

Abstract
There is a fundamental conflict between two different views of how proteins fold. Kinetic experiments and theoretical calculations are often interpreted in terms of different population fractions folding through different intermediates in independent unrelated pathways (IUP model). However, detailed structural information indicates that all of the protein population folds through a sequence of intermediates predetermined by the foldon substructure of the target protein and a sequential stabilization principle. These contrary views can be resolved by a predetermined pathway—optional error (PPOE) hypothesis. The hypothesis is that any pathway intermediate can incorporate a chance misfolding error that blocks folding and must be reversed for productive folding to continue. Different fractions of the protein population will then block at different steps, populate different intermediates, and fold at different rates, giving the appearance of multiple unrelated pathways. A test of the hypothesis matches the two models against extensive kinetic folding results for hen lysozyme which have been widely cited in support of independent parallel pathways. The PPOE model succeeds with fewer fitting constants. The fitted PPOE reaction scheme leads to known folding behavior, whereas the IUP properties are contradicted by experiment. The appearance of a conflict with multipath theoretical models seems to be due to their different focus, namely on multitrack microscopic behavior versus cooperative macroscopic behavior. The integration of three well-documented principles in the PPOE model (cooperative foldons, sequential stabilization, optional errors) provides a unifying explanation for how proteins fold and why they fold in that way.
Keywords: protein folding, misfolding, foldons, sequential stabilization, optional error, predetermined pathway, lysozyme
Detailed structural information obtained from hydrogen exchange (HX) studies of cytochrome c (Cyt c) and related studies with other proteins support a classical view of protein folding in which all of the molecules in a refolding population fold through essentially the same intermediate structures (Chamberlain and Marqusee 2000; Englander 2000; Maity et al. 2005). The pathway constructs the native protein by the stepwise addition of the cooperative folding units of the native structure, called foldons. The order of pathway steps is guided by a sequential stabilization process in which prior native-like structure templates the formation of subsequent complementary structure. Thus the folding pathway is determined by the same cooperative interactions that determine the target native structure. The pathway may be linear or may branch at any step as directed by the logic of the sequential stabilization process (Krishna et al. 2006).
By contrast, other experiments reveal a wide range of kinetic protein folding behavior that has been interpreted quite differently, in terms of independent unrelated pathways (IUP model). Proteins can fold in a simple two-state way with only the unfolded (U) and native (N) states significantly populated, or in a multistate way with one or more intermediates (I) transiently populated, or heterogeneously with some population fraction folding in a two-state way and others in a three-state way. Common conclusions are that specific intermediates need not exist (fast two-state folding), that populated intermediates are detrimental (slower multistate fractions), and that different population fractions fold through independent parallel pathways (heterogeneous folding). These varied behaviors have been described mainly by spectroscopic methods which can track kinetic folding in real time but provide almost no structural detail. Nevertheless, these views draw credibility from theoretical calculations that often portray folding in terms of different population fractions traversing different parts of the energy landscape, generally interpreted as multiple independent pathways (Baldwin 1995; Bryngelson et al. 1995; Wolynes et al. 1995; Dill and Chan 1997; Brooks 1998; Plotkin and Onuchic 2002a,b).
The choice between the predetermined pathway view and the multiple independent pathway view is fundamental to the protein folding problem. These grossly discrepant interpretations of accepted experimental results can be resolved by modifying the predetermined pathway model to include probabilistic misfolding errors that block the forward progress of normally occurring intermediates (Sosnick et al. 1994; Krishna et al. 2004). We term this the predetermined pathway–optional error (PPOE) model. Chance misfolding errors can act to corrupt different naturally occurring intermediates and insert optional error-repair barriers at different points in a pathway. When the error probability is zero at all pathway steps, folding appears to be a two-state process. When it is unity at one particular step, three-state folding occurs. Any other values or combinations will produce mixed behavior in which different population fractions display different naturally occurring but partially corrupted intermediates, or none at all, and fold at different rates. This heterogeneous behavior, when detected by the usual spectroscopic observations of kinetic phases, will appear to represent multiple alternative pathways.
The optional error hypothesis is grounded in the observation that folding errors are ubiquitous. Well-known misfolding errors include prolyl and non-prolyl peptide bond misisomerization, transient aggregation, nonnative hydrophobic clusters, disulfide shuffling, heme misligation, and perhaps nonnative domain docking modes. These errors are optional, not intrinsic to the folding process, and they can often be inserted or removed by the manipulation of folding conditions. In vitro, misfolding-dependent aggregation obstructs laboratory and industrial protein expression (Chi et al. 2003). In vivo, folding errors appear to result in the loss of 30% or more of synthesized polypeptides (Yewdell 2005), and they account for a large fraction of human pathologies. In response, biology finds it cost-effective to elaborate multiple helper proteins, error repair systems, and turnover machineries.
As one test of the PPOE model, we re-examine results for the highly studied hen egg white lysozyme (HEWL) protein. Dobson and Radford and their coworkers (Dobson et al. 1994) found that HEWL folds in a three-state way so that a transient intermediate accumulates. The HEWL intermediate that has been most thoroughly studied contains native-like structural elements but also some seriously misfolded structure. In comprehensive experiments and analysis, Kiefhaber and coworkers found and quantified additional HEWL folding phases and intermediates, and they matched possible folding models against their extensive kinetic data (Kiefhaber 1995; Kiefhaber et al. 1997; Wildegger and Kiefhaber 1997; Bieri et al. 1999; Bieri and Kiefhaber 2001). All of these results have been interpreted in terms of multiple independent parallel pathways, as has been done also for many other proteins.
