

Abstract
The mechanisms linking human immunodeficiency virus replication to the progressive immunodeficiency of acquired immune deficiency syndrome are controversial, particularly the relative contribution of CD4+ T cell destruction. Here, we used the simian immunodeficiency virus (SIV) model to investigate the relationship between systemic CD4+ T cell dynamics and rapid disease progression. Of 18 rhesus macaques (RMs) infected with CCR5-tropic SIVmac239 (n = 14) or CXCR4-tropic SIVmac155T3 (n = 4), 4 of the former group manifested end-stage SIV disease by 200 d after infection. In SIVmac155T3 infections, naive CD4+ T cells were dramatically depleted, but this population was spared by SIVmac239, even in rapid progressors. In contrast, all SIVmac239-infected RMs demonstrated substantial systemic depletion of CD4+ memory T cells by day 28 after infection. Surprisingly, the extent of CD4+ memory T cell depletion was not, by itself, a strong predictor of rapid progression. However, in all RMs destined for stable infection, this depletion was countered by a striking increase in production of short-lived CD4+ memory T cells, many of which rapidly migrated to tissue. In all rapid progressors (P < 0.0001), production of these cells initiated but failed by day 42 of infection, and tissue delivery of new CD4+ memory T cells ceased. Thus, although profound depletion of tissue CD4+ memory T cells appeared to be a prerequisite for early pathogenesis, it was the inability to respond to this depletion with sustained production of tissue-homing CD4+ memory T cells that best distinguished rapid progressors, suggesting that mechanisms of the CD4+ memory T cell generation play a crucial role in maintaining immune homeostasis in stable SIV infection.
Keywords: AIDS, CD4+ T lymphocytes, lymphocyte depletion, immunologic memory, rhesus macaque
Introduction
Progressive cellular immunodeficiency clearly underlies the clinical disease known as AIDS (1, 2), but even after decades of investigation, the mechanisms by which HIV replication disables the immune system are poorly understood (3). The early association of clinical AIDS with CD4+ T lymphopenia and the subsequent determination that the causative agent of AIDS, HIV, directly targets CD4+ T cells led to the hypothesis that CD4+ T cell destruction results in the loss of critical immune effector and/or regulatory functions, ultimately degrading immunity below the threshold necessary to keep opportunistic pathogens at bay (1). Although this original model has been refined in recent years to include (a) coreceptor targeting of specific CD4+ T cell subpopulations, (b) indirect mechanisms of CD4+ T cell killing (e.g., apoptosis) related to chronic activation or interactions with viral products, and (c) a more dynamic view of the process in which direct or indirect CD4+ T cell destruction is countered by regenerative mechanisms (3–9), the mechanisms responsible for this depletion, its precise dynamic nature, its impact on host immunity, and its relationship to overt immune deficiency have not yet been well characterized.
Simian immunodeficiency virus (SIV) infection of rhesus macaques (RMs) provides a useful model to explore the linkage between viral replication, CD4 T cell depletion, and disease. Although the tempo of both viral replication and disease progression in this model is significantly accelerated compared with human HIV infection, both the pathobiologic characteristics of SIV (SIVmac and SIVsm derivatives) and the clinical syndrome associated with its infection are highly analogous to HIV and human AIDS (6, 10–16). This model allows use of cloned viruses of defined characteristics, study of the entire course of infection, as well as repeated access to tissue compartments, and thus provides for the longitudinal, systemic analysis of a primate immune system transitioning from normality to clinically overt insufficiency as a result of infection with an AIDS-causing lentivirus. Here, we have used this model to investigate the impact of CCR5- and CXCR4-tropic SIV infection on the composition and dynamics of the CD4+ T cell compartment, and to determine which, if any, of these parameters are associated with the development of early pathogenesis (overt AIDS within 200 d of infection). Our data strongly associate the early onset of symptomatic disease with the following two factors: (a) an initial profound depletion of preexistent CD4+ memory T cells in effector sites, and (b) in those animals with such depletion, a subsequent inability to maintain production and tissue delivery of short-lived CD4+ memory T cells. These results specifically link CD4+ memory T cell depletion in tissue with immunodeficiency, and provide the first direct evidence that the sustained increase in CD4+ memory T cell proliferation and turnover associated with lentiviral infection (a component of infection-associated chronic “hyperactivation”) may contribute to survival, allowing RMs to partially compensate for acute CD4+ memory T cell loss.
Materials and Methods
Animals and Veterinary Procedures.
34 purpose-bred male RMs (Macaca mulatta) of Indian genetic background and free of Cercopithicine herpesvirus 1, D-type simian retrovirus, simian T lymphotrophic virus type 1, and SIV infection were used in this study. 14 were infected with WT SIVmac239, 4 with SIVmac239 (Δnef), 4 with SIVmac155T3, 6 with rhesus CMV (RhCMV) strain 68.1, and 6 were studied as uninfected controls. SIV infections were initiated with intravenous injection of 5-ng equivalents of SIV p27 (2.7–3.0 × 104 infectious centers). RhCMV infections were initiated with subcutaneous injection of 106 PFU. Bronchoalveolar lavage was performed by infusion and aspiration of 10-ml aliquots of sterile, pyrogen-free saline using a 3-mm pediatric fiberoptic endoscope (Olympus) gently wedged into a subsidiary bronchus. For in vivo 5-bromo-2′-deoxyuridine (BrdU) pulse labeling, 10 mg/ml of filter-sterilized BrdU, pH 7.2 (Sigma-Aldrich), was prepared in HBSS (Ca2+/Mg++-free; Fisher Scientific) and administered intravenously at 30 mg/kg body weight at 24-h intervals for 4 d. All RMs were housed at the Oregon National Primate Research Center in accordance with the standards of the Center's Animal Care and Use Committee and the “NIH Guide for the Care and Use of Laboratory Animals” (17). RMs that developed disease states that were not reasonably clinically manageable were killed in accordance with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association (18).
Viruses and Viral Quantification.
The WT SIVmac239 (nef open), SIVmac239(Δnef), and SIVmac155T3 clonal virus stocks were prepared from pBR-SIVmac239, pBR-SIVmac239 Δnef, and pBR-SIVmac155T3 plasmids, respectively. The full-length pBR-SIVmac239 plasmid was provided by S. Wong (Oregon Health & Science University, Beaverton, OR; reference 19). The sequence of pBR-SIVmac239 Δnef (20) was modified to contain an Mlu1 site at position 9503 that spans the 181-bp nef deletion and results in a frame shift of downstream nef-coding elements (insuring stable nef inactivation). The pBR-SIVmac155T3 env recombinant was derived by replacing the env sequence in pBR-SIVmac239 from the Sph1 (nucleotide 6450) to Cla1 (nucleotide 8073) sites with the PCR-derived SIVmac155T3 sequence (21). The SIVmac155T3 recombinant clone has 22 amino acid differences compared with SIVmac239 env, and is a novel T cell–tropic SIVmac variant that uses CXCR4 as the primary coreceptor for virus entry (CCR5 use is reduced by 10-fold; unpublished data). Plasmid DNAs were transfected into COS1 cells using TransIT-LT1 (Mirus Corp.). The culture medium was collected from the cells 72 h after transfection, sterile filtered, and frozen in aliquots in liquid nitrogen. The virus stocks were assayed for p27-gag concentration by SIV core antigen assay (Beckman Coulter) and for infectious particles by sMAGI assay (22). Plasma SIV RNA was assessed using a real-time RT-PCR assay, essentially as described previously (threshold sensitivity = 100 SIV gag RNA copy equivalents/ml of plasma; interassay CV ≤25%; reference 23). RhCMV strain 68.1 was obtained from the American Type Culture Collection and propagated and titered in RM fibroblasts (24).
Immunofluorescent Staining and Flow Cytometric Analysis.
PBMCs, peripheral lymph node cells, and bronchoalveolar lymphocytes (BALs) were obtained and stained for flow cytometric analysis as described previously (25). Six-parameter flow cytometric analysis was performed on a two-laser FACSCalibur instrument using FITC, PE, peridinin chlorophyll protein-Cy5.5 (True Red), and allophycocyanin (APC) as the four fluorescent parameters. List mode multiparameter data files were analyzed using the PAINT-A-GATE Plus software program (BD Biosciences). Each analysis included a lineage-defining marker, CD4 or CD8β (with or without CD3ɛ), and two to three phenotyping markers. Memory and naive T cell subsets in peripheral blood and lymph node were delineated based on CD28 and CD95 expression patterns using previously described criteria (25). The specificity of staining and criteria for setting positive versus negative markers for CCR5, Ki-67, and BrdU expression were determined using isotype-matched negative control mAbs, and in the case of BrdU, BrdU analysis of study animals before BrdU pulsing.
mAbs.
mAbs L200 (CD4; True Red and APC conjugated), SP34 (CD3; FITC, PE, True Red, and APC), CD28.2 (CD28; FITC, PE, True Red, and APC), B56 (Ki-67; FITC and PE), B44 (anti-BrdU; FITC), DX2 (CD95; PE and APC), 12G5 (CXCR4; PE), 3A9 (CCR5; PE), and IgG1 and IgG2 isotype-matched controls were obtained from BD Biosciences. mAb 2ST8.5h7 (CD8β; PE, unconjugated) was obtained from Beckman Coulter. Purified 2ST8.5h7 was custom conjugated to True Red by BD Biosciences.
Statistical Analysis.
Between-group differences in quantitative dependent variables (except BrdU) were tested at all time points (days 0–205 after infection) and during acute (days 0–42) and plateau (days 42–205) phases of infection. BrdU decay rates were compared for 70 d after the last BrdU pulse. Given the small sample size and reported simulation results (26), we elected not to use mixed model ANOVA procedures. Instead, we took the mean of all time points for each animal within each interval, insuring that for each animal, a single value was modeled and, therefore, that data were independently distributed. Given the small and unequal sample sizes across groups in some tests, we conducted further sensitivity analyses to evaluate the robustness of our findings by redoing all tests using the nonparametric Wilcoxin rank sum test. In all cases but one, the conclusion with respect to statistical significance for the t test was confirmed by nonparametric testing, lending substantial confidence to the results. In the one case in which they were different (see Fig. 4), the difference was modest.
