectra. We also thank Drs. David C.P. Tu, M.F. Tam, and Michael M.T. Lee for constructive discussions and critical reading of the manuscript, and Dr. L.Y. Lin for the generous gift of Escherichia coli methionine amino-peptidase gene.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04679104.
References
- Abe, A., Saeki, K., Yasunaga, T., and Wakabayashi, T. 2000. Acetylation at the N-terminus of actin strengthens weak interaction between actin and myosin. Biochem. Biophys. Res. Commun. 268 14–19. [DOI] [PubMed] [Google Scholar]
- Adachi, K., Yamaguchi, T., Yang, Y., Konitzer, P.T., Pang, J., Reddy, K.S., Ivanova, M., Ferrone, F., and Surrey, S. 2000. Expression of functional soluble human β-globin chains of hemoglobin in bacteria. Protein Expr. Purif. 20 37–44. [DOI] [PubMed] [Google Scholar]
- Belagaje, R.M., Reams, S.G., Ly, S.C., and Prouty, W.F. 1997. Increased production of low molecular weight recombinant proteins in Escherichia coli. Protein Sci. 6 1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Bassat, A., Bauer, K., Chang, S.Y., Myambo, K., Boosman, A., and Chang, S. 1987. Processing of the initiation methionine from proteins: Properties of the Escherichia coli methionine aminopeptidase and its gene structure. J. Bacteriol. 169 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boix, E., Wu, Y., Vasandani, V.M., Saxena, S.K., Ardelt, W., Ladner, J., and Youle, R.J. 1996. Role of the N terminus in RNase A homologues: Differences in catalytic activity, ribonuclease inhibitor interaction and cytotoxicity. J. Mol. Biol. 257 992–1007. [DOI] [PubMed] [Google Scholar]
- Busby Jr., W.H., Quackenbush, G.E., Humm, J., Youngblood, W.W., and Kizer, J.S. 1987. An enzyme(s) that converts glutaminyl-peptides into pyroglutamyl-peptides. Presence in pituitary, brain, adrenal medulla, and lymphocytes. J. Biol. Chem. 262 8532–8536. [PubMed] [Google Scholar]
- Chang, S.Y., McGary, E.C., and Chang, S. 1989. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J. Bacteriol. 171 4071–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S., Vetro, J.A., and Chang, Y.H. 2002. The specificity in vivo of two distinct methionine aminopeptidases in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 398 87–93. [DOI] [PubMed] [Google Scholar]
- Endo, S., Yamamoto, Y., Sugawara, T., Nishimura, O., and Fujino, M. 2001. The additional methionine residue at the N-terminus of bacterially expressed human interleukin-2 affects the interaction between the N- and C-termini. Biochemistry 40 914–919. [DOI] [PubMed] [Google Scholar]
- Fischer, W.H. and Spiess, J. 1987. Identification of a mammalian glutaminyl cyclase converting glutaminyl into pyroglutamyl peptides. Proc. Natl. Acad. Sci. 84 3628–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirel, P.H., Schmitter, M.J., Dessen, P., Fayat, G., and Blanquet, S. 1989. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc. Natl. Acad. Sci. 86 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, H.C., Wang, S.C., Leu, Y.J., Lu, S.C., and Liao, Y.D. 1998. The Rana catesbeiana rcr gene encoding a cytotoxic ribonuclease. Tissue distribution, cloning, purification, cytotoxicity, and active residues for RNase activity. J. Biol. Chem. 273 6395–6401. [DOI] [PubMed] [Google Scholar]
- Hwang, D.D., Liu, L.F., Kuan, I.C., Lin, L.Y., Tam, T.C., and Tam, M.F. 1999. Co-expression of glutathione S-transferase with methionine aminopeptidase: A system of producing enriched N-terminal processed proteins in Escherichia coli. Biochem.J. 338 (Pt. 2): 335–342. [PMC free article] [PubMed] [Google Scholar]
- Ishitani, M., Liu, J., Halfter, U., Kim, C.S., Shi, W., and Zhu, J.K. 2000. SOS3 function in plant salt tolerance requires N-myristoylation and calcium binding. Plant Cell 12 1667–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu, Y.J., Chern, S.S., Wang, S.C., Hsiao, Y.Y., Amiraslanov, I., Liaw, Y.C., and Liao, Y.D. 2003. Residues involved in the catalysis, base specificity, and cytotoxicity of ribonuclease from Rana catesbeiana based upon mutagenesis and X-ray crystallography. J. Biol. Chem. 278 7300–7309. [DOI] [PubMed] [Google Scholar]