We find that the PPOE model quantitatively accounts for the measured folding and unfolding of the lysozyme intermediates and native state and their dependence on denaturant with fewer fitting parameters than the multiple pathway models considered before. More importantly, the fitted PPOE model exhibits folding properties that are common to proteins very broadly, whereas the fitted IUP model exhibits properties that are contradicted by experiment. The PPOE formalism seems able to explain the varied folding behavior of proteins quite generally.
Results
Previous work
Hen egg white lysozyme (HEWL) is a 129-residue protein stabilized by four disulfide bonds. It has two domains, rich in α and β structure, respectively. When HEWL is unfolded in concentrated denaturant and then diluted into folding conditions, it rapidly collapses (<1 msec) to form a compact C state (Chaffotte et al. 1992; Itzhaki et al. 1994; Segel et al. 1999), evidently dependent on its disulfide cross-links since the disulfide-reduced form (Roux et al. 1997) and proteins more generally (Plaxco et al. 1999; Jacob et al. 2004) do not collapse in this way. The chain collapse may or may not reflect a distinctly structured intermediate; hydrogen exchange protection seems to occur only at residues closely adjacent to the pre-existing disulfide bridges (Gladwin and Evans 1996; see also Goldberg and Guillou 1994; Roux et al. 1997).
The major folding sequence then proceeds from the C state. Spectroscopic experiments show that an intermediate, called I, forms and accumulates (∼30 msec), and then folds more slowly (∼400 msec) to a nearly native N′ state (Dobson et al. 1994; Kulakarni et al. 2006). Finally, the native substrate binding site forms in an optically silent N′-to-N step (Kulakarni et al. 2006). Like other workers before, we focus on the major folding behavior that carries the protein from the early C state to the near-final N′ state, which is pertinent to the folding problem most generally.
The hypothesis pursued here is that proteins fold through predetermined native-like intermediates that transiently accumulate only when their forward progress is blocked by an optional misfolding error (Sosnick et al. 1994; Englander et al. 1998; Krishna et al. 2004). The following discussion formulates an “error” as a step that removes the protein from the productive folding pathway and must be reversed for forward folding to resume. As expected from this hypothesis, the populated HEWL intermediate has much native-like structure, seen by HX pulse labeling (Radford et al. 1992; Miranker et al. 1993), but also some obvious misfolding (Fig. 1). The intermediate has 1.5 times the CD225 of the N state (Dobson et al. 1994), lower and blue-shifted fluorescence compared to both U and N (Segel et al. 1999), some proline misisomerization that affects late-stage kinetics (Kiefhaber 1995), and a nonnative tryptophan cluster (Rothwarf and Scheraga 1996; Klein-Seetharaman et al. 2002). The blocking error is clearly optional. The deletion of one disulfide bond removes the nonnative CD and fluorescence (Eyles et al. 1994); the slow phase can be relieved, at least in part, by destabilizing the nonnative tryptophan cluster (Rothwarf and Scheraga 1996), and a significant fraction of the refolding population (∼15%) folds in a fast two-state manner with no blocking at all (Kiefhaber 1995; Wildegger and Kiefhaber 1997).
Figure 1.

Lysozyme folding followed by CD225 (A) and fluorescence (B). The smoothed traces shown are from Dobson et al. (1994; reprinted with permission from Elsevier). The dashed lines show the native signal.
These results are as expected from the PPOE model, but they do not provide a quantitative test of the hypothesis. We therefore turn to an analysis of the extensive and coherent data sets for HEWL folding and unfolding collected by the Kiefhaber group (Kiefhaber et al. 1997; Wildegger and Kiefhaber 1997; Bieri et al. 1999; Bieri and Kiefhaber 2001). First we compare the measured folding and unfolding data to the multipath IUP and PPOE reaction schemes (Fig. 2). As for any kinetic process, the measured macroscopic rates can represent complex mixtures of the microscopic rate constants that connect the various states in ways that depend critically on the reaction scheme considered. The usual kinetic fitting exercise is intended to match alternative reaction schemes against the data, rule out those that do not fit, and find one that does, ideally with the fewest fitting parameters.
Figure 2.

The reaction schemes invoked here. Schemes 1–3 represent the independent unrelated pathway (IUP) models found by Kiefhaber and coworkers (Kiefhaber et al. 1997; Wildegger and Kiefhaber 1997; Bieri et al. 1999; Bieri and Kiefhaber 2001). Scheme 4 represents a predetermined pathway–optional error (PPOE) model. Diagonal steps may also occur. The pathway may branch when the sequential stabilization principle allows two or more alternative next steps. (Similar models have been used to explain the complex folding kinetics of cytochrome c and staphylococcal nuclease [Elöve et al. 1994; Sosnick et al. 1994; Colón et al. 1996; Walkenhorst et al. 1997; Englander et al. 1998].) Schemes 5–7 show the limiting cases used here. Ii and Iix are pictured to have essentially the same native-like structured regions, but Iix has in addition some misfolding error that interferes with the formation and docking of an incoming foldon unit. It may act immediately or can be incipient and block at a later step (e.g., misisomerized proline).