Figure 4.
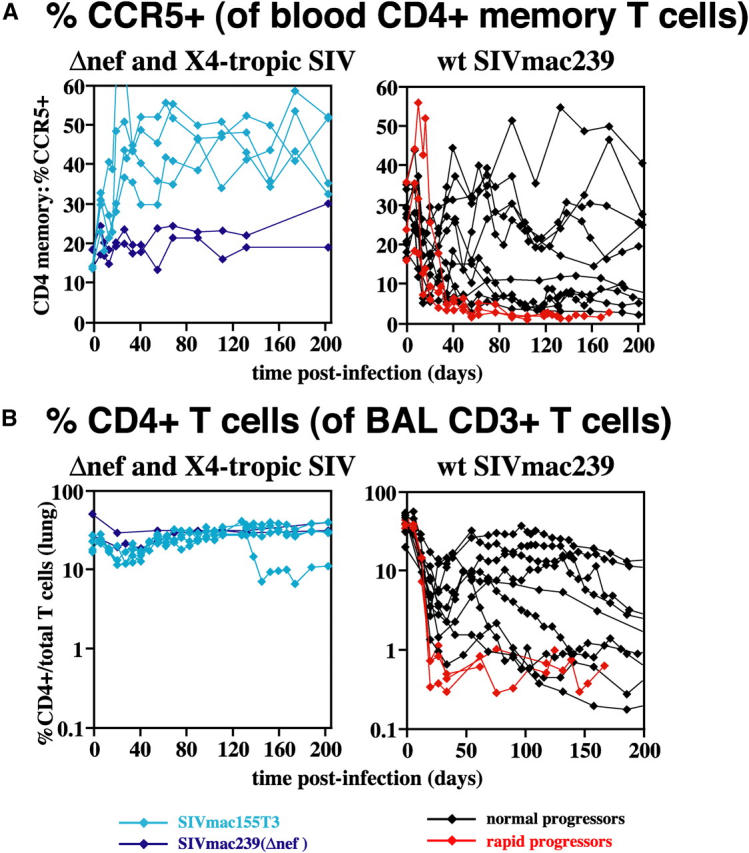
Depletion patterns of CCR5+ CD4+ memory T cell “targets” in blood (A) and tissue (B) in CCR5-tropic, CXCR4-tropic, and attenuated SIV infections. (A) The percent of cells expressing CCR5 within the peripheral blood CD4+ memory subset was determined as shown in Fig. 2. Data on this parameter were not available for one WT SIVmac239-infected, rapid progressor and two SIVmac239(Δnef)-infected RMs; all other study RMs are included in the figure. The difference in CCR5+ CD4+ memory T cell frequencies between rapid and normal progressors was significant in plateau phase. P = 0.004 and 0.01 for after day 42 after infection by t and Wilcoxin rank sum tests, respectively. (B) The percentage of CD4+ cells within the total population of CD3+ BAL T cells is shown for 18 of the 22 SIV-infected RMs. In the four RMs not shown, BAL data was not obtained until day 77 after infection or later. The two of these RMs infected with SIVmac239(Δnef) demonstrated normal CD4+ T cell representation in BALs (33–39%) at these later time points (consistent with no depletion). In contrast, the two of these RMs infected with WT SIVmac239, one a rapid progressor and one a normal progressor, both demonstrated CD4+ T cell frequencies in BALs of <1% after day 77 after infection (consistent with profound depletion). Statistical comparison of the degree of depletion in SIVmac239-infected rapid versus normal progressors was equivocal. In acute infection (before day 42 after infection), the difference in CD4+ T cell percentages between WT SIVmac239-infected rapid and normal progressors was significant at P = 0.02 by the Wilcoxin rank sum test, but not by the t test. After day 42 after infection, the reverse was true: the t test demonstrated a p-value of 0.008, whereas the Wilcoxin rank sum test was not significant.
Results
Clinical and Virologic Characteristics of WT and Variant SIVmac239 Infection.
22 RMs were monitored for 205 d after IV infection with (a) WT (CCR5-tropic) SIVmac239 (n = 14), (b) the (CXCR4-tropic) SIVmac239 env recombinant SIVmac155T3 (n = 4), or (c) the attenuated SIVmac239(Δnef; n = 4). 10 of the 14 WT SIVmac239-infected RMs remained free of SIV-related disease during the first 200 d of infection, and all 10 of these RMs survived at least 1 yr after infection. In contrast, the remaining four WT SIVmac239-inoculated RMs developed progressive disease requiring euthanasia at days 107–175 after infection. Necropsy revealed one or more AIDS-defining opportunistic infections in all four of these animals (Table I). Viral replication in the 14 RMs inoculated with WT SIVmac239 was quite high, with peak viral loads (almost invariably at day 10 after infection) ranging from 2 × 107 to 2 × 108 copies/ml, and plateau phase viral loads from 105 to >108 copies/ml (Fig. 1 A). One RM with the protective Mamu A*O1/B*17 MHC alleles (27) manifested spontaneous, temporary control of viral replication to <100 copy equivalents/ml at day 56 after infection, but reverted to a high replicative state (viral loads > 4 × 105) ∼40 d later. In keeping with previous studies (16, 28, 29), the plasma viral loads in the four rapid progressors, particularly the post-acute phase (>day 42 after infection) viral levels, were significantly higher than in the normal progressor group (Fig. 1, legend). However, it is noteworthy that two of that latter group manifested viral replication kinetics overlapping that of the rapid progressors, yet maintained immune competence for >1 yr after infection.
Table I.
Clinical and Pathologic Features of WT SIVmac239-infected Rapid Progressor RMs
| RM no. | Clinical findings | Pathologic findings at necropsy | |
|---|---|---|---|
| 20165 | Day 95a: | Diarrhea, dehydration, electrolyte imbalance | Disseminated CMV infection: small intestine, prostate, epididymis, and leptomeninges |
| Day 126: | Diarrhea, dehydration, electrolyte imbalance, thrombocytopenia, weight loss (15%), euthanasia |
Cryptosporidiosis: cholecystitis, cholangitis, and cholangiohepatitis Mesangioproliferative glomerulonephritis Lymphoid depletion: lymph nodes, spleen, and thymus |
|
| 20322 | Day 111: | Diarrhea, vomiting | Lymphocytic interstitial pneumonia |
| Day 120: | Diarrhea, dehydration, anorexia | Protozoal (flagellate) gastritis | |
| Day 126: | Weight loss (13%), euthanasia | Cryptosporidiosis: cholecystitis, cholangitis, cholangiohepatitis, and enteritis |
|
| Ulcerative colitis | |||
| Mesangioproliferative glomerulonephritis | |||
| Lymphoid depletion: lymph nodes, spleen, and thymus | |||
| 20418 | Day 125: | Diarrhea, dehydration | Pneumocystis carinii pneumonia |
| Day 172: | Diarrhea, dehydration, anorexia | Disseminated CMV infection: right atrioventricular valve, spinal cord leptomeninges, nerves, and ganglia |
|
| Day 175: | Weight loss (14%), euthanasia | Diffuse nonsuppurative gastritis | |
| Cryptosporidiosis: cholecystitis, cholangitis, and cholangiohepatitis |
|||
| Mesangioproliferative glomerulonephritis | |||
| Lymphoid depletion: lymph nodes, spleen, and thymus | |||
| 21070 | Day 56: | Thrombocytopenia | CMV pneumonia |
| Day 93: | Diarrhea, Campylobacter coli and flagellate protozoa |
Aspiration pneumonia Granulomatous cholangiohepatitis |
|
| Day 104: | Diarrhea, dehydration, electrolyte imbalance, anorexia; |
Hemorrhagic, ulcerative bacterial gastritis and enteritis Lymphoid depletion: lymph nodes, spleen, and thymus |
|
| Elevated LDH, SGPT/OT, BUN, creatinine | |||
| Day 107: | Diarrhea, dehydration, electrolyte imbalance, anorexia, anemia, weight loss (12%), euthanasia |
Days are all after infection.
Figure 1.
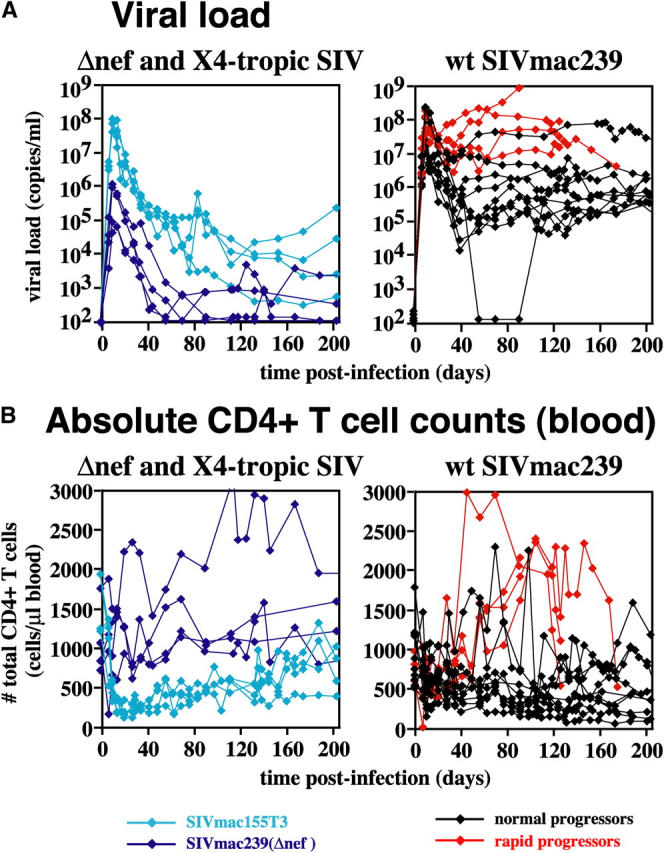
Plasma viral loads (A) and absolute peripheral blood CD4+ T cell counts (B) in CCR5-tropic, CXCR4-tropic, and attenuated SIV infections. Data from all 22 SIV-infected RMs included in this work are shown: 4 each with SIVmac239(Δnef) (dark blue traces) and SIVmac155T3 (light blue), and 14 with WT SIVmac239, of which 4 were rapid (red) and 10 (black) were normal progressors. Both acute and plateau phase viral loads were significantly higher in the rapid progressor RMs than in their clinically stable SIVmac239-infected counterparts. P = 0.004 and 0.001 (t test), and P = 0.02 and 0.01 (Wilcoxin rank sum) for on or before, or after, day 42 after infection, respectively. The absolute CD4 counts of these two groups were significantly different after day 42 after infection (P = 0.001 and 0.01 for t and Wilcoxin rank sum tests, respectively), but paradoxically, although the normal progressors showed the expected slow decline in CD4 counts, rapid progressors showed a pronounced CD4+ T lymphocytosis.
