

Abstract
Stimulation of CD4+ helper T lymphocytes by antigen-presenting cells requires the degradation of exogenous antigens into antigenic peptides which associate with major histocompatibility complex (MHC) class II molecules in endosomal or lysosomal compartments. B lymphocytes mediate efficient antigen presentation first by capturing soluble antigens through clonally distributed antigen receptors (BCRs), composed of membrane immunoglobulin (Ig) associated with Ig-α/Ig-β heterodimers which, second, target antigens to MHC class II–containing compartments. We report that antigen internalization and antigen targeting through the BCR or its Ig-α–associated subunit to newly synthesized class II lead to the presentation of a large spectrum of T cell epitopes, including some cryptic T cell epitopes. To further characterize the intracellular mechanisms of BCR-mediated antigen presentation, we used two complementary experimental approaches: mutational analysis of the Ig-α cytoplasmic tail, and overexpression in B cells of dominant negative syk mutants. Thus, we found that the syk tyrosine kinase, an effector of the BCR signal transduction pathway, is involved in the presentation of peptide– MHC class II complexes through antigen targeting by BCR subunits.
Keywords: B cell receptor, syk tyrosine kinase, antigen presentation, major histocompatibility complex class II, immunoglobulin
Antigen-presenting cells (APCs), to stimulate CD4+ helper T lymphocytes, must capture and process exogenous antigens into antigenic peptides which associate with MHC class II molecules in the endocytic pathway (1, 2). The formation of peptide–class II complexes classically occurs in endolysosomal compartments (called MIIC or CIIV), which accumulate newly synthesized class II molecules (3–5), or in recycling endosomes, which contain mature class II molecules internalized from the cell surface and then recycled back to the plasma membrane (6, 7). Therefore, peptide–class II complexes may be generated at any point throughout the endocytic pathway (8). This study delineates how antigen receptors might determine the delivery of antigens to different compartments and thus could modulate MHC class II–restricted antigen presentation.
B lymphocytes mediate efficient class II–restricted antigen presentation first by facilitating the uptake of soluble antigens through clonally distributed receptors (BCRs)1 (9). These receptors are multichain complexes composed of a ligand-binding module, the membrane immunoglobulin (mIg), and a transducing module, the Ig-α/Ig-β heterodimer (10, 11), both chains containing a conserved peptidic motif located in their cytoplasmic tails. This immunoreceptor tyrosine–based activation motif (ITAM [12, 13]) consists of conserved amino acid residues (D2xY2xL7xY2xL) which couple receptors to intracellular effectors, leading to cell activation (11) and antigen internalization (14–16). Antigen recognition by mIg triggers the activation of Src family phosphotyrosine kinases (PTKs), resulting in tyrosine phosphorylation of Ig-α/Ig-β ITAM (17) and the recruitment of PTK Syk by phosphorylated ITAM (18, 19), which turns on different signaling pathways (11). Concomitantly, the BCR facilitates the endocytosis of soluble antigens and their access to endosomal/lysosomal compartments, where the antigens are degraded into peptides which then associate to MHC class II molecules to be presented to T lymphocytes (20). Therefore, another important function of the BCR is to address soluble antigens to the intracellular sites of peptide loading on class II molecules (5, 15). To understand the role of the BCR in antigen presentation, a critical question should therefore be addressed: Do signal transduction effectors, involved in BCR-mediated B cell activation, determine the presentation of peptide–MHC class II complexes through BCR-mediated antigen internalization?
The functions of BCR subunits in the intracellular targeting of the BCR partially answer this question. Indeed, the BCR is involved in the delivery of mIg-bound antigens to newly synthesized MHC class II molecules accumulating in endosomal compartments (5). The association of the Ig-α/Ig-β sheath to mIg may account for the ability of the BCR to induce both specific endosomal targeting and efficient antigen presentation (14, 15). In the complex structure of the BCR, both Ig-α and Ig-β cytoplasmic domains contain internalization signals (16), but only the Ig-α cytoplasmic tail was able to target antigens to newly synthesized MHC class II, whereas Ig-β cytoplasmic tail contains targeting signals that direct antigens towards recycling class II (16). In addition, the cytoplasmic tails of Ig-α and Ig-β were reported to interact with distinct cytoplasmic effectors (21) and to trigger different signaling pathways (22). Thus, the cytoplasmic domain of Ig-α interacts with specific cytoplasmic proteins that might be necessary for the intracellular targeting of the antigen-bound BCR to MHC class II compartments. Two related questions were addressed specifically: Does BCR-mediated antigen targeting to class II compartments have any immunological consequences for the selection of specific T cell epitopes, and what are the cytoplasmic effectors of this particular endosomal sorting of antigen-bound BCR to class II compartments? We show that the delivery of mIg-bound antigens, through Ig-α, to newly synthesized MHC class II molecules induced the presentation of a large spectrum of T cell epitopes derived from λ repressor (C1), hen egg lysozyme (HEL), or ovalbumin (OVA). In contrast, antigen targeting to recycling class II through the Ig-β cytoplasmic tail led to the presentation of only some of these epitopes. We then identified a signal in the Ig-α cytoplasmic tail that was responsible for the presentation of these T cell epitopes and the activation of syk tyrosine kinase. Endogenous syk activation and presentation of T cell epitopes, induced through the Ig-α cytoplasmic tail, were abolished by the overexpression of a syk mutant devoid of kinase domain, which had no effect on antigen presentation induced by the Ig-β cytoplasmic tail. Therefore, the recruitment of syk tyrosine kinase by the Ig-α BCR subunit in the endosomal pathway may be an important step in potentiating secondary B cell immune response by addressing antigens to MHC class II compartments.
Materials and Methods
B Lymphoma Cell Lines.
The B lymphoma IIA1.6 is an FcγR-defective variant of A20 B lymphoma cells (23). The anti-TNP A20 cells were obtained by transfection of genomic clones encoding the light and the heavy μ chains of the BCR specific for TNP (24). These cell lines were cultured in RPMI 1640 containing 10% FCS, 10 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-ME, and 5 mM sodium pyruvate (GIBCO BRL, Paisley, UK). FcγR/Ig-α and FcγR/Ig-β chimeras were constructed by adding the DNA sequences encoding the cytoplasmic domain of the Ig-α or Ig-β subunits to cDNA encoding the extracellular and transmembrane domains of mouse FcγRII (22). cDNAs encoding FcγR/Ig-α chimeras with simple or double mutations of tyrosine residues, contained in the ITAM, were constructed using PCR with alanine in place of tyrosine. All of these constructs have been described previously (25). The resulting constructions were inserted in an expression vector bearing a neomycin gene resistance, sequenced, and stably expressed in the mouse B cell line IIA1.6. Cell surface expression of FcγR chimeras was measured with the rat anti–mouse FcγR mAb 2.4G2, and was revealed by FITC-coupled mouse anti–rat antibodies (26). The samples were analyzed with a FACScan® flow cytometer (Becton Dickinson, San Jose, CA).
Stable Expression of Syk Dominant Negative Mutant.
The syk dominant negative mutant was constructed by joining the EcoRI-KpnI fragment of rat syk cDNA (27) to a KpnI-XbaI PCR fragment amplified from a hemagglutinin (HA)-tag–containing plasmid. The sequence coding for the HA-tag is recognized by the mAb 12CA5. The resulting truncated syk has been sequenced, and corresponded to the first 260 amino acids of the rat syk cDNA fused to the sequence GSGYSYDVPDYA. This construct was then inserted in an Sr-driven expression vector bearing the puromycin gene resistance. After linearization with ScaI restriction enzyme, 50 μg of DNA was used to electroporate B cells expressing FcγR/Ig-α or FcγR/Ig-β chimeras as described (26). After 48 h, the transfected cells were resuspended at 103 cells/ml in a culture medium containing 2 μg/ml puromycin (Sigma Chemical Co., St. Louis, MO). This cell suspension was distributed in 96-well plates at 100 μl/well and kept at 37°C for several weeks. Growing cells were selected for the expression of the truncated syk using FITC-coupled 12CA5 anti–HA-tag antibody (Boehringer Mannheim Corp., Indianapolis, IN). Cells were fixed with 3% paraformaldehyde in PBS, then incubated for 10 min in 100 mM glycine in PBS. Cells were permeabilized with 0.05% saponin (Sigma Chemical Co.) in PBS, then further incubated for 30 min at room temperature with 20 μg/ml FITC-coupled 12CA5 in 0.05% saponin, 0.2% BSA in PBS. After washing, the cells were resuspended in PBS, and the samples were analyzed by FACScan® (Becton Dickinson). Positive cells were further characterized by Western blotting using an affinity-purified rabbit antibody raised against the amino acids 13–31, mapping in the first src homology 2 (SH2) domain of the human syk (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). The sequence of this peptide is identical in rat and mouse syk. 5 × 105 cells were lysed with 0.5% Triton X-100, then run on 10% polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Millipore Corp., Bedford, MA). The filters were incubated for 1 h with 0.1 μg/ml of the anti-syk antibody, washed three times, then further incubated with a horseradish peroxidase (HRP)-coupled goat anti–rabbit antiserum (Amersham Pharmacia Biotech, Inc., Piscataway, NJ). Chemiluminescence was detected using a commercial kit (Boehringer Mannheim Corp.). The G2 and C2 clones for the FcγR/Ig-α–expressing cells, and the E8 and E10 clones for the FcγR/Ig-β–expressing cells were selected because they expressed high levels of truncated syk (30 kD), and because they expressed similar levels of FcγR chimera (detected with the rat anti–mouse FcγR antibody 2.4G2) as parental cells.
T Cell Hybridomas.
T cell hybridomas and cells used in antigen presentation assays were cultured in RPMI 1640 containing 10% FCS, 10 mM glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml), and 2β-ME (5 × 10−5 M). The specificity of all CD4+ T cell hybridomas is shown in Table 1. The C1 λ repressor–specific hybridomas 24.4, A128, 26.1, and 4G2 were characterized previously (28–30). The anti-HEL T cell hybridomas B9.1 and CAB43 and the anti-OVA T cell hybridomas 3DO 54.8 and 3DO18.3 were described previously (31, 32).
