

Abstract
To provide insight into the dynamics and source of residual viremia in human immunodeficiency virus (HIV) patients successfully treated with antiretroviral therapy, 14 intensely monitored patients treated with indinavir and efavirenz sustaining HIV RNA at <50 copies/ml for >5 years were studied. Abacavir was added to the regimen of eight patients at year 5. After the first 9 months of therapy, HIV RNA levels had reached a plateau (“residual viremia”) that persisted for over 5 years. Levels of residual viremia differed among patients and ranged from 3.2 to 23 HIV RNA copies/ml. Baseline HIV DNA was the only significant pretreatment predictor of residual viremia in regression models including baseline HIV RNA, CD4 count, and patient age. In the four of five patients with detectable viremia who added abacavir to their regimen after 5 years, HIV RNA levels declined rapidly. The estimated half-life of infected cells was 6.7 days. Decrease in activated memory cells and a reduction in gamma interferon production to HIV Gag and p24 antigen in ELISpot assays were observed, consistent with a decrease in HIV replication. Thus, in patients treated with efavirenz plus indinavir, levels of residual viremia were established by 9 months, were predicted by baseline proviral DNA, and remained constant for 5 years. Even after years of highly suppressive therapy, HIV RNA levels declined rapidly after the addition of abacavir, suggesting that productive infection contributes to residual ongoing viremia and can be inhibited with therapy intensification.
Treatment with potent antiretroviral therapy leads to rapid and sustained reductions in human immunodeficiency virus (HIV) replication but does not eradicate the virus from an infected patient (23, 27, 46). Long-lived, latently infected resting CD4+ lymphocytes persist in treated patients, ensuring an ongoing source of viral particles (9, 18, 47). Other cellular and anatomic reservoirs can also contribute to the persistent viral pool, with quasispecies exhibiting distinct evolutionary pathways (8, 39, 50). Contemporary therapeutic antiretroviral regimens reduce HIV RNA levels below 50 copies/ml, the limit of detection of commercially available assays (43). However, in most patients studied to date, HIV RNA can still be detected in the plasma by more-sensitive assays in patients even after years of therapy (15, 45).
The dynamics and source of residual viremia in patients treated with antiretroviral therapy are not well characterized. The viral particles present in plasma may be a result of ongoing productive infection, release of viral particles from latently infected cells, or both. Indirect evidence for ongoing productive infection includes the presence of HIV RNA in cells (20, 32, 35), viral evolution (24), and rapid rebound of virus after therapy interruption (13, 14, 49). Conversely, the presence of drug-sensitive, archival virus in patients responding to potent antiretroviral regimens suggests that productive infection contributes little to residual viremia (26). Some patients who achieve sustained reductions in HIV RNA below 50 copies/ml exhibit intermittent viremic episodes or “blips” above 50 copies/ml; these patients retain more viremia between blips than do patients without intermittent viremia (25). In these same patients, intensification of therapy results in reduction in frequency of blips and accelerates the decay of the latent cellular reservoir, suggesting that residual replication in these patients replenishes latent reservoirs (B. Ramratnam, R. Ribeiro, T. He, P. Cauldwell, N. Ruiz, A. Hurley, L. Zhang, A. S. Perelson, D. D. Ho, and M. Markowitz, 8th Conf. Retrovir. Opportunistic Infect., abstr. 502, 2001).
We conducted a longitudinal analysis of patients with sustained suppression of HIV RNA below 50 copies/ml of plasma for 5 years. By utilizing an assay achieving sensitivity of HIV RNA to 2.5 copies/ml, we were able to determine if HIV RNA levels were stable or decreasing over a period of 5 years. We found that, by 9 months after initiation of treatment, HIV RNA levels reached a steady state in the plasma that persisted for over 5 years. By adding the reverse transcriptase inhibitor abacavir to the regimen and measuring the HIV RNA levels prospectively, we determined that productive infection is contributing to residual replication and thus that activation of latently infected cells is not the sole source of virus production.
MATERIALS AND METHODS
Study subjects.
All study subjects were HIV type 1 (HIV-1)-infected participants of a phase II dose-finding study of efavirenz (600 mg daily) and indinavir. Indinavir was administered at a dose of 800 to 1,200 mg three times daily until month 4, when all patients received 1,200 mg. All patients received indinavir plus efavirenz at the onset of the study, except for two patients who started indinavir first for 6 months and then added efavirenz. Patients 2 and 5 (Table 1) also received lamivudine and stavudine, respectively, 6 months after therapy initiation. Subject 5 discontinued study therapy for drug toxicity (nephrolithiasis) after 5 years. Therapy intensification was offered to all patients after 5 years and consisted of abacavir in all subjects except patient 3, who requested ritonavir. Patients 2, 11, and 12 stopped abacavir at week 10 and 3 and day 2, respectively, for mild side effects. Viral kinetics were not evaluated for subject 15, who had transferred from another institution.
TABLE 1.