The peak viremias observed in the SIVmac239(Δnef)-infected RMs, as is characteristic of these infections (30), was blunted compared with WT SIVmac239, and post-acute viral loads stabilized in the 2–3 log range (Fig. 1 A). SIVmac155T3 is a novel variant of SIVmac239 with 22 amino acid substitutions in gp120 that confer dominant CXCR4 tropism. Peak viral loads with this clone were similar to those of WT SIVmac239, but plateau phase viremia was considerably reduced, ranging from 103 to 105 copies/ml (Fig. 1 A). All of the SIVmac239(Δnef)- and SIVmac155T3-infected RMs studied here remained healthy for >1 yr after infection.
SIV Coreceptor Tropism, Patterns of CD4+ T Cell Depletion, and Early Disease Progression.
Although total peripheral blood CD4 counts have long been the benchmark of immunologic assessment of disease progression in both HIV and SHIV infection (1, 2, 31, 32), this parameter was not predictive of early pathogenesis in the SIV-infected RMs studied here (Fig. 1 B). Although the healthy SIVmac239(Δnef)-infected RMs in this work showed the expected stability of these counts (indeed, on average, CD4+ T cell numbers rose 39% in these RMs), the clinically indistinguishable SIVmac155T3-infected cohort showed rapid and profound initial depletion (>90%), followed by a delayed, slow rebound. Although the postinfection course of total CD4 counts in the clinically stable WT SIVmac239-infected RMs showed considerable variability, overall, these animals manifested a slow decline in total CD4+ T cell counts, averaging 44% over the 200 d of observation. Paradoxically, the WT SIVmac239-infected RMs destined for early pathogenicity (rapid progressors) uniformly demonstrated a striking (approximately threefold) CD4+ T lymphocytosis, starting at about day 42 after infection, and extending until the onset of overt disease (usually 2–3 wk before they were killed).
CD4+ T cells in peripheral blood are a composite of two phenotypically and functionally distinct subpopulations: the naive and memory/effector subsets (25), each with a characteristic pattern of coreceptor expression. As illustrated in Fig. 2, A and B (top), the primary coreceptor for WT SIVmac239, CCR5, is almost exclusively expressed by a subset of memory CD4+ T cells (mean CCR5+ ± SD for 21 uninfected RMs = 21.8 ± 7.7% of CD4+ memory T cells in peripheral blood). The alternative coreceptor CXCR4 is robustly expressed by both subsets, with the intensity of expression highest on naive cells (Fig. 2 B, top). Infection with CCR5- versus CXCR4-tropic SIV resulted in dramatically different changes in CD4+ T cell subset composition, in keeping with coreceptor-directed targeting. As shown in Fig. 2 A, a typical CCR5-tropic WT SIVmac239 infection induced a 75% loss of CD4+ memory T cells by day 49 after infection, and within the memory population, a 75% depletion of the CCR5-expressing population. Concomitant with this striking depletion of CCR5-expressing, CD4+ memory T cells, we observed a prominent change in the proliferation status of this subset, as measured by the cell cycle marker Ki-67 (25). Whereas before infection, CCR5+ CD4+ memory T cells were predominantly Ki-67−, after infection-associated depletion, this subset was predominantly Ki-67bright. Thus, circulating CCR5+ CD4+ memory T cells are markedly depleted in SIVmac239 infection with the small, residual CCR5+ subset appearing to be maintained by a proliferative process. In contrast, a typical CXCR4-tropic SIVmac155T3 infection profoundly and preferentially depleted the naive CD4+ subset in the same timeframe (Fig. 2 B). CXCR4-expressing CD4+ memory T cells were not resistant to this virus, but this subset appeared to down-regulate CXCR4 expression so as to escape wholesale depletion. The frequencies of CCR5+ cells within the CD4+ memory T cell population actually increased in SIVmac155T3-infected RMs due to CCR5 expression by the proliferating subset (see below).
Figure 2.

CD4+ T cell depletion patterns in CCR5- (A) versus CXCR4-tropic (B) SIV infection. (A) PBMCs from a typical WT SIVmac239-infected normal progressor were examined for their correlated expression of cell surface CD4 versus CCR5 versus CD28 versus CD95 or CD4 versus CCR5 versus Ki-67 versus CD95, both before and 49 d after infection (viral load = 7.4 × 106 copies/ml and absolute blood CD4 counts = 500 cells/μl at day 49 after infection). 5,000 events, gated on total CD4+ T cells (left and middle) or CD4+ (CD95high) memory T cells (right) are shown. Overall, naive (blue) and memory (red) T cell frequencies are shown in the upper right corner of the left panels. The percent CCR5+ within the memory subset is shown in the middle panels, and the memory subsets defined by CCR5 versus Ki-67 are shown in the right panels. Note the striking relative depletion of CD4+ memory T cells, and within the memory population, the CCR5-expressing subset, with this subset going from predominantly Ki-67− preinfection to predominantly Ki-67bright+ postinfection (arrows). (B) PBMCs from a representative SIVmac155T3-infected RM were examined for their correlated expression of cell surface CD4 versus CCR5 versus CD28 versus CD95 or CD4 versus CXCR4 versus CD28 versus CD95, both before and 49 d after infection (viral load = 1.2 × 105 copies/ml and absolute blood CD4 count = 440 cells/μl at day 49 after infection). 5,000 events, gated on total CD4+ T cells are shown with events within the naive and memory cell clusters colored blue and red, respectively. Note the profound loss of naive CD4+ T cells, increased frequencies of CCR5+ cells among CD4+ memory T cells, and loss of CD4+ T cell CXCR4 staining intensity, particularly within the memory subset.
Analysis of absolute peripheral blood naive and memory T cell numbers in the different cohorts confirms and extends these findings (Fig. 3). In SIVmac239(Δnef)-infected RMs, absolute numbers of naive and memory CD4+ T cells in blood fluctuated over time, but overall, preinfection levels of both subsets were maintained for >200 d after infection. In SIVmac155T3 infection, the aforementioned rapid loss of circulating CD4+ T cells can be seen to be primarily the result of naive T cell depletion (essentially complete depletion was also observed in lymph nodes). Circulating CD4+ memory T cell numbers declined modestly during acute infection with this X4-tropic virus, but rebounded, and remained stable thereafter. In contrast, among WT SIVmac239-infected normal progressors, naive CD4+ T cell numbers were stable, whereas memory CD4+ T cell numbers demonstrated an abrupt decline in acute infection (after a brief CD4+ memory T lymphocytosis at day 7 after infection), followed by a slow decline thereafter. 4 of the 10 normal progressors showed some fluctuating or transient rebound in these CD4+ memory T cell numbers, but even for these RMs, the overall trend was downward. Among the SIVmac239-infected rapid progressors, the above-described CD4+ T lymphocytosis is clearly attributable to the naive subset, which given the low turnover of this population (25), likely reflects a redistribution of these cells from tissue to blood. Circulating CD4+ memory T cells uniformly decline in these RMs, but the kinetics and extent of this loss are largely overlapping with that of the normal progressors.
Figure 3.

Absolute peripheral blood CD4+ memory (A) and naive (B) T cell counts in CCR5-tropic, CXCR4-tropic, and attenuated SIV infections. Absolute memory (A) and naive (B) CD4+ T cell counts were determined using absolute total CD4 counts (see Fig. 1) and naive/memory fractions based on CD28 versus CD95 staining criteria (reference 25). Data for all 22 SIV-infected RMs are shown. Rapid progressors did not differ from normal progressors in regard to memory CD4+ T cell numbers (P = 0.11 and 0.18 for days 0–200 after infection by t and Wilcoxin rank sum tests, respectively). In contrast, with the onset of the plateau phase of infection, rapid progressors manifested a pronounced CD4+ naive T cell lymphocytosis, whereas naive CD4+ T cell numbers in normal progressors were largely stable (P < 0.0001 and P = 0.006 for after day 42 after infection by t and Wilcoxin rank sum tests, respectively).
Representation of the CCR5-expressing component of the CD4+ memory population briefly increased in blood at day 7 after infection in all WT SIVmac239-infected RMs, but then rapidly declined (Fig. 4 A). In the clinically stable WT SIVmac239-infected RMs, the decline during acute infection was accompanied, as indicated above, by the appearance of Ki-67+ CCR5+ CD4+ T cells, which either maintained this fraction at a low level (at least 5% of the memory fraction; 5 of 10 RMs), or increased it back toward normal levels (the other 5 normal progressors). The initial decline of this population was similar in rapid progressors, but in contrast to the normal progressors, there was little evidence of rebound, and after day 42 after infection, the frequency of CCR5-expressing cells among CD4+ memory T cells was <3%, significantly lower than the normal progressor group (Fig. 4). The CCR5-expressing memory fraction increased in RMs infected with CXCR4-tropic SIV, and did not change in attenuated (Δnef) SIV infection.
