

Abstract
The effects of the adenylyl cyclase activator forskolin on steady-state and transient currents generated by the Na+-K+ pump were studied in guinea-pig ventricular myocytes by means of whole-cell voltage clamp at 30 °C.
In external solution containing 144 mM Na+ () and 10 mM K+ (), steady-state Na+-K+ pump current (Ip) activated by 5 mM pipette Na+ () at -20 mV was reversibly augmented by forskolin (4 μM) to 133 ± 4 % of the control current (n = 15). The forskolin analogue 1,9-dideoxyforskolin (10 μM), which does not activate adenylyl cyclases, did not increase Ip (n = 2). Application of the protein kinase A (PKA) inhibitor H-89 (10 μM) in the continued presence of forskolin reversed the forskolin-induced elevation of Ip (n = 3).
The forskolin effect on Ip persisted in the presence of 50 mM which ensured that the internal Na+-binding sites of the Na+-K+ pump were nearly saturated. Under these conditions, the drug increased Ip to 142 ± 3 % of the control Ip when the pipette free Ca2+ concentration ([Ca2+]pip) was 0·013 nM (n = 5) and to 138 ± 4 % of the control Ip when free [Ca2+]pip was 15 nM (n = 9).
In Na+-free external solution, Ip activated by 50 mM and 1·5 mM was likewise increased by forskolin but to a lesser extent than in Na+-containing medium (116 ± 3 % of control, n = 10).
In order to investigate exclusively partial reactions in the Na+ limb of the pump cycle, transient pump currents under conditions of electroneutral Na+-Na+ exchange were studied. Transient pump currents elicited by voltage jumps displayed an initial peak and then decayed monoexponentially. Moved charge (Q) and the rate constant of current decay varied with membrane potential (V). The Q-V relationship followed a Boltzmann distribution characterized by the midpoint voltage (V0·5) and the maximum amount of movable charge (ΔQmax). Forskolin (2-10 μM) shifted V0·5 to more negative values while ΔQmax was not affected (n = 11). The effects of forskolin on transient pump currents were mimicked by 8-bromo-cAMP (500 μM; n = 2) and abolished by a peptide inhibitor of PKA (PKI, 10 μM; n = 5).
We conclude that activation of the cAMP-PKA pathway in guinea-pig ventricular myocytes increases Na+-K+ pump current at least in part by modulating partial reactions in the Na+ limb of the pump cycle. Under physiological conditions, the observed stimulation of the cardiac Na+-K+ pump may serve to shorten the action potential duration and to counteract the increased passive sarcolemmal Na+ and K+ fluxes during sympathetic stimulation of the heart.
The sympathetic nervous system exerts its effects on cardiac cells via activation of both α- and β-adrenergic receptors present in the sarcolemma. β-Adrenoceptors couple to Gs-proteins which stimulate adenylyl cyclases catalysing the formation of cAMP from ATP. The resulting increase in intracellular cAMP levels activates the cAMP-dependent protein kinase (PKA) which in turn phosphorylates various target proteins in the cell, among them several ionic channels in the sarcolemma such as the L-type Ca2+ channel (review: McDonald et al. 1994), K+ channels (e.g. Huang et al. 1994) or the cAMP-dependent Cl− channel (Bahinski et al. 1989; Harvey & Hume, 1989). Regulation of channel activity by this pathway is an important mechanism underlying the positive chronotropic, positive dromotropic and positive inotropic effects of β-adrenergic agonists. By contrast, much less is known about the modulation of the cardiac Na+-K+ pump via the cAMP-PKA pathway. The Na+-K+ pump links the export of three Na+ out of and the import of two K+ into the cell to the hydrolysis of one molecule of ATP. It thereby establishes and maintains the electrochemical gradients for Na+ and K+ across the sarcolemma, which are essential for cardiac excitation. Furthermore, due to its electrogenic nature the Na+-K+ pump also contributes directly to the resting potential and to the shape of the action potential. Therefore, it is of physiological significance to investigate whether the activity of the cardiac Na+-K+ pump is regulated by the cAMP-PKA cascade, and if so, how.
Early studies on the effects of β-adrenergic agonists on the Na+-K+ pump in multicellular cardiac preparations revealed a stimulatory action (reviewed in Glitsch et al. 1989). It remained unclear, however, whether stimulation of pump activity by these drugs was direct or indirect. In recent years, several investigators have examined the modulation of the cardiac Na+-K+ pump current (Ip) via the cAMP-PKA pathway in isolated mammalian ventricular cells, with somewhat inconsistent results. From measurements of the Na+ activity in rabbit ventricular myocytes, Desilets & Baumgarten (1986) concluded that the Na+-K+ pump is directly stimulated by isoprenaline. Conflicting observations were published regarding the β-adrenergic regulation of the rat cardiac Na+-K+ pump. Whereas Ishizuka & Berlin (1993) found no effect of β-agonists on Ip, recently Dobretsov et al. (1998) reported that the Na+-K+ pump current in rat ventricular cells is increased by noradrenaline or isoprenaline. The latter authors also investigated the mechanism underlying the catecholamine-induced stimulation of the Na+-K+ pump. They suggested that an accelerated deocclusion/ release of K+ at intracellular sites is responsible for the observed increase in pump current. According to Gao et al. (1992), stimulation or inhibition of Ip by isoprenaline in guinea-pig ventricular myocytes is a function of [Ca2+]i. The partial reactions of the pump cycle involved, however, remained unknown. Thus, important questions concerning the mechanism of β-adrenergic regulation of the cardiac Na+-K+ pump in various mammalian species remain unanswered.
In order to contribute to a better understanding of the sympathetic regulation of cardiac Ip, we investigated the modulation of steady-state and transient pump currents by forskolin in guinea-pig ventricular myocytes. Our results reveal that activation of the cAMP-PKA cascade in these cells causes an increase in Na+-K+ pump current at nanomolar and subnanomolar concentrations of intracellular free Ca2+. Furthermore, the increase in the steady-state pump current can be attributed, at least in part, to a modulation of partial reactions in the Na+ limb of the pump cycle. Some of the results have been published in abstract form (Kockskämper et al. 1999a,b).
METHODS
Cell isolation
Isolation of ventricular myocytes has been described in detail before (Kockskämper et al. 1997) and was conducted according to national guidelines. Briefly, adult guinea-pigs (200-450 g) were killed by cervical dislocation. Following thoracotomy, hearts were cannulated via the aorta and quickly excised, then mounted on a Langendorff apparatus and perfused at 37°C with oxygenated Ca2+-free solution containing (mM): sucrose, 204; NaCl, 35; KCl, 5.4; MgCl2, 1.0; EGTA, 2.0; Hepes, 10; pH 7.4 (adjusted with NaOH). After 2 min, the perfusion was changed to enzyme-containing solution including 0.1 mM instead of 2.0 mM EGTA, collagenase B (0.4 mg ml−1; Boehringer Mannheim), protease (Type XIV, 0.3 mg ml−1; Sigma, Deisenhofen, Germany), elastase (10 μl ml−1; Serva, Heidelberg, Germany), DNase (0.15 mg ml−1; Sigma) and bovine serum albumin (BSA, 0.5 mg ml−1; Sigma). Digested ventricles were cut into small pieces and transferred to nominally Ca2+-free, protease-free solution containing BSA and DNase. This solution was then exchanged for cell culture medium (1.3 mM Ca2+; Hanks’ medium 199; PAA, Linz, Austria) supplemented with 105 i.u. l−1 penicillin and 100 mg l−1 streptomycin (both from Sigma). Isolated ventricular myocytes were plated on culture dishes and kept in the incubator (37°C, 3 % CO2) until use for experimentation on the same day.
Electrical recording
Membrane currents of single ventricular cells were recorded in the whole-cell mode of the patch clamp technique (Hamill et al. 1981) using an EPC-7, EPC-7B (both from List-Medical, Darmstadt, Germany) or an EPC-8 (HEKA, Lambrecht, Germany) patch clamp amplifier. Currents were low-pass filtered at 200 Hz for recording of steady-state Na+-K+ pump currents and at 2 kHz for measurements of transient pump currents. The sampling rate was at least 4 times the filter rate. Voltage protocols were generated and membrane currents were recorded by the program ISO2 (MFK, Niedernhausen, Germany) running on a personal computer. The computer was connected to the amplifier via 12-bit A/D and D/A converters.
Experimental procedure and solutions
A culture dish containing isolated ventricular myocytes was placed on the stage of an inverted microscope (Diaphot, Nikon). The bath solution was pre-warmed to ∼35°C by means of a Peltier device. The medium contained (mM): NaCl, 144; NiCl2, 5.0; BaCl2, 2.0 or 4.0; CaCl2, 1.8; MgCl2, 0.5; Hepes, 10; pH 7.4 (adjusted with NaOH). For measurements of steady-state Na+-K+ pump currents 10 mM KCl was added.