The Triangular model: Two independent folding pathways
Kiefhaber and coworkers (Kiefhaber 1995; Wildegger and Kiefhaber 1997) showed that the kinetics of fluorescence-detected HEWL folding includes a small faster phase (∼15%) that carries C to N′ without the population of an intermediate state, in agreement with the fast phase seen by Radford et al. (1992) in HX pulse-labeling experiments. In an extensive analysis, Kiefhaber and coworkers (Wildegger and Kiefhaber 1997) used stopped-flow fluorescence experiments to measure folding and unfolding rates of HEWL as a function of denaturant (chevron curves in Fig. 3A). They also measured the time-dependent populations of I and N′ at one GdmCl concentration (0.6 M GdmCl) (Fig. 3B) by the interrupted refolding method (start folding, delay, unfold at higher denaturant, measure the rate and amplitude of each unfolding phase) (see Schmid 1983 for experimental details).
Figure 3.

Lysozyme folding data (fluorescence) obtained in the low-salt condition (from Wildegger and Kiefhaber 1997). (A,D) The chevron plots show measured macroscopic relaxation rates as a function of GdmCl concentration. (B,E) The data show the U, I, and N populations as a function of folding time (in 0.6 M GdmCl), measured in interrupted folding experiments. (C,F) The folding models and their fitted microscopic rate constants (at 0.6 M GdmCl); m-values needed for the chevrons are in parentheses; m = d(RT ln k)/d[GdmCl]). The parameters fully constrained by the data fitting are in boldface. Also shown are χR 2 values for the global fit and the fluorescence signal strength (S) of the populated intermediates calculated relative to the previous results (from Fig. 1 of Wildegger and Kiefhaber 1997; see Materials and Methods). The red curves in panels A and B fit the Triangular model to the data using the rate constants given in Figure 3 of Wildegger and Kiefhaber (1997). Black curves show the global refitting done here. (D) The T model predicts another faster phase whose rates are, however, very poorly determined because of the minimal population of I, as suggested by the multiple predicted chevrons shown (dotted lines). Note that S for the populated intermediate, the folding flux away from U, the unfolding flux away from N, and the flux into the blocked intermediate populations is the same in the two different models. The rate constants (seconds−1) and m-values (kilocalories/mole per molar GdmCl) used in constructing the red lines in the Triangular model are: k UN = 14 (−1.9), k NU = 9E − 7 (0.7), k UI = 19 (−1.2), k IU = 3.5 (0.3), k IN = 2.5 (−0.5), k NI = 9E − 7 (0.7), where E indicates powers of 10.
Wildegger and Kiefhaber (1997) then considered possible folding models that could explain all of these results. Several considerations led to a Triangular model with folding through two independent parallel pathways, C to N′ and C to I to N′, written as Scheme 1 (Fig. 2) in terms of U, I, and N for generality. The larger population fraction (85%) folds in a three-state manner, U to I to N. In an independent parallel pathway, the faster fraction folds directly to N in a two-state manner (U to N).
The three-species Triangular model predicts the presence of two kinetic phases, shown in the form of chevron plots in Figure 3A. The excellent kinetic fitting to the chevron data found by Wildegger and Kiefhaber is indicated by the red curves (data and fit from Fig. 3 of Wildegger and Kiefhaber 1997). This set of fitted rate constants matches the time-dependent population data less well (Fig. 3B; data from Fig. 1 of Wildegger and Kiefhaber 1997). We refit the Triangular model to the data globally. The modified rate constants (Fig. 3C, m-values in parentheses) yield a good fit to both data sets (black in Fig. 3A,B). Global χR 2 falls from 42 to 1.6. Four of the six rate constants are well constrained by the implicitly measured mass flow at critical pathway points.
The PPOE model: A predetermined pathway with optional errors
The PPOE hypothesis leads to a different kind of model. In the PPOE model, proteins fold through a predetermined sequence of native-like intermediates that can be corrupted and transiently blocked by chance misfolding errors. Given the obvious misfolding of the populated lysozyme intermediate (Fig. 1), other workers have previously considered that its forward folding might be rate-limited by slow error repair (Dobson et al. 1994; Eyles et al. 1994; Rothwarf and Scheraga 1996; Matagne et al. 1997, 1998; Klein-Seetharaman et al. 2002).
A fairly general PPOE model is shown as Scheme 4 (Fig. 2). The minimal PPOE pathway diagrammed as the T model in Scheme 5 (Fig. 2) can serve to illustrate the issues. Here the pathway is represented by a single normally occurring on-pathway intermediate, I, and a corrupted Ix form that has in addition some misfolding. The probability for error formation is k II x/(k II x + k IN + k IU). If the error-repair step required to return Ix to the productive pathway is slow, Ix will accumulate and kinetic folding will appear to be three-state.
The T model provides an excellent fit to all of the HEWL data (solid lines in Fig. 3D,E) with the same number of fitting rate constants required for the Triangular model (six) and an equivalent χR 2. The three rate constants that lead to I (boldface in Fig. 3F) and their m-values (in parentheses in Fig. 3F) are fully constrained because they are dominated by well-measured folding and unfolding rates (Fig. 4B). The others are underdetermined and are free to vary over a wide range without degrading the goodness of fit. Only their ratios are constrained. They control the partitioning of I into three competing reactions.
Figure 4.

Comparison of the fitted macroscopic relaxation rates (solid lines) with the calculated microscopic rate constants (black dashed lines) determined for the (A) Triangular and the (B,C) T models. Data are from Wildegger and Kiefhaber (1997). The dashed red lines show that the extrapolation of the fast-phase unfolding arm intersects the measured slow-phase folding data, which was used by Wildegger and Kiefhaber (1997) to reject the conventional off-pathway intermediate possibility, assumed as an Ix ↔ U ↔ N sequence. In comparison, the Ix-to-I curves (k I x I in panels B and C) show that the measured data are not inconsistent with the T model.