Baseline characteristics and viral kinetics of patients receiving indinavir and efavirenz (n = 15) and HIV RNA pre- and post-therapy intensification (n = 9)a
| Patient | Baseline characteristic
|
Clearance half-life in days
|
Residual viremia
|
Intensification
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Log HIV RNA (c/ml) | HIV DNA (c/ml) | CD4 (cells/mm3) | HIV RNA, phase I | HIV RNA, phase II | HIV DNA, yr 1 | HIV RNA, blips (no.) | Set point log HIV RNA ± SD (n) | Set point log HIV RNA (c/ml) | Postintensi- fication log HIV RNA ± SD (n) | Postintensi- fication HIV RNA (c/ml) | |
| Intensifying therapy | |||||||||||
| 1 | 4.79 | 9,379 | 512 | 0.89 (0.56, 2.01) | 23.2 (14.5, 35.2) | 43.8 (129, ∞) | 2 | 1.4 ± 0.3 (12) | 23.0 | 1.0 ± 0.1 (6) | 9.7* |
| 2 | 4.84 | 2,744 | 291 | 0.83 (0.15, 1.78) | 27.9 (16.6, 32.2) | 13.4 (77, 484) | 0 | 0.8 ± 0.3 (12) | 7.8 | 0.9 ± 0.2 (5) | 7.9 |
| 3 | 5.66 | 1,029 | 174 | 1.47 (1.14, 2.06) | 21.2 (17.9, 26.3) | 11.0 (80, 177) | 0 | 0.5 ± 0.2 (13) | 3.2 | <0.4 (6) | <2.5 |
| 4 | 5.48 | 5,610 | 262 | 1.70 (1.39, 2.58) | 16.6 (12.3, 23.6) | ∞ (142, ∞) | 1 | 1.0 ± 0.4 (14) | 9.9 | <0.4 (6) | <2.5* |
| 9 | 4.50 | 1,229 | 259 | 1.16 (0.59, 2.17) | 32.6 (25.8, 42.5) | ∞ (209, ∞) | 0 | 0.8 ± 0.3 (11) | 5.9 | 0.4 ± 0.1 (6) | 2.7* |
| 10 | 4.68 | 313 | 422 | 0.89 (0.35, 2.27) | 35.6 (25.9, 49.0) | 1,416 (253, ∞) | 0 | 0.6 ± 0.3 (11) | 4.4 | 0.5 ± 0.2 (6) | 3.4 |
| 11 | 4.99 | 1,668 | 210 | 1.42 (0.91, 1.81) | 11.6 (7.6, 18.2) | ∞ (698, ∞) | 0 | 0.9 ± 0.3 (11) | 7.6 | 0.7 ± 0.3 (4) | 5.4 |
| 12 | 4.54 | 546 | 472 | 1.20 (0.61, 2.6) | 5.2 (3.8, 7.0) | 493 (117, ∞) | 0 | 0.7 ± 0.2 (11) | 5.1 | 0.4 ± 0.04 (5) | 2.7 |
| 14 | 4.54 | 466 | 294 | 1.00 (0.73, 1.91) | 13.5 (9.8, 19.2) | 212 (82, ∞) | 1 | 0.7 ± 0.3 (11) | 5.6 | <0.4 (6) | <2.5* |
| Not intensifying therapy | |||||||||||
| 6 | 4.91 | 22,000 | 81 | 0.64 (0.46, 1.10) | 72.3 (29.6, 129) | 264 (102, ∞) | 0 | NA | NA | 0.5 ± 0.2 (3) | 3.1 |
| 7 | 5.56 | 8,103 | 243 | 1.46 (1.01, 2.03) | 10.4 (7.7, 13.6) | 466 (103, ∞) | 0 | 0.8 ± 0.3 (13) | 6.1 | 0.6 ± 0.4 (3) | 5.5 |
| 8 | 5.02 | 1,957 | 349 | 1.16 (0.59, 2.17) | 30.2 (23.8, 39.5) | ∞ (478, ∞) | 1 | 0.7 ± 0.2 (12) | 4.8 | 0.7 ± 0.3 (3) | 7.9 |
| 13 | 4.56 | 2,850 | 104 | 2.03 (0.81, 3.34) | 23.4 (11.4, 45.5) | ∞ (204, ∞) | 0 | 0.6 ± 0.3 (13) | 3.9 | <0.4 (3) | <2.5 |
| 15 | 4.77 | NA | 469 | NA | NA | NA | 0 | NA | NA | 0.7 ± 0.3 (3) | 7.2 |
| 5 | 4.95 | 4,100 | 247 | NA | NA | 198 (99, ∞) | 0 | 1.1 ± 0.2 (11) | 13.1 | NA | NA |
Residual HIV RNA set point values are geometric means of all measurements (11 to 14 time points per patient) after the end of second-phase RNA clearance and before intensification. Blips are HIV RNA values of >50 copies/ml occurring after the first year of therapy. Postintensification values are geonetric means of values obtained from six samples drawn at 4-week intervals beginning 4 weeks after initiation of abacavir. Assay sensitivity prevented statistical determination of the effect of intensification in four patients; four of the five remaining patients (indicated by asterisks) experienced a significant (P < 0.05, Wilcoxon test) decrease in viral load after abacavir intensification. NA, not available. c/ml, copies per milliliter.
Assay of residual viremia.
Assays for residual viremia between 2.5 and 50 copies/ml were performed according to published methods (25). The Amplicor Ultrasensitive assay was modified to permit the nominal detection of 2.5 copies of HIV RNA (Roche Molecular Diagnostics, Branchburg, N.J.)/ml. Two milliliters (versus 0.5 ml) of frozen plasma was thawed and centrifuged at 23,600 × g for 2 h (versus 1 h) at 4°C to pellet virus. One-half the normal quantitation standard (QS) volume was added to the sample prior to the lysis step, and the RNA pellet was resuspended in 50 μl of diluent volume (versus 100 μl for the Ultrasensitive assay). The entire volume (50 μl) of resuspended RNA was assayed by reverse transcription and PCR amplification. These and subsequent detection steps followed the manufacturer's protocol exactly. Because only half the QS volume is added prior to the lysis and RNA extraction steps but the extracted RNA is resuspended in half the usual volume, normalization for the detected QS value could be performed without modification. The computed value of copies per milliliter was corrected for the protocol modifications by dividing by 8. This included a fourfold correction factor for the larger volume of plasma assayed (2 versus 0.5 ml) and a twofold correction factor for the volume of diluent used to resuspend the extracted RNA (50 versus 100 μl).
To establish the limit of detection and reproducibility of the assay, plasma from two HIV-infected patients with viral RNA concentrations previously determined by the Amplicor or Amplicor Ultrasensitive assays were serially diluted in control plasma from healthy HIV-negative donors to a calculated concentration of 50 copies/ml down to 1.25 copies/ml. Replicate samples at each dilution were assayed by the modified 2.5-copies/ml assay. The correlation of the results based on Amplicor and the modified 2.5-ml assay was excellent with an r2 of 0.92 (P = 0.003). Coefficients of variation at individual concentrations ranged from 121% at 1.25 copies/ml to 9% at 50 copies/ml. At 2.5 copies/ml, the coefficient of variation was 37%.
HIV DNA assays.
Peripheral blood mononuclear cell (PBMC) DNA quantitation was performed using a PCR-based system with colorimetric detection according to published methods (6). Based on genomic DNA input into PCRs, the lower limit for reliable quantitation was 5 HIV DNA copies/μg of PBMC DNA. Viral DNA values presented here are normalized to total blood volume by multiplying copy numbers per purified lymphocyte by simultaneous peripheral blood lymphocyte counts. In a natural history study, values computed in this normalization were more stable over the course of HIV disease, including a range of CD4 counts extending below 200 cells/mm3, than when normalized to number of PBMC or CD4+ T cells (12). However, normalization of HIV DNA copies to micrograms of PBMC DNA or CD4 DNA gave qualitatively similar results (data not shown).
Viral kinetics.
Initial decline of HIV RNA obeyed biphasic exponential decay to a nonzero constant. Plasma HIV RNA values V(t) from the first year of therapy were fitted to the form
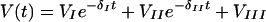 |
(1) |
assuming multiplicative Gaussian noise g(0, s) of zero mean and standard deviation σ.