Na+-free solution contained (mM): choline chloride, 144; KCl, 1.5; NiCl2, 5.0; BaCl2, 4.0; CaCl2, 1.8; MgCl2, 0.5; Hepes, 10; atropine, 0.002-0.005; pH 7.4 (adjusted with tetraethylammonium hydroxide (TEA-OH)). All solutions, including the pipette solutions (see below), were selected so as to isolate Na+-K+ pump-mediated currents and the cAMP-dependent chloride current (ICl(cAMP)).
The cell under study was additionally superfused by means of a multibarrelled, solenoid-operated pipette with a tip diameter of 500 μm. Gravity-fed solution flow was ∼0.4 ml min−1 and solution exchange near the cell was complete within 1 s. Under these conditions, the temperature in the vicinity of the myocyte was ∼30°C. Ventricular cells were whole-cell voltage clamped with low resistance patch pipettes made from borosilicate glass capillaries (GC150TF-10, Clark Electromedical Instruments, Reading, UK). Pipettes were pulled on a horizontal puller (DMZ, Zeitz, Munich, Germany) and had initial resistances between 1 and 2 MΩ when backfilled with the respective pipette solution. Two pipette solutions were used, a high- and a low-chloride solution. The high-chloride pipette solution contained (mM): TEA-Cl, 130; TEA-OH, 20; NaCl, 5; MgCl2, 3; EGTA, 6; Hepes, 16; MgATP, 10; pH 7.2-7.4 (adjusted with TEA-OH). Assuming a contamination of 1 μM Ca2+, free Ca2+ and Mg2+ concentrations were calculated to be 0.013 nM and 2.4 mM, respectively (according to Fabiato & Fabiato, 1979). The low-chloride pipette solution contained only 36 mM Cl− instead of 141 mM Cl−, with chloride being replaced by aspartate. Thus, when the high- or low-chloride pipette solution was used, the chloride equilibrium potential (ECl) equalled -4 or -40 mV, respectively. When the pipette Na+ or K+ concentration ([Na+]pip and [K+]pip) was increased to 50 or 90 mM, respectively, [TEA+] was reduced accordingly. For some experiments, the pipette free Ca2+ concentration ([Ca2+]pip) was increased to 15 nM by addition of 0.7 mM CaCl2 to the pipette solution.
Potential differences between the pipette and superfusion solution were nulled immediately before patch formation. After establishing a gigaohm seal between the pipette tip and the cell membrane, gentle suction was applied to the pipette to achieve the whole-cell configuration. Cell membrane capacitance (Cm) was estimated using at least one of the following procedures: (i) integration of the capacitative current transient during a hyperpolarizing voltage step from 0 to -10 mV, (ii) compensation of the capacitative current transient by means of the built-in devices of the amplifier, or (iii) a program routine applying ± 10 mV voltage ramps from a holding potential (Vh) of 0 mV. All estimates gave nearly identical results. Cm of the myocytes used in this study was 137 ± 6 pF (n = 78).
Steady-state pump currents in Na+-containing external solution were estimated as external K+ ()-activated currents. Control experiments revealed that these currents were almost identical to cardiac glycoside-sensitive currents. When external Na+ ()-free solution was used, however, Ip was measured as cardiac glycoside-sensitive current (1 mM dihydro-ouabain; DHO) due to the very high apparent affinity of the pump under these conditions (K0.5, ∼0.08 mM; Bielen et al. 1993).
Transient pump currents were recorded as cardiac glycoside-sensitive currents (200 μM DHO) in response to a voltage step (100 ms) under conditions of electroneutral Na+-Na+ exchange (see Läuger, 1991). The superfusion medium contained 144 mM Na+ and zero K+. The pipette solution included 5 mM Na+ and 10 mM ATP. Vh was either 0 or -40 mV. We refer to these transient pump currents as Na+-dependent transient charge movements or Na-TCM. To speed up the voltage clamp, Na-TCM were recorded after cancellation of Cm and compensation of series resistance (Rs; 30–80 %). The remaining capacitative transient decayed with a time constant, τ, of 0.02-0.10 ms, much faster than the fastest Na-TCM (τ = 1.5-2.0 ms). The amount of moved charge (Q) and the rate constant of current decay (k) of Na-TCM were analysed as a function of membrane potential (V). k values were obtained as the reciprocal of the time constant of a monoexponential function fitted to the declining part of the transient pump current beginning 1–3 ms after the voltage step. Q values were then calculated by integration of the monoexponential function between t = 0 ms and t = 100 ms. The sigmoid Q-V relationship was fitted according to the following Boltzmann equation:
where ΔQmax is the maximum amount of movable charge, Qmin is the bottom of the curve, V0.5 is the midpoint voltage, and α is a steepness factor corresponding to the equivalent charge. F is Faraday's constant, R is the universal gas constant and T is absolute temperature.
Drugs
DHO, a specific inhibitor of the Na+-K+ pump, was purchased from Roth (Karlsruhe, Germany). It was either diluted from a 10−2 M aqueous stock solution (final concentration, 2 × 10−4 M) or directly dissolved in the bath solution (final concentration, 10−3 M). Forskolin and 1,9-dideoxyforskolin (both from Sigma) were prepared as 10−2 M stock solutions in ethanol. 8-Bromo-cAMP (Sigma) was directly dissolved in the bath solution, and PKI(5-24) (PKI; Alexis, Grünberg, Germany) and microcystin LR (Alexis) were directly dissolved in the pipette solution. H-89 dihydrochloride ([N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide.2 HCl]; H-89; Alexis) was prepared as a 10−2 M and atropine sulphate monohydrate (Sigma) as a 10−3 M aqueous stock solution. Ethanol at a concentration of 0.1 %, the maximal concentration used, did not affect membrane current (n = 3).
Statistics
Data are presented as means ±s.e.m. Error bars are only shown when they exceed the size of the symbols. n indicates the number of cells studied. Differences between data points were checked by Student's t test and considered significant if P < 0.05.
RESULTS
Steady-state Na+-K+ pump currents of isolated guinea-pig ventricular myocytes were studied at a Vh of -20 mV using low-chloride pipette solutions. ECl was -40 mV under these conditions. Thus, an increase in intracellular [cAMP] would result in an outward current shift due to activation of ICl(cAMP). Ip was measured at near physiological [Na+] of 5 mM pipette Na+ () and 144 mM . To saturate external K+-binding sites of the pump, 10 mM was used for maximal Ip activation. The amplitude of Ip was estimated as the inward current shift upon application of -free solution (see Methods). Current recording was begun after the holding current had reached a steady state, usually 2–3 min after access to the cell interior was achieved. Figure 1A shows an original current trace. At the beginning, Ip under control conditions was estimated by application of -free solution, as indicated by the short horizontal bars above the trace. Its amplitude was 34 pA. The respective trace designated a is shown on an enlarged scale in Fig. 1B. Superfusion of the myocyte with 10 μM 1,9-dideoxyforskolin, a forskolin analogue that does not stimulate adenylyl cyclases (Seamon & Daly, 1985), neither activated ICl(cAMP) nor increased Ip. In the presence of 1,9-dideoxyforskolin Ip was slightly reduced to 30 pA (Fig. 1Ab and Bb). By contrast, application of 4 μM forskolin, a concentration previously shown to maximally stimulate ICl(cAMP) in these cells (Traebert et al. 1998), activated, after a lag of approximately 20 s, an outward current which was identified as ICl(cAMP) (see Fig. 8). Furthermore, as is evident from the repeated application of -free solution, the drug induced a delayed stimulation of Ip that reached maximal amplitude roughly 3 min after the start of the superfusion with forskolin-containing solution. At this point, Ip amplitude was 59 pA(Fig. 1Ac and Bc) or 174 % of the control. Upon wash-out of the adenylyl cyclase activator both the activation of ICl(cAMP) and the stimulation of the Na+-K+ pump current were reversed. Thus, the holding current shifted in the inward direction and the Ip amplitude was reduced to 28 pA (Fig.1Ad and Bd). In a total of 15 ventricular cells, 4 μM forskolin augmented Ip to 133 ± 4 % of the control (see also Fig. 3). Stimulation of Ip was almost always delayed compared with activation of ICl(cAMP). Maximal Ip in the presence of forskolin occurred about 2–5 min after superfusion of the myocytes with the drug was begun. Application of H-89, a specific inhibitor of PKA, in the continued presence of forskolin, reduced Na+-K+ pump current to values below the control. In three cells, in which forskolin had increased Ip to 123 ± 9 % of the control, H-89 (10 μM) reduced Ip to 64 ± 8 % of the control (not shown). The results presented so far suggest that forskolin increased Ip at near physiological concentrations of intra- and extracellular Na+ by activation of the cAMP- PKA pathway. Next, we sought to elucidate the mechanism of the forskolin-induced stimulation of Ip.