The T model has four species including the hidden on-pathway I state, and therefore predicts the presence of three kinetic phases. The additional third phase is described by the underdetermined rate constants, as shown by the dotted lines in Figure 3D obtained from multiple independent fittings. It is very poorly determined by the kinetic data because the on-pathway uncorrupted intermediate goes forward rapidly, barely accumulates, and is hardly detectable. The kinetic parameters for the hidden on-pathway intermediate could be specified by additional unfolding experiments (see below).
The rates predicted by both models perfectly match the measured data. Both produce the same total flux away from U, the same flux into their respective populated intermediate, the same flux away from N, and also the same S value (relative fluorescence signal; see Materials and Methods) for the intermediate. Figure 4 shows how the measured macroscopic relaxation rates (data points) compare with the microscopic rate constants (dashed lines) calculated for the two different models.
Rejection criteria and the Triangular model
The two-pathway Triangular model seemed to be required by three qualitative constraints (Wildegger and Kiefhaber 1997).
-
(1) The major constraint that seemed to require independent parallel pathways relates to the absence of a lag phase for reaching N. (The upsweep in N formation in Fig. 3B is due to the logarithmic time scale and not to a kinetic lag.) If an intermediate accumulates and then folds to N in a sequential pathway (U to I to N), N will form with a kinetic lag that keys to the time scale for populating I. The two-pathway Triangular model accounts for the absence of a lag in N formation by the fast formation of N in a parallel U to N pathway with sufficient flux. Kinetic coupling due to the fact that both pathways deplete the pool of U ensures that both of these steps occur on the same time scale.
However, the absence of a lag can also be satisfied by a single pathway that contains an optional off-pathway misfolding-repair step (T model), as described below.
-
(2) A second constraint rejected the possibility that the populated intermediate might be misfolded and off-pathway. The off-pathway model conventionally written as Ix to U to N predicts a reverse denaturant effect in the folding arm of the slow-phase chevron at very low denaturant. Here the observed folding rate should initially increase with added denaturant because the rate for folding to N becomes dominated by an unfolding step, Ix to U. As a result of multipathway kinetic coupling, the same Ix-to-U step also determines the measured unfolding arm of the faster phase. Therefore, the expected reverse denaturant effect in the slow-phase folding arm can be predicted by back extrapolation from the fast-phase unfolding arm. The extrapolation shown as a red dashed line in Figure 4, A and B, crosses rather than merging with the measured data, indicating that the reverse denaturant effect expected for the off-pathway model is absent. This test rules out the conventional model for an off-pathway misfolded intermediate. The test was made more convincing by repeating the kinetic experiments in added Na2SO4, which stabilizes the intermediate and elongates the rollover region (Fig. 4C).
However, this test does not reject the type of off-pathway misfolding that enters the PPOE model, as described below.
(3) The possibility that the multiple kinetic phases might originate because of some pre-existing dichotomy of the U state (U and Ux) was tested in a double jump experiment (unfold, delay time, jump to refolding conditions). The results ruled out the usual U-to-Ux option because of proline isomerism, which occurs more slowly than the 30-msec experimental deadtime.
These rejection criteria led, by elimination of all apparent alternatives, to the requirement for independent parallel pathways. The simplest version, with two independent pathways and one intermediate that goes forward slowly and accumulates, is the Triangular model.
Rejection criteria and the PPOE model
Analysis shows that the T reaction scheme satisfies the same criteria, as follows.
(1) N is formed with no kinetic lag. In the T model, the formation of N through the major blocked route alone (U through Ix to N) would show a lag in N formation on the time scale of the U-to-Ix step. The lag is filled in by the unblocked faster U-to-I-to-N route. As before, kinetic coupling inherent in the reaction scheme ensures that N formation occurs on the unblocked route at the same rate as the U-to-Ix step.
(2) Although the T model contains an off-pathway partially misfolded intermediate, it is not rejected by the off-pathway test applied before. The issue can be understood from Figure 4. In the T reaction scheme, the slow Ix-to-I rate plays the role of the previous Ix-to-U rate in the conventional off-pathway scheme. However, unlike the previous Ix-to-U rate (red line in Fig. 4), Ix to I is not coincident with the measured fast-phase unfolding arm, as shown by the k I x I line in Figure 4, B and C. Back extrapolation of the Ix-to-I line to low denaturant does not cross the slow-phase folding data in the region measured; therefore the off-pathway misfolding is not rejected by the data measured. It would ultimately rate-limit the slow-phase folding rate and produce a reverse denaturant effect but only in an unreachable “negative [GdmCl]” region.
(3) The third rejection test rules out a slowly produced (>30 msec) U-to-Ux form. The T model does not require a Ux form to fit the present lysozyme data, although some population fraction of HEWL does contain pre-existing Ux forms (nonnative Trp cluster, misisomerized prolines) that cause slow folding phases (Eyles et al. 1994; Kiefhaber 1995; Rothwarf and Scheraga 1996; Klein-Seetharaman et al. 2002). The more general PPOE model in Scheme 4 (in Fig. 2) includes this possibility.
A hidden intermediate detected by unfolding
Kiefhaber et al. (1997) used an interrupted folding experiment to populate the slowly folding lysozyme intermediate and measured its unfolding by a jump to high denaturant. The unfolding of the populated intermediate could not be fit with a single exponential. The results define an additional faster kinetic phase (filled circles in Fig. 5).
Figure 5.