Previous work has established that CCR5 is expressed by the vast majority of CD4+ memory T cells in mucosal tissues such as the intestinal and vaginal mucosae and the tissue–air interface of the lung, and that these populations are subject to extensive, acute depletion during CCR5-tropic SIV infection (4, 13, 33–35). Using the latter (BALs) as a repeatedly accessible “window” into the mucosal tissue compartment, we determined the relationship between the rate and extent of “effector” site CD4+ T cell depletion and clinical course in our study cohorts. As shown in Fig. 4 B, CD4+ T cell numbers in BALs were stable during early infection with either attenuated SIVmac239(Δnef) or CXCR4-tropic SIVmac155T3 infections, but rapidly declined with CCR5-tropic WT SIVmac239 infection. Normal progressors lost, on average, 82% of their lung CD4+ T cells by day 21 after infection (range: 63–96%), and 86% by day 28 after infection (range: 71–97%). Thereafter, about half of these RMs were able to transiently and partially regenerate CD4+ T cell percentages in the lung, whereas in the others, the CD4+ T cell fraction declined to ≤1%. WT SIVmac239-infected rapid progressors demonstrated a uniform 98–99% decline in CD4+ T cell frequencies by day 21 after infection, and showed no significant regeneration. Statistical assessments of the differences in the extent and kinetics of CD4+ (memory) T cell depletion in BALs in WT SIVmac239-infected normal versus rapid progressors were equivocal, with t tests suggesting significance after, but not before, day 42 after infection and nonparametric testing indicating the reverse (Fig. 4 B).
CD4+ Memory T Cell Regenerative Failure Precedes and Predicts Rapid Progression.
Next, we assessed whether differences in CD4+ memory T cell proliferation and turnover correlated with clinical outcome, particularly in animals with substantial acute-phase CD4+ memory T cell depletion in tissue. Two approaches were used to investigate this issue. First, we serially determined the fraction of CD4+ (and for comparison, CD8+) memory T cells expressing the proliferation marker Ki-67 (Fig. 5). Ki-67 expression provides a running average of the fraction of cells that have recently gone through S phase of the cell cycle with moderate/bright and dim staining reflecting S phase in the 3–4 and 5–8 d before sampling, respectively (25). Second, we labeled proliferating cells in vivo at strategic time points after infection with the thymidine analogue BrdU, followed by serial measurement of the fraction of CD4+ memory cells retaining this DNA label. Peak BrdU incorporation immediately after the last BrdU dose provides an independent measurement of the proliferating fraction, and the relative kinetics of BrdU decay during the wash-out period provides insight into the relative fate of the proliferating component over that time period. To facilitate interpretation of BrdU decay kinetics in SIV-infected RMs, we included two control groups in the study: healthy uninfected RMs (n = 6) and healthy RMs with primary RhCMV infection (n = 6).
Figure 5.

CD4+ and CD8+ memory T cell proliferation in CCR5-tropic, CXCR4-tropic, and attenuated SIV infections. PBMCs were examined for their correlated expression of Ki-67 versus CD28 versus CD95 versus either CD4 or CD8β, and the percent Ki-67+ was determined for the memory T cell subset of each lineage. The figure includes CD4+ lineage data from all 22 SIV-infected RMs, and CD8 lineage data for all 14 RMs infected with WT SIVmac239. The plateau phase differences in CD4+ memory T cell proliferation (e.g., percent Ki-67+) between the SIVmac239-infected normal and rapid progressors was highly significant with P < 0.0001 and P = 0.006 for after day 42 after infection by t and Wilcoxin rank sum tests, respectively. In contrast, CD8+ memory T cell proliferation during the same time period did not significantly differ between these two groups.
These analyses revealed that SIV infection, in general, results in profound changes in CD4+ memory T cell turnover. In the first 7–10 d after infection with any of the SIV variants, coincident with peak viral replication, most animals showed a transient drop in the fraction of Ki-67+ cells within the circulating CD4+ memory T cell population. However, starting on days 14–21 after infection, the frequency of Ki-67+ cells within this population dramatically increased (Fig. 5). For WT SIVmac239-infected normal progressors, the fraction of CD4+ memory T cells expressing Ki-67 on day 42 after infection increased from preinfection values by an average (±SEM) of 2.6 (±0.3)-fold (an average increase of 27 ± 2.1 percentage points). A similar increase in Ki-67 expression was observed in CD4+ memory T cells in lymph nodes (not depicted) and in CD8+ memory T cells (Fig. 5). Most noteworthy, however, was the observation that the infection-associated increase in CD4+ memory T cell proliferation was sustained in all SIV-infected animals except those destined for rapid progression. In all four rapid progressors, the CD4+ memory proliferative response initiated with normal kinetics (peak percent Ki-67 within the CD4+ memory subset in blood during the acute phase, day 14–30 after infection, was 4.1- ± 0.9-fold higher than preinfection levels), but failed by day 42 after infection, with CD4+ memory T cell proliferative frequencies returning back to or toward baseline levels (Fig. 5). After day 42 after infection, the difference in CD4+ memory T cell proliferative frequencies between RMs destined for normal versus rapid progression was highly significant (P < 0.0001 by t test). Significantly, proliferative failure was CD4 memory T cell specific. There was no difference in the postinfection proliferative kinetics of CD8+ memory T cells between rapid and normal progressing WT SIVmac239-infected RMs.
Peak BrdU labeling in the acute phase of infection (days 10–13 or 24–27 after infection) confirmed the CD4+ memory T cell proliferative “burst,” with mean ± SEM of maximum percent BrdU+ of blood CD4+ memory T cells as 5.4 ± 0.5% for uninfected RMs, 20.6 ± 1.7% for WT SIVmac239-infected normal progressors, 16.2 ± 5.1% for WT SIVmac239-infected rapid progressors, 21.9 ± 5.3% for SIVmac155T3-infected RMs, and 14.7 ± 1.2% for SIVmac239(Δnef)-infected RMs (all SIV cohorts were significantly different from uninfected controls with t test p-values from 0.02 to <0.001). During the post-BrdU pulse “washout” period, BrdU+ memory T cells decline in frequency in a multiphasic process (25). In the initial 4 d of washout (phase 1), most BrdU+ cells are Ki-67moderate–high, indicating many are still cycling, and labeled cells decline in frequency because of proliferative dilution (>5 postlabel divisions), as well as loss from the circulating pool (Fig. 6, A and B). From washout days 4–14 (phase 2), labeled cells have largely left the cell cycle (manifested by loss of Ki-67 expression and stabilization of BrdU staining intensity), and display a relatively rapid removal from the recirculating pool, as compared with the considerably slower, steady loss at later time points (phase 3). In SIV-infected RMs, loss of labeled cells in phases 1 and 2 was dramatically increased versus uninfected RMs (80 to >90% vs. ∼50%), consistent with a great predominance of high turnover/short-lived cells within the CD4+ memory proliferative burst. This CD4+ memory BrdU decay pattern was observed in acute SIV infection of all types, as well as primary RhCMV infection (Fig. 6 C), suggesting it is a common response to viral infection. Importantly, there was no significant difference between the CD4+ memory BrdU decay kinetics of the WT SIVmac239-infected RMs destined for normal versus rapid progression, or between either of these groups and the SIVmac155T3-infected RMs. Thus, the rate and extent to which proliferating CD4+ memory T cells are lost from the recirculating pool in acute SIV infection does not correlate with rapid progression.
Figure 6.

BrdU decay kinetics in circulating CD4+ memory T cells in normal RMs, primary RhCMV infections, and CCR5-tropic, CXCR4-tropic, and attenuated SIV infections. SIV-infected and control RMs (either uninfected or 1° RhCMV infected) were administered 30 mg/kg BrdU IV for 4 consecutive days, with the first sampling occurring 24 h after the last BrdU dose. In the SIV infections, acute phase labeling was performed either on days 10–13 after infection or days 24–27 after infection. Plateau phase labeling was performed after day 99 after infection. In the 1° RhCMV infections, labeling was performed on days 10–13 after infection. (A and B) The correlated expression of BrdU versus Ki-67 on CD4+ CD95high (memory) T cells from representative healthy, uninfected (A) and WT SIVmac239-infected normal progressor (B) RMs are shown (each profile with 5,000 gated events). The percentages in the upper right corners of the profiles represent the total BrdU+ fraction of the gated CD4+ memory cells. The dotted horizontal lines delineate the upper extent of the negative population in the Ki-67 channel and serve as a reference for evaluating the progressive loss of Ki-67 expression by BrdU+ cells. (C) PBMCs were stained for BrdU versus CD28 versus CD95 versus CD4 at the designated time points, and percent BrdU+ was determined for the CD4+ memory subset. The decay kinetics of BrdU+ (CD4+ memory) cells in all acutely infected cohorts were significantly different from uninfected animals (P < 0.0001 by t test for normal vs. 1° RhCMV-, WT SIVmac239-, SIVmac155T3-, or SIVmac239(Δnef)-infected RMs), but these kinetics did not significantly differ between WT SIVmac239-infected rapid versus normal progressors (P = 0.84 by t test), or between either of these cohorts and RMs infected with SIVmac155T3 (P = 0.15 and 0.06, respectively).
As mentioned above, Ki-67 frequencies in the CD4+ memory population of WT SIVmac239-infected normal progressors remain high throughout the period of observation (Fig. 5), and plateau phase (after day 100 after infection) BrdU analysis of this population (Fig. 6 C, far right) demonstrates continued rapid decay, suggesting that the high turnover state is similarly maintained. Significantly, in the one WT SIVmac239-infected rapid progressor that survived long enough for a plateau phase BrdU decay analysis (i.e., after the onset of proliferative failure), CD4+ memory T cell BrdU decay kinetics was similar to that of uninfected RMs, suggesting that collapse of the infection-associated proliferative burst abrogates the high turnover/short-lived component of CD4+ memory proliferation, leaving a low turnover/long-lived component behind.