Figure 1. Forskolin reversibly increases Ip in -containing medium.
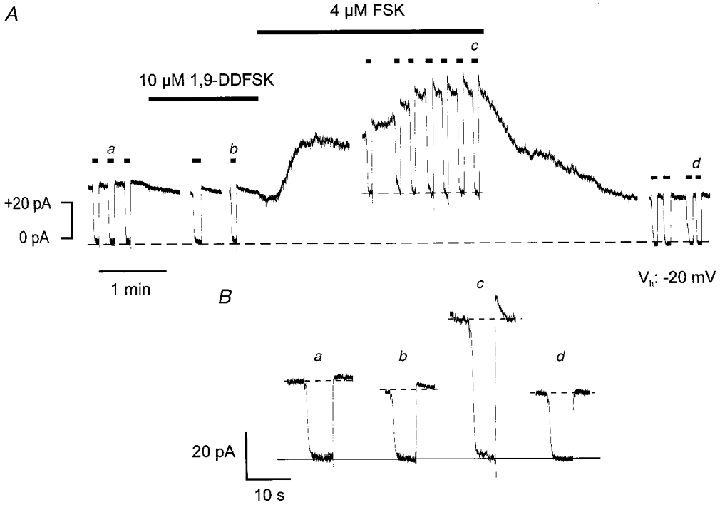
A, membrane current of a guinea-pig ventricular myocyte (Cm, 131 pF) in Na+-containing solution at -20 mV. [Na+]pip is 5 mM. The short horizontal bars above the current trace indicate application of -free medium for estimation of Ip. The longer horizontal bars indicate application of 1,9-dideoxyforskolin (1,9-DDFSK) or forskolin (FSK). The former drug is ineffective whereas the latter induces an outward current (ICl(cAMP)) and a delayed stimulation of Ip. Both effects are reversible. Dashed lines represent membrane current during blockade of Ip before and following forskolin application. The difference between these two current levels represents ICl(cAMP). B, Ip estimates in -free medium at various times as indicated in A. Ip amplitudes are the differences between the dashed and the continuous lines. Gaps between the current traces in A represent omissions of 2, 0.5, 0.5 and 1 min of recording, respectively.
Figure 8. Forskolin activates ICl(cAMP) in addition to its stimulation of Ip.
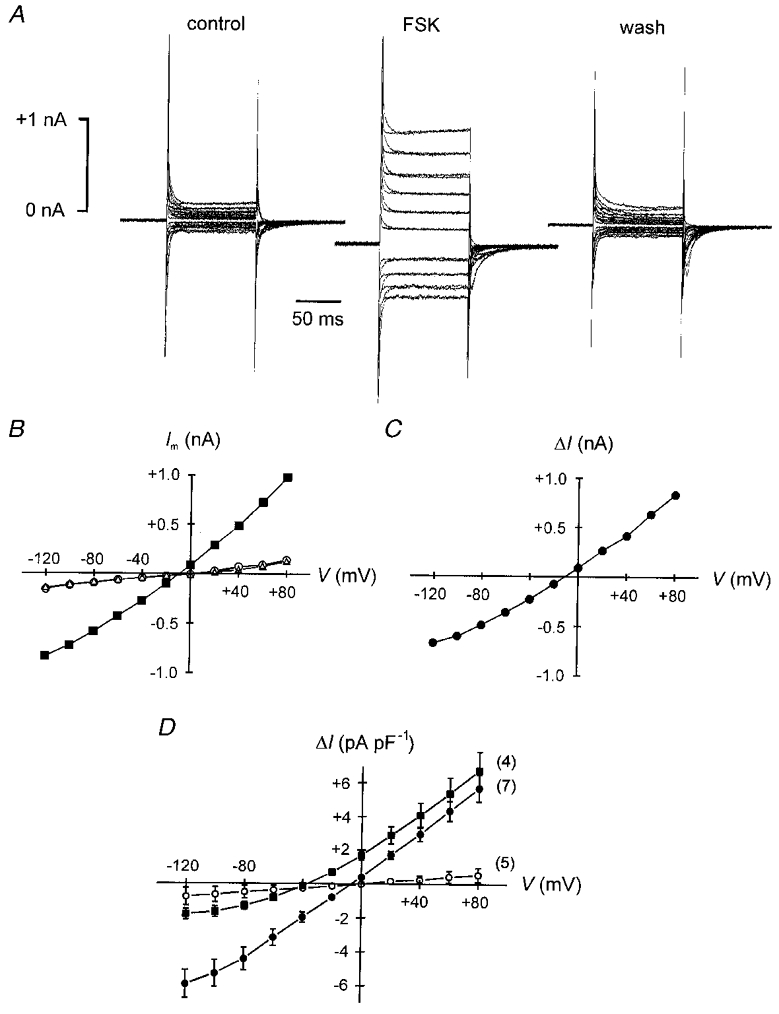
A, membrane currents of a ventricular myocyte dialysed with a pipette solution containing 141 mM Cl− during voltage steps to potentials between -120 and +80 mV from a Vh of -40 mV. Same cell as in Fig. 5. Current traces obtained in the absence and in the presence of 200 μM DHO are shown superimposed. Left panel: control in forskolin-free solution. Middle panel: forskolin (2 μM) activates a conductance and shifts the holding current in the inward direction. Right panel: the effect of the adenylyl cyclase activator is completely reversible. B, steady-state membrane currents (Im) derived from the data in A as a function of clamp potential (V). ^ and ▵, Im before and after application of forskolin, respectively; ▪, Im in the presence of the drug. C, voltage dependence of the difference current (ΔI) from the I–V curves in B (▪ minus ^ and ▵). The forskolin-activated current exhibits shallow outward rectification and reverses near the ECl of -4 mV. D, mean I–V relationships of the forskolin-induced current. Decreasing [Cl−]pip from 141 mM (•) to 36 mM (▪) shifts ECl from -4 to -40 mV and, correspondingly, the reversal potential of the drug-induced current. Note the stronger outward rectification at 36 mM . Inclusion of PKI (10 μM) in the pipette solution (141 mM Cl−) almost completely abolishes the forskolin-activated current (^). The properties of this current are characteristic for ICl(cAMP). Numbers in parentheses denote the number of myocytes studied.
Figure 3. Forskolin-induced increase in Ip under various ionic conditions.
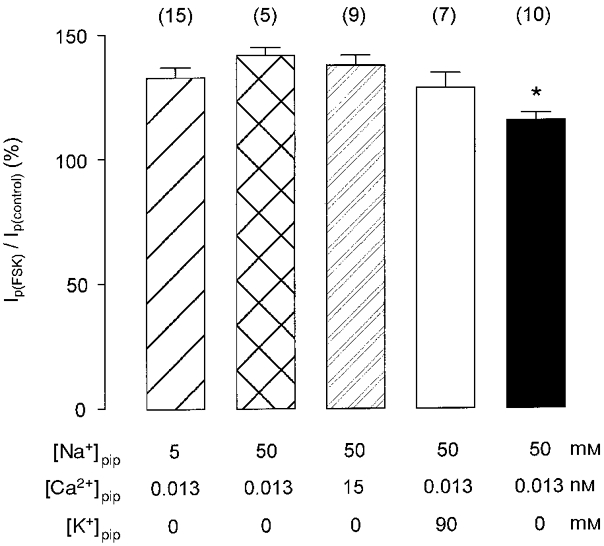
Bar graph showing the mean (±s.e.m.) forskolin-induced increase in Ip as a percentage of control under various conditions. The right bar (▪) represents results from measurements in -free solution, whereas the other bars show data obtained in -containing medium. Forskolin augments Ip significantly less in Na+-free than in Na+-containing solution (*P < 0.05). The effect of forskolin in the latter medium is similar irrespective of the Na+, Ca2+, and K+ concentrations of the pipette solution, which are specified below the respective bars. Numbers in parentheses indicate the number of cells studied.
Corresponding experiments using 50 mM to nearly saturate internal Na+-binding sites of the pump (Nakao & Gadsby, 1989) revealed that forskolin stimulated Ip to 142 ± 3 % of the control (n = 5; see Fig. 3), i.e to nearly the same extent as with a low [Na+]pip of 5 mM. Once again, Ip stimulation by the adenylyl cyclase activator occurred with a considerable time lag compared with ICl(cAMP) activation. Whereas ICl(cAMP) was completely activated after about 60 s, Ip became maximal only after 2–5 min. Almost identical results were obtained when free [Ca2+]pip was increased from 0.013 to 15 nM. Under these conditions, Ip amplitude was increased by the drug to 138 ± 4 % of the control (n = 9; see Fig. 3). Similarly, when 90 mM pipette K+ () was used to alter intracellular K+ deocclusion/release, Ip stimulation by forskolin was unaffected (129 ± 6 % of the control, n = 7; see Fig. 3). In addition, the activation of ICl(cAMP) was nearly identical under all conditions (0.39 ± 0.05 pA pF−1 at 5 mM (n = 19), 0.34 ± 0.08 pA pF−1 at 50 mM (n = 4), 0.29 ± 0.06 pA pF−1 at 50 mM /15 nM (n = 9), and 0.29 ± 0.04 pA pF−1 at 50 mM /90 mM (n = 5); all P values > 0.05).