Extension of Figure 3 (open symbols) to include the additional data for two-phase unfolding for the populated intermediate (filled symbols) (from Figs. 3 and 4A of Kiefhaber et al. 1997). For the Triangular model, the additional fast unfolding phase requires an added intermediate (Nu) that was not measured in the folding direction. The unmodified PPOE model already predicts the additional fast unfolding phase, and the new unfolding data fix its previously underdetermined parameters. In this case, the fitting adjusts the signal proportionality constants (S-values) for the silent intermediates differently in the two models so that the fluxes at corresponding points do not match. The two models require eight and six fitting rate constants, respectively.
In principle, the three-species Triangular model can only produce two relaxation phases, and unfolding of the populated intermediate can only exhibit single exponential two-state I-to-U kinetics. To explain the results, another species is required. Kiefhaber et al. (1997) proposed an on-pathway intermediate, called Nu (for nucleating), placed before the populated I in the folding direction. Figure 5, A–C, includes these new data (from Figs. 3 and 4A of Kiefhaber et al. 1997) and globally fits all of the data to the modified Triangular + Nu model (Scheme 2 in Fig. 2).
The unmodified T model naturally predicts the non-two-state unfolding of the populated intermediate and the additional fast kinetic folding phase. Some fraction of Ix unfolds directly (Ix through I to U), whereas another fraction recycles back from I to Ix and reaches U more slowly. When the unfolding data are included, kinetic fitting of the T model retains the previously determined rate constants and fixes the previously underdetermined ones (χR 2 = 0.7) (Fig. 5D–F).
The modified Triangular model approaches the T model. Both have three populated species and a kinetically hidden intermediate. However, the Triangular model still requires in addition the straight-through U-to-N pathway in order to satisfy the first rejection test (no lag in N formation). The extended Triangular model then requires eight rate constants to explain the available lysozyme data compared to six for the more minimal T model.
Multipathway model for two populated intermediates
Bieri and coworkers (Bieri et al. 1999; Bieri and Kiefhaber 2001) repeated these experiments at 10°C (Fig. 6) and 20°C (data not shown) in the presence of added NaCl (0.85 M NaCl at pH 5.2, called the high salt condition), and also at higher pH (pH 9.2, 20°C) (Fig. 7). Each of these conditions made it possible to detect an additional folding phase, which requires the population of another intermediate. Several kinetic models were tested, in this case with four populated species (U, N, I1, I2) (Bieri et al. 1999). The rejection criteria appeared to require an additional independent pathway and ruled out several possible models. The inability to fit all of the data ruled out some other models. The successful scheme with three independent pathways is represented as a Diamond model (Scheme 3 in Fig. 2).
Figure 6.

Three phase folding data obtained at the high-salt condition (from Bieri et al. 1999), which requires two populated intermediates. Fits obtained for the Diamond model are shown in red for the rate constants given in Figure 7 of Bieri et al. (1999) (m-values adjusted for best fit) and in black when globally refit. The Diamond and PPOE models require 12 and eight fitting rate constants, respectively. Other notes are as for Figure 3. The rate constants used in constructing the red lines in the Diamond model are (at 0.6 M GdmCl; m-values in parenthesis): k UN = 1.2 (−1.5), k NU = 9E − 9 (1.0), k UI1 = 9.7 (−1.4), k I1U = 4.3 (0.2), k UI2 = 6.9 (−1.6), k I2U = 0.9 (2E − 5), k I1I2 = 1.4 (−4E−5), k I2I1 = 1.8 (1E−5), k I1N = 0.3 (−6E − 5), k NI1 = 5E−8 (0.9), k I2N = 0.2 (−7E − 3), k NI2 = 2E − 9 (0.1).
Figure 7.

Three-phase folding data obtained at the high pH condition (from Bieri and Kiefhaber 2001), which requires two populated intermediates. Fits obtained for the Diamond model are shown in red for the rate constants given in Figure 3 of Bieri and Kiefhaber (2001) (m-values adjusted for best fit) and in black when globally refit. The Diamond and PPOE models require 12 and eight fitting rate constants, respectively. Other notes are as for Figure 3. The rate constants used in constructing the red lines in the Diamond model are (at 0.6 M GdmCl; m-values in parenthesis): k UN = 1.8 (−1.6), k NU = 2E − 6 (0.7), k UI1 = 46 (−1.4), k I1U = 4.0 (0.2), k UI2 = 13 (−1.4), k I2U = 0.1 (0.4), k I1I2 = 6.9 (−0.13), k I2I1 = 1.7 (6E − 6), k I1N = 0.8 (−0.1), k NI1 = 3E − 6 (0.6), k I2N = 2E − 6 (−0.03), k NI2 = 2E − 6 (0.7).
As before, the published fitting factors (from Fig. 7 of Bieri et al. 1999 and Fig. 3 of Bieri and Kiefhaber 2001) matched one of the data sets less well (red curves in Figs. 6 and 7), in this case the chevron data. The agreement could be improved by a global fitting (black). As before, only some of the kinetic constants shown for the Diamond model in Figures 6 and 7 are well constrained by the data (shown in boldface).
PPOE model with two populated intermediates
The T model contains four species (U, N, I, Ix) and therefore predicts three kinetic phases, with the third phase coming from the minimally populated on-pathway I, as noted before. However, the simple T model does not produce a good global fit to the results in Figures 6 and 7. As for the multipathway model, another well-populated intermediate is necessary. An extended T model adds the required intermediate in either a Plus or a Double T configuration, shown as Schemes 6 and 7 (Fig. 2), which turn out to be kinetically equivalent.