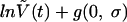 |
(2) |
by Bayesian methods. The clearance rates δI and δII are ln(2) times the reciprocal of the first-phase half-life TI and the second phase half-life TII, respectively. Simulations were performed using the Markov chain Monte Carlo technique in WinBugs. Estimates for decay half-lives computed by a least-squares fit to equation 1 were not statistically different.
The duration of phase II HIV RNA clearance was defined as the period during which more virus was produced from this pool than from longer-lived (residual) sources, based on the optimal biphasic fit of HIV RNA dynamics. HIV RNA dynamics in patient 8 differed significantly in several regards. In this patient, the measured baseline DNA was higher (22,000 copies/ml), baseline CD4 was lower (81 cells/mm3), and second-phase RNA clearance was slower. In addition, HIV DNA continued to decay significantly (P < 0.05) in this patient throughout the study period. Residual HIV RNA and DNA could therefore not be appropriately defined in this patient, and he was excluded from the analysis of predictors of residual viremia. The inclusion in our cohort of one patient who failed to reach steady states of HIV RNA or HIV DNA raises the possibility that some patients exhibit different long-term viral clearance kinetics; however, much larger cohorts would be necessary to study this intriguing minority.
Decay rates after abacavir intensification were computed by least-squares, treating undetectable values as 2.5 copies/ml. They therefore represent an underestimate of the true decay rate, except in patient 1, who retained detectable viremia. To determine whether transient elevations of HIV RNA at the time of intensification were responsible for the rapid kinetics, clearance rates were also computed, substituting the residual viremia over the previous 4 years as the baseline value.
Semiquantitative microculture assays.
Fifty milliliters of fresh acid citrate dextrose anticoagulated whole blood was processed for CD4 lymphocyte enrichment or CD8 lymphocyte depletion using the Rossette-sep procedure according to the manufacturer's directions (Stem Cell Technologies, Vancouver, British Columbia, Canada). Purity of CD4 fractions was >90% with fewer than 2% CD8+ or NK cells or monocytes by fluorescence-activated cell sorting (FACS) analysis. Semiquantitative microculture assays were performed according to previously described methods except for replicate wells at concentrations of 1 × 105, 3 × 105, and 1 × 106 activated patient CD4 cells depending on cell yields (44).
FACS analyses.
Leukocyte differentiation was performed using a Coulter counter (Coulter Electronics, Inc., Miami Lakes, Fla.). Lymphocyte subsets were evaluated by flow cytometric analysis, using 50 μl of EDTA peripheral blood incubated for 30 min at 4°C with fluorochrome-labeled monoclonal antibodies. After incubation, erythrocyte lysis and fixation of marked cells were performed using the Immuno-Prep EPICS Kit (Coulter Electronics).
IFN-γ ELISpot assays.
Ninety-six-well nitrocellulose plates were first precoated with a layer of gamma interferon (IFN-γ) monoclonal antibodies (MABTECH, Nacka, Sweden). PBMC were then added in duplicate wells either with a pool of five previously described synthetic peptides from the gp160 envelope of HIV-1 (20 μM final concentration), with HIV-1 p24 (Protein Sciences Co., Meriden, Conn.) (p24) (0.1 μg/ml) in the presence or the absence of neutralizing anti-CD4 monoclonal antibody (see below), or with no peptide (negative control) (11). The five peptides used in the stimulation are promiscuous as they are recognized by multiple HLA class I molecules (including HLA A1, A2, A3, A9, A25, A26, A29, etc.) and are mostly conserved between different HIV clades. Because these epitopes can also be recognized by HLA class II molecules (10), IFN-γ production by CD4+ was blocked by preincubating PBMC with 100 ng of neutralizing recombinant human CD4 monoclonal antibody (R&D Systems, Minneapolis, Minn.)/ml. Plates were incubated overnight at 37°C in 7% CO2, then the cells were discarded, and the plates were incubated at room temperature for 3 h. A second biotinylated anti-IFN-γ monoclonal antibody (7-B6-1 biotin; MABTECH), followed by streptavidin-conjugated alkaline phosphatase (MABTECH) for 2 h, was subsequently used. Individual IFN-γ-producing cells were detected as dark blue spots using an alkaline phosphatase conjugate substrate kit (Bio-Rad Laboratories, Hercules, Calif.). The spots were counted using a dissecting microscope (×40). HIV-specific responses were reported as number of spot-forming units per 106 mononuclear cells. Baseline and week 24 values were compared using a paired, two-tailed Student t test.
RESULTS
Kinetics of HIV RNA in the plasma.
To characterize kinetics of residual viremia, we examined the decay of HIV RNA in the plasma in our 14 patients sustaining HIV RNA levels of <50 copies/ml for 5 years after initiation of treatment with indinavir plus efavirenz. Prior to treatment, these patients had a median CD4 cell count of 261 cells/mm3 and HIV RNA level of 4.9 log copies/ml (Table 1). HIV RNA values from the first year were fitted assuming biphasic exponential decay to a constant nonzero value. The estimated clearance half-lives of infected cells were 1.2 ± 0.8 days during phase I decay and 24 ± 16 days during phase II decay, consistent with previous reports. We were also able to estimate the duration of phase II decay as between 33 and 260 days (Fig. 1). Prior estimates of the dynamics of the end of phase II have been limited by the sensitivity of the viral RNA assays used. Although the likely complex mixture of cell populations contributing to the pool of plasma virus prevents the association of this time interval with the lifetimes of a particular cellular phenotype, this first direct estimate of the duration of phase II decay provides a quantitative basis for defining phases of HIV treatment.
FIG. 1.

Initial decline of HIV RNA obeys biphasic exponential decay to a nonzero constant. Viral HIV RNA decayed initially with a phase I half-life of 1.2 ± 0.8 days and then with a phase II half-life of 24 ± 16 days. This decay is shown in patient 1 (top) (first-phase half-life, 0.89 [0.41, 1.9] days and second-phase half-life, 23.9 [16.3, 35.7] days) and in patient 9 (bottom) (first-phase half-life, 1.21 [0.59, 2.2] days and second-phase half-life, 30.6 [23.8, 39.5] days). Phase II ended within 6 months of treatment in 13 of 14 patients and within 9 months in the remaining patient.