Next, we investigated whether forskolin-dependent stimulation of Ip persisted in the absence of extracellular Na+ using choline as a Na+ substitute. is known to cause a voltage-dependent inhibition of steady-state Na+-K+ pump current (Nakao & Gadsby, 1989) due to voltage-dependent rebinding to the pump molecule in a putative external access channel in which part of the electrical field across the membrane drops (Gadsby et al. 1993). This inhibitory effect of is abolished in -free solution. Figure 2 illustrates an experiment conducted under these conditions. Vh was -20 mV, as before. Switching to a superfusate containing 1 mM DHO shifted the holding current in the inward direction by 80 pA under control conditions (Ip(control)), as shown in the left-hand part of the figure. During wash-out of the cardiac glycoside (not shown) membrane current slowly returned to its initial value. The cell was then challenged with 4 μM forskolin. Forskolin caused an outward shift of the holding current by 35 pA. Application of DHO in the presence of the adenylyl cyclase activator revealed that Ip was increased to 96 pA (Ip(FSK)) or to 120 % of the control. Afterwards, first DHO and then forskolin were washed out (not shown). Four minutes after switching to drug-free solution, the holding current had shifted in the inward direction and Ip was only 44 pA (Ip(wash)) demonstrating that the forskolin effects on ICl(cAMP) and Ip were completely reversible. In a total of 10 ventricular myocytes studied using this protocol, forskolin elevated Ip to 116 ± 3 % of the control. It should be noted that application of DHO in the presence of forskolin was always started about 2–3 min after superfusion of the cell with forskolin-containing solution had begun, since the measurements in -containing superfusate had revealed that Ip stimulation was maximal only after this time lag (see above). The fact that the Ip amplitude after wash-out of forskolin was lower than the control value before forskolin was a consistent finding. This was observed under all experimental conditions (see also Fig. 1) and may be attributed to a rundown of pump current, as described previously in cardiac myocytes (Gadsby & Nakao, 1989; Glitsch & Tappe, 1993).
Figure 2. Forskolin reversibly stimulates Ip in -free solution.
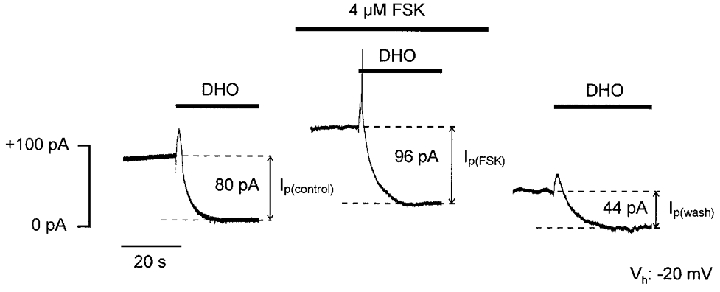
Effects of forskolin on Ip of a ventricular cell (Cm, 99 pF) in -free medium at -20 mV. [Na+]pip is 50 mM. Bars directly above the current trace indicate application of 1 mM DHO to measure Ip amplitudes visualized as distances between the dashed lines. Wash-out of the cardiac glycoside is not shown. Superfusion of the myocyte with forskolin-containing solution induces an outward shift of the holding current and increases Ip. Both drug effects are reversible. Outward current spikes in response to DHO application are artifactual. Gaps between the current traces represent omissions of 3.5 min (left) and 4 min (right) of recording.
Figure 3 summarizes the effects of forskolin on Ip in -free or -containing superfusate and at various concentrations of pipette Na+, K+, and Ca2+. In -containing solution, Ip was increased by forskolin to ∼130-140 % of the control, regardless of the composition of the pipette solution. Since the magnitude of Ip stimulation by the drug was almost identical at low and high [Na+]pip, binding of intracellular Na+ to the pump seems to be unaltered. Hence, an increase in the apparent affinity of the Na+-K+ pump to intracellular Na+ can be ruled out as the mechanism underlying the stimulation of Ip by the cAMP-PKA pathway. In addition, intracellular K+ in the range 0–90 mM and intracellular free Ca2+ in the nanomolar to subnanomolar range both do not affect the cAMP-PKA-dependent regulation of Ip.
In -free superfusate, Ip was still increased by forskolin. The increase, however, was less pronounced than in -containing solution. Whereas in the presence of extracellular Na+ the Ip amplitude was augmented by forskolin to ∼130-140 % of the control, this effect was only about one-half as large (116 %) in the absence of extracellular Na+.
To examine whether partial reactions in the Na+ limb of the pump cycle are involved in the stimulation of the Na+-K+ pump current by forskolin, we studied the effects of the drug on transient pump currents under conditions of electroneutral Na+-Na+ exchange (Na-TCM). Na-TCM indicate voltage-dependent alterations of the equilibrium between the two main conformations of the pump, E1 and E2. In the E1 conformation Na+-binding sites face the cytosol, whereas in the E2 conformation they are orientated towards the extracellular space. Figure 4A illustrates original current traces of Na-TCM of a ventricular myocyte. Vh was 0 mV. Na-TCM were obtained in response to a voltage step (indicated by the vertical arrows) to +60 mV (upper traces) or -80 mV (lower traces) 5 min (left), 10 min (middle) and 15 min (right) after access to the cell interior was achieved. At -80 mV (generally at negative potentials) transient inward currents were observed indicating that the E1-E2 equilibrium was shifted in favour of E1 (see Nakao & Gadsby, 1986). The opposite was true for the voltage jump to +60 mV (generally to positive voltages). The currents declined monoexponentially with voltage-dependent time constants as shown above or below the traces. Current decay occurred much faster at -80 than at +60 mV. In addition, prolonged cell dialysis resulted in an increase in the time constants at both potentials. The respective k-V relationships are shown in Fig. 4B (^, 5 min; □, 10 min; ▵, 15 min). Rate constants increased at negative potentials and were almost voltage independent at positive voltages. Furthermore, after 10 or 15 min k values were decreased at all potentials investigated. The relative decrease in the rate constants, however, was more pronounced at positive potentials. The Q-V relationships are displayed in Fig. 4C (symbols as in Fig. 4B). Five minutes after establishment of the whole-cell configuration, the midpoint voltage, which is a measure of the voltage-dependent equilibrium between the E1 and the E2 conformation, was -43.1 mV. It was shifted to the more positive values of -9.2 and -7.9 mV after 10 and 15 min, respectively, indicating that the E1-E2 transition was decelerated. Simultaneously, ΔQmax was reduced by ∼25 % from 39.0 fC pF−1 after 5 min to 30.5 fC pF−1 after 15 min. On average, V0.5 and ΔQmax, respectively, after 5 min were -42.6 ± 2.7 mV and 44.4 ± 2.4 fC pF−1 and after 10 min -24.8 ± 4.4 mV and 37.2 ± 2.8 fC pF−1 (n = 8). Whereas the drop in V0.5 was statistically significant (P < 0.01) the difference in ΔQmax was not (P = 0.07). Assuming a specific membrane capacitance of 1 μF cm−2 and translocation of one elementary charge per pump molecule during Na-TCM, a ΔQmax of 35–45 fC pF−1 translates into 2200–2800 Na+-K+ pumps per square micrometre. This number is close to previous estimates for guinea-pig (1200-3200 μm−2, Nakao & Gadsby, 1986; Collins et al. 1992) or rat ventricular myocytes (2600 μm−2, Dobretsov et al. 1998).
Figure 4. Time course of Na-TCM under control conditions.