In the Plus reaction scheme, the on-pathway I appears to generate two different error-dependent Ix forms. This PPOE model survives the rejection tests, and it provides excellent global fits to the data, as shown in Figures 6 and 7. As before, only the rate constants (and their m-values) leading to the on-pathway I are fixed by the data available. Only the ratios of the other four rate constants are fixed. They determine the partitioning of I into the four competing pathways and the time-dependent populations of the individual species.
As before, the fitted Diamond and Plus models both produce the same S-values (relative fluorescence signals) for the intermediates and hence the same total flux away from U, the same flux into their respective blocked intermediates, and the same flux away from N. As before, the Plus model fits all of the data with fewer fitting rate constants than the Diamond model (8 vs. 12) and an equivalent χR 2 parameter in each case.
We also tested Scheme 7 (Fig. 2), called the Double T model, with two on-pathway intermediates and their misfolded analogs. This scheme survives the rejection tests and globally fits the chevron and population data sets at all conditions. It formally has two more kinetic fitting constants than the Plus model, but it is kinetically equivalent and reduces to the Plus model because I1 and I2 interconvert rapidly and effectively play the role of a single I.
Discussion
Early work in protein folding was guided by the conjecture that proteins could find their predestined native state within the vast space of possible folds only by traveling through some predetermined pathway (Levinthal 1968, 1969). Kinetic experiments detected pathway intermediates that were interpreted in these terms (Kim and Baldwin 1982, 1990). In agreement, a range of structural experiments characterized partially folded forms, molten globules, and folded protein fragments believed to mimic specific kinetic folding intermediates (Oas and Kim 1988; Staley and Kim 1990; Wu et al. 1993; Ptitsyn 1995; Chamberlain et al. 1999; Feng et al. 2005; Maity et al. 2005). Structural information from hydrogen exchange and related methods found and characterized partially native-like intermediates and showed that they construct classical folding pathways (Roder et al. 1988; Bai et al. 1995; Chamberlain et al. 1996; Fuentes and Wand 1998a,b; Xu et al. 1998; Milne et al. 1999; Chamberlain and Marqusee 2000; Chu et al. 2002; Hoang et al. 2002; Silverman and Harbury 2002; Yan et al. 2002, 2004; Feng et al. 2003, 2005; Krishna et al. 2003a,b, 2004, 2006; Maity et al. 2004, 2005; Cecconi et al. 2005; Weinkam et al. 2005). The direct NMR structure determination of two engineered variants of apocytochrome b 562 (Feng et al. 2005) showed them to be closely equivalent to the on-pathway kinetic intermediates previously defined by native state HX methods under two-state folding conditions (Fuentes and Wand 1998a,b; Chu et al. 2002). These observations provide the experimental basis for a classical predetermined pathway.
However, developments in the theoretical area fostered a different view, namely, that proteins can fold through any number of unrelated paths, directed only by a random energetically downhill search for the native state (Zwanzig et al. 1992; Bryngelson et al. 1995; Wolynes et al. 1995; Brooks 1998; Plotkin and Onuchic 2002a,b). Partly on this basis, the varied folding behavior observed for many proteins by spectroscopic methods has more recently been widely interpreted in terms of independent parallel pathways through unrelated intermediate states (Baldwin 1995; Dobson et al. 1998; Bilsel and Matthews 2000; Wallace and Matthews 2002) (even though experimental results find only a small number of alternative parallel pathways rather than the many implied by theoretical simulations).
The independent pathways and predetermined pathway views project fundamentally different representations of how proteins actually fold. These sharply different interpretations concerning the most fundamental modalities of protein folding pathways can be rationalized by the optional error hypothesis, suggested by the well-known tendency of proteins to misfold. Chance misfolding within a single pathway will mimic multipathway behavior.
Lysozyme folding
Can the predetermined pathway–optional error model provide a quantitative match to real protein behavior? Hen egg lysozyme has been most extensively characterized and analyzed in previous kinetic folding studies. The results have been interpreted in terms of multiple independent pathways. Analysis shows that the single-pathway PPOE model can fit the HEWL data as well as the multiple independent pathway model, and it does so with fewer fitting parameters.
Both models satisfy the rejection criteria developed by the Kiefhaber group. Both quantitatively fit the heterogeneous folding of lysozyme under disparate conditions with different sets of measured relaxation rates, and therefore seem able to fit any other accurate self-consistent data set for the two-state, multistate, or heterogeneous folding of any protein. In the general case, additionally measured kinetic phases could be accommodated by either model, undoubtedly with equally good fit. It is difficult to conceive of any fundamentally different kind of model that can meet these requirements.
Comparison with experimental information
Can one go past a data-fitting exercise and choose between the multipath IUP and the PPOE models on more substantial grounds? The derived parameters endow the two models with distinctively different folding characteristics. Their validity may be judged by comparing them with known folding information.
Two-state folding
In the IUP model, population fractions that fold in a two-state manner are explained by a direct U-to-N pathway. Intermediates do not accumulate, apparently because they are either absent or unstable. In the PPOE model, pathway intermediates are obligatory (except for proteins that are smaller than two foldon units). Two-state folding occurs because intermediates inherently fold forward much faster than the rate-limiting initial nucleation step and do not significantly populate.