Table 1.
Presentation of Different T Cell Epitopes after Antigen Internalization through BCR Ig-α or Ig-β Subunits
| T cell hybridoma and epitopes | FcγR/Ig-α | FcγR/Ig-β | FcγR/Ig-α Y23-A | FcγR/Ig-α Y34-A | FcγR/Ig-α Y23-A Y34-A | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ repressor | ||||||||||||
| 24.4 | IAd 12-26 | + | + | + | + | + | ||||||
| 26.1 | IEd 12-26 | + | − | − | − | − | ||||||
| A128 | IAd 48-64 | + | + | ND | ND | ND | ||||||
| 4G2 | IAd 80-102 | + | − | ND | ND | ND | ||||||
| HEL | ||||||||||||
| B9.1 | IEd 108-116 | ++ | + | + | + | + | ||||||
| CAB43 | IAd 44-62 | + | − | − | − | − | ||||||
| OVA | ||||||||||||
| 3DO 54.8 | IAd | + | + | ND | ND | ND | ||||||
| 3DO 18.3 | IAd | + | − | ND | ND | ND | ||||||
The data reported in this table summarize the results of five independent experiments for the C1 hybridomas and three experiments for the HEL and OVA hybridomas. +, Increase (of at least 30-fold) in the efficiency of antigen presentation of IC compared with free antigen. −, A similar efficiency of antigen presentation of free antigen and IC.
Assays for Antigen Presentation.
Antigen presentation was assessed by culturing transfected IIA1.6 cells together with specific T cell hybridomas for 18–20 h in the presence of various concentrations of antigens complexed or not with specific antibodies. The λ repressor was complexed with the two mAbs 22D and 51F (33), the HEL (Sigma Chemical Co.) was complexed with the mAbs F10.6.14 and F9.13.7 (34), and the OVA was complexed with the IgG fraction of a rabbit anti-OVA antiserum (Sigma Chemical Co.). The complexes were preformed by incubating different concentrations of purified λ repressor, HEL, or OVA (from 30,000 to 0.5 ng) with 15 μg/ml of each mAb or 50 μg/ml of rabbit IgG at 37°C for 15 min. The release of IL-2 by the T cell hybridoma was determined by a CTL.L2 proliferation assay (35). Each point represents the average of duplicate samples, which varied by <5%.
Kinetics analysis was performed as described previously (16). In brief, a dose of antigen (3 μg/ml) was used for which antigen presentation was strictly dependent on immune complex (IC) formation with the mAbs. IC were preformed at 37°C by mixing the purified λ repressor with 51F and 22D mAbs to make a 10× mix in the culture medium. 30 μl of preformed IC was added to 270 μl of APCs adjusted to 2 × 106 cells/ml and incubated at 37°C for different times. To assess antigen presentation by A20 cells expressing TNP-specific IgM, TNP-coupled λ repressor (10 TNP per molecule of λ repressor) were incubated under similar conditions. The cells were then washed twice with PBS, fixed with 0.05% glutaraldehyde for 20 min on ice, and washed again twice. Fixed cells in duplicate samples of 100 μl were added to 50 μl of T cell hybridomas and adjusted to 2 × 106 cells/ml. After 24 h, IL-2 production was tested as above. When indicated, APCs were preincubated for 3 h at 37°C with 10 μg/ml cycloheximide diluted from a stock solution at 10 mg/ml in water. In this case, cycloheximide was also present during incubation with preformed IC before fixation with glutaraldehyde.
The binding of OVA IC to FcγRs was confirmed by immunofluorescence and FACScan® analysis. The complexes were made by mixing OVA (15 μg/ml; Sigma Chemical Co.) and the IgG fraction of a rabbit anti-OVA antiserum (50 μg/ml; Sigma Chemical Co.). For costimulation experiments, APCs were incubated with or without OVA IC for 18 h and then fixed as described above. Fixed cells in duplicate samples of 100 μl were added to 50 μl of T cell hybridomas 24.4 and 26.1 (adjusted to 2 × 106 cells/ml) and various concentrations of C1 λ repressor 12–26 peptides. In another set of experiments, APCs were incubated either with λ repressor (30 μg/ml) and preformed OVA IC to stimulate FcγR, or with preformed λ repressor IC (as described above) and F(ab′)2 fragments of specific goat anti–mouse IgG2a antibodies (15 μg/ml; Southern Biotechnology Associates, Inc., Birmingham, AL), which do not cross-react with IgG1 anti–λ repressor mAbs 51F and 22D, and specifically stimulate endogenous membrane IgG2a on IIA1.6 cells. After 18 h, the cells were fixed and incubated with C1 λ repressor–specific T cell hybridomas 24.4 and 26.1. T cell stimulation was assessed using a CTL.L2 proliferation assay.
Detection of Tyrosine Kinase Activation.
Cells were preincubated at 4°C with or without 20 μg/ml of 2.4G2 for 30 min, and then washed twice with RPMI 1640. 5 × 106 cells were then stimulated with prewarmed F(ab′)2 fragments of mouse anti–rat antiserum (50 μg/ml) at 37°C for 2 min. As a positive control, untreated cells were stimulated with prewarmed F(ab′)2 fragments of anti-IgG antibodies (30 μg/ml) for 2 min at 37°C. The stimulated cells were washed and resuspended in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.5% Triton X-100) containing a cocktail of protease inhibitors and tyrosine phosphatase inhibitors (5 mM NaF, 5 mM EDTA, 1 mM Orthovanadate). Phosphoproteins were immunoprecipitated with agarose-coupled antiphosphotyrosine antibodies PT-66 (Sigma Chemical Co.) or a rabbit antiserum raised against the 11 COOH-terminal amino acids from human syk coupled with KLH. The phosphoproteins were then run on 10% SDS-PAGE gels and transferred onto polyvinylidene difluoride membrane (Millipore Corp.) to be detected with the antiphosphotyrosine mAb Py20 coupled with HRP (ICN Biomedicals, Orsay, France). Chemiluminescence was detected with a commercial kit (Boehringer Mannheim Corp.) by exposure of the filters to X-omat film (Eastman Kodak Co., Rochester, NY). The filters were then stripped by a 30-min incubation at 50°C in 50 mM Tris, pH 6.8, 2% SDS, 100 mM β-mercaptoethanol, incubated for 1 h with 0.1 μg/ml of the anti-syk antibody (Santa Cruz Biotechnology, Inc.), washed three times, then further incubated with an HRP-coupled goat anti– rabbit antiserum. For the kinase assay, 1 μg of glutathione S-transferase (GST)-HS1 fusion protein served as a specific substrate for syk immunoprecipitated (36) in 30 μl of kinase buffer (30 mM Hepes, pH 7, 10 mM MgCl2, 5 mM MnCl2, 70 mM Orthovanadate). It was constructed by joining a BamHI-EcoRI PCR fragment containing the HSI peptide motif (EQEDEPEGDYEEVLE-Stop) in-frame into the polylinker of the pGEK-2TK vector.
IC Internalization.
The cells were washed once in internalization buffer (RPMI 1640, 5% FCS, 10 mM glutamine, 5 mM sodium pyruvate, 50 mM 2-ME, and 20 mM Hepes, pH 7.4) and incubated with HRP–anti-HRP IC for 2 h at 4°C (107 cells/ml). IC were prepared as a 10× solution in internalization buffer (HRP 50 μg/ml, and a polyclonal rabbit anti-HRP antibody at 400 μg/ml) for 30 min at 37°C. After fixation of HRP IC, the cells were washed three times in internalization buffer and incubated at 37°C for various times (2 × 106 cells/ml). Internalization was stopped by adding cold internalization buffer, and the cells were washed once in PBS. Duplicates for each time point were either left in PBS at 4°C to measure cell surface HRP IC, or incubated in Triton X-100 (0.1%) for 5 min at room temperature to measure the total amount of HRP IC. The HRP was revealed by adding substrate buffer (0.5 mg/ml OPD [Sigma Chemical Co.] and 0.12% H2O2 in 0.05 M phospho-citrate buffer, pH 5.0) at 4°C. The reaction was stopped with 6 N HCl, and the change in color was determined spectrophotometrically at 492 nm.
Results
BCR-mediated Antigen Uptake Induces the Presentation of Numerous T Cell Epitopes.
The binding of exogenous antigens to the BCR potentiates antigen uptake and degradation in B lymphocytes. To determine if BCR-mediated antigen uptake has any consequences for the presentation of various T cell epitopes, we compared antigen presentation after fluid phase or BCR-mediated antigen uptake using A20 B cells expressing or not expressing TNP-specific IgM. The λ phage repressor (C1) was used as antigen. TNP-coupled (3 μg/ml) or -uncoupled C1 (30 μg/ml) was incubated for various times with A20 cells expressing anti-TNP IgM (A20 anti-TNP). The cells were then fixed, and C1 presentation was detected with two T hybridomas specific for the same peptide in association with IA or IE of the H2d haplotype: the hybridoma 24.4, which recognized a dominant T cell epitope (IAd 12-26), and the hybridoma 26.1, which recognized a cryptic T cell epitope (IEd 12-26) (28). These two T cell epitopes were detected on anti-TNP A20 cells after 2 h of incubation with TNP-coupled antigens (Fig. 1 a). The incubation of presenting cells with the protein synthesis inhibitor cycloheximide for 3 h before and during incubation with TNP-coupled C1 completely abolished BCR-mediated antigen presentation (Fig. 1 a). In contrast, fluid phase uptake of uncoupled C1 (Fig. 1 a) only led to the presentation of the IAd 12-26 epitope after a lag time of 4 h. Under these conditions, antigen presentation was partially resistant to the cycloheximide treatment. Therefore, BCR-mediated antigen internalization has a dual function in antigen presentation: by addressing antigens to newly synthesized MHC class II molecules, BCR induces the presentation of T cell epitopes which are not presented after fluid phase antigen uptake.
Figure 1.