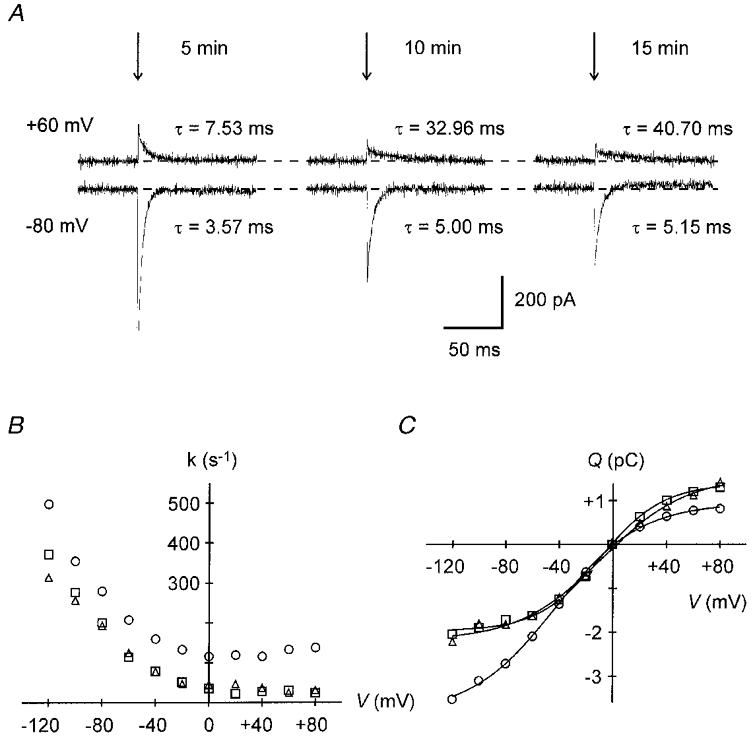
A, pump current transients elicited by voltage steps (arrows) to +60 mV (upper traces) or -80 mV (lower traces) from 0 mV holding potential. The pairs of current traces were recorded 5 min (left), 10 min (middle) and 15 min (right) after accessing the cell interior. The time constant, τ, of the monoexponential current decline is indicated near the respective current trace. Note the slowing of current decay with time. B, variation of the Na-TCM rate constants with time. Rate constants, k = 1/τ, of current decline are plotted versus membrane potential, 5 (^), 10 (□) and 15 min (▵) after establishing the whole-cell configuration. The decrease in k with time is more distinct at positive potentials. C, charge (Q) moved during Na-TCM as a function of membrane voltage and time of cell dialysis. Symbols as in B. Note the positive shift of the midpoint potential with time. Data in B and C are from the experiment partially shown in A (Cm, 123 pF).
Figure 5 shows the effect of 2 μM forskolin on Na-TCM. In this experiment, Vh was -40 mV. Original current traces of Na-TCM during voltage steps to +20 mV (upper traces) and -100 mV (lower traces) are illustrated in Fig. 5A. First, Na-TCM were recorded under control conditions (left). Then, the myocyte was challenged with forskolin. Na-TCM in the presence of the drug were obtained 10 min after access to the cell interior was achieved (middle). Finally, Na-TCM were recorded after wash-out of forskolin (right). In the presence of the adenylyl cyclase activator the time constants were hardly changed compared with the control before application of forskolin. This can also be seen in the respective k-V relationships in Fig. 5B. Rate constants in the presence of forskolin (▪) were similar to the values obtained before application of the drug (^). Hence, compared with control conditions (Fig. 4B) the k values 10 min after establishment of the whole-cell configuration were increased by the drug. At the same time, the Q-V relationship (Fig. 5C) was shifted to the left by forskolin (▪), i.e. in favour of E2, from a V0.5 of -52.1 mV to a V0.5 of -59.4 mV. The drug effect was reversible as is evident both from the slowing of the rate constants (▵, Fig. 5B) and the positive shift of the Q-V curve (V0.5: -21.1 mV, ▵, Fig. 5C) after wash-out.
Figure 5. Effect of forskolin on the time course of Na-TCM.
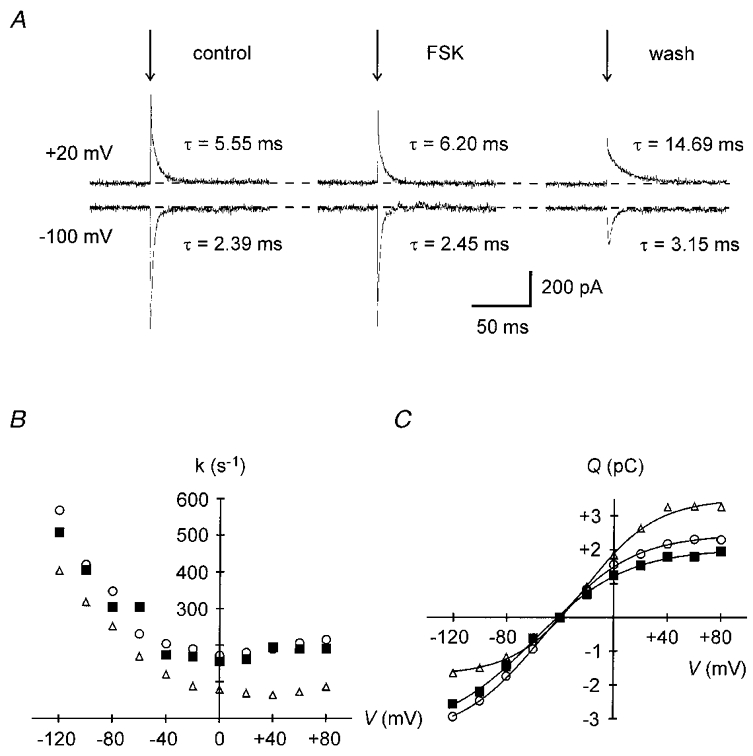
A, transient pump currents elicited by voltage jumps (arrows) to +20 mV (upper traces) or -100 mV (lower traces) from a Vh of -40 mV before (left, 5 min after establishing the whole-cell configuration), during (middle, 10 min), and after (right, 15 min) application of 2 μM forskolin. The time constant τ is indicated near the respective current trace. Forskolin inhibits the slowing of current decline with time. B, effect of forskolin on the k-V relationship. ^, control; ▪, 2 μM forskolin; ▵, after wash-out. The progressive decrease in k with time is prevented by forskolin. C, effect of forskolin on the Q-V relationship. Symbols as in B. The drug induces a negative shift of the midpoint voltage. Data in B and C are from the experiment partially shown in A (Cm, 116 pF).
The forskolin effect on Na-TCM was due to activation of the cAMP-PKA pathway, as illustrated in Fig. 6. Midpoint voltages are displayed as a function of time under various experimental conditions. Under control conditions (Fig. 6A), V0.5 became progressively more positive with the duration of cell dialysis: -42.2 mV after 5 min and -10.0 mV after 15 min. By contrast, application of 2 μM forskolin (Fig. 6B) shifted V0.5 from -52.1 to -59.4 mV. After wash-out of the drug the midpoint voltage was much more positive amounting to -21.1 mV (values taken from Fig. 5). When 10 μM PKI was present in the pipette solution to inhibit PKA, forskolin had no effect (Fig. 6C). Under these conditions, a marked positive shift of V0.5 from -63.7 to -30.5 mV was observed despite superfusion of the cell with a medium containing 2 μM forskolin. Finally, application of 500 μM 8-bromo-cAMP mimicked the forskolin effect (Fig. 6D). The cAMP analogue caused a negative shift of V0.5 from -41.6 mV under control conditions to -52.0 and -46.6 mV after 10 and 15 min, respectively. Mean values for V0.5 obtained after 10 min of cell dialysis under various conditions are shown in Fig. 6E. In the absence of any drugs V0.5 was -24.8 ± 4.4 mV (n = 8). In the presence of forskolin (2-10 μM), the midpoint voltage was -53.7 ± 2.3 mV (n = 11, P < 0.01 compared with the control). The significant negative shift of V0.5 induced by the adenylyl cyclase activator was counteracted by the inclusion of PKI in the pipette solution. Under these conditions, V0.5 reached -37.7 ± 4.4 mV (n = 5, P < 0.01 compared with forskolin). Mean values of ΔQmax for the same batches of cells were 37.2 ± 2.8 fC pF−1 under control conditions, 35.8 ± 2.4 fC pF−1 in the presence of forskolin and 34.8 ± 3.9 fC pF−1 in the additional presence of PKI (not shown, P values > 0.05). Thus, forskolin did not change the number of Na+-K+ pumps involved in Na+-Na+ exchange. Figures 5C and 6B demonstrate that forskolin shifted the V0.5 of the Q-V curve to potentials more negative than the initial control values. Thus, the drug facilitates the E1-E2 transition in the Na+ limb of the pump cycle by a mechanism which must be different from a simple prevention of rundown of V0.5 with time. However, the results presented in Fig. 5B may be interpreted to mean that forskolin inhibits the rundown of pump activity. To address this point, the time-dependent changes in V0.5 were compared in drug-free and drug-containing media. In this series of experiments, forskolin was applied 2 min after access to the cell interior was achieved to facilitate comparison of the V0.5 values obtained during the whole course of the measurements. Furthermore, to increase PKA-dependent phosphorylation some experiments were conducted in the additional presence of 10 μM microcystin in the pipette solution. Microcystin is a specific inhibitor of protein phosphatases 1 and 2A (MacKintosh et al. 1990). Thus, if forskolin affected the V0.5 rundown via activation of PKA, this effect would be expected to increase in the additional presence of microcystin. Figure 7A–C shows plots of V0.5versus time. Each circle represents a V0.5 measurement. In order to quantify the rundown of V0.5, regression lines were arbitrarily fitted to the data. Irrespective of the conditions chosen, the midpoint voltage monotonously increased towards more positive potentials. The slopes of the fitted lines were almost identical. Extrapolation to zero time yields midpoint voltages of -45 mV under control conditions (Fig. 7A) and of approximately -65 mV in media containing forskolin (Fig. 7B and C). After 5 min of whole-cell recording the mean value of V0.5 was -42.6 ± 2.7 mV (n = 8) under control conditions. The corresponding values from forskolin-treated myocytes were calculated to be -60.4 ± 4.5 mV (forskolin, n = 9) and -60.5 ± 3.5 mV (forskolin + microcystin, n = 6), both being significantly more negative than the control value (P < 0.05). Therefore, forskolin facilitates the E1-E2 transition by a mechanism different from that responsible for the rundown of V0.5.