Delivery of CD4+ Memory T Cells to the Pulmonary Mucosal Surface in Stable versus Rapidly Progressive Infection.
The potential impact of CD4+ memory T cell proliferative failure on CD4+ memory populations in effector sites, and its possible link with rapid disease progression, was first revealed by comparison of BrdU versus Ki-67 staining patterns and BrdU kinetics in the blood, lymph node, and lung of (plateau phase) WT SIVmac239-infected normal progressors. As shown in Fig. 7 A, 24 h after the last dose of a 4-d BrdU pulse, the vast majority of BrdU-labeled CD4+ memory T cells in the blood and lymph node are also Ki-67+, indicating that they are relatively close to their last S phase. In contrast, at the exact same time, BrdU-labeled CD4+ memory T cells in the lung are predominantly Ki-67− or dim, strongly suggesting that these cells are more remote from their most recent S phase, and did not go through S phase in the lung itself. This impression is strengthened by the observation that although maximum BrdU labeling in the blood and lymph node occurs 24 h after the last BrdU dose, maximum BrdU labeling in the lung occurs 7 d later (Fig. 7 C). These data lead to the conclusion that the pulmonary mucosal surface in these RMs is continually supplied by an influx of new CD4+ memory T cells that are the products of prior proliferative events occurring elsewhere.
Figure 7.

BrdU labeling and decay kinetics in the blood versus lymph node versus lung in a plateau phase WT SIVmac239-infected normal progressor. (A and B) 1 d after BrdU administration on days 99–102 after infection, PBMCs, lymph node cells, and BALs from a clinically stable WT SIV mac239-infected RM were examined for correlated expression of CD4, BrdU, and either Ki-67 (A) or CCR5 (B), with CD95 also used in PBMC and lymph node analyses to delineate the CD4+ memory subset. 5,000 events are shown, gated on CD4+ CD95high memory T cells in PBMC and lymph node preparations, and on total CD4+ T cells (which are all memory) in BALs. In A, the percentages of BrdU+ cells within these gated populations are shown in the upper right hand corner of each profile, with the percentages adjacent to the arrows indicating the fraction of these BrdU+ cells that are also Ki-67+. In B, the percentages adjacent to the arrows indicate the fraction of BrdU+ cells that are also CCR5+. (C) The percent of CD4+ memory T cells demonstrating BrdU reactivity are plotted for each site over the time course shown. The results shown are representative of three plateau phase SIVmac239-infected normal progressors and four SIVmac155T3-infected RMs.
Thus, the postinfection CD4+ memory proliferative response appears to provide a steady influx of new CD4+ memory T cells in mucosal effector sites, potentially countering the severe CD4+ T cell depletion in these sites and preventing overt immunodeficiency. Although accurate direct measurement of BrdU-labeled CD4+ T cell influx into the lungs was not possible in severely depleted SIV-infected animals, the phenotypic signatures of lung CD4+ T cells in normal versus rapid progressors strongly support this hypothesis. As shown in Fig. 7 B, newly produced, BrdU-labeled memory CD4+ T cells in the blood and lymph node might be either CCR5+ or CCR5−, but those arriving in the lung are almost all CCR5+. Moreover, although BrdU+ cells arriving in lung have undergone S phase elsewhere, many of them retain low level Ki-67 reactivity (Fig. 7 A). Taken together, these observations suggest that most new emigrants to lung will express Ki-67, CCR5, or both markers. As illustrated in Fig. 8 A, BAL CD4+ T cells of all WT SIVmac239-infected normal progressors did indeed predominantly express either or both CCR5 and Ki-67 at all plateau phase time points (mean ± SEM for BAL CD4+ T cells lacking both Ki-67 and CCR5 = 12.1 ± 4.5%), notably including those RMs with profound CD4+ T cell depletion in the lung (e.g., <1% CD4+ T cells). In striking contrast (Fig. 8 B), the residual BAL CD4+ T cells of the rapid progressors were almost entirely negative for both CCR5 and Ki-67 (mean ± SEM for BAL CD4+ T cells lacking both Ki-67 and CCR5 = 84.6 ± 2.4%). These differences were highly significant (P < 0.0001 by t test). Thus, even though normal and rapid progressors may have had identical overall frequencies of lung CD4+ T cells, the phenotypes of these T cells strongly suggest that the lungs of the clinically stable RMs were receiving a continuous influx of newly produced CD4+ effector T cells, whereas the lungs of animals destined for rapid progression were not.
Figure 8.
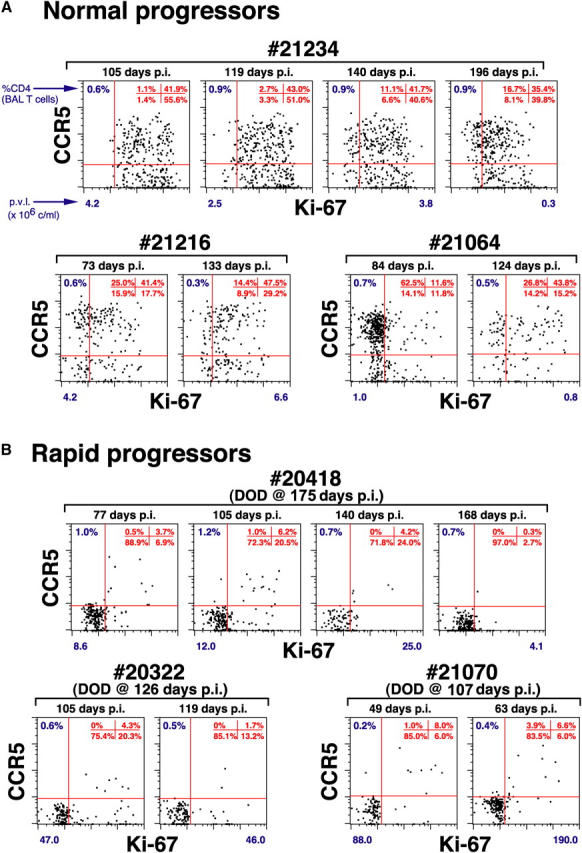
Pulmonary CD4+ T cell phenotype in WT SIVmac239-infected rapid versus normal progressors. BAL preparations were examined for correlated expression of CD4, CD8β, Ki-67, and CCR5 at different plateau phase time points in all 14 WT SIVmac239-infected RMs. The profiles are gated on CD4+ T cells with all available events shown (>200). The percent of CD4+ T cells (of total T cells) in each BAL specimen is indicated in the upper left of each profile. The quadrant statistics for CCR5 versus Ki-67 expression are provided in the upper right of each profile. The plasma viral loads (p.v.l. × 106 copies/ml) for each time point represented are shown beneath each profile. The profiles of WT SIVmac239-infected normal progressors are representative of the patterns observed during plateau phase in all 10 RMs in this cohort with the salient, common feature being that the majority of lung CD4+ T cells in these RMs expressed either CCR5, Ki-67, or both. In contrast, in all rapid progressors, >80% of residual CD4+ T cells in the BALs of rapid progressors lacked both CCR5 and Ki-67 expression (see Results).
Discussion
In the setting of RM infection with pathogenic CCR5-tropic SIV, there is little question that viral replication drives pathogenesis (16, 28, 29), and that the origin of rapid progression clearly lies in the relatively high levels of viral replication during early infection, most likely resulting from (a) intrinsic host differences in cellular susceptibility to productive infection with the virus in question (28, 36), (b) host immune factors (37, 38), and/or (c) concomitant infection (39, 40). However, some CCR5-tropic SIV-infected RMs can maintain immune homeostasis for long periods despite viral loads as high as, or nearly as high as, rapid progressors (Fig. 1 A), suggesting that high viral replication produces disease (e.g., overt immune deficiency) through differentially regulated host mechanisms. Here, we sought to critically examine the role of CD4+ T cell depletion in this “downstream” pathogenetic sequence by analyzing in detail the dynamics of this population in rapidly progressive, normally progressive, and non- or slowly progressive SIV infection, as well as a control non-SIV infection, and then determining which, if any, of the dynamic parameters are common to viral infection in general or to SIV infection alone, and of the latter, which are closely linked to early pathogenesis.
The first significant observation is that total CD4+ T cell depletion is not required for acute pathogenesis, an observation in agreement with other reports demonstrating rapid progression in the absence of severe CD4+ T cell depletion in blood and lymphoid tissues (41–43). The more detailed analyses of this work unambiguously demonstrate that fundamental differences in coreceptor-mediated SIV targeting of the major peripheral CD4+ T cell subpopulations, naive versus memory, underlie this seemingly incongruous observation. CD4+ T cell expression of the CCR5 coreceptor is largely restricted to memory T cells, including a relatively small subset in blood and secondary lymphoid tissues, but the vast majority of such cells in tissue effector sites (intestinal lamina propria, vaginal mucosa, lung, liver, skin, and synovium; reference 44). CCR5-tropic SIV selectively depletes this population (this paper and references 13, 33, and 35), and all rapid progressors manifested profound memory depletion before the onset of overt disease. In contrast, naive CD4+ T cells, which lack CCR5 expression, appear to escape destruction in acute/early CCR5-tropic SIV infection, even with the high viral replication of rapid progressors. Naive cells account for the paradoxical CD4+ T lymphocytosis associated with rapid progression, and comprise the vast preponderance of the “preserved” CD4+ T cell compartment (41) in the lymphoid tissues of rapid progressors found at necropsy (not depicted). Moreover, naive CD4+ T cells are selectively depleted in (CXCR4-tropic) SIVmac155T3-infected RM; yet, these animals remain healthy as long as memory T cell numbers are maintained (particularly in tissues).