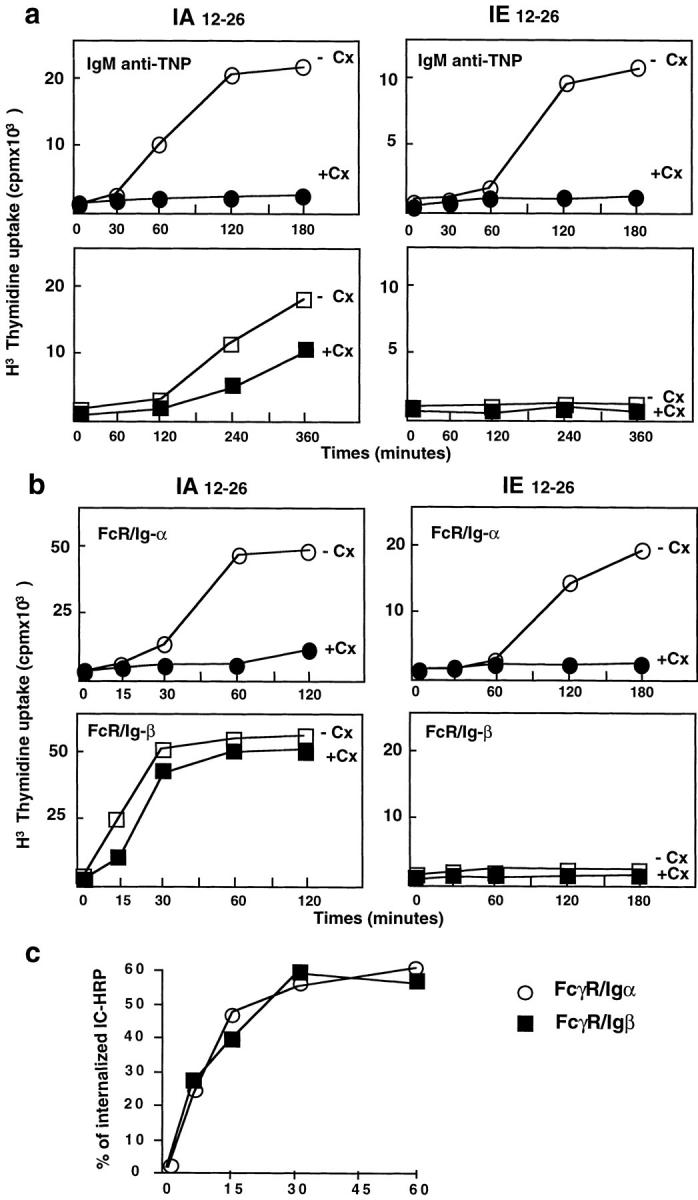
The BCR and BCR Ig-α subunit determined antigen presentation by newly synthesized class II molecules. (a) A20 cells expressing anti-TNP–specific mIg were incubated with TNP-coupled (top, circles) or uncoupled (bottom, squares) λ repressor (3 and 30 μg/ml, respectively) for various times with (+) or without (−) cycloheximide (Cx; 10 μg/ml). The cells were then fixed with glutaraldehyde before incubation with the T cell hybridoma 24.4, specific for the IAd 12-26 dominant epitope, or with the T cell hybridoma 26.1, specific for the IEd 12-26 cryptic epitope. (b) IIA1.6 cells expressing either FcγR/Ig-α (top) or FcγR/Ig-β chimera (bottom) were incubated with C1 λ repressor–IgG complexes for various times with (+) or without (−) cycloheximide (Cx; 10 μg/ml). The cells were then fixed with glutaraldehyde before incubation with the same two T cell hybridomas for 24 h. T cell stimulation was evaluated by a CTL.L2 proliferation assay. Similar results were obtained in three independent experiments. (c) Internalization of the HRP–anti-HRP IC by FcγR/Ig-α or FcγR/Ig-β chimera was measured as described in Materials and Methods.
The cytoplasmic tail of the BCR Ig-α subunit is able to target antigen to newly synthesized MHC class II (16). Therefore, we investigated whether the direct targeting of C1 through the cytoplasmic tail of Ig-α is able to induce the presentation of the dominant, IAd 12-26, and the cryptic, IEd 12-26, T cell epitopes. IIA1.6 B cells (FcγR-defective variant of A20 B cells) expressing FcγR chimeras containing the cytoplasmic tail of Ig-α or Ig-β (FcγR/Ig-α and FcγR/Ig-β, respectively [22]) were incubated with C1 (3 μg/ml) complexed with two anti-C1 mAbs (15 μg/ml each) to target C1 on FcγR chimeras. After various periods of culturing at 37°C, the presenting cells were fixed and incubated with the two T cell hybridomas 24.4 and 26.1. The presentation of IAd 12-26 and IEd 12-26 T cell epitopes occurred with similar kinetics on FcγR/Ig-α–expressing cells (Fig. 1 b) and A20 anti-TNP (Fig. 1 a). In addition, the incubation of presenting cells with cycloheximide inhibited antigen presentation induced by direct targeting of C1 through the Ig-α cytoplasmic tail (Fig. 1 b). Surprisingly, similar experiments with FcγR/Ig-β–expressing cells revealed that endosomal targeting of C1 to recycling class II by the Ig-β cytoplasmic tail (16) was not able to induce presentation of the cryptic IEd 12-26 T cell epitope, whereas the dominant IAd 12-26 T cell epitope was efficiently and quickly presented by recycling class II molecules (no effect of cycloheximide treatment; Fig. 1 b). Using HRP–anti-HRP rabbit IgG IC as a multivalent ligand of FcR chimeras, we observed that both Ig-α and Ig-β chimeras internalized IC with similar efficiency and kinetics (Fig. 1 c). Therefore, the incapacity of the Ig-β chimera to induce presentation of the IEd 12-26 epitope was not due to inefficient ligand internalization. These results suggested that the BCR induced presentation of a dominant and a cryptic T cell epitope by newly synthesized class II molecules through a targeting signal contained in the cytoplasmic tail of Ig-α.
Therefore, a function of the BCR in antigen presentation might be to induce the presentation of a large spectrum of T cell epitopes by targeting antigens to newly synthesized class II. To address this question, we used a panel of T cell hybridomas specific for various peptides deriving from C1, HEL, or OVA, restricted by the IA or IE H2d class II molecules. The efficiency of presentation of IgG-complexed antigens was compared in FcγR/Ig-α– and FcγR/Ig-β–expressing cells (Table 1). The targeting of these different proteins through the cytoplasmic tail of Ig-α induced the presentation of all epitopes tested, i.e., C1 IAd 48-64 and IAd 80-102, HEL IEd 108-116 and IAd 44-62, and the IAd-restricted OVA epitopes recognized by the 3DO54.8 and the 3DO18.3 T cell hybridomas. In contrast, the processing of the same IC by FcγR/Ig-β–expressing cells led only to the presentation of some of these T cell epitopes (Table 1). Antigen targeting by the Ig-α or Ig-β cytoplasmic tails through newly synthesized or recycling class II molecules, respectively, did not lead to the presentation of the same peptides. These results raised the question, What is the intracellular mechanism of specific presentation of these T cell epitopes by Ig-α and BCR-mediated antigen internalization?
B Cell Activation Is Not Sufficient for Presentation of the Cryptic Epitope.
The Ig-α and Ig-β cytoplasmic tails induce the activation of different signaling pathways (22) and interact with distinct cytoplasmic effectors (21). Therefore, the selective presentation of epitopes by B cells expressing anti-TNP–specific IgM or FcγR/Ig-α chimera might be either a direct consequence of a different intracellular targeting of antigen, or an indirect effect of B cell activation which might induce surface expression of putative costimulatory molecules required for the efficient activation of T cell hybridomas.
To distinguish between these possibilities, the FcγR/ Ig-α– and FcγR/Ig-β–expressing cells were incubated with or without irrelevant IC (OVA, 15 μg/ml; anti-OVA rabbit IgG, 50 μg/ml), to induce B cell activation (not shown). After 24 h, the cells were then fixed and incubated with increasing concentrations of 12-26 peptide and either the 24.4 or the 26.1 T cell hybridoma. The peptide was presented to both T cells with similar efficiency by the unstimulated and the stimulated cells (Fig. 2 a). Similar results were obtained when the cells were fixed after 6 or 12 h incubation with IC (not shown). Therefore, the signaling pathways induced through Ig-α and Ig-β did not modify the efficiency of presentation of the 12-26 peptide by IAd and IEd molecules.
Figure 2.

B cell activation per se does not induce presentation of the IEd-restricted epitope. (a) FcγR/Ig-α and FcγR/Ig-β chimera–expressing cells were incubated for 24 h with OVA–anti-OVA IC. The cells were then fixed with glutaraldehyde and incubated at increasing concentrations of 12-26 C1 λ repressor peptide and the T cell hybridomas specific for the dominant (IAd 12-26) or cryptic (IEd 12-26) epitopes. The secretion of IL-2 in the cell culture supernatants was measured. Activation of chimera-expressing cells with IC did not modify the efficiency of presentation of the 12-26 peptide. Similar results were obtained in two independent experiments. (b) Cell activation through FcγR/Ig-α chimera does not induce presentation of the cryptic epitope after antigen internalization by fluid phase uptake. FcγR/Ig-α–expressing cells were incubated overnight in the presence of 30 μg/ml C1 λ repressor with or without irrelevant OVA–anti-OVA IC, in order to engage FcγR/Ig-α chimeric receptors. The cells were then fixed and reincubated overnight with T cell hybridomas specific for the dominant or the cryptic epitopes. Engagement of FcγR/Ig-α chimeric receptors did not induce presentation of the IEd-restricted cryptic epitope. (c) Cell activation through surface IgG does not induce the presentation of the C1 λ repressor cryptic epitope after IC internalization through FcγR/Ig-β. Cells expressing FcγR/Ig-β were incubated continuously in the presence of C1 λ repressor IC in the absence or presence of specific goat anti–mouse IgG2a F(ab′)2 fragments, in order to induce cell activation through endogenous surface IgG2a. The cells were then fixed and reincubated overnight with T cell hybridomas specific for the dominant or the cryptic epitopes. Cell activation through surface IgG2a did not induce presentation of the cryptic epitope by FcγR/ Ig-β –expressing cells. Errors bars in b and c indicate the mean variation of T cell stimulation of three experiments.