Figure 6. Forskolin induces a negative shift of the midpoint voltage, V0.5, which is mediated by the cAMP-PKA pathway.
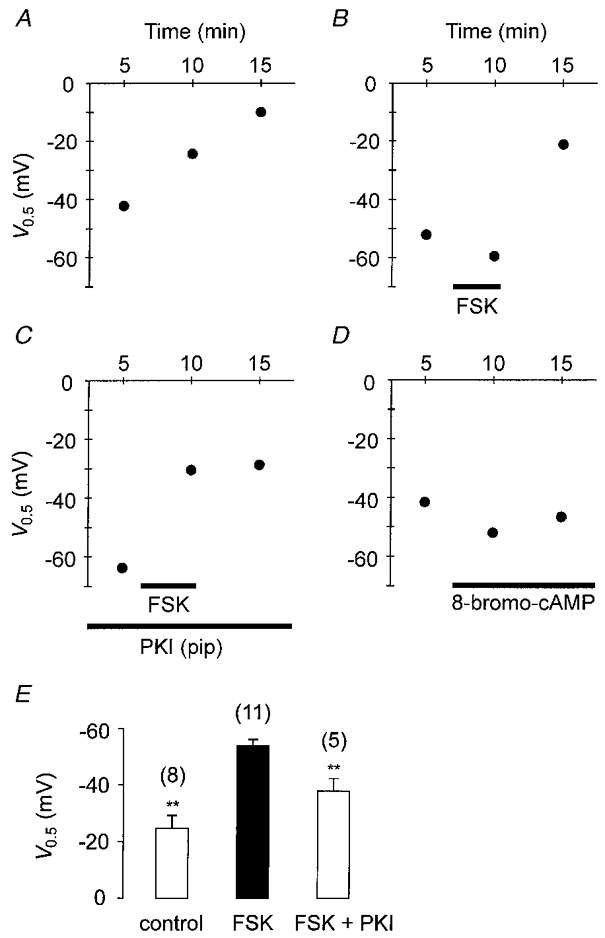
Variations of V0.5 as a function of time. A-D illustrate data from four ventricular myocytes. Under control conditions (A) V0.5 declines continuously with time. Forskolin (2 μM, B) induces a negative shift of V0.5, an effect that is mimicked by 8-bromo-cAMP (500 μM, D) and prevented by inclusion of PKI in the pipette solution (10 μM, C). E, mean values of V0.5 as affected by forskolin (2-10 μM) and PKI (10 μM). Data were obtained 10 min after establishing the whole-cell configuration. V0.5 of forskolin-treated myocytes (middle) is significantly more negative than V0.5 of control (left, **P < 0.01) or of cells containing PKI (right, **P < 0.01). Numbers in parentheses denote the number of cells studied.
Figure 7. Time-dependent shift of V0.5 in the absence and presence of forskolin.
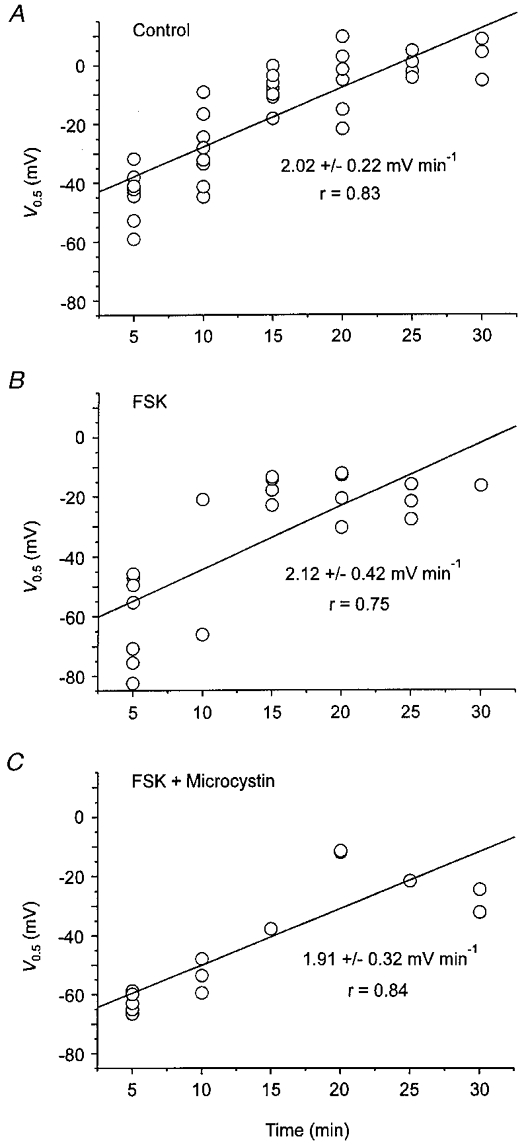
A, variation of V0.5 with time in myocytes superfused with drug-free solution (Control). B, time course of V0.5 in forskolin-treated cells. C, changes in V0.5 with time in myocytes treated with forskolin plus microcystin (10 μM). In B and C, forskolin (4 μM) was applied to the cells 2 min after establishing the whole-cell configuration. Each circle in A-C represents a single measurement of V0.5.
Apart from accelerating the E1-E2 transition of the pump cycle forskolin activated a membrane conductance that was identified as the cAMP-dependent chloride conductance, as shown in Fig. 8 (see also Figs 1 and 2). Figure 8A displays original membrane currents in response to voltage jumps to potentials between +80 and -120 mV. Vh was -40 mV (same recording as in Fig. 5). Currents obtained in the absence and presence of 200 μM DHO are shown superimposed. They differed immediately after the voltage step (most easily seen in the middle panel) but were identical in the steady state, as expected for Na-TCM. It is clear that application of forskolin reversibly increased the membrane conductance. Steady-state membrane currents measured at the end of each voltage jump before (^), during (▪) and after (▵) application of forskolin are plotted versus membrane potential in Fig. 8B. The I–V relationship of the forskolin-activated membrane conductance is illustrated in Fig. 8C. The values shown represent the difference between the currents measured in the presence and in the absence of forskolin (from Fig. 8B). The drug-induced current showed slight outward rectification and reversed its sign at approximately -7 mV. Similarly, the mean I–V curve obtained from seven cells under these conditions (• in Fig. 8D) exhibited slight outward rectification and a reversal potential of -5 mV, which was close to the calculated ECl of -4 mV. When 10 μM PKI was included in the pipette solution (^ in Fig. 8D, n = 5) almost no current was activated by forskolin implying that it was dependent on PKA activity. Finally, when ECl was set to -40 mV (▪ in Fig. 8D, 36 mM , n = 4) the forskolin-induced current reversed at -37 mV and displayed even more pronounced outward rectification than at near symmetrical chloride concentrations. These properties are characteristic for the cAMP-dependent chloride current known to be present in these cells.
DISCUSSION
The main findings of the present study are that (i) the Na+-K+ pump current of guinea-pig ventricular myocytes is increased by forskolin via activation of the cAMP-PKA pathway and (ii) this increase is mediated, at least in part, by a modulation of one or more partial reactions in the Na+ limb of the pump cycle.
The first conclusion is based on the observation that forskolin, which is known to raise intracellular cAMP levels by activation of adenylyl cyclases, reversibly increased Ip under various experimental conditions (Fig. 3). In contrast, 1,9-dideoxyforskolin, which does not activate adenylyl cyclases, was unable to mimic the forskolin effect. Furthermore, a cAMP analogue produced forskolin-like effects on Na-TCM (Fig. 6D) and two inhibitors of PKA, H-89 and PKI, abolished the forskolin action on Ip or Na-TCM, respectively. The results, therefore, demonstrate that a cAMP-dependent activation of PKA was responsible for the observed effects of forskolin on both steady-state and transient pump currents in these cells.