Taken together, these observations implicate the CD4+ memory T cell compartment as a primary participant in SIV pathogenesis. However, when we explicitly asked whether faster and/or greater depletion predicted the accelerated disease course in rapid progressors, we found, somewhat surprisingly, that the rate and extent of CD4+ memory T cell depletion in rapid progressors either overlapped with (total memory T cells in blood) or was only very marginally greater/faster than (CCR5+ CD4+ memory T cells in blood and lung) the lower end of the normal progressor spectrum for the same parameters. Thus, even the relatively reduced viral replication level characteristic of normal progression is frequently sufficient to massively deplete CD4+ memory T cells in tissue, suggesting that although such depletion may ultimately be required for overt immunodeficiency, it is not, by itself, sufficient to trigger its onset. However, one intriguing difference between the CD4+ memory depletion patterns of WT SIVmac239 normal and rapid progressors was observed: the latter cohort showed little or no rebound in CD4+ memory T cell populations after approximately day 42 after infection, suggesting that this subset's regenerative potential might be compromised in the rapid progressors.
Direct analysis of CD4+ memory T cell dynamics supported this hypothesis. As has been previously reported (45–49), SIV infection is associated with a striking increase in CD4+ memory T cell proliferation (as measured by both Ki-67 expression and in vivo BrdU uptake) and turnover (as measured by BrdU decay), starting ∼2–3 wk after infection. This increased memory proliferation and turnover occurred in the absence of overt depletion (e.g., SIVmac239(Δnef)-infected RMs) and among CD8+ cells, suggesting that the primary inciting stimulus for this response is infection-mediated immune stimulation, rather than a homeostatic response to T cell depletion. Moreover, given that high proliferative/high turnover state was strongly manifest in the CD4+ memory compartment of SIVmac155T3-infected RMs, which almost completely lacked a naive CD4+ T cell compartment from weeks 2–10 or longer, the increased proliferation/turnover would appear to primarily involve preestablished CD4+ memory T cells, rather than recent naive to memory converts responding to antigen for the first time. In WT SIVmac239-infected normal progressors, and in all SIVmac239(Δnef)- and SIVmac155T3-infected RMs, the increased CD4+ memory T cell proliferative rates were maintained throughout the entire 200 d of observation. However, although WT SIVmac239-infected rapid progressors initiated the memory T cell proliferative response with similar kinetics, the CD4+ component uniformly failed in these animals by day 42 after infection (P < 0.0001), and CD4+ memory T cell proliferative and turnover rates returned to or approached baseline values thereafter.
The potential impact of this CD4+ memory proliferative collapse on peripheral tissues was revealed by another series of observations. First, as explained in Results, both the kinetics of BrdU labeling of blood, lymph node, and BAL T cells, and the pattern of Ki-67 expression by these labeled cells, firmly establish that the pulmonary tissue–air interface of SIV-infected normal progressors is constantly being seeded by recently divided CD4+ memory T cells, originating elsewhere (likely organized lymphoid tissues). Thus, SIV infection increases CD4+ memory T cell proliferation in peripheral lymphoid tissues, producing progeny that directly disperse to extralymphoid effector sites. Because there is both a paucity of Ki-67high T cells and minimal immediate BrdU uptake by T cells in BALs, these cells do not appear to further proliferate in these sites, but given the rapid decline in BrdU labeling observed in our BAL samples (Fig. 7), likely die in situ, only to be continuously replaced by subsequent rounds of proliferation/migration. Second, in other studies, we have used cytokine flow cytometry and pan-SIV genome consecutive peptide mixes, as well as RhCMV whole viral preps and selected RCMV peptide mixes, to investigate the antigen specificity of the post-SIV infection memory T cell proliferative response. These analyses revealed that total SIV-specific responses could account, at most, for 25% of the postinfection memory T cell proliferating (Ki-67+ or immediate BrdU+) subset (usually much less), and that responses to RhCMV were invariably a component of the increased memory T cell proliferative activity (unpublished data). Moreover, we have recently found that provision of IL-15 to uninfected RMs elicits a burst of CD4+ and CD8+ memory T cell production and tissue homing similar to that observed in SIV infection (unpublished data), and published reports suggest that IL-7 has related properties (50–53), indicating the potential of infection-induced pro-proliferative cytokines to broadly stimulate CD4+ and CD8+ memory T cell proliferation. Taken together, these data suggest that the postinfection CD4+ memory T cell proliferative response is composed of diverse antigen specificities, including cells responsive to opportunistic pathogens.
With this background, a potential linkage between the CD4+ memory T cell proliferative response and rapid progression in CCR5-tropic SIV infection can be proposed. We posit that in the setting of profound CD4+ memory T cell depletion, the increased production and tissue emigration of broadly reactive CD4+ memory T cells are essential for maintenance of immune competence. These cells are short-lived, and with failure of the CD4+ memory T cell proliferative response, their continuous flow into potential effector sites (especially environmental interface tissues such as the lung, gastrointestinal tract mucosa, and skin) ceases, depriving these sites of critically needed immunologic function. In SIVmac239-infected RMs destined for normal progression, even those with profound tissue CD4+ T cell depletion, we demonstrated that BAL CD4+ T cells express either or both CCR5 and Ki-67, consistent with their recent production and migration (the new CCR5+ effector cell influx explaining the apparent paradox of why, in most CCR5-tropic SIV infections, the majority of CD4+ T cells in markedly depleted effector sites still retain CCR5 expression). In sharp contrast, the CD4+ T cells remaining in the BALs of rapid progressors after day 42 after infection (e.g., after the failure of the postinfection proliferative response) were almost all CCR5− and Ki-67−, consistent with a lack of influx of newly produced cells. Although the critical functions provided by these newly produced, short-lived, tissue-homing CD4+ memory T cells remain speculative, the strong mutual correlation between CD4+ memory T cell production (e.g., proliferative failure), BAL CD4+ T cell phenotype, and clinical outcome strongly suggests that the dual loss of preexistent CD4+ memory T cells in tissue and the ability to produce replacements result in local immunodeficiency, setting up such animals for the stochastic occurrence of opportunistic infection.
If loss of tissue-homing, activated CD4+ memory T cells does, as we suggest, play a crucial role in the pathogenesis of rapid progression, then it follows that normal progressors avoid early pathogenesis by the sustained production of such cells. It is important to note that this CD4+ memory T cell production response is likely a major component of the persistent immune hyperactivation state that has been associated with chronic pathogenesis in HIV infection (3–5). Thus, persistent immune activation might not be solely pathologic. At least for the CD4+ T cell lineage in the SIV-RM model, its memory T cell production component appears to be critical for the maintenance of immune competence in the face of tissue memory T cell depletion. This benefit might very well be “accidental,” arising from a generalized response to viral infection that evolved for purposes other than offsetting massive CD4+ T cell destruction. Moreover, continuous high level production of short-lived CD4+ memory T cells would likely represent only a temporary solution to the ongoing destructive effects of viral replication, and may introduce or exacerbate pathological processes as well. For example, CD4+ memory T cell hyperproliferation clearly provides a continuous source of new target cells for CCR5-tropic SIV (Fig. 2 A), likely facilitating viral replication and evolution. In addition, the long-term maintenance of markedly increased CD4 and/or CD8+ memory T cell production/turnover may ultimately result in cellular hypofunctionality and/or deleterious alterations in memory differentiation and repertoire, factors that would contribute to the development of immunodeficiency in chronic infection.
The strong coreceptor dependence of CD4+ T depletion in acute SIV infection strongly suggests that viral infection, either directly or via CTL-mediated killing, mediates this process. The observation that SIVmac155T3-infected RMs experience a similar elevation of memory T cell activation/turnover as SIVmac239-infected animals, yet preserve BAL CD4+ memory T cell populations, suggests that indirect killing via activation-induced apoptosis does not play a major role in the early loss of CD4+ memory T cells in SIVmac239 infections. The CD4+ memory T cell proliferative failure in SIVmac239-infected rapid progressors may have a similar origin. Sustained high viral replication might directly destroy proliferating CD4+ CCR5+ memory T cells and/or their immediate precursors or progeny, leaving their CD8+ counterparts intact. It is noteworthy, however, that (a) the CD4+ memory T cell proliferative response does initiate in rapid progressors in the face of very high viral replication; (b) not all the proliferating/high turnover cells are CCR5+; (c) the fate of these cells in acute infection, as determined by BrdU labeling decay, appears to be no different than the fate of similarly labeled cells in normal progressors (Fig. 6 B); and (d) the collapse of CD4+ memory T cell proliferation in rapid progression is not total, as a relatively long-lived CD4+ memory proliferative component remains intact (Figs. 5 and 6 C). These observations suggest mechanisms other than overwhelming, direct, virus-mediated destruction might also be operative; perhaps a concomitant destruction of supporting lymphoid microenvironments (e.g., IL-15– or IL-7–producing macrophages/stromal cells). Given the relative preservation of the CD8+ memory and long-lived CD4+ memory T cell proliferative compartments in rapid progressors, this mechanism would entail an increased sensitivity of the short-lived, tissue-homing CD4+ memory compartment to the putative microenvironmental defect (4). The relative contribution of these mechanisms and whether the operative mechanisms are reversible with viral suppression or immunotherapy (e.g., provision of pro-proliferative cytokines) remains to be determined in future studies.
Rapid progression is relatively uncommon in human HIV infection (54), and thus, the applicability of these findings to the pathogenesis of most human AIDS cases remains uncertain. However, the immune deficiency syndrome manifest by our rapid progressor RM is highly analogous to human AIDS (1, 2, 55), and our data unequivocally demonstrate that such a clinical state can be strongly associated with the combination of profound mucosal CD4+ memory T cell depletion and loss of an apparently compensating, infection-associated proliferative response. These findings persuasively argue that tissue CD4+ memory/effector T cell dynamics can be a key arbitrator of progressive disease. In addition, the CD4 memory T cell depletion and hyperproliferation/turnover initiated in acute infection almost certainly has a prolonged impact on immune physiology thereafter, and thus our results provide a framework for further analysis of chronic phase T cell memory dynamics in SIV-infected monkeys that escape rapid progression. As evidence is accumulating that mucosal CD4+ memory T cell depletion and increased CD4+ memory T cell proliferation and turnover also occur in HIV infection (3, 4, 56–60), it is highly likely that the mechanisms operating in SIV-infected RMs will have many direct parallels in human AIDS.