Nevertheless, the BCR or Ig-α cytoplasmic tail could induce signaling events that modified the composition of processing compartments, leading to the presentation of the IEd 12-26 epitope. The effect of Ig-α–induced cell activation on intracellular processing of C1 was assessed by coincubating FcγR/Ig-α–expressing cells with irrelevant IC (as shown in Fig. 2 a) in order to trigger cell activation through the Ig-α cytoplasmic tail, and with soluble C1 (30 μg/ml), which typically only induced the presentation of the IAd 12-26 epitope (see Fig. 1 a and Fig. 3 b). After 24 h, the cells were fixed and incubated either with 24.4 or 26.1 T cells (Fig. 2 b). B cell signaling through Ig-α did not lead to the presentation of the IEd 12-26 epitope and did not increase the presentation of the IAd 12-26 epitope after the fluid phase uptake of the C1 (Fig. 2 b).
Figure 3.
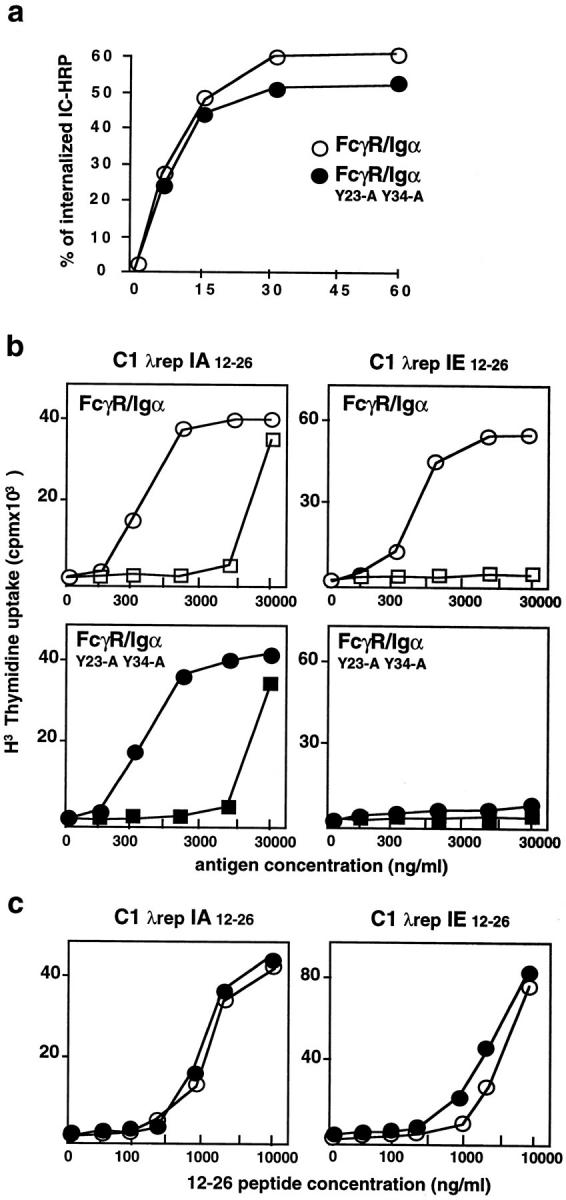
Tyrosine residues of Ig-α ITAM are involved in the selective presentation of a cryptic epitope. (a) Internalization of the HRP–anti-HRP IC by FcγR/Ig-α chimera or FcγR/Ig-α Y23A Y34A mutant was measured as described in Materials and Methods. (b) IIA1.6 cells expressing either FcγR/Ig-α chimera or FcγR/Ig-α Y23A Y34A mutant were cultured in the presence of increasing concentrations of C1 λ repressor (fluid phase uptake, squares) or C1 λ repressor–IgG complexes (FcγR-mediated endocytosis, circles). T cell hybridomas specific for a dominant epitope (IAd 12-26) or a cryptic epitope (IEd 12-26) from the C1 λ repressor were also included in the cultures. The secretion of IL-2 by the T cell hybrids was measured after an overnight incubation. (c) Both transfected cell lines presented the 12-26 peptide on IAd or IEd with identical efficiency. The data presented are representative of results obtained in three independent experiments.
However, fluid phase antigen uptake is not very efficient in B cells, and could be a limiting step that leads to failure to generate enough peptides to induce the presentation of the IEd 12-26 epitope. To test this possibility, the FcγR/ Ig-β–expressing cells were simultaneously incubated with IgG1-complexed C1 (to allow efficient generation of 12-26 peptide) and with specific anti-IgG2a antibodies (to induce B cell activation through endogenous mIg). The cells were then fixed and incubated with the two T cell hybridomas. BCR stimulation did not induce FcγR/Ig-β–expressing cells to present IEd 12-26 epitope and did not increase presentation of the IAd 12-26 epitope (Fig. 2 c). Therefore, the stimulation of the whole BCR was not sufficient to allow presentation of the cryptic IEd 12-26 epitope after C1 internalization through the Ig-β cytoplasmic tail; however, the 12-26 peptide was efficiently generated since it was presented by IAd molecules, although the direct targeting of C1 to the TNP-specific BCR was able to induce presentation of the same T cell epitope (Fig. 1 a). In addition, the failure of the Ig-β chimera to induce presentation of cryptic IEd 12-26 epitope was not due to a lower sensitivity of the T cell hybridoma, as suggested by the results obtained for fixed presenting cells (shown in Fig. 2 a), since similar results were obtained with unfixed cells (Table 1). Indeed, on unfixed cells, the two T cell hybridomas detected the IAd 12-26 and IEd 12-26 T cell epitopes with similar sensitivity (see Fig. 3 c, and data not shown). These data suggested that the BCR Ig-α subunit contains a signal that can recruit proteins involved in the antigen targeting toward endosomal compartments, where the 12-26 peptide may be generated and associated to IAd and IEd MHC class II molecules.
Tyrosine Residues Contained in the Ig-α Cytoplasmic Tail Are Required for Receptor-mediated Presentation of the Cryptic IEd 12-26 T Cell Epitope.
The two major functions assigned to the Ig-α/Ig-β heterodimer are intracellular signaling and receptor internalization, both dependent on ITAMs contained in their cytoplasmic tails. To discriminate between these two functions of the BCR subunits, we analyzed antigen presentation through FcγR/Ig-α chimeras containing mutated tyrosine residues, previously shown to prevent the activation of tyrosine kinases (25).
First, we analyzed IC internalization by the FcγR/Ig-α chimera in which tyrosine residues of Ig-α ITAM were replaced by alanines (FcγR/Ig-α Y23-A Y34-A). HRP– anti-HRP rabbit IgG IC were bound to wild-type and mutated FcγR/Ig-α–expressing cells at 4°C, then the cells were cultured at 37°C for various times to allow receptor internalization. Internal and cell surface fractions of IC were determined by an enzymological assay. As shown in Fig. 3 a, both wild-type and mutated Ig-α chimeras internalized IC with similar kinetics. Tyrosine residues of Ig-α ITAM, as for Ig-β (37), did not appear to be required for the internalization of multivalent ligands.
Therefore, we evaluated whether mutation of these tyrosine residues, which inhibit B cell activation, altered the presentation of the dominant, IAd 12-26, and the cryptic, IEd 12-26, T cell epitopes. Cells were incubated with various concentrations of C1 complexed or not with two anti-C1 mAbs (15 μg/ml each) and the two T cell hybridomas. Although wild-type and double-mutated FcγR/Ig-α chimeras were able to induce presentation of the IAd 12-26 epitope at low concentrations of IgG-complexed C1, no presentation of the cryptic IEd 12-26 epitope was detected when IC were internalized through the FcγR/Ig-α Y23-A Y34-A chimera (Fig. 3 b). We verified that stimulation of the 24.4 and 26.1 T cell hybridomas was obtained at similar concentrations of 12-26 peptides with FcγR/Ig-α– or FcγR/Ig-α Y23-A Y34-A–expressing cells (Fig. 3 c). In addition, we obtained similar results with B cells expressing Ig-α chimera where the first (FcγR/Ig-α Y23-A) or the second (FcγR/Ig-α Y34-A) tyrosine residue of Ig-α ITAM was mutated to alanine (Table 1). The mutation of tyrosine residues of the Ig-α ITAM also affected the presentation of IAd 48-62 HEL epitopes, but had no effect on the IEd 108-116 T cell epitope derived from HEL when the antigen was complexed with specific antibodies (Table 1). Therefore, we concluded that mutations that abolished the ability of the Ig-α cytoplasmic tail to induce B cell activation also blocked the intracellular targeting of antigens, determining the presentation of some, but not all, T cell epitopes.
The Recruitment of Syk Tyrosine Kinase Is Required for BCR-mediated Antigen Presentation.
After BCR stimulation, tyrosine kinases phosphorylate ITAM tyrosine residues, enabling them to associate with the two SH2 domains of syk tyrosine kinase (18, 19). Since mutation of the tyrosine of Ig-α ITAM inhibits the generation of some T cell epitopes, we further evaluated the role of syk tyrosine kinase in antigen presentation through the BCR Ig-α subunit.
First, we tested whether the double mutation of both tyrosine residues contained in Ig-α ITAM affected syk activation through the cytoplasmic tail of Ig-α. Wild-type and mutated FcγR/Ig-α–expressing cells were stimulated for 2 min at 37°C through FcγRs (using the anti-FcγR mAb, 2.4G2), or through endogenous mIgG2a (using F(ab′)2 fragments of rabbit anti–mouse IgG), or, in a control experiment, with irrelevant F(ab′)2 fragments of mouse anti– rat IgG. Cell lysates were prepared, and phosphotyrosine proteins were immunoprecipitated with agarose-coupled PT66 and analyzed by immunoblotting with HRP-coupled antiphosphotyrosine mAb Py20 or rabbit anti-syk antisera. As shown in Fig. 4 a, B cell stimulation through endogenous BCR or the Ig-α chimera induced the phosphorylation of similar proteins, among them the syk tyrosine kinase (Fig. 4 b). Mutated Ig-α chimera lacked the ability to induce phosphorylation of syk as well as of other kinases substrates, although endogenous mIgs were functional in these cells (Fig. 4). In addition, the kinase activity of syk was increased after FcγR/Ig-α stimulation but not after cross-linking of the tyrosine-mutated Ig-α chimera as measured by phosphorylation of a specific substrate of syk kinase, a GST fusion protein containing HS1 polypeptide (reference 36; Fig. 4 b). Therefore, the Ig-α cytoplasmic tail is able to activate syk tyrosine kinase and to mediate the presentation of numerous T cell epitopes via a targeting signal located in the Ig-α ITAM.