We next examined whether there was further HIV RNA decline in the plasma after phase II decay during subsequent years of therapy. Prior longitudinal studies of patients with chronic infection have measured HIV RNA using assays with a limit of detection of 50 copies/ml and thus not addressed this issue. Studies using more sensitive HIV RNA assays have detected virus in the plasma but have not examined patients frequently with longitudinal samples (15, 45). In this cohort of patients, using an assay with a limit of detection of 2.5 copies/ml, we were able to measure virus longitudinally and found no decline in levels of residual viremia in 13 of 14 patients over 5 years of observation (Fig. 2). Patients had a median of 13 measures of residual viremia. Of note, intermittent viremia (HIV RNA greater than 50 copies/ml after 9 months of therapy) was infrequent in this cohort. Only four of our patients had any evidence of intermittent viremia after the first 9 months of therapy. The overall frequency of viremia greater than 50 copies/ml after 9 months of antiretroviral therapy was 0.02 episodes per patient-year.
FIG. 2.

Plateau of plasma HIV RNA. The second-phase RNA decay depicted in Fig. 1 ended in all patients during the first 9 months of treatment. Over the subsequent 4 years of treatment, RNA dynamics in 13 of 14 patients were statistically consistent with a plateau. Levels of residual viremia are shown for patient 1 (top) as 23 copies/ml and for patient 9 (bottom) as 5.9 copies/ml. This low-level virus production is consistent with establishment of a new steady state on antiretroviral therapy.
Predictors of residual viremia.
Levels of residual viremia in the plasma varied among patients. Geometric mean RNA values varied from 3.2 to 23 copies/ml, with a median of 6.0 copies/ml (Table 1). When we examined the predictors of the level of residual viremia in these patients, we found that pretreatment levels of total HIV DNA correlated with residual viremia (Fig. 3). Interestingly, baseline HIV RNA and CD4 cell count were not predictors of residual viremia.
FIG. 3.

HIV DNA before treatment with indinavir plus efavirenz correlates with residual viremia. Residual HIV RNA was defined as the geometric mean of all measured values after 9 months of therapy. Baseline DNA was the only significant pretreatment predictor of residual viremia in either univariate (R2 = 0.51, P = 0.003) or multivariate (P = 0.002) regression including baseline HIV RNA, CD4 counts, and patient age. These correlations remained when HIV DNA was normalized to cell counts, when residual viremia was computed using a maximum likelihood approach with censoring, and when nonparametric statistical methods were used. While the correlation does not define the mechanism connecting HIV DNA and residual RNA, the predictive value of pretreatment DNA levels suggests that long-lived cellular reservoirs of viral DNA activate to kindle HIV RNA production.
Kinetics of HIV DNA.
The kinetics of viral DNA were also examined in this cohort of patients. Pretreatment levels ranged from 313 to 22,000 HIV DNA copies per ml of blood. These levels were not associated with pretreatment HIV RNA measurements, CD4 cell count, prior treatment history, or estimated duration of HIV infection. We measured HIV DNA at 2-month intervals during the first year of treatment and at 6-month intervals over the remainder of the study period. During the first year of antiretroviral therapy, HIV DNA declined by a median of twofold (interquartile range, 1.7- to 3.5-fold). The median half-life during this period (Table 1) was 466 days. This is somewhat longer than previously reported, in part due to the normalization of DNA copy numbers to blood volume, which eliminates the dilutional decrease seen when normalizing to rising total lymphocyte numbers (1, 29, 30). After the first year of treatment, the HIV DNA showed no significant decline. DNA copy numbers remained measurable in all patients throughout the course of the study.
Viral kinetics after treatment intensification.
Given that HIV RNA and DNA remained constant for years after the initial 9 months of therapy, we sought to determine if there was a dynamic steady state of viral production or if plasma virus resulted exclusively from release of particles from latently infected cells. A nucleoside reverse transcriptase inhibitor would block infection of new cells but would have no effect on the release of virus directly from latently infected cells. Therefore, we measured plasma HIV RNA and DNA levels and cell-associated infectivity in patients at frequent intervals for 24 weeks in patients from this cohort who chose to add the nucleoside reverse transcriptase inhibitor abacavir (eight patients) or ritonavir (one patient) to their regimen after 5 years of therapy. The five patients from this cohort who did not add abacavir were also monitored prospectively.
Plasma HIV RNA levels rapidly declined by 4 weeks in four of five patients with detectable HIV RNA who received abacavir (Fig. 4) (Table 1). We were unable to quantify the magnitude of decline because of the limit of detection of our assay. The estimated half-life of infected cells was 6.7 days (range, 4.4 to 11 days). In two of the five patients (subjects 7 and 8) who did not add abacavir, HIV RNA levels did not decrease. Steady-state levels below the limit of detection or missing data precluded evaluation in the other three patients Total HIV DNA levels did not decline over the 24 weeks of observation (data not shown). There was no change in semiquantitative cultures of HIV infectivity (data not shown).
FIG. 4.

Effect of abacavir intensification on viremia and cell-associated infectivity. Addition of abacavir resulted in a significant decrease in plasma viremia in four of five patients with sufficient residual viremia to allow a change to be detected. Among these four individuals, only patient 1 (a) retained detectable viremia (>2.5 copies/ml) after intensification. In all others, intensification resulted in suppression of viremia to below this detection threshold (b to d).
Cellular activation and HIV immune responses after therapy intensification.
Reductions in HIV RNA levels in patients initiating therapy are associated with rises in CD4 cell counts, reduction in HIV-associated immune responses, and decline in cellular activation (2). Initial CD4 cell increases are attributed to redistribution of memory cells (36). In this group of patients already treated for 5 years, we did not observe increases in CD4 cell counts after therapy intensification (data not shown). However, we did observe a reduction in some markers of cellular activation in both CD4+ and CD8+ cells (Table 2). CD8-mediated HIV Gag and p24 antigen responses were also lower in patients receiving abacavir, although the differences between the groups were not statistically different when the lower background activation rates in the abacavir group were included in the analyses. These observations provide support for a reduction in HIV antigen load with therapy intensification, suggesting perturbation of a dynamic equilibrium by the addition of an additional antiretroviral agent.
TABLE 2.
Significant changes in cellular activation markers observed for nine patients undergoing intensification of antiretroviral therapy with abacavir and four control patients not receiving abacavira
| Patient group and marker or antigen | Median (range)
|
P | |
|---|---|---|---|
| Baseline | Wk 24 | ||
| Abacavir patients | |||
| Cell marker (FACS analysis) | |||
| CD4-DRII | 0.99 (0.66-1.26) | 0.64 (0.21-1.09) | 0.03 |
| CD8-CD38 | 2.41 (0.94-3.62) | 1.12 (0.28-1.67) | 0.01 |
| HIV antigen (ELISpot) | |||
| None | 216 (180-265) | 105 (75-150) | <0.001 |
| p24 | 266 (200-325) | 153 (125-205) | <0.001 |
| Env | 275 (235-335) | 161 (120-220) | <0.001 |
| Control patients | |||
| Cell marker | |||
| CD4-DRII | 0.74 (0.42-1.33) | 0.67 (0.29-1.03) | NS |
| CD8-CD38 | 0.93 (0.25-3.34) | 0.87 (0.22-1.96) | NS |
| HIV antigen (ELISpot) | |||
| None | 146 (95-210) | 120 (100-155) | NS |
| p24 | 200 (125-255) | 167 (140-215) | NS |
| Env | 193 (125-290) | 164 (125-210) | NS |
For abacavir patients, values are before and after intensification. ELISpot responses are expressed as spot-forming units per 106 cells. NS, not significant.