The forskolin-induced increase in Ip was ∼130-140 % of the control in Na+-containing extracellular solution, irrespective of the composition of the pipette solutions used. Similar increases in Ip upon activation of β-adrenergic receptors by catecholamines were described in ventricular myocytes (140-145 %, Dobretsov et al. 1998) and skeletal myoballs (150 %, Li & Sperelakis, 1994) of the rat. Also, Gao et al. (1994) found that at micromolar concentrations of Na+-K+ pump current in guinea-pig ventricular myocytes was augmented by cAMP-increasing drugs to about 125 % of the control. Thus, a general picture seems to emerge that activation of the cAMP-PKA cascade in striated muscle, at least in rat and guinea-pig, results in an increased Ip. However, the molecular mechanism underlying this increased pump current may be different between the two species, as discussed below.
A major difference between the results presented above and those of Gao and co-workers is the [Ca2+]pip dependence of the Ip regulation by the cAMP-PKA pathway. Whereas our measurements clearly showed that Ip was stimulated in a cAMP-dependent manner at nanomolar and subnanomolar concentrations of (Fig. 3), Gao et al. (1992, 1996, 1998) reported that at concentrations < 150 nM Ip was decreased in the very same cell type under otherwise comparable experimental conditions. Only at concentrations > 150 nM did they observe an increased Ip upon isoprenaline application (Gao et al. 1992, 1994, 1996). The reason for this discrepancy remains unknown at present and clearly needs to be investigated further. It should be noted, however, that the catecholamine-induced stimulation of Ip in rat ventricular myocytes also occurred at nanomolar [Ca2+]pip (Dobretsov et al. 1998) and that an elevation of [Ca2+]pip up to 1 μM likewise resulted in an increased Ip amplitude in response to isoprenaline (Stimers & Dobretsov, 1998).
Besides increasing Ip, forskolin also activated the cAMP-dependent chloride current (ICl(cAMP)), which was identified by its dependence on activation of the cAMP-PKA pathway and its I–V characteristics (Fig. 8). ICl(cAMP) served as a positive control for the effects on Ip of agents intervening with the cAMP signalling cascade. Thus, when the forskolin action on Ip was blocked by PKA inhibitors, activation of ICl(cAMP) was also abolished. In our measurements of steady-state pump current (e.g. Fig. 1), activation of ICl(cAMP) due to forskolin application was visible as an outward shift of the holding current. This outward current developed about 15–25 s after starting superfusion of the myocyte with forskolin and was complete within another minute, very similar to the time course previously reported by Pelzer and co-workers (Pelzer et al. 1997). Interestingly, stimulation of Ip only occurred once this activation of ICl(cAMP) had commenced. In most cases, Ip amplitude began to rise after a time lag of about 50–120 s and was maximal roughly 2–5 min after application of forskolin was begun. The reason for these different time courses remains unknown, but it can be speculated that one or more components of the cAMP-PKA pathway may have preferential access to the chloride channels. Further studies to clarify this issue are underway.
In principle, several (sufficiently slow) partial reactions in the Na+-K+ pump cycle may be affected by forskolin to cause the observed increase in Ip. According to Dobretsov et al. (1998), the relative increase in Ip in rat ventricular myocytes depends on [K+]pip. At zero , isoprenaline augmented Ip amplitude to ∼120 % of the control while at 100 mM Ip was elevated to ∼160 % of the control. The data were fitted with a cyclic model of the Na+-K+ pump and it was concluded that enhanced intracellular deocclusion/ release of K+ was responsible for the observed stimulation of the pump current. A similar mechanism, however, does not seem to be operative in guinea-pig ventricular myocytes, since our measurements showed that the forskolin-induced increase in Ip amplitude was almost identical at 0 and 90 mM (Fig. 3). In addition, although not studied in detail, an increased apparent affinity of the pump towards extracellular K+ can probably be excluded as the reason for the augmented Ip, since saturating concentrations of this external activator cation of the pump were used in all our experiments. This conclusion is further strengthened by the observation of Gao et al. (1992) that an increase in [K+]o from 4.6 to 20 mM did not affect the β-adrenergic stimulation of Ip in the same cell type. Hence, neither intra- nor extracellular K+ binding/release seem to be involved in the forskolin-induced increase in Ip. We cannot, however, exclude the possibility that other partial reactions in the K+ limb of the pump cycle are modified by the cAMP-PKA cascade.
To isolate partial reactions in the Na+ limb of the pump cycle transient pump currents under conditions of electroneutral Na+-Na+ exchange were recorded. Under these conditions, and the E2 conformation of the Na+-K+ pump exists. Stepping a voltage-dependent equilibrium between the E1 membrane voltage to positive potentials shifts the equilibrium in favour of E2 while clamping the membrane voltage to negative potentials shifts it towards E1. Five minutes after establishment of the whole-cell configuration a midpoint voltage, V0.5, of approximately -45 mV was obtained, which is in good agreement with previous measurements on rat (-25 to -30 mV, Dobretsov et al. 1998) or guinea-pig ventricular myocytes (-20 mV, Nakao & Gadsby, 1986) or giant membrane patches thereof (-45 mV, Hilgemann, 1994; -37 to -52 mV, Friedrich & Nagel, 1997). During sustained cell dialysis this value became more positive and was -25 mV after 10 min. This rightward shift of the Q-V curve indicates that the E1-E2 transition was impaired. The phenomenon is reminiscent of the rundown of Ip as described in this and other studies on cardiac myocytes (Gadsby & Nakao, 1989; Glitsch & Tappe, 1993). By contrast, forskolin caused a significant leftward shift of the Q-V curve. In its presence, V0.5 was significantly more negative than the initial control value (Figs 6B and 7) indicating that the E1-E2 transition was facilitated by a mechanism different from prevention of V0.5 rundown, which was not affected by forskolin (Fig. 7). In line with this finding rate constants under forskolin were larger than the respective controls at the same time (compare Figs 4B and 5B). The fact that the rate constants of transient pump current decline in the presence of forskolin did not exceed the initial values in its absence (Fig. 5B) is not inconsistent with this conclusion. One has to take into account that the measured rate constants k are the sum of the rate constants of the forward and backward reaction of the E1-E2 transition, k1-2 and k2-1. It is easily possible that forskolin increases k1-2 and simultaneously decreases k2-1 without large changes in k. The effects of forskolin on Na-TCM thus demonstrate that activation of the cAMP-PKA pathway facilitates the E1-E2 transition in the Na+ limb of the pump cycle, a result that may well explain the observed increase in Ip.
A further important conclusion can be drawn from these measurements. Since forskolin did not change the number of pump molecules involved in Na+-Na+ exchange, as judged from the identical ΔQmax values in its presence or absence, respectively, the recruitment of additional Na+-K+ pumps to the plasma membrane can be excluded as the underlying cause for the augmented Ip. Such a mechanism has recently been shown to be responsible for the cAMP-dependent increased cation transport and hydrolytic activity of the Na+-K+-ATPase in rat kidney (Carranza et al. 1998).
From the above reasoning it is clear that a modulation of partial reactions in the Na+ limb of the pump cycle is probably at least one cause for the increase in Ip observed upon activation of the cAMP-PKA cascade in guinea-pig ventricular myocytes. The question, then, concerning which partial reaction(s) is (are) involved remains. Although from our results no definite answer can be given to that question, some limitations may apply. Firstly, an increase in the apparent affinity of the Na+-K+ pump for intracellular Na+ cannot account for the augmented Ip, since the quantitative rise in pump current at both low (5 mM) and high (50 mM) was almost identical. The same conclusion was drawn previously by Gao et al. (1992). Secondly, extracellular Na+ deocclusion/release may be altered and could be at least one mechanism for the increased Ip. This hypothesis is based on the finding that forskolin stimulated Ip significantly less in the absence of extracellular Na+. One possible mechanism involved may be, for example, that the voltage-dependent rebinding of Na+ in the putative external access channel, which is absent in -free solution, is reduced by forskolin. Consequently, Ip would be increased by the drug in Na+-containing solution. This interpretation must, however, be considered with caution for the following two reasons. (i) Ip -free solution was measured as DHO-sensitive current. Wash-out of DHO occurred rather slowly (∼1.5-3 min) so that there was a considerable delay between the estimation of Ip before and following forskolin application. During this time rundown of Ip may have proceeded and Ip amplitude in the presence of the adenylyl cyclase activator may have been underestimated. (ii) Because of this time-consuming wash-out of DHO only one determination of Ip magnitude during forskolin treatment of a cell was possible. This estimation occurred 2–3 min after forskolin application had started. However, maximal Ip in the presence of the drug was reached at quite variable times, usually 2–5 min from the beginning of application. Thus, maximal stimulation may not have been reached at that time resulting in an (additional) underestimate of Ip. Taking this into account, the only firm conclusion from our experiments is that (a) partial reaction(s) in the Na+ limb is (are) modified by the cAMP-PKA pathway leading to a facilitated E1-E2 transition.