Figure 4.
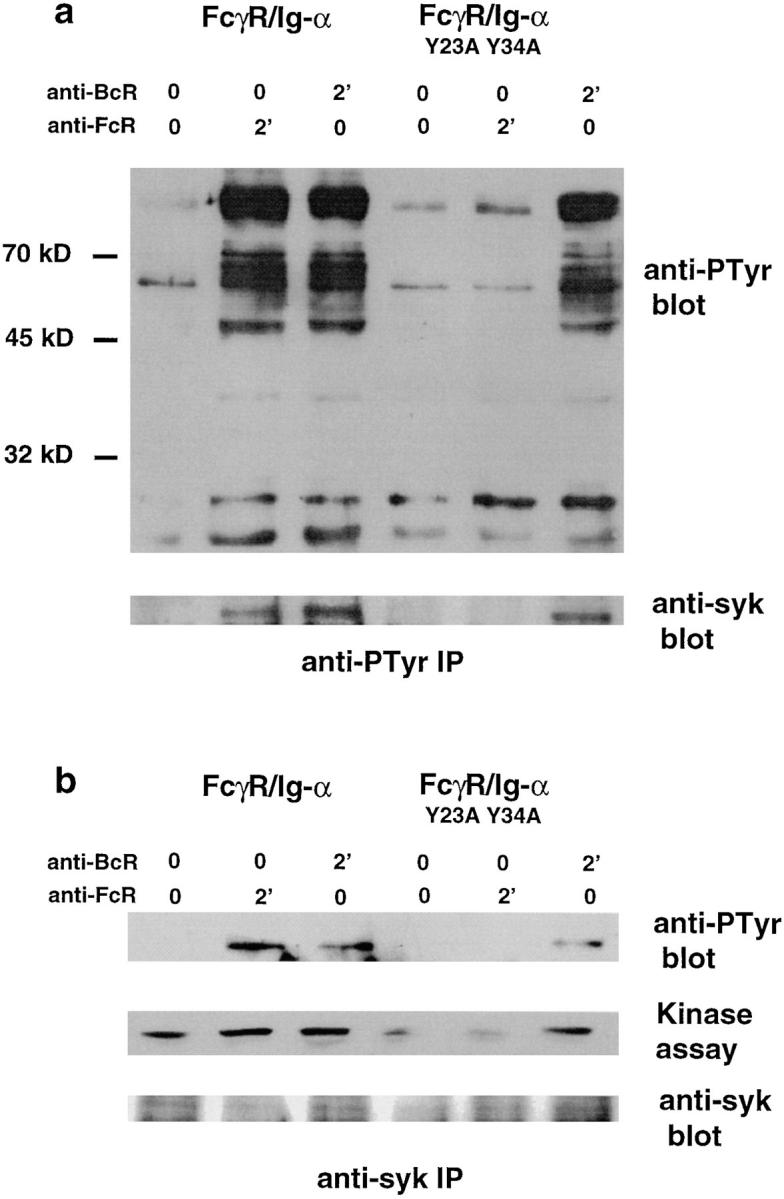
Tyrosine residues of Ig-α ITAM are required for activation of the syk tyrosine kinase through the BCR. IIA1.6 cells expressing either FcγR/Ig-α chimera or FcγR/Ig-α Y23A Y34A mutant were left unstimulated or stimulated through endogenous BCR with F(ab′)2 fragments of rabbit anti–mouse IgG antibodies (15 μg/ml), or through Fc chimera with the rat anti–mouse FcγR antibody 2.4G2 (20 μg/ml) then F(ab′)2 fragments of mouse anti–rat IgG (30 μg/ml). In control lanes, the cells were incubated for the same time with only F(ab′)2 fragments of mouse anti–rat IgG (30 μg/ml). The cells were then washed and lysed with 0.5% Triton X-100. Cell lysates were immunoprecipitated with antiphosphotyrosine antibodies (a) or rabbit anti-syk antibodies (b). (a) Phosphoproteins were detected by Western blotting using HRP-coupled antiphosphotyrosine antibody Py20 (top), or a rabbit antiserum specific for the NH2-terminal end of the syk tyrosine kinase (bottom). (b) The phosphorylation of syk in immunoprecipitates was detected by Western blotting using HRP-coupled antiphosphotyrosine antibody Py20 (top). The syk tyrosine kinase activity was measured in a kinase assay using a specific substrate, the GST–HS1 fusion proteins. The phosphorylation of GST– HS1 fusion proteins was detected by Western blotting using HRP-coupled antiphosphotyrosine antibody Py20 (middle). The rate of immunoprecipitated syk tyrosine kinase was controlled for each point on the same filter using a rabbit antiserum specific for the NH2-terminal end of the syk tyrosine kinase (bottom).
To further evaluate the role of syk tyrosine kinase in antigen presentation by B cells, we devised a strategy to specifically block the recruitment and activation of syk by the Ig-α subunit of the BCR. For this purpose, an inactive form of syk was stably expressed in FcγR/Ig-α–expressing B cells. A cDNA encoding the two SH2 domains of syk tagged with an HA peptide but lacking its kinase domain was inserted in an expression vector bearing the puromycin resistance gene. After transfection, expressing cells were selected with puromycin and cloned. The clones were screened for the expression of HA-tag by intracellular FACScan® analysis using FITC-coupled anti-HA mAb (not shown). Positive clones were further characterized by Western blotting using a rabbit antiserum specific for a peptide contained in the NH2 portion of the syk SH2 domains present both in dominant negative mutant (32 kD) and in endogenous syk (70 kD; Fig. 5 a). The activation of endogenous syk kinase through the Ig-α cytoplasmic tail was then analyzed in the A2 and G2 clones.
Figure 5.
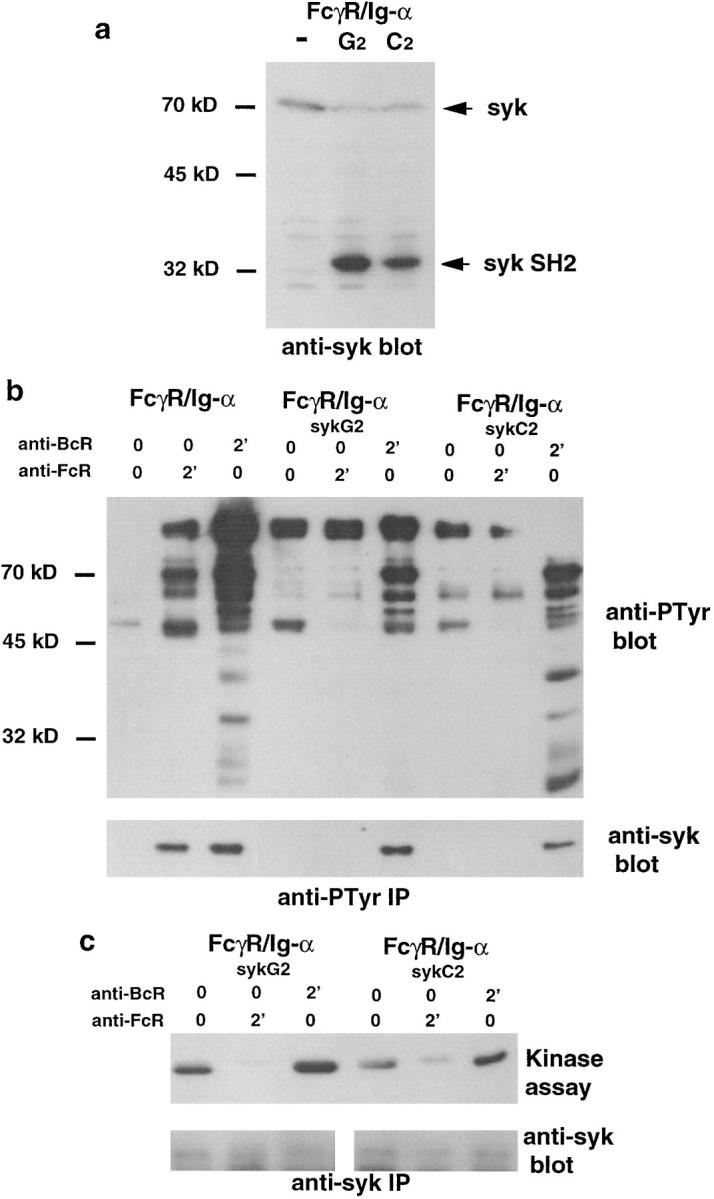
A dominant negative mutant of syk inhibits the activation of endogenous syk tyrosine kinase through the BCR Ig-α subunit. (a) A cDNA encoding the two SH2 domains of the rat syk tyrosine kinase was stably overexpressed in FcγR/Ig-α chimera cells. The clones G2 and C2 are representative of a series of 20 independent clones characterized by Western blotting using a rabbit antiserum specific for the NH2-terminal end of syk. Arrowheads, Endogenous syk tyrosine kinase (70 kD) and the dominant negative mutant (32 kD). FcγR/Ig-α–positive cells overexpressing or not the dominant negative syk (G2 and C2 clones) mutant were left unstimulated or stimulated through the endogenous BCR or Fc chimera as described in the legend to Fig. 4. The cells were then washed and lysed with 0.5% Triton X-100. Cell lysates were immunoprecipitated with antiphosphotyrosine antibodies (b) or rabbit anti-syk antibodies (c). (b) Phosphoproteins (top) or syk tyrosine kinase (bottom) was detected by Western blotting as in Fig. 4. (c) The phosphorylation of GST–HS1 fusion proteins was detected, after a kinase assay, by Western blotting using HRP-coupled antiphosphotyrosine antibody Py20 (top). The quantity of immunoprecipitated syk was controlled for each point on the same filter using a rabbit antiserum specific for the NH2-terminal end of syk (bottom).