DISCUSSION
We sought to address two main questions in this longitudinal study of patients receiving potent antiretroviral therapy for 6 years. First, is there a measurable reduction in plasma levels of HIV RNA over time in patients with high-level viral suppression (<50 copies/ml)? Second, is there evidence for ongoing productive infection in these patients after 5 years? We found that levels of HIV RNA in the plasma reached a steady state within 9 months of therapy initiation that we defined as residual viremia. Levels of residual viremia varied among patients and were predicted by pretreatment DNA levels. HIV RNA levels declined rapidly after intensification with abacavir, providing direct evidence for ongoing productive infection. Supporting evidence for a reduction in viral burden after treatment intensification was provided by studies of immune activation and HIV immune responses. These data support a model of residual replication in treated patients in which both localized activation of latently infected cells and productive infection contribute to residual replication.
The variability of residual replication and correlation with viral DNA levels and residual replication could be interpreted in a number of ways. Adherence and drug levels are known to influence the antiviral effect of antiretroviral regimens (28, 34, 37). Variability in these parameters among our patients could produce differing levels of residual replication, which would affect the pool of residual cellular DNA. However, these mechanisms would not explain a correlation between pretreatment proviral DNA levels and residual replication. The simplest interpretation of our data is that HIV DNA levels reflect the size of the pool of long-lived latently infected cells that determine the level of residual viremia in patients receiving chronic treatment. Cell-associated DNA is comprised of both integrated and unintegrated HIV DNA, and a substantial proportion of the HIV DNA in PBMC represents replication-defective genomes (7, 33, 42). Linear unintegrated DNA is labile, and this subset of HIV DNA present before treatment is therefore unlikely to produce replication-competent virus after several years of therapy (4, 5, 48). We did not directly measure integrated DNA or distinguish replication-competent from defective genomes. Thus, our interpretation that the number of latently infected lymphocytes determines residual viral replication relies on the assumption that the proportion of viral DNA consisting of integrated genomes with the capacity to sustain viral replication is relatively constant across patients. Other interpretations of our findings are also possible. Total HIV DNA measured in our patients at baseline may reflect a net susceptibility of lymphocytes to infection, a characteristic maintained even years after initiation of combination therapy and reflected in levels of residual viral replication.
After an initial reduction associated with therapy initiation, HIV DNA levels, like RNA levels, did not decrease measurably over the 6 years of observation. Most of the cellular reservoir of HIV occurs in resting memory lymphocytes (7), but resting naïve cells have also recently been recognized as a smaller but detectable reservoir (38). T lymphocytes specific for HIV are overrepresented among all HIV-infected memory T cells from untreated patients (16), and the expected rapid turnover of such cells in the presence of abundant HIV antigens may in part account for the higher HIV DNA clearance rate over the first year of treatment. Once such cells are depleted and antigen is cleared, the residual infected cells might be expected to clear more slowly. Our observation of the apparent long-term stability of the pool of latently infected lymphocytes is consistent with findings reported by Douek et al. (16) and corroborates some previous reports based on both PBMC DNA measurements (20) and quantitative coculture assays.
An alternative reason for the slow clearance of latently infected cells may be the contribution of replenishment of the latent cellular reservoir by ongoing residual replication. The bidirectional interaction between ongoing replication during antiretroviral therapy and the dynamics of the latent reservoir has been the subject of speculation. Replenishment of the cellular reservoir is suggested by the lower decay rates of the latent pool in patients with intermittent viremia, an effect that can be modulated by therapy intensification (25, 41). We studied patients with an extraordinarily low frequency of intermittent viremia, a patient profile that has, to date, not been demonstrated to support replenishment of latent reservoirs. In the present study, reduction of the observed residual viremia by addition of abacavir was not associated with a decrease in HIV DNA or cell-associated infectivity, although continued follow-up of these patients will determine whether slow declines in this reservoir are occurring over time. In a related study using drug resistance mutations to track subpopulations of persistently infected lymphocytes likely to be sustained by persistent replication, we observed that reseeding of the reservoir, while demonstrable, was not the principal reason for the durability of cells carrying HIV DNA in the absence of intermittent viremia (44). Nevertheless, we cannot exclude the possibility with these data that some reseeding can occur even in the absence of intermittent viremia of >50 copies/ml.
The decline of HIV RNA levels observed after therapy intensification with abacavir provides insight into the source of virus contributing to residual viremia. If activation of latently infected cells without productive infection were the sole source of residual viremia, the HIV RNA level would not have declined with the addition of abacavir. The rapid decline of HIV RNA levels suggest that either continuous productive infection, for example in gut lymphoid tissue, or small bursts of self-limited infection triggered by activation of latently infected cells contribute to residual viral replication. In one model, HIV replication is depicted as proximal activation and transmission events that depend on long-lived infected lymphocytes to sustain infection by bridging spatially and temporally discontinuous foci of viral replication (22). This model is supported by two of our observations. The correlation between the size of the stable HIV DNA reservoir and residual viremia implicates long-lived cells in ongoing replication. The rapid decay of HIV RNA after intensification suggests that residual viremia is being produced primarily in cells that turn over rapidly, perhaps in activated, productively infected lymphocytes. Further, the idea of repeated but discontinuous activation events as a source of HIV RNA is supported indirectly by the durable viral suppression in the face of selective pressure of a drug (efavirenz) that requires only a single mutation to confer drug resistance. Thus, our data support previous suggestions of a complex interaction between rapid viral replication during treatment and long-lived cellular reservoirs of HIV.
Both the effectiveness and limits of present antiretroviral therapy are illustrated by the findings of this study. Therapy reduces viral replication to a level at which the virus is unable to escape and rebound, despite the fact that productive infection maintains virus at a steady-state level detectable even in the plasma. We acknowledge that indinavir plus efavirenz is not currently recommended as initial therapy for HIV infection and that the generalizability of our findings requires examinations of patients treated with different regimens. However, it is important to note that these patients achieved high, well-documented levels of viral suppression with only rare episodes of intermittent viremia. We doubt that the added antiviral potency demonstrated by abacavir is due to inhibition of drug-resistant virus and instead speculate that abacavir inhibits virus production from lymphocytes not accessible to indinavir or efavirenz for pharmacological reasons such as drug penetration, access, or drug resistance transporters (31).