- Li, X. and Chang, Y.H. 1995. Amino-terminal protein processing in Saccharomyces cerevisiae is an essential function that requires two distinct methionine aminopeptidases. Proc. Natl. Acad. Sci. 92 12357–12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y.D. and Wang, J.J. 1994. Yolk granules are the major compartment for bullfrog (Rana catesbeiana) oocyte-specific ribonuclease. Eur. J. Biochem. 222 215–220. [DOI] [PubMed] [Google Scholar]
- Liao, Y.D., Huang, H.C., Leu, Y.J., Wei, C.W., Tang, P.C., and Wang, S.C. 2000. Purification and cloning of cytotoxic ribonucleases from Rana catesbeiana (bullfrog). Nucleic Acids Res. 28 4097–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y.D., Wang, S.C., Leu, Y.J., Wang, C.F., Chang, S.T., Hong, Y.T., Pan, Y.R., and Chen, C. 2003. The structural integrity exerted by N-terminal pyroglutamate is crucial for the cytotoxicity of frog ribonuclease from Rana pipiens. Nucleic Acids Res. 31 5247–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther, W.T. and Matthews, B.W. 2000. Structure and function of the methionine aminopeptidases. Biochim. Biophys. Acta 1477 157–167. [DOI] [PubMed] [Google Scholar]
- Lowther, W.T., Orville, A.M., Madden, D.T., Lim, S., Rich, D.H., and Matthews, B.W. 1999. Escherichia coli methionine aminopeptidase: Implications of crystallographic analyses of the native, mutant, and inhibited enzymes for the mechanism of catalysis. Biochemistry 38 7678–7688. [DOI] [PubMed] [Google Scholar]
- Moerschell, R.P., Hosokawa, Y., Tsunasawa, S., and Sherman, F. 1990. The specificities of yeast methionine aminopeptidase and acetylation of amino-terminal methionine in vivo. Processing of altered iso-1-cytochromes c created by oligonucleotide transformation. J. Biol. Chem. 265 19638–19643. [PubMed] [Google Scholar]
- Notomista, E., Cafaro, V., Fusiello, R., Bracale, A., D’Alessio, G., and Di Donato, A. 1999. Effective expression and purification of recombinant on-conase, an antitumor protein. FEBS Lett. 463 211–215. [DOI] [PubMed] [Google Scholar]
- Prchal, J.T., Cashman, D.P., and Kan, Y.W. 1986. Hemoglobin Long Island is caused by a single mutation (adenine to cytosine) resulting in a failure to cleave amino-terminal methionine. Proc. Natl. Acad. Sci. 83 24–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick, S.L. and Matthews, B.W. 1993. Structure of the cobalt-dependent methionine aminopeptidase from Escherichia coli: A new type of proteolytic enzyme. Biochemistry 32 3907–3912. [DOI] [PubMed] [Google Scholar]
- Shapiro, R., Harper, J.W., Fox, E.A., Jansen, H.W., Hein, F., and Uhlmann, E. 1988. Expression of Met-(−1) angiogenin in Escherichia coli: Conversion to the authentic less than Glu-1 protein. Anal. Biochem. 175 450–461. [DOI] [PubMed] [Google Scholar]
- Tahirov, T.H., Oki, H., Tsukihara, T., Ogasahara, K., Yutani, K., Ogata, K., Izu, Y., Tsunasawa, S., and Kato, I. 1998. Crystal structure of methionine aminopeptidase from hyperthermophile, Pyrococcus furiosus. J. Mol. Biol. 284 101–124. [DOI] [PubMed] [Google Scholar]
- Tobias, J.W., Shrader, T.E., Rocap, G., and Varshavsky, A. 1991. The N-end rule in bacteria. Science 254 1374–1377. [DOI] [PubMed] [Google Scholar]
- Varshavsky, A. 1996. The N-end rule: Functions, mysteries, uses. Proc. Natl. Acad. Sci. 93 12142–12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetro, J.A. and Chang, Y.H. 2002. Yeast methionine aminopeptidase type 1 is ribosome-associated and requires its N-terminal zinc finger domain for normal function in vivo. J. Cell. Biochem. 85 678–688. [DOI] [PubMed] [Google Scholar]
- Walker, K.W. and Bradshaw, R.A. 1999. Yeast methionine aminopeptidase I. Alteration of substrate specificity by site-directed mutagenesis. J. Biol. Chem. 274 13403–13409. [DOI] [PubMed] [Google Scholar]
- Wilkins, M.R., Gasteiger, E., Bairoch, A., Sanchez, J.C., William, K.L., Appel, R.D., and Hochstrsser, D.F. 1998. Protein identification and analysis tools in the ExPASy Server. Humana Press, Totowa, NJ. [DOI] [PubMed]
- Wu, S., Gupta, S., Chatterjee, N., Hileman, R.E., Kinzy, T.G., Denslow, N.D., Merrick, W.C., Chakrabarti, D., Osterman, J.C., and Gupta, N.K. 1993. Cloning and characterization of complementary DNA encoding the eukaryotic initiation factor 2-associated 67-kDa protein (p67). J. Biol. Chem. 268 10796–10801. [PubMed] [Google Scholar]