For this purpose, the two clones were left unstimulated or stimulated through the Ig-α chimera or endogenous mIgG2a, then phosphoproteins were immunoprecipitated and analyzed by Western blotting using Py20 and anti-syk antibodies. Overexpression of the syk dominant negative mutant prevented the phosphorylation of various intracellular proteins, as well as endogenous syk kinase, induced through the Ig-α cytoplasmic tail. Although global tyrosine phosphorylation was affected, syk phosphorylation was only marginally decreased after B cell stimulation through endogenous mIg (Fig. 5 b). Similarly, we found that overexpression of syk SH2 domains led to a substantial inhibition of kinase activity in syk immunoprecipitates obtained after cell stimulation through the Ig-α cytoplasmic tail, whereas only a marginal effect was found after endogenous mIg stimulation, although the same quantity of syk protein was immunoprecipitated (Fig. 5 c). These results suggested that syk could be activated through an Ig-α–dependent and –independent pathway.
To verify this hypothesis, we examined the effect of overexpression of syk dominant negative mutant on syk activation through the cytoplasmic tail of Ig-β. Clones overexpressing the syk dominant negative mutant were obtained in B cells expressing the FcγR/Ig-β chimera. The E8 and E10 clones, shown in Fig. 6 a, were analyzed. Syk kinase activity and phosphorylation could not be inhibited by overexpression of syk SH2 domains after stimulation of the Ig-β chimera (Fig. 6 b). In conclusion, both subunits of the Ig-α/Ig-β heterodimer were able to activate syk tyrosine kinase, but only syk activation through Ig-α was inhibited by the syk dominant negative mutant. This model allowed us to evaluate the role of syk kinase in MHC class II–restricted antigen presentation through the subunits of the BCR.
Figure 6.

The syk dominant negative mutant did not affect endogenous syk activation of endogenous syk tyrosine kinase through the BCR Ig-β subunit. (a) A cDNA encoding the two syk SH2 domains was stably overexpressed in FcγR/Ig-β chimera–positive cells. The E8 and E10 clones are representative of a series of 10 independent clones characterized by Western blotting using a rabbit antiserum specific for the NH2-terminal end of syk. Arrowheads, Endogenous syk tyrosine kinase (70 kD) and the dominant negative mutant (32 kD). (b) FcγR/Ig-β–positive cells overexpressing the dominant negative syk (E8 and E10 clones) mutant were left unstimulated or stimulated through the endogenous BCR or Fc chimera as in the legend to Fig. 4. The cells were then washed and lysed with 0.5% Triton X-100. Cell lysates were immunoprecipitated with rabbit anti-syk antibodies (top; the kinase activity was measured and controlled as in the legend to Fig. 4) or with antiphosphotyrosine antibodies (bottom; phosphorylated syk was blotted with an anti-syk antiserum as in the legend to Fig. 4).
Using HRP–anti-HRP rabbit IgG IC, we first established that the overexpression of the syk dominant negative mutant did not affect the internalization of cross-linked Ig-α chimera (Fig. 7 a) or Ig-β chimera (not shown). The presentation of C1 was next analyzed in cells overexpressing the syk dominant negative mutant. No presentation of the cryptic IEd 12-26 epitope was detected when IC were internalized through the FcγR/Ig-α chimera in cells overexpressing the two syk SH2 domains, whereas presentation of the dominant IAd 12-26 epitope was only slightly reduced (Fig. 7 b). No effect of the syk dominant negative mutant was observed with FcγR/Ig-β–expressing cells (Fig. 7 b), although the cells stimulated the 24.4 and 26.1 T cell hybridomas at similar concentrations of 12-26 peptide (Fig. 7 c). In addition, overexpression of the syk dominant negative mutant slowed down presentation of the IAd 12-26 epitope after the internalization of IgG-complexed C1 through the Ig-α chimera (Fig. 7 d ). Therefore, the overexpression of the syk dominant negative mutant seemed able to modulate the presentation of T cell epitopes which are specifically generated by antigen targeting to newly synthesized, such as the cryptic IEd 12-26, T epitope issuing from the C1. The syk tyrosine kinase, a major effector of the BCR-induced signaling pathway, therefore, is also involved in BCR-mediated antigen presentation by newly synthesized MHC class II molecules.
Figure 7.
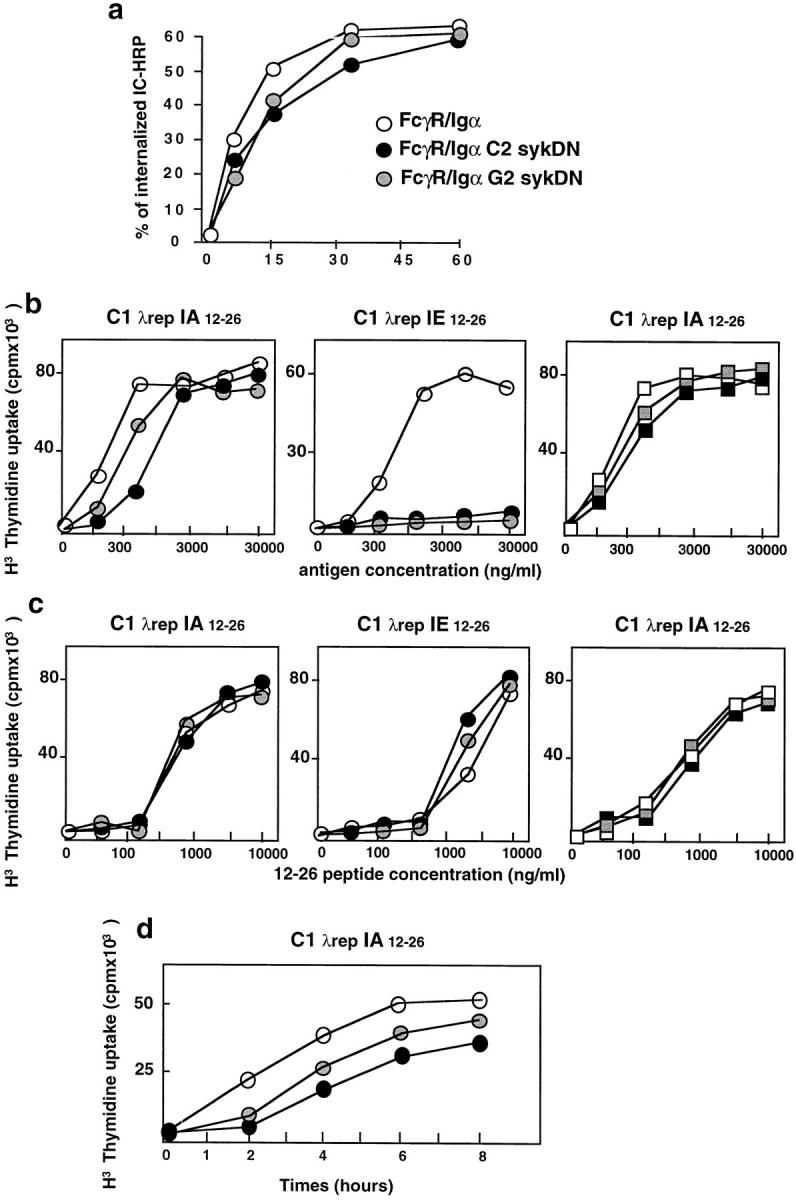
Overexpression of syk dominant negative mutant selectively affected presentation of the cryptic T cell epitope through the BCR Ig-α subunit. (a) Internalization of the HRP–anti-HRP IC by the FcγR/Ig-α chimera was measured in G2 ( gray circles) and C2 (black circles) clones overexpressing the syk dominant negative mutant. (b) FcγR/Ig-α (circles) and FcγR/Ig-β (squares) chimera–positive cells overexpressing (black and gray) or not (white) syk dominant negative mutant were cultured in the presence of increasing concentrations of C1–IgG complexes (FcγR-mediated endocytosis). T cell hybridomas specific for a dominant epitope (IAd 12-26) or a cryptic epitope (IEd 12-26) from the C1 λ repressor were also included in the cultures. The secretion of IL-2 by the T cell hybrids was measured after an overnight incubation. (c) All the transfected cell lines presented the 12-26 peptide on IAd or IEd with identical efficiency. (d) The FcγR/Ig-α–positive presenting cells, overexpressing (black and gray) or not (white) syk dominant negative mutant, were incubated with C1– IgG complexes for various times, then fixed with glutaraldehyde before incubation with the IAd 12-26–specific T cell hybridomas for 24 h. T cell stimulation was evaluated by a CTL.L2 proliferation assay. Similar results were obtained in three independent experiments.
Discussion
Capture of antigens by BCRs is the first step in antigen entry into the endosomal pathway, where antigenic peptides are generated and associated with newly synthesized MHC class II molecules. We report here that antigen targeting to newly synthesized class II molecules through the BCR or Ig-α–associated subunit induces the presentation of a larger spectrum of T cell epitopes than antigen targeting to recycling class II molecules. Using mutagenesis analysis of the Ig-α cytoplasmic tail and stable overexpression in B cells of a syk dominant negative mutant, we describe evidence that signal transduction effectors of B lymphocyte activation are involved in the generation of peptide–MHC class II complexes.