Our observations have implications for long-term HIV therapeutic strategies. We have identified pretreatment DNA as a potential predictor of residual HIV replication and shown that residual viremia can be reduced even in patients with effective and sustained viral suppression. Recent demonstrations of the value of HIV DNA in predicting untreated disease course (21; C. Rouzioux, 1st IAS Conf. HIV Pathogenesis Treatment, abstr. 23, 2001), together with our results, suggest that this marker may be an informative marker of viral burden. Innovative approaches to eradication that rely on activation of latently infected cells have already identified ongoing residual replication as a major obstacle to the success of this strategy (19, 40). Therapeutic vaccines may be most likely to produce virologic remission in patients with the lowest levels of residual replication (3). Likewise, structured intermittent therapy may be most successful in patients with maximal suppression prior to therapy interruption (17). Finally, the ability to predict levels of residual replication may permit assessments of antiviral potency required to sustain virologic suppression and minimize drug toxicity.
Acknowledgments
We thank the patients for their participation in this study; Trevor Scott, Betsy Dusak, and Dorothy Brey for their encouragement and support; Lee Bacheler for helpful discussions; Linda Meixner for patient management; Gary Dyak, Ruby Lam, Theresa Macaranas, and Otto Daly for laboratory assistance; and Simon Frost, Doug Richman, Zvi Grossman, and Steven Deeks for their comments on the manuscript.
This work was supported in part under NIH grants A136214, A147745, and A151982 (D.V.H.); by CM07198 and a La Jolla Interfaces in Sciences Fellowship (M.C.S.); by Istituto Superiore di Sanita II Programma Nazionale di Ricerca sull' AIDS 1999 (M.C.); and by PHS AI 43752, AI 47745, and AI 38858 and a VA Research Merit Award (J.K.W.). Abacavir and partial support for immune studies were provided by Glaxo Smith Kline. HIV DNA assay kits were kindly provided by Shirley Kwok, Karen Young, and Cindy Christopherson, Roche Molecular Diagnostics.
REFERENCES
- 1.Andreoni, M., S. G. Parisi, L. Sarmati, E. Nicastri, L. Ercoli, G. Mancino, G. Sotgiu, M. Mannazzu, M. Trevenzoli, G. Tridente, E. Concia, and A. Aceti. 2000. Cellular proviral HIV-DNA decline and viral isolation in naive subjects with <5,000 copies/ml of HIV-RNA and >500 × 106/l CD4 cells treated with highly active antiretroviral therapy. AIDS 14:23-29. [DOI] [PubMed] [Google Scholar]
- 2.Autran, B., G. Carcelain, T. S. Li, C. Blanc, D. Mathez, R. Tubiana, C. Katlama, P. Debre, and J. Leibowitch. 1997. Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science 277:112-116. [DOI] [PubMed] [Google Scholar]
- 3.Barouch, D. H., S. Santra, J. E. Schmitz, M. J. Kuroda, T. M. Fu, W. Wagner, M. Bilska, A. Craiu, X. X. Zheng, G. R. Krivulka, K. Beaudry, M. A. Lifton, C. E. Nickerson, W. L. Trigona, K. Punt, D. C. Freed, L. Guan, S. Dubey, D. Casimiro, A. Simon, M. E. Davies, M. Chastain, T. B. Strom, R. S. Gelman, D. C. Montefiori, M. G. Lewis, E. A. Emini, J. W. Shiver, and N. L. Letvin. 2000. Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science 290:486-492. [DOI] [PubMed] [Google Scholar]
- 4.Blankson, J. N., D. Finzi, T. C. Pierson, B. P. Sabundayo, K. Chadwick, J. B. Margolick, T. C. Quinn, and R. F. Siciliano. 2000. Biphasic decay of latently infected cD4+ T cells in acute human immunodeficiency virus type 1 infection. J. Infect. Dis. 182:1636-1642. [DOI] [PubMed]
- 5.Bukrinsky, M. I., T. L. Stanwick, M. P. Dempsey, and M. Stevenson. 1991. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 254:423-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christopherson, C., Y. Kidane, B. Conway, J. Krowka, H. Sheppard, and S. Kwok. 2000. PCR-based assay to quantify human immunodeficiency virus type 1 DNA in peripheral blood mononuclear cells. J. Clin. Microbiol. 38:630-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chun, T. W., L. Carruth, D. Finzi, X. Shen, J. A. DiGiuseppe, H. Taylor, M. Hermankova, K. Chadwick, J. Margolick, T. C. Quinn, Y. H. Kuo, R. Brookmeyer, M. A. Zeiger, P. Barditch-Crovo, and R. F. Siliciano. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183-188. [DOI] [PubMed] [Google Scholar]
- 8.Chun, T. W., R. T. Davey, Jr., M. Ostrowski, J. S. Justement, D. Engel, J. I. Mullins, and A. S. Fauci. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 6:757-761. [DOI] [PubMed] [Google Scholar]
- 9.Chun, T. W., L. Stuyver, S. B. Mizell, L. A. Ehler, J. A. Mican, M. Baseler, A. L. Lloyd, M. A. Nowak, and A. S. Fauci. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 94:13193-13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clerici, M., D. R. Lucey, R. A. Zajac, R. N. Boswell, H. M. Gebel, H. Takahashi, J. A. Berzofsky, and G. M. Shearer. 1991. Detection of cytotoxic T lymphocytes specific for synthetic peptides of gp160 in HIV-seropositive individuals. J. Immunol. 146:2214-2219. [PubMed] [Google Scholar]
- 11.Clerici, M., N. I. Stocks, R. A. Zajac, R. N. Boswell, D. C. Bernstein, D. L. Mann, G. M. Shearer, and J. A. Berzofsky. 1989. Interleukin-2 production used to detect antigenic peptide recognition by T-helper lymphocytes from asymptomatic HIV-seropositive individuals. Nature 339:383-385. [DOI] [PubMed] [Google Scholar]
- 12.Cone, R. W., P. Gowland, M. Opravil, P. Grob, and B. Ledergerber. 1998. Levels of HIV-infected peripheral blood cells remain stable throughout the natural history of HIV-1 infection. Swiss HIV Cohort Study. AIDS 12:2253-2260. [DOI] [PubMed] [Google Scholar]