Experiment favors the PPOE view. Intermediates that fold forward rapidly and are kinetically silent, even though they are on-pathway and stable (relative to U), have been demonstrated for several proteins by native state HX (Bai et al. 1995; Chamberlain et al. 1996; Fuentes and Wand 1998a,b; Xu et al. 1998; Milne et al. 1999; Chamberlain and Marqusee 2000; Chu et al. 2002; Hoang et al. 2002; Yan et al. 2002, 2004; Feng et al. 2003, 2005; Krishna et al. 2003b, 2006; Maity et al. 2004, 2005), and by a related experiment using sulfhydril reactivity (Silverman and Harbury 2002), and also by NMR relaxation dispersion (Korzhnev et al. 2004). Kinetic considerations support sequential on-pathway intermediates for many other proteins (Sánchez and Kiefhaber 2003a,b).
The possibility that intermediates are absent in the two-state folding of sizeable proteins like lysozyme (129 residues) seems unlikely in view of the fact that the foldon size in the best worked out case of Cyt c is 15–20 residues. Even the small fyn SH3 protein with only 59 residues is seen to exhibit a stable but kinetically silent on-pathway intermediate (Korzhnev et al. 2004). Two-state folding kinetics is seen especially for small proteins, where one expects the probability for folding errors to be minimal. As protein size increases, the probability of error formation increases and one sees an increasing non-two-state fraction, as expected.
According to the PPOE parameters, uncorrupted on-pathway intermediates are inherently fast. This makes the first pathway step inherently rate-limiting and folding inherently two-state, unlike the multipathway model. This property is consistent with the success of the contact order formalism (Plaxco et al. 1998, 2000) and with nucleation models in general (Wetlaufer 1973; Fersht 1995; Sosnick et al. 1996). The contact order formulation implies that an initial whole molecule conformational search for a collapsed chain conformation with sufficient native-like topology is the inherently slow step (Sosnick et al. 1996; Plaxco et al. 1998). The PPOE model provides a physical rationale. The small-scale templated conformational search necessary to build onto pre-existing native-like structure should be naturally faster than the initial whole-molecule untemplated search.
According to the PPOE parameters, non-two-state folding occurs because of off-pathway errors. Fast two-state folding seems biologically favorable insofar as it avoids the lengthy occupation of partially folded states that would promote in vivo proteolysis and aggregation. This is desirable both for initial folding and subsequently since native proteins repeatedly unfold and refold even under native conditions (Bai et al. 1995). Intermediates that are inherently slow as in the multipathway model would exacerbate the problem. The problem is relieved by an initial rate-limiting barrier that requires the polypeptide to find a near-native topology, which maximizes the probability that subsequent steps go forward rapidly without blocking. One evolutionary strategy is suggested by the prevalence of the N-terminal to C-terminal docking motif. Half of all single-domain proteins in the Protein Data Bank and close to 100% of all known kinetically two-state proteins have this motif, and it tends to form as an initial step, which seems favorable for minimizing subsequent misfolding errors (Krishna and Englander 2005).
Three-state folding
In the fitted IUP model, three-state folding occurs because stable on-pathway intermediates naturally fold forward more slowly than the initial untemplated whole-molecule search. In contradiction, stable on-pathway intermediates that fold forward rapidly and are kinetically silent have been demonstrated for several proteins, as just listed.
In the PPOE model, the native protein is built in foldon-sized steps. Three-state folding occurs when a native-like intermediate additionally contains some slowly repaired misfolding error. In agreement, folding intermediates for many proteins are seen to be partial replicas of the native protein, whether they are kinetically populated or not, and the kinetically blocked forms are also seen to contain some significant misfolding (Kiefhaber et al. 1992; Dobson et al. 1994; Elöve et al. 1994; Muñoz et al. 1994; Sosnick et al. 1994, 1996; Weissman and Kim 1995; Silow and Oliveberg 1997; Bai 1999; Bilsel et al. 1999; Bhuyan and Udgaonkar 2001; Capaldi et al. 2002; Wallace and Matthews 2002; Krishna et al. 2003a, 2004; Bollen et al. 2004; Rojsajjakul et al. 2004; Religa et al. 2005; Wintrode et al. 2005; Nishimura et al. 2006).
It can be noted that partially folded intermediates are likely to minimize energy by exploiting adventitious nonnative interactions (Feng et al. 2003). Their reversal may or may not be rate-limiting. One needs to distinguish naturally occurring but innocuous nonnative interactions from slowly repaired blocking errors (Feng et al. 2005). Within the terms of the PPOE model, misfolding errors interfere with the sequential stabilization of a succeeding foldon unit.
Heterogeneous folding
In the IUP model, folding is heterogeneous because different molecules naturally fold in different tracks through different transition states and intermediates in different parts of the folding landscape. The PPOE model, based on cooperative native-like units and their interactions, pictures that all of the molecules naturally fold by way of the same barriers and native-like intermediates. The probabilistic introduction of misfolding errors can produce a heterogeneous mix of two-state and three-state population fractions.
Experimental observations favor the predetermined pathway view. In Cyt c, an early intermediate with the same native-like structure and stability was characterized under two-state conditions (native state HX) (Bai et al. 1995; Milne et al. 1999) and in three-state folding (HX pulse labeling) (Roder et al. 1988; Krishna et al. 2003a). The entire protein population folds through the same intermediate (Sosnick et al. 1996; Krishna et al. 2003a) whether it is blocked by an added misfolding error or not. An analogous demonstration was obtained for ribonuclease H (Raschke and Marqusee 1997; Chamberlain and Marqusee 2000). In many proteins, it appears that much or all of the population folds to and blocks at the same intermediate step (85% in lysozyme). The multiple apparent tracks seen in the folding of α-Trp synthase appear to result from the chance insertion of different barriers due to proline misisomerization (Wu and Matthews 2002, 2003).