How do Ig-α and Ig-β cytoplasmic tails induce presentation of different T cell epitopes? Quantitative differences in the rate of antigen internalization can be excluded, since the Ig-α chimera was expressed at a lower level than the Ig-β chimera (22), and since both chimeric receptors mediated internalization of IC with similar kinetics and efficiency (Fig. 1 c [16]). In contrast, the kinetics of antigen presentation induced by the Ig-α and Ig-β cytoplasmic tails demonstrated that they target antigens to newly synthesized or recycling pools of MHC class II molecules, respectively. Analysis of various T cell epitopes provides new evidence to further distinguish these two antigen-presentation pathways. Thus, antigen internalization via the Ig-α, but not the Ig-β, chimera stimulated the DO18.3 hybridoma (Table 1), which recognizes an IAd-restricted OVA T cell epitope specifically generated in invariant chain–positive presenting cells (32, 38). In contrast, the invariant chain– independent epitope recognized by DO54.8 hybridoma (32) is presented after antigen internalization through the Ig-α or Ig-β chimera. These data suggest that the Ig-α cytoplasmic tail targets antigens to endosomal compartments, where class II molecules transiently accumulate through an invariant chain–dependent mechanism which characterizes newly synthesized class II molecules. In addition, the data also supported the hypothesis that some T cell epitopes cannot be generated in an alternative antigen-presentation pathway defined by compartments where class II molecules cycle with the cell surface (6, 7). Indeed, antigen targeting to recycling class II molecules through Ig-β did not induce the stimulation of DO18.3 hybridoma or other T cell hybridomas specific for cryptic or subdominant epitopes derived from the λ repressor and HEL. Therefore, the two MHC class II–restricted antigen pathways can be functionally distinguished on the basis of T cell epitopes they are able to efficiently generate. However, the complete BCR as well as the Ig-α chimera address antigen to the newly synthesized pool of MHC class II molecules, presenting a large spectrum of peptides. Therefore, the Ig-α subunit is dominant over Ig-β in this process and accounts for the ability of the BCR to induce efficient antigen presentation during secondary in vivo immune responses. This conclusion raised a new question: What is the intracellular mechanism of BCR- or Ig-α–mediated antigen presentation?
Two distinct functions have been assigned to Ig-α and Ig-β: first, to address antigen to different pools of MHC class II molecules (16), and second, to induce different signaling pathways (22, 25). Therefore, the overall cell activation induced by Ig-α or the complete BCR might be responsible for the ability of the Ig-α chimera to induce stimulation of numerous T cells. However, this hypothesis is unlikely because, first, the stimulation of 12-26 IAd– and IEd–restricted T cell hybridomas by Ig-α chimera–expressing cells was not modified by chimera cross-linking, and second, the overall cell activation induced by BCR cross-linking was not able to increase presentation of the IAd 12-26 epitope or to induce presentation of the IEd 12-26 epitope by Ig-β chimera–expressing cells. These data indicated that potential alterations in antigen processing during cell activation do not account for induction of presentation of these T cell epitopes. In addition, we excluded the possibility that BCR or Ig-α chimera cross-linking increased cell surface expression of MHC class II or costimulatory molecules such as B.7 or CD40 ligand (not shown). In contrast, mutagenesis analysis identifies, in the Ig-α cytoplasmic tail, a targeting signal able to induce efficient antigen presentation of numerous peptides. Indeed, mutation of tyrosine residues contained in the Ig-α ITAM, which completely abolish signal transduction (25, 39, 40), affects Ig-α–mediated antigen presentation, whereas similar mutation in Ig-β ITAM did not affect BCR internalization and BCR-mediated antigen presentation (37). These results indicate that tyrosine residues of Ig-β and Ig-α ITAM are differently involved in the recruitment of cytoplasmic effectors mediating antigen presentation.
Tyrosine mutants of the Ig-α ITAM also provide a new tool to dissociate the two major functions of the BCR, ligand internalization and signal transduction, since these mutations block activation of most of the tyrosine kinases but had no effect on receptor internalization. In addition, these mutations specifically alter the presentation of some, but not all, T cell epitopes and inhibited the phosphorylation and activation of syk tyrosine kinase. The role of the kinase activity of syk in BCR-mediated antigen presentation was investigated using B cells overexpressing the two SH2 domains of syk. This mutant protein may be recruited by tyrosine-phosphorylated ITAM and should act as a dominant negative mutant of ITAM function. Indeed, in cells overexpressing the syk mutant, the activation of endogenous syk kinase was blocked after Ig-α chimera stimulation and was inhibited after BCR stimulation, whereas syk activation through Ig-β was not affected by overexpression of the syk mutant. It is probable that the ITAMs of both BCR subunits activated syk by different mechanisms as suggested by previous data indicating that the Ig-α and Ig-β cytoplasmic tails interact with distinct cytoplasmic effectors (21) and activate different signaling pathways (22). An attractive hypothesis is that only the phosphorylated ITAM of Ig-α directly interacts with the SH2 domains of syk and induce syk activation, whereas the activation of syk through the Ig-β cytoplasmic tail is determined by the interaction of another molecule. Whatever the intracellular mechanism of syk activation through the two BCR subunits, our data clearly show that the overexpression of the syk mutant differentially affects antigen presentation through Ig-α and Ig-β. This is in agreement with previous data showing that the mutation of Ig-β ITAM tyrosine residues did not affect Ig-β–mediated antigen presentation (37), whereas similar mutations in Ig-α ITAM only affected the presentation of some, but not all, T cell epitopes (Table 1). Therefore, there is compelling evidence that functionally distinguishes classic and alternative pathways of antigen presentation. Different peptides associate with recycling and newly synthesized pools of class II molecules which are specifically targeted by signals contained in the cytoplasmic tails of antigen receptors. Although these signals are located in a similar ITAM, they can be functionally distinguished. Mutation of tyrosine residues in ITAM and overexpression of the syk dominant negative mutant did not affect antigen presentation using recycling class II molecules through the Ig-β cytoplasmic tail, whereas both alter antigen presentation using newly synthesized class II through the Ig-α cytoplasmic tail. These data indicate that although both Ig-α– and Ig-β–phosphorylated ITAMs activate syk, the kinase domain of syk seems to be involved only in the targeting of antigen receptors to newly synthesized class II molecules which accumulated, before cell surface arrival, in endocytic compartments called CIIV (for Class II vesicles [3]).
What is the function of the kinase domain of syk in BCR-mediated antigen presentation? The phosphorylated ITAMs of the Ig-α/Ig-β sheath interact and activate syk as well as other cytoplasmic proteins (21). Recently, syk was shown to associate with or to activate the regulatory subunit of the phosphatidylinositol 3-kinase (PI3 kinase), p85 (41), which activates the catalytic subunit, p110 (42). The target of this enzyme is the inositol ring bond to the fatty acids constituting most cell membranes. The role of PI3 kinases in intracellular transport has been demonstrated by analysis of yeast mutants (vps 34, yeast equivalent of the PI3 kinase) deficient for transport to the vacuole (yeast equivalent of lysosome), and by using wortmannin, inhibitor of endosomal transport (42). The sequential recruitment of syk and PI3 kinase, after receptor aggregation and ITAM phosphorylation, could be a crucial step in endosomal sorting of the BCR, as demonstrated previously for PTK receptors. The kinase domain of the epidermal growth factor receptor (EGFR), is required in receptor sorting toward multivesicular endosomal compartments (43). In the case of the platelet growth factor receptor (PGFR), the sorting of the receptor toward lysosomes is determined by the interaction of its kinase domain with PI3 kinase (44). Although the precise mechanism by which syk kinase determines antigen presentation remains unclear, our results allow us to propose two nonexclusive hypotheses for syk kinase activity in endosomal sorting of antigen-bound BCR. Since the syk dominant negative mutant specifically affected antigen presentation using newly synthesized class II molecules, syk might be required for the endosomal sorting of antigen-bound BCR to class II compartments by the direct activation of the kinase domain of syk or by the recruitment of cytoplasmic effectors, such as PI3 kinases. In a second mechanism, the recruitment and activation of syk kinase might alter the structure and composition of endosomal compartments involved in antigen processing. In either case, it appears likely that syk has a novel and important function in intracellular trafficking and antigen presentation.
Acknowledgments
We are grateful to R. Grynsteen for critical reading of the manuscript, and to all of the members of INSERM CJF 95-01 for useful discussions.
This work was supported by grants from INSERM, Institut Curie, the Association de Recherche contre le Cancer (ARC), and the Ligue Nationale Contre le Cancer. V. Briken was supported by a European Community fellowship.
Abbreviations used in this paper
- BCR
B cell antigen receptor
- GST
glutathione S-transferase
- HA
hemagglutinin
- HEL
hen egg lysozyme
- HRP
horseradish peroxidase
- IC
immune complex(es)