- 13.Davey, R. T., Jr., N. Bhat, C. Yoder, T. W. Chun, J. A. Metcalf, R. Dewar, V. Natarajan, R. A. Lempicki, J. W. Adelsberger, K. D. Miller, J. A. Kovacs, M. A. Polis, R. E. Walker, J. Falloon, H. Masur, D. Gee, M. Baseler, D. S. Dimitrov, A. S. Fauci, and H. C. Lane. 1999. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 96:15109-15114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Jong, M. D., R. J. de Boer, F. de Wolf, N. A. Foudraine, C. A. Boucher, J. Goudsmit, and J. M. Lange. 1997. Overshoot of HIV-1 viraemia after early discontinuation of antiretroviral treatment. AIDS 11:F79-F84. [DOI] [PubMed] [Google Scholar]
- 15.Dornadula, G., H. Zhang, B. VanUitert, J. Stern, L. Livornese, Jr., M. J. Ingerman, J. Witek, R. J. Kedanis, J. Natkin, J. DeSimone, and R. J. Pomerantz. 1999. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 282:1627-1632. [DOI] [PubMed] [Google Scholar]
- 16.Douek, D. C., J. M. Brenchley, M. R. Betts, D. R. Ambrozak, B. J. Hill, Y. Okamoto, J. P. Casazza, J. Kuruppu, K. Kunstman, S. Wolinsky, Z. Grossman, M. Dybul, A. Oxenius, D. A. Price, M. Connors, and R. A. Koup. 2002. HIV preferentially infects HIV-specific CD4+ T cells. Nature 417:95-98. [DOI] [PubMed] [Google Scholar]
- 17.Dybul, M., T. W. Chun, C. Yoder, B. Hidalgo, M. Belson, K. Hertogs, B. Larder, R. L. Dewar, C. H. Fox, C. W. Hallahan, J. S. Justement, S. A. Migueles, J. A. Metcalf, R. T. Davey, M. Daucher, P. Pandya, M. Baseler, D. J. Ward, and A. S. Fauci. 2001. Short-cycle structured intermittent treatment of chronic HIV infection with highly active antiretroviral therapy: effects on virologic, immunologic, and toxicity parameters. Proc. Natl. Acad. Sci. USA 98:15161-15166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finzi, D., M. Hermankova, T. Pierson, L. M. Carruth, C. Buck, R. E. Chaisson, T. C. Quinn, K. Chadwick, J. Margolick, R. Brookmeyer, J. Gallant, M. Markowitz, D. D. Ho, D. D. Richman, and R. F. Siliciano. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295-1300. [DOI] [PubMed] [Google Scholar]
- 19.Fraser, C., N. M. Ferguson, A. C. Ghani, J. M. Prins, J. M. Lange, J. Goudsmit, R. M. Anderson, and F. de Wolf. 2000. Reduction of the HIV-1-infected T-cell reservoir by immune activation treatment is dose-dependent and restricted by the potency of antiretroviral drugs. AIDS 14:659-669. [DOI] [PubMed] [Google Scholar]
- 20.Furtado, M. R., D. S. Callaway, J. P. Phair, K. J. Kunstman, J. L. Stanton, C. A. Macken, A. S. Perelson, and S. M. Wolinsky. 1999. Persistence of HIV-1 transcription in peripheral-blood mononuclear cells in patients receiving potent antiretroviral therapy. N. Engl. J. Med. 340:1614-1622. [DOI] [PubMed] [Google Scholar]
- 21.Gottlieb, G. S., P. S. Sow, S. E. Hawes, I. Ndoye, M. Redman, A. M. Coll-Seck, M. A. Faye-Niang, A. Diop, J. M. Kuypers, C. W. Critchlow, R. Respess, J. I. Mullins, and N. B. Kiviat. 2002. Equal plasma viral loads predict a similar rate of CD4+ T cell decline in human immunodeficiency virus (HIV) type 1- and HIV-2-infected individuals from Senegal, West Africa. J. Infect. Dis. 185:905-914. [DOI] [PubMed] [Google Scholar]
- 22.Grossman, Z., M. Polis, M. B. Feinberg, I. Levi, S. Jankelevich, R. Yarchoan, J. Boon, F. de Wolf, J. M. Lange, J. Goudsmit, D. S. Dimitrov, and W. E. Paul. 1999. Ongoing HIV dissemination during HAART. Nat. Med. 5:1099-1104. [DOI] [PubMed] [Google Scholar]
- 23.Gulick, R. M., J. W. Mellors, D. Havlir, J. J. Eron, A. Meibohm, J. H. Condra, F. T. Valentine, D. McMahon, C. Gonzalez, L. Jonas, E. A. Emini, J. A. Chodakewitz, R. Isaacs, and D. D. Richman. 2000. 3-year suppression of HIV viremia with indinavir, zidovudine, and lamivudine. Ann. Intern. Med. 133:35-39. [DOI] [PubMed] [Google Scholar]
- 24.Gunthard, H. F., S. D. Frost, A. J. Leigh-Brown, C. C. Ignacio, K. Kee, A. S. Perelson, C. A. Spina, D. V. Havlir, M. Hezareh, D. J. Looney, D. D. Richman, and J. K. Wong. 1999. Evolution of envelope sequences of human immunodeficiency virus type 1 in cellular reservoirs in the setting of potent antiviral therapy. J. Virol. 73:9404-9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Havlir, D. V., R. Bassett, D. Levitan, P. Gilbert, P. Tebas, A. C. Collier, M. S. Hirsch, C. Ignacio, J. Condra, H. F. Gunthard, D. D. Richman, and J. K. Wong. 2001. Prevalence and predictive value of intermittent viremia with combination HIV therapy. JAMA 286:171-179. [DOI] [PubMed] [Google Scholar]
- 26.Hermankova, M., S. C. Ray, C. Ruff, M. Powell-Davis, R. Ingersoll, R. T. D'Aquila, T. C. Quinn, J. D. Siliciano, R. F. Siliciano, and D. Persaud. 2001. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. JAMA 286:196-207. [DOI] [PubMed] [Google Scholar]
- 27.Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123-126. [DOI] [PubMed] [Google Scholar]
- 28.Hoetelmans, R. M., R. P. van Heeswijk, M. Profijt, J. W. Mulder, P. L. Meenhorst, J. M. Lange, P. Reiss, and J. H. Beijnen. 1998. Comparison of the plasma pharmacokinetics and renal clearance of didanosine during once and twice daily dosing in HIV-1 infected individuals. AIDS 12:F211-F216. [DOI] [PubMed] [Google Scholar]
- 29.Ibanez, A., T. Puig, J. Elias, B. Clotet, L. Ruiz, and M. A. Martinez. 1999. Quantification of integrated and total HIV-1 DNA after long-term highly active antiretroviral therapy in HIV-1-infected patients. AIDS 13:1045-1049. [DOI] [PubMed] [Google Scholar]
- 30.Izopet, J., G. Salama, C. Pasquier, K. Sandres, B. Marchou, P. Massip, and J. Puel. 1998. Decay of HIV-1 DNA in patients receiving suppressive antiretroviral therapy. J. Acquired Immune Defic. Syndr. Hum. Retrovir. 19:478-483. [DOI] [PubMed] [Google Scholar]