Similarly, the PPOE model implies that protein molecules in a refolding population should tend to use the same initial nucleation strategy, encoded in the same initial U-to-I1 step, whether folding is two-state or three-state or heterogeneous. In many two-state folding proteins, ϕ and ψ analysis show that all of the molecules fold through the same initial rate-limiting transition state. The initial barrier has been measured to be identical for two-state and three-state folding in the case of Cyt c (same rate, temperature dependence, and denaturant dependence) (Sosnick et al. 1996). The analysis of Kuwajima and coworkers (Kamagata et al. 2004) suggests that the first barrier in three-state folding proteins obeys a contact order relationship similar to that for the initial barrier in two-state folding proteins.
In the PPOE model, the three-state discrimination occurs after the initial step when some molecules happen to encounter an error-repair barrier. In Cyt c, the insertion of a misligation barrier can be turned on or off simply by adjusting the pH in the unfolded condition (Elöve et al. 1994; Sosnick et al. 1994, 1996). For the proline-dependent misfolding of many proteins, barrier insertion can be turned on or off simply by adjusting the time spent in the unfolded condition (Nall 1994). Earlier folding kinetics are unaffected.
Summary
Known folding behavior for many proteins parallels the PPOE model and not the multipathway model.
Comparison with theoretical models
A comparison of the PPOE model with theoretical energy landscape models suggests some similarities and some differences. The major concern for present purposes is the apparent conflict between the independent unrelated pathways view and the foldon-determined view. The conflict is more apparent than real.
Pathway multiplicity
Energy landscape theory generally pictures that proteins can fold through many unrelated paths (Bryngelson et al. 1995; Wolynes et al. 1995; Dill and Chan 1997; Brooks 1998; Plotkin and Onuchic 2002a,b). Trajectories are often pictured to disperse at the initiation of folding because of the different positions in unfolded space occupied by the different molecules. Copious branching occurs in subsequent pathway steps because the protein grows by adding one amino acid at a time in no predictable order. Different molecules move downhill through different regions of the funnel-shaped landscape on which every point corresponds to a different three-dimensional structure.
Analogous to funnel landscapes, the logic of the sequential stabilization mechanism allows the folding pathway to branch when a given intermediate is able to template the addition of more than one subsequent foldon. However, the degree of branching is severely limited by the fact that alternative pathway steps must use the same few foldon units and foldon–foldon interactions. For example, Cyt c is experimentally observed to fold in five discrete foldon steps (Bai et al. 1995; Xu et al. 1998; Milne et al. 1999; Hoang et al. 2002; Krishna et al. 2003a,b, 2004, 2006; Maity et al. 2004, 2005). It follows a linear pathway for its first three on-pathway steps, then branches and regroups to N in the final two steps, all as dictated by its native structure and the sequential stabilization principle (Krishna et al. 2006). Branching would be vastly more complex if folding proceeded in 104 unordered steps, the number of its amino acids. Other proteins are also seen to fold one foldon unit at a time (Chamberlain et al. 1996; Fuentes and Wand 1998a,b; Chamberlain and Marqusee 2000; Chu et al. 2002; Silverman and Harbury 2002; Yan et al. 2002, 2004; Feng et al. 2003, 2005).
Intermediate multiplicity
In the energy landscape view, intermediates become visible in kinetic folding because some of the molecules chance to fall into one or more of the many misfolding traps that dot the folding landscape. This view agrees with the present hypothesis that errors (traps) are optional and error repair can be rate-limiting.
However, a critical experimental observation is that all of the three-state HEWL fraction, 85% of the total population, occupies the same intermediate state. All of the molecules in the populated intermediate have essentially the same native-like structure, the same misfolding errors, and the same rates for formation and decay (Radford et al. 1992; Miranker et al. 1993; Dobson et al. 1994). Structural information shows a similar result for the populated intermediates of other proteins even when they are early, high in the not-yet-converged funnel (see, e.g., Krishna et al. 2003a, 2004).
These experimental observations are hard to understand if different population fractions descend the landscape through several unrelated paths. It is, however, just what is required by the PPOE model in the limit of complete pathway linearity.
Amino acids versus cooperative foldons
The major difference in the behavior expected from the foldon and funnel views is due to the different levels of structural behavior that are considered. Theoretical simulations generally focus on folding behavior at the microscopic amino acid level. The assembly of a given foldon, for example, a single helix, does occur at the amino acid level with many nucleation and growth alternatives, which is well represented by the multitrack funnel model. However, experiment is able to detect protein assembly only at a more macroscopic, cooperative foldon level where folding pathways are assembled by foldon units of the native protein and their stepwise native-like association. The micro and macro views are each pertinent to their own domain. They appear to conflict when either is inappropriately applied to the domain of the other.
It seems intuitively likely that theoretical simulations that understand proteins as accretions of a small number of cooperative foldon units rather than many independent amino acids would calculate pathways that proceed in macroscopic steps through a sequence of predetermined native-like intermediates in the growing native format. The role of foldons may also bear on related issues that are usually visualized in terms of amino acid level interactions (Go models, landscape smoothness, contact order, ϕ and ψ analysis) but might be better understood in the context of cooperative native-like foldon structure and interactions.
Misconceptions
Discussions of possible folding models have been handicapped by some common misconceptions. (1) Intermediates if stable must accumulate. (2) If intermediates don't affect measured folding kinetics, they are irrelevant. (3) Populated intermediates