Sympathetic stimulation of the heart leads, via activation of β-adrenoceptors, to increased action potential frequencies and passive Na+ and K+ fluxes. In order to maintain the Na+ and K+ gradients across the sarcolemma, which are essential for cardiac excitation, active Na+-K+ transport would also have to be increased under these conditions. The stimulation of the Na+-K+ pump by the cAMP-PKA pathway, as documented in this study, may be one means by which large changes in intracellular Na+ and K+ are prevented.
Acknowledgments
We thank Kirsty Brebner for critically reading the manuscript. The technical help of A. Balzer-Ferrai and U. Müller is gratefully acknowledged. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG, Gl 72/7-3).
References
- Bahinski A, Nairn AC, Greengard P, Gadsby DC. Chloride conductance regulated by cyclic AMP-dependent protein kinase in cardiac myocytes. Nature. 1989;340:718–721. doi: 10.1038/340718a0. [DOI] [PubMed] [Google Scholar]
- Bielen FV, Glitsch HG, Verdonck F. Na+ pump current-voltage relationships of rabbit cardiac Purkinje cells in Na+-free solution. The Journal of Physiology. 1993;465:699–714. doi: 10.1113/jphysiol.1993.sp019701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carranza ML, Rousselot M, Chibalin AV, Bertorello AM, Favre H, Féraille E. Protein kinase A induces recruitment of active Na+,K+-ATPase units to the plasma membrane of rat proximal convoluted tubule cells. The Journal of Physiology. 1998;511:235–243. doi: 10.1111/j.1469-7793.1998.235bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins A, Somlyo AV, Hilgemann DW. The giant cardiac membrane patch method: stimulation of outward Na+-Ca2+ exchange current by MgATP. The Journal of Physiology. 1992;454:27–57. doi: 10.1113/jphysiol.1992.sp019253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desilets M, Baumgarten CM. Isoproterenol directly stimulates the Na+-K+ pump in isolated cardiac myocytes. American Journal of Physiology. 1986;251:H218–225. doi: 10.1152/ajpheart.1986.251.1.H218. [DOI] [PubMed] [Google Scholar]
- Dobretsov M, Hastings SL, Stimers JR. Na+-K+ pump cycle during β-adrenergic stimulation of adult rat cardiac myocytes. The Journal of Physiology. 1998;507:527–539. doi: 10.1111/j.1469-7793.1998.527bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. Journal de Physiologie. 1979;75:463–505. [PubMed] [Google Scholar]
- Friedrich T, Nagel G. Comparison of Na+/K+-ATPase pump currents activated by ATP concentration or voltage jumps. Biophysical Journal. 1997;73:186–194. doi: 10.1016/S0006-3495(97)78059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Nakao M. Steady-state current-voltage relationship of the Na/K pump in guinea pig ventricular myocytes. Journal of General Physiology. 1989;94:511–537. doi: 10.1085/jgp.94.3.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Rakowski RF, De Weer P. Extracellular access to the Na,K pump: pathway similar to ion channel. Science. 1993;260:100–103. doi: 10.1126/science.7682009. [DOI] [PubMed] [Google Scholar]
- Gao J, Cohen IS, Mathias RT, Baldo GJ. Regulation of the β-stimulation of the Na+-K+ pump current in guinea-pig ventricular myocytes by a cAMP-dependent PKA pathway. The Journal of Physiology. 1994;477:373–380. doi: 10.1113/jphysiol.1994.sp020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Cohen IS, Mathias RT, Baldo GJ. The inhibitory effect of β-stimulation on the Na/K pump current in guinea pig ventricular myocytes is mediated by a cAMP-dependent PKA pathway. Pflügers Archiv. 1998;435:479–484. doi: 10.1007/s004240050542. [DOI] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Baldo GJ. Isoprenaline, Ca2+ and the Na+-K+ pump in guinea-pig ventricular myocytes. The Journal of Physiology. 1992;449:689–704. doi: 10.1113/jphysiol.1992.sp019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Shi J, Baldo GJ. The effects of β-stimulation on the Na+-K+ pump current-voltage relationship in guinea-pig ventricular myocytes. The Journal of Physiology. 1996;494:697–708. doi: 10.1113/jphysiol.1996.sp021525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch HG, Krahn T, Pusch H, Suleymanian M. Effect of isoprenaline on active Na transport in sheep cardiac Purkinje fibres. Pflügers Archiv. 1989;415:88–94. doi: 10.1007/BF00373145. [DOI] [PubMed] [Google Scholar]
- Glitsch HG, Tappe A. The Na+/K+ pump of cardiac Purkinje cells is preferentially fuelled by glycolytic ATP production. Pflügers Archiv. 1993;422:380–385. doi: 10.1007/BF00374294. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Harvey RD, Hume JR. Autonomic regulation of a chloride current in heart. Science. 1989;244:983–985. doi: 10.1126/science.2543073. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW. Channel-like function of the Na,K pump probed at microsecond resolution in giant membrane patches. Science. 1994;263:1429–1432. doi: 10.1126/science.8128223. [DOI] [PubMed] [Google Scholar]
- Huang X-Y, Morielli AD, Peralta EG. Molecular basis of cardiac potassium channel stimulation by protein kinase A. Proceedings of the National Academy of Sciences of the USA. 1994;91:624–628. doi: 10.1073/pnas.91.2.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka N, Berlin JR. β-Adrenergic stimulation does not regulate Na pump function in voltage-clamped ventricular myocytes of the rat heart. Pflügers Archiv. 1993;424:361–363. doi: 10.1007/BF00384364. [DOI] [PubMed] [Google Scholar]
- Kockskämper J, Erlenkamp S, Glitsch HG. Forskolin modulates partial reactions in the Na+ limb of the Na+/K+ pump cycle. Pflügers Archiv. 1999a;437:R60. [Google Scholar]
- Kockskämper J, Gisselmann G, Glitsch HG. Comparison of ouabain-sensitive and -insensitive Na/K pumps in HEK293 cells. Biochimica et Biophysica Acta. 1997;1325:197–208. doi: 10.1016/s0005-2736(96)00259-3. [DOI] [PubMed] [Google Scholar]
- Kockskämper J, Glitsch HG, Pusch H. Effects of forskolin on Na+-dependent transient charge movements by the Na+/K+ pump in single guinea-pig ventricular myocytes. Biophysical Journal. 1999b;76:A452. [Google Scholar]
- Läuger P. Electrogenic Ion Pumps. Sunderland, MA, USA: Sinauer Associates, Inc.; 1991. [Google Scholar]
- Li K-X, Sperelakis N. Electrogenic Na-K pump current in rat skeletal myoballs. Journal of Cellular Physiology. 1994;159:181–186. doi: 10.1002/jcp.1041590122. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- MacKintosh C, Beattie KA, Klumpp S, Cohen P, Codd GA. Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Letters. 1990;264:187–192. doi: 10.1016/0014-5793(90)80245-e. [DOI] [PubMed] [Google Scholar]
- Nakao M, Gadsby DC. Voltage dependence of Na translocation by the Na/K pump. Nature. 1986;323:628–630. doi: 10.1038/323628a0. [DOI] [PubMed] [Google Scholar]
- Nakao M, Gadsby DC. [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea pig ventricular myocytes. Journal of General Physiology. 1989;94:539–565. doi: 10.1085/jgp.94.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelzer S, You Y, Shuba YM, Pelzer DJ. β-Adrenoceptor-coupled Gs protein facilitates the activation of cAMP-dependent cardiac Cl− current. American Journal of Physiology. 1997;273:H2539–2548. doi: 10.1152/ajpheart.1997.273.6.H2539. [DOI] [PubMed] [Google Scholar]
- Seamon KB, Daly JW. High-affinity binding of forskolin to rat brain membranes. Advances in Cyclic Nucleotide and Protein Phosphorylation Research. 1985;19:125–135. [PubMed] [Google Scholar]
- Stimers JR, Dobretsov M. Adrenergic stimulation of Na/K pump current in adult rat cardiac myocytes in short-term culture. Journal of Membrane Biology. 1998;163:205–216. doi: 10.1007/s002329900384. [DOI] [PubMed] [Google Scholar]
- Traebert M, Trebeß I, Erlenkamp S, Hüser J, Kockskämper J, Glitsch HG, Hartung C, Welzel P. High affinity regulation of cardiac Cl− and Ca2+ conductances by (13R)-spiroforskolin. Naunyn-Schmiedeberg's Archives of Pharmacology. 1998;358:538–546. doi: 10.1007/pl00005290. [DOI] [PubMed] [Google Scholar]