- 31.Jones, K., P. G. Bray, S. H. Khoo, R. A. Davey, E. R. Meaden, S. A. Ward, and D. J. Back. 2001. P-glycoprotein and transporter MRP1 reduce HIV protease inhibitor uptake in CD4 cells: potential for accelerated viral drug resistance? AIDS 15:1353-1358. [DOI] [PubMed] [Google Scholar]
- 32.Lewin, S. R., M. Vesanen, L. Kostrikis, A. Hurley, M. Duran, L. Zhang, D. D. Ho, and M. Markowitz. 1999. Use of real-time PCR and molecular beacons to detect virus replication in human immunodeficiency virus type 1-infected individuals on prolonged effective antiretroviral therapy. J. Virol. 73:6099-6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, Y., H. Hui, C. J. Burgess, R. W. Price, P. M. Sharp, B. H. Hahn, and G. M. Shaw. 1992. Complete nucleotide sequence, genome organization, and biological properties of human immunodeficiency virus type 1 in vivo: evidence for limited defectiveness and complementation. J. Virol. 66:6587-6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Brien, T. R., N. S. Padian, T. Hodge, J. J. Goedert, S. J. O'Brien, and M. Carrington. 1998. CCR-5 genotype and sexual transmission of HIV-1. AIDS 12:444-445. [PubMed] [Google Scholar]
- 35.Padow, M., L. Lai, R. J. Fisher, Y. C. Zhou, X. Wu, J. C. Kappes, and E. M. Towler. 2000. Analysis of human immunodeficiency virus type 1 containing HERV-K protease. AIDS Res. Hum. Retrovir. 16:1973-1980. [DOI] [PubMed] [Google Scholar]
- 36.Pakker, N. G., D. W. Notermans, R. J. de Boer, M. T. Roos, F. de Wolf, A. Hill, J. M. Leonard, S. A. Danner, F. Miedema, and P. T. Schellekens. 1998. Biphasic kinetics of peripheral blood T cells after triple combination therapy in HIV-1 infection: a composite of redistribution and proliferation. Nat. Med. 4:208-214. [DOI] [PubMed] [Google Scholar]
- 37.Paterson, D. L., S. Swindells, J. Mohr, M. Brester, E. N. Vergis, C. Squier, M. M. Wagener, and N. Singh. 2000. Adherence to protease inhibitor therapy and outcomes in patients with HIV infection. Ann. Intern. Med. 133:21-30. [DOI] [PubMed] [Google Scholar]
- 38.Pierson, T., T. L. Hoffman, J. Blankson, D. Finzi, K. Chadwick, J. B. Margolick, C. Buck, J. D. Siliciano, R. W. Doms, and R. F. Siliciano. 2000. Characterization of chemokine receptor utilization of viruses in the latent reservoir for human immunodeficiency virus type 1. J. Virol. 74:7824-7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pomerantz, R. J. 2002. Reservoirs of human immunodeficiency virus type 1: the main obstacles to viral eradication. Clin. Infect. Dis. 34:91-97. [DOI] [PubMed] [Google Scholar]
- 40.Prins, J. M., S. Jurriaans, R. M. van Praag, H. Blaak, R. van Rij, P. T. Schellekens, I. J. ten Berge, S. L. Yong, C. H. Fox, M. T. Roos, F. de Wolf, J. Goudsmit, H. Schuitemaker, and J. M. Lange. 1999. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS 13:2405-2410. [DOI] [PubMed] [Google Scholar]
- 41.Ramratnam, B., J. E. Mittler, L. Zhang, D. Boden, A. Hurley, F. Fang, C. A. Macken, A. S. Perelson, M. Markowitz, and D. D. Ho. 2000. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 6:82-85. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez, G., X. Xu, J. C. Chermann, and I. Hirsch. 1997. Accumulation of defective viral genomes in peripheral blood mononuclear cells of human immunodeficiency virus type 1-infected individuals. J. Virol. 71:2233-2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Staszewski, S., J. Morales-Ramirez, K. T. Tashima, A. Rachlis, D. Skiest, J. Stanford, R. Stryker, P. Johnson, D. F. Labriola, D. Farina, D. J. Manion, N. M. Ruiz, et al. 1999. Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. N. Engl. J. Med. 341:1865-1873. [DOI] [PubMed] [Google Scholar]
- 44.Strain, M. C., H. F. Gunthard, D. V. Havlir, C. C. Ignacio, D. M. Smith, A. J. Leigh-Brown, T. R. Macaranas, R. Y. Lam, O. A. Daly, M. Fischer, M. Opravil, H. Levine, L. Bacheler, C. A. Spina, D. D. Richman, and J. K. Wong. 2003. Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: intrinsic stability predicts lifelong persistence. Proc. Natl. Acad. Sci. USA 100:4819-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Praag, R. M., F. W. Wit, S. Jurriaans, F. de Wolf, J. M. Prins, and J. M. Lange. 2002. Improved long-term suppression of HIV-1 replication with a triple-class multidrug regimen compared with standard of care antiretroviral therapy. AIDS 16:719-725. [DOI] [PubMed] [Google Scholar]
- 46.Wei, X., S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. H. Hahn, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117-122. [DOI] [PubMed] [Google Scholar]
- 47.Wong, J. K., M. Hezareh, H. F. Gunthard, D. V. Havlir, C. C. Ignacio, C. A. Spina, and D. D. Richman. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291-1295. [DOI] [PubMed] [Google Scholar]
- 48.Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Chen. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213-222. [DOI] [PubMed] [Google Scholar]
- 49.Zhang, L., C. Chung, B. S. Hu, T. He, Y. Guo, A. J. Kim, E. Skulsky, X. Jin, A. Hurley, B. Ramratnam, M. Markowitz, and D. D. Ho. 2000. Genetic characterization of rebounding HIV-1 after cessation of highly active antiretroviral therapy. J. Clin. Investig. 106:839-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang, L., B. Ramratnam, K. Tenner-Racz, Y. He, M. Vesanen, S. Lewin, A. Talal, P. Racz, A. S. Perelson, B. T. Korber, M. Markowitz, and D. D. Ho. 1999. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N. Engl. J. Med. 340:1605-1613. [DOI] [PubMed] [Google Scholar]