Acknowledgments
The authors would like to thank Drs. Z. Grossman and D. Douek for critical review of the manuscript, Dr. D. Watkins for providing MHC typing information, and Mr. A. Legasse for expert animal care.
This work was supported by National Institutes of Health (NIH) grants RO1-AI054292, P51-RR00163, U42-RR016025, and U24-RR018107, and from NCI funds under contract number NO1-CO-124000.
The authors have no conflicting financial interests.
Abbreviations used in this paper: APC, allophycocyanin; BAL, bronchoalveolar lymphocyte; BrdU, 5-bromo-2′-deoxyuridine; RhCMV, rhesus CMV; RM, rhesus macaque; SIV, simian immunodeficiency virus.
References
- 1.Levy, J.A. 1993. Pathogenesis of human immunodeficiency virus infection. Microbiol. Rev. 57:183–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen, O.J., and A.S. Fauci. 2001. Pathogenesis and medical aspests of HIV-1 infection. Fields Virology. D.M. Knipe and P.M. Howley, editors. Lippincott Williams & Wilkins, Philadelphia. 2043–2094.
- 3.McCune, J.M. 2001. The dynamics of CD4+ T-cell depletion in HIV disease. Nature. 410:974–979. [DOI] [PubMed] [Google Scholar]
- 4.Douek, D.C., L.J. Picker, and R.A. Koup. 2003. T cell dynamics in HIV-1 infection. Annu. Rev. Immunol. 21:265–304. [DOI] [PubMed] [Google Scholar]
- 5.Hazenberg, M.D., D. Hamann, H. Schuitemaker, and F. Miedema. 2000. T cell depletion in HIV-1 infection: how CD4+ T cells go out of stock. Nat. Immunol. 1:285–289. [DOI] [PubMed] [Google Scholar]
- 6.Doms, R.W. 2001. Chemokine receptors and HIV entry. AIDS. 15:S34–S35. [DOI] [PubMed] [Google Scholar]
- 7.Grivel, J.C., M.L. Penn, D.A. Eckstein, B. Schramm, R.F. Speck, N.W. Abbey, B. Herndier, L. Margolis, and M.A. Goldsmith. 2000. Human immunodeficiency virus type 1 co-receptor preferences determine target T-cell depletion and cellular tropism in human lymphoid tissue. J. Virol. 74:5347–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grossman, Z., and R.B. Herberman. 1997. T-cell homeostasis in HIV infection is neither failing nor blind: modified cell counts reflect an adaptive response of the host. Nat. Med. 3:486–490. [DOI] [PubMed] [Google Scholar]
- 9.Grossman, Z., M. Meier-Schellersheim, A.E. Sousa, R.M. Victorino, and W.E. Paul. 2002. CD4+ T-cell depletion in HIV infection: are we closer to understanding the cause? Nat. Med. 8:319–323. [DOI] [PubMed] [Google Scholar]
- 10.Kestler, H., T. Kodama, D. Ringler, M. Marthas, N. Pedersen, A. Lackner, D. Regier, P. Sehgal, M. Daniel, and N. King. 1990. Induction of AIDS in rhesus monkeys by molecularly cloned simian immunodeficiency virus. Science. 248:1109–1112. [DOI] [PubMed] [Google Scholar]
- 11.Lewis, M.G., S. Bellah, K. McKinnon, J. Yalley-Ogunro, P.M. Zack, W.R. Elkins, R.C. Desrosiers, and G.A. Eddy. 1994. Titration and characterization of two rhesus-derived SIVmac challenge stocks. AIDS Res. Hum. Retroviruses. 10:213–220. [DOI] [PubMed] [Google Scholar]
- 12.O'Connor, D.H., T.M. Allen, T.U. Vogel, P. Jing, I.P. DeSouza, E. Dodds, E.J. Dunphy, C. Melsaether, B. Mothe, H. Yamamoto, et al. 2002. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat. Med. 8:493–499. [DOI] [PubMed] [Google Scholar]
- 13.Veazey, R.S., M. DeMaria, L.V. Chalifoux, D.E. Shvetz, D.R. Pauley, H.L. Knight, M. Rosenzweig, R.P. Johnson, R.C. Desrosiers, and A.A. Lackner. 1998. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 280:427–432. [DOI] [PubMed] [Google Scholar]
- 14.Horton, H., T.U. Vogel, D.K. Carter, K. Vielhuber, D.H. Fuller, T. Shipley, J.T. Fuller, K.J. Kunstman, G. Sutter, D.C. Montefiori, et al. 2002. Immunization of rhesus macaques with a DNA prime/modified vaccinia virus Ankara boost regimen induces broad simian immunodeficiency virus (SIV)-specific T-cell responses and reduces initial viral replication but does not prevent disease progression following challenge with pathogenic SIVmac239. J. Virol. 76:7187–7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirsch, V.M., and J.D. Lifson. 2000. Simian immunodeficiency virus infection of monkeys as a model system for the study of AIDS pathogenesis, treatment, and prevention. Adv. Pharmacol. 49:437–477. [DOI] [PubMed] [Google Scholar]
- 16.Smith, S.M., B. Holland, C. Russo, P.J. Dailey, P.A. Marx, and R.I. Connor. 1999. Retrospective analysis of viral load and SIV antibody responses in rhesus macaques infected with pathogenic SIV: predictive value for disease progression. AIDS Res. Hum. Retroviruses. 15:1691–1701. [DOI] [PubMed] [Google Scholar]
- 17.Institute for Laboratory Animal Research. 1996. Guide for the Care and Use of Laboratory Animals. National Academic Press, Washington, D.C.
- 18.Panel on Euthanasia, American Veterinary Medical Association. 2001. 2000 report of the AVMA panel on euthanasia. J. Am. Vet. Med. Assoc. 218:669–696. [DOI] [PubMed]
- 19.Endres, C.L., E. Bergquam, M.K. Axthelm, and S.W. Wong. 1995. Assessing genetic-based therapies for AIDS using the simian immunodeficiency virus. J. Med. Primatol. 24:141–144. [DOI] [PubMed] [Google Scholar]
- 20.Gibbs, J.S., D.A. Regier, and R.C. Desrosiers. 1994. Construction and in vitro properties of SIVmac mutants with deletions in “nonessential” genes. AIDS Res. Hum. Retroviruses. 10:607–616. [DOI] [PubMed] [Google Scholar]
- 21.Means, R.E., T. Matthews, J.A. Hoxie, M.H. Malim, T. Kodama, and R.C. Desrosiers. 2001. Ability of the V3 loop of simian immunodeficiency virus to serve as a target for antibody-mediated neutralization: correlation of neutralization sensitivity, growth in macrophages, and decreased dependence on CD4. J. Virol. 75:3903–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chackerian, B., N.L. Haigwood, and J. Overbaugh. 1995. Characterization of a CD4-expressing macaque cell line that can detect virus after a single replication cycle and can be infected by diverse simian immunodeficiency virus isolates. Virology. 213:386–394. [DOI] [PubMed] [Google Scholar]
- 23.Lifson, J.D., J.L. Rossio, M. Piatak Jr., T. Parks, L. Li, R. Kiser, V. Coalter, B. Fisher, B.M. Flynn, S. Czajak, et al. 2001. Role of CD8(+) lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J. Virol. 75:10187–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang, W.L., V. Kirchoff, G.S. Pari, and P.A. Barry. 2002. Replication of rhesus cytomegalovirus in life-expanded rhesus fibroblasts expressing human telomerase. J. Virol. Methods. 104:135–146. [DOI] [PubMed] [Google Scholar]
- 25.Pitcher, C.J., S.I. Hagen, J.M. Walker, R. Lum, B.L. Mitchell, V.C. Maino, M.K. Axthelm, and L.J. Picker. 2002. Development and homeostasis of T cell memory in rhesus macaque. J. Immunol. 168:29–43. [DOI] [PubMed] [Google Scholar]
- 26.Catellier, D.J., and K.E. Muller. 2000. Tests for gaussian repeated measures with missing data in small samples. Stat. Med. 19:1101–1114. [DOI] [PubMed] [Google Scholar]
- 27.O'Connor, D.H., B.R. Mothe, J.T. Weinfurter, S. Fuenger, W.M. Rehrauer, P. Jing, R.R. Rudersdorf, M.E. Liebl, K. Krebs, J. Vasquez, et al. 2003. Major histocompatibility complex class I alleles associated with slow simian immunodeficiency virus disease progression bind epitopes recognized by dominant acute-phase cytotoxic-T-lymphocyte responses. J. Virol. 77:9029–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lifson, J.D., M.A. Nowak, S. Goldstein, J.L. Rossio, A. Kinter, G. Vasquez, T.A. Wiltrout, C. Brown, D. Schneider, L. Wahl, et al. 1997. The extent of early viral replication is a critical determinant of the natural history of simian immunodeficiency virus infection. J. Virol. 71:9508–9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staprans, S.I., P.J. Dailey, A. Rosenthal, C. Horton, R.M. Grant, N. Lerche, and M.B. Feinberg. 1999. Simian immunodeficiency virus disease course is predicted by the extent of virus replication during primary infection. J. Virol. 73:4829–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Connor, R.I., D.C. Montefiori, J.M. Binley, J.P. Moore, S. Bonhoeffer, A. Gettie, E.A. Fenamore, K.E. Sheridan, D.D. Ho, P.J. Dailey, and P.A. Marx. 1998. Temporal analyses of virus replication, immune responses, and efficacy in rhesus macaques immunized with a live, attenuated simian immunodeficiency virus vaccine. J. Virol. 72:7501–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steger, K.K., M. Dykhuizen, J.L. Mitchen, P.W. Hinds, B.L. Preuninger, M. Wallace, J. Thomson, D.C. Montefiori, Y. Lu, and C.D. Pauza. 1998. CD4+-T-cell and CD20+-B-cell changes predict rapid disease progression after simian-human immunodeficiency virus infection in macaques. J. Virol. 72:1600–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Igarashi, T., C.R. Brown, R.A. Byrum, Y. Nishimura, Y. Endo, R.J. Plishka, C. Buckler, A. Buckler-White, G. Miller, V.M. Hirsch, and M.A. Martin. 2002. Rapid and irreversible CD4+ T-cell depletion induced by the highly pathogenic simian/human immunodeficiency virus SHIV(DH12R) is systemic and synchronous. J. Virol. 76:379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veazey, R.S., K.G. Mansfield, I.C. Tham, A.C. Carville, D.E. Shvetz, A.E. Forand, and A.A. Lackner. 2000. Dynamics of CCR5 expression by CD4(+) T cells in lymphoid tissues during simian immunodeficiency virus infection. J. Virol. 74:11001–11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kewenig, S., T. Schneider, K. Hohloch, K. Lampe-Dreyer, R. Ullrich, N. Stolte, C. Stahl-Hennig, F.J. Kaup, A. Stallmach, and M. Zeitz. 1999. Rapid mucosal CD4(+) T-cell depletion and enteropathy in simian immunodeficiency virus-infected rhesus macaques. Gastroenterology. 116:1115–1123. [DOI] [PubMed] [Google Scholar]