- ITAM
immunoreceptor tyrosine–based activation motif
- mIg
membrane immunoglobulin
- PI3 kinase
phosphatidylinositol 3-kinase
- PTK
phosphotyrosine kinase
- SH2
src homology 2 domain
Footnotes
D. Lankar and V. Briken contributed equally to this work.
References
- 1.Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 2.Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 3.Amigorena S, Drake JR, Webster P, Mellman I. Transient accumulation of new class II MHC molecules in a novel endocytic compartment in B lymphocytes. Nature. 1994;369:113–120. doi: 10.1038/369113a0. [DOI] [PubMed] [Google Scholar]
- 4.Tulp A, Verwoerd D, Dobberstein B, Ploegh HL, Pieters J. Isolation and characterization of the intracellular MHC class II compartment. Nature. 1994;369:120–126. doi: 10.1038/369120a0. [DOI] [PubMed] [Google Scholar]
- 5.West MA, Lucocq JM, Watts C. Antigen processing and class II MHC peptide-loading compartments in human B-lymphoblastoid cells. Nature. 1994;369:147–151. doi: 10.1038/369147a0. [DOI] [PubMed] [Google Scholar]
- 6.Pinet V, Vergelli M, Martin R, Bakke O, Long EO. Antigen presentation mediated by recycling of surface HLA-DR molecules. Nature. 1995;375:603–606. doi: 10.1038/375603a0. [DOI] [PubMed] [Google Scholar]
- 7.Guangming Z, Romagnoli P, Germain RN. Related leucine-based cytoplasmic targeting signals in invariant chain and major histocompatibility complex class II molecules control endocytic presentation of distinct determinants in a single protein. J Exp Med. 1997;185:429–438. doi: 10.1084/jem.185.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castellino F, Germain RN. Extensive trafficking of MHC class II-invariant chain complexes in the endocytic pathway and appearance of peptide-loaded class II in multiple compartments. Immunity. 1995;2:73–88. doi: 10.1016/1074-7613(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 9.Lanzavecchia A. Antigen uptake and accumulation in antigen-specific B cells. Immunol Rev. 1987;99:39–51. doi: 10.1111/j.1600-065x.1987.tb01171.x. [DOI] [PubMed] [Google Scholar]
- 10.Neuberger MS, Patel KJ, Dariavach P, Nelms K, Peaker CJ, Williams GT. The mouse B-cell antigen receptor: definition and assembly of the core receptor of the five immunoglobulin isotypes. Immunol Rev. 1993;132:147–161. doi: 10.1111/j.1600-065x.1993.tb00841.x. [DOI] [PubMed] [Google Scholar]
- 11.DeFranco AL. The complexity of signaling pathways activated by the BCR. Curr Opin Immunol. 1997;9:296–308. doi: 10.1016/s0952-7915(97)80074-x. [DOI] [PubMed] [Google Scholar]
- 12.Reth M. Antigen receptor tail clue. Nature. 1989;338:383–384. [PubMed] [Google Scholar]
- 13.Cambier JC. New nomenclature for the Reth motif (or ARH1/TAM/ARAM/YXXL) Immunol Today. 1995;16:110. doi: 10.1016/0167-5699(95)80105-7. [DOI] [PubMed] [Google Scholar]
- 14.Shaw AC, Mitchell RN, Weaver YK, Campos TJ, Abbas AK, Leder P. Mutations of immunoglobulin transmembrane and cytoplasmic domains: effects on intracellular signaling and antigen presentation. Cell. 1990;63:381–392. doi: 10.1016/0092-8674(90)90171-a. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell RN, Barnes KA, Grupp SA, Sanchez M, Misulovin Z, Nussenzweig MC, Abbas AK. Intracellular targeting of antigens internalized by membrane immunoglobulin in B lymphocytes. J Exp Med. 1995;181:1705–1714. doi: 10.1084/jem.181.5.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonnerot C, Lankar D, Hanau D, Spehner D, Davoust J, Salamero J, Fridman WH. Role of B cell receptor Ig alpha and Ig beta subunits in MHC class II-restricted antigen presentation. Immunity. 1995;3:335–347. doi: 10.1016/1074-7613(95)90118-3. [DOI] [PubMed] [Google Scholar]
- 17.Gold MR, Matsuuchi L, Kelly RB, DeFranco AL. Tyrosine phosphorylation of components of the B-cell antigen receptors following receptor crosslinking. Proc Natl Acad Sci USA. 1991;88:3436–3440. doi: 10.1073/pnas.88.8.3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutchcroft JE, Harrison ML, Geahlen RL. Association of the 72-kDa protein-tyrosine kinase PTK72 with the B cell antigen receptor. J Biol Chem. 1992;267:8613–8619. [PubMed] [Google Scholar]
- 19.Yamada T, Taniguchi T, Yang C, Yasue S, Saito H, Yamamura H. Association with B-cell-antigen receptor with protein-tyrosine kinase p72syk and activation by engagement of membrane IgM. Eur J Biochem. 1993;213:455–459. doi: 10.1111/j.1432-1033.1993.tb17781.x. [DOI] [PubMed] [Google Scholar]
- 20.Lanzavecchia A. Receptor-mediated antigen uptake and its effect on antigen presentation to class II-restricted T lymphocytes. Annu Rev Immunol. 1990;8:773–793. doi: 10.1146/annurev.iy.08.040190.004013. [DOI] [PubMed] [Google Scholar]
- 21.Clark MR, Campbell KS, Kazlauskas A, Johnson SA, Hertz M, Potter TA, Pleiman C, Cambier JC. The B cell antigen receptor complex: association of Ig-alpha and Ig-beta with distinct cytoplasmic effectors. Science. 1992;258:123–126. doi: 10.1126/science.1439759. [DOI] [PubMed] [Google Scholar]
- 22.Choquet D, Ku G, Cassard S, Malissen B, Korn H, Fridman WH, Bonnerot C. Different patterns of calcium signaling triggered through two components of the B lymphocyte antigen receptor. J Biol Chem. 1994;269:6491–6497. [PubMed] [Google Scholar]
- 23.Jones B, Tite JP, Janeway CA. Different phenotypic variants of the mouse B cell tumor A20/2J are selected by antigen- and mitogen-triggered cytotoxicity of L3T4-positive, IA-restricted T cell clones. J Immunol. 1986;136:348–356. [PubMed] [Google Scholar]
- 24.Watanabe M, Watts TH, Gariepy J, Hozumi N. Function and behavior of surface immunoglobulin receptors in antigen-specific T cell-B cell interaction. Cell Immunol. 1988;112:226–235. doi: 10.1016/0008-8749(88)90291-2. [DOI] [PubMed] [Google Scholar]
- 25.Cassard S, Choquet D, Fridman WH, Bonnerot C. Regulation of ITAM signaling by specific sequences in Ig-beta B cell antigen receptor subunit. J Biol Chem. 1996;271:23786–23791. doi: 10.1074/jbc.271.39.23786. [DOI] [PubMed] [Google Scholar]
- 26.Bonnerot C, Amigorena S, Choquet D, Pavlovich R, Choukroun V, Fridman WH. Role of associated gamma-chain in tyrosine kinase activation via murine Fc gamma RIII. EMBO (Eur Mol Biol Organ) J. 1992;11:2747–2757. doi: 10.1002/j.1460-2075.1992.tb05340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benhamou M, Ryba NJ, Kihara H, Nishikata H, Siraganian RP. Protein-tyrosine kinase p72syk in high affinity IgE receptor signaling. Identification as a component of pp72 and association with the receptor gamma chain after receptor aggregation. J Biol Chem. 1993;268:23318–23324. [PubMed] [Google Scholar]
- 28.Guillet JG, Lai MZ, Briner TJ, Smith JA, Gefter ML. Interaction of peptide antigens and class II major histocompatibility complex antigens. Nature. 1986;324:260–262. doi: 10.1038/324260a0. [DOI] [PubMed] [Google Scholar]
- 29.Lai MZ, Ross DT, Guillet JG, Briner TJ, Gefter ML, Smith JA. T lymphocyte response to bacteriophage lambda repressor cI protein. Recognition of the same peptide presented by Ia molecules of different haplotypes. J Immunol. 1987;139:3973–3980. [PubMed] [Google Scholar]
- 30.Li WF, Fan MD, Pan CB, Lai MZ. T cell epitope selection: dominance may be determined by both affinity for major histocompatibility complex and stoichiometry of epitope. Eur J Immunol. 1992;22:943–949. doi: 10.1002/eji.1830220410. [DOI] [PubMed] [Google Scholar]
- 31.Cibotti R, Cabaniols JP, Pannetier C, Delarbre C, Vergnon I, Kanellopoulos JM, Kourilsky P. Public and private V beta T cell receptor repertoires against hen egg white lysozyme (HEL) in nontransgenic versus HEL transgenic mice. J Exp Med. 1994;180:861–872. doi: 10.1084/jem.180.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peterson M, Miller J. Invariant chain influences the immunological recognition of MHC class II molecules. Nature. 1990;345:172–174. doi: 10.1038/345172a0. [DOI] [PubMed] [Google Scholar]
- 33.Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, Webster P, Sautes C, Mellman I, Fridman WH. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science. 1992;256:1808–1812. doi: 10.1126/science.1535455. [DOI] [PubMed] [Google Scholar]
- 34.Harper M, Lema F, Boulot G, Poljack RJ. Antigen specificity and cross-reactivity of monoclonal anti-lysozyme antibodies. Mol Immunol. 1987;2:97–108. doi: 10.1016/0161-5890(87)90081-2. [DOI] [PubMed] [Google Scholar]
- 35.Amigorena S, Salamero J, Davoust J, Fridman WH, Bonnerot C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature. 1992;358:337–341. doi: 10.1038/358337a0. [DOI] [PubMed] [Google Scholar]
- 36.Brunati AM, Donella-Deana A, Ruzzene M, Marin O, Pinna LA. Site specificity of p72syk protein tyrosine kinase: efficient phosphorylation of motifs recognized by Src homology 2 domains of the Src family. FEBS Lett. 1995;367:149–152. doi: 10.1016/0014-5793(95)00555-n. [DOI] [PubMed] [Google Scholar]
- 37.Patel KJ, Neuberger MS. Antigen presentation by the B cell antigen receptor is driven by the α/β sheath and occurs independently of its cytoplasmic tyrosines. Cell. 1993;74:939–946. doi: 10.1016/0092-8674(93)90473-4. [DOI] [PubMed] [Google Scholar]
- 38.Peterson M, Miller J. Antigen presentation enhanced by the alternatively spliced invariant chain product p41. Nature. 1992;357:596–598. doi: 10.1038/357596a0. [DOI] [PubMed] [Google Scholar]
- 39.Sanchez M, Misulovin Z, Burkhardt AL, Mahajan S, Costa T, Franke R, Bolen JB, Nussenzweig M. Signal transduction by immunoglobulin is mediated through Igα and Igβ. J Exp Med. 1993;178:1049–1055. doi: 10.1084/jem.178.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flaswinkel H, Weiser P, Kim K-M, Reth M. Signaling and internalisation function of the B cell antigen receptor complex. Adv Exp Med Biol. 1994;135:1–8. doi: 10.1007/978-1-4899-0987-9_1. [DOI] [PubMed] [Google Scholar]
- 41.Deckert M, Tartare-Deckert S, Couture C, Mustelin T, Altman A. Functional and physical interactions of Syk family kinases with the Vav proto-oncogene product. Immunity. 1996;5:591–604. doi: 10.1016/s1074-7613(00)80273-3. [DOI] [PubMed] [Google Scholar]
- 42.Liscovitch M, Cantley LC. Signal transduction and membrane traffic: the PITP/phosphoinositide connection. Cell. 1995;81:659–662. doi: 10.1016/0092-8674(95)90525-1. [DOI] [PubMed] [Google Scholar]
- 43.Felder S, Miller K, Moehren G, Ullrich A, Schlessinger J, Hopkins CR. Kinase activity controls the sorting of the epidermal growth factor receptor within the multivesicular body. Cell. 1990;61:623–634. doi: 10.1016/0092-8674(90)90474-s. [DOI] [PubMed] [Google Scholar]
- 44.Joly M, Kazlauskas A, Corvera S. Phosphatidylinositol 3-kinase activity is required at a postendocytic step in platelet-derived growth factor receptor trafficking. J Biol Chem. 1995;270:13225–13230. doi: 10.1074/jbc.270.22.13225. [DOI] [PubMed] [Google Scholar]