- 35.Veazey, R.S., P.A. Marx, and A.A. Lackner. 2003. Vaginal CD4+ T cells express high levels of CCR5 and are rapidly depleted in simian immunodeficiency virus infection. J. Infect. Dis. 187:769–776. [DOI] [PubMed] [Google Scholar]
- 36.Goldstein, S., C.R. Brown, H. Dehghani, J.D. Lifson, and V.M. Hirsch. 2000. Intrinsic susceptibility of rhesus macaque peripheral CD4(+) T cells to simian immunodeficiency virus in vitro is predictive of in vivo viral replication. J. Virol. 74:9388–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans, D.T., L.A. Knapp, P. Jing, J.L. Mitchen, M. Dykhuizen, D.C. Montefiori, C.D. Pauza, and D.I. Watkins. 1999. Rapid and slow progressors differ by a single MHC class I haplotype in a family of MHC-defined rhesus macaques infected with SIV. Immunol. Lett. 66:53–59. [DOI] [PubMed] [Google Scholar]
- 38.Garber, D.A., G. Silvestri, A.P. Barry, A. Fedanov, N. Kozyr, H. McClure, D. Montefiori, C.P. Larsen, J.D. Altman, S.I. Staprans, and M.B. Feinberg. 2004. Blockade of T cell costimulation reveals interrelated actions of CD4+ and CD8+ T cells in control of SIV replication. J. Clin. Invest. 113:836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sequar, G., W.J. Britt, F.D. Lakeman, K.M. Lockridge, R.P. Tarara, D.R. Canfield, S.S. Zhou, M.B. Gardner, and P.A. Barry. 2002. Experimental co-infection of rhesus macaques with rhesus cytomegalovirus and simian immunodeficiency virus: pathogenesis. J. Virol. 76:7661–7671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou, D., Y. Shen, L. Chalifoux, D. Lee-Parritz, M. Simon, P.K. Sehgal, L. Zheng, M. Halloran, and Z.W. Chen. 1999. Mycobacterium bovis bacille Calmette-Guerin enhances pathogenicity of simian immunodeficiency virus infection and accelerates progression to AIDS in macaques: a role of persistent T cell activation in AIDS pathogenesis. J. Immunol. 162:2204–2216. [PubMed] [Google Scholar]
- 41.Hirsch, V.M., S. Santra, S. Goldstein, R. Plishka, A. Buckler-White, A. Seth, I. Ourmanov, C.R. Brown, R. Engle, D. Montefiori, et al. 2004. Immune failure in the absence of profound CD4+ T-lymphocyte depletion in simian immunodeficiency virus-infected rapid progressor macaques. J. Virol. 78:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dykhuizen, M., J.L. Mitchen, D.C. Montefiori, J. Thomson, L. Acker, H. Lardy, and C.D. Pauza. 1998. Determinants of disease in the simian immunodeficiency virus-infected rhesus macaque: characterizing animals with low antibody responses and rapid progression. J. Gen. Virol. 79:2461–2467. [DOI] [PubMed] [Google Scholar]
- 43.Ryzhova, E., J.C. Whitbeck, G. Canziani, S.V. Westmoreland, G.H. Cohen, R.J. Eisenberg, A. Lackner, and F. Gonzalez-Scarano. 2002. Rapid progression to simian AIDS can be accompanied by selection of CD4-independent gp120 variants with impaired ability to bind CD4. J. Virol. 76:7903–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kunkel, E.J., J. Boisvert, K. Murphy, M.A. Vierra, M.C. Genovese, A.J. Wardlaw, H.B. Greenberg, M.R. Hodge, L. Wu, E.C. Butcher, and J.J. Campbell. 2002. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am. J. Pathol. 160:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaur, A., C.L. Hale, S. Ramanujan, R.K. Jain, and R.P. Johnson. 2000. Differential dynamics of CD4(+) and CD8(+) T-lymphocyte proliferation and activation in acute simian immunodeficiency virus infection. J. Virol. 74:8413–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monceaux, V., R. Ho Tsong Fang, M.C. Cumont, B. Hurtrel, and J. Estaquier. 2003. Distinct cycling CD4(+)- and CD8(+)-T-cell profiles during the asymptomatic phase of simian immunodeficiency virus SIVmac251 infection in rhesus macaques. J. Virol. 77:10047–10059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sopper, S., D. Nierwetberg, A. Halbach, U. Sauer, C. Scheller, C. Stahl-Hennig, K. Matz-Rensing, F. Schafer, T. Schneider, V. ter Meulen, and J.G. Muller. 2003. Impact of simian immunodeficiency virus (SIV) infection on lymphocyte numbers and T-cell turnover in different organs of rhesus monkeys. Blood. 101:1213–1219. [DOI] [PubMed] [Google Scholar]
- 48.Rosenzweig, M., M.A. DeMaria, D.M. Harper, S. Friedrich, R.K. Jain, and R.P. Johnson. 1998. Increased rates of CD4(+) and CD8(+) T lymphocyte turnover in simian immunodeficiency virus-infected macaques. Proc. Natl. Acad. Sci. USA. 95:6388–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Boer, R.J., H. Mohri, D.D. Ho, and A.S. Perelson. 2003. Turnover rates of B cells, T cells, and NK cells in simian immunodeficiency virus-infected and uninfected rhesus macaques. J. Immunol. 170:2479–2487. [DOI] [PubMed] [Google Scholar]
- 50.Fry, T.J., M. Moniuszko, S. Creekmore, S.J. Donohue, D.C. Douek, S. Giardina, T.T. Hecht, B.J. Hill, K. Komschlies, J. Tomaszewski, et al. 2003. IL-7 therapy dramatically alters peripheral T-cell homeostasis in normal and SIV-infected nonhuman primates. Blood. 101:2294–2299. [DOI] [PubMed] [Google Scholar]
- 51.Geginat, J., F. Sallusto, and A. Lanzavecchia. 2001. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4+ T cells. J. Exp. Med. 194:1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nugeyre, M.T., V. Monceaux, S. Beq, M.C. Cumont, R. Ho Tsong Fang, L. Chene, M. Morre, F. Barre-Sinoussi, B. Hurtrel, and N. Israel. 2003. IL-7 stimulates T cell renewal without increasing viral replication in simian immunodeficiency virus-infected macaques. J. Immunol. 171:4447–4453. [DOI] [PubMed] [Google Scholar]
- 53.Muthukumar, A., A. Wozniakowski, M.C. Gauduin, M. Paiardini, H.M. McClure, R.P. Johnson, G. Silvestri, and D.L. Sodora. 2004. Elevated interleukin-7 levels not sufficient to maintain T-cell homeostasis during simian immunodeficiency virus-induced disease progression. Blood. 103:973–979. [DOI] [PubMed] [Google Scholar]
- 54.Munoz, A., M.C. Wang, S. Bass, J.M. Taylor, L.A. Kingsley, J.S. Chmiel, and B.F. Polk. 1989. Acquired immunodeficiency syndrome (AIDS)-free time after human immunodeficiency virus type 1 (HIV-1) seroconversion in homosexual men. Multicenter AIDS Cohort Study Group. Am. J. Epidemiol. 130:530–539. [DOI] [PubMed] [Google Scholar]
- 55.Hirsch, V.M. 2004. What can natural infection of African monkeys with simian immunodeficiency virus tell us about the pathogenesis of AIDS? AIDS Rev. 6:40–53. [PubMed] [Google Scholar]
- 56.Guadalupe, M., E. Reay, S. Sankaran, T. Prindiville, J. Flamm, A. McNeil, and S. Dandekar. 2003. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J. Virol. 77:11708–11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clayton, F., G. Snow, S. Reka, and D.P. Kotler. 1997. Selective depletion of rectal lamina propria rather than lymphoid aggregate CD4 lymphocytes in HIV infection. Clin. Exp. Immunol. 107:288–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hellerstein, M.K., R.A. Hoh, M.B. Hanley, D. Cesar, D. Lee, R.A. Neese, and J.M. McCune. 2003. Subpopulations of long-lived and short-lived T cells in advanced HIV-1 infection. J. Clin. Invest. 112:956–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brenchley, J.M., T.W. Schacker, L.E. Ruff, D.A. Price, J.H. Taylor, G.J. Beilman, P.L. Nguyen, A. Khoruts, M. Larson, A.T. Haase, and D.C. Douek. 2004. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 200:749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mehandru, S., M.A. Poles, K. Tenner-Racz, A. Horowitz, A. Hurley, C. Hogan, D. Boden, P. Racz, and M. Markowitz. 2004. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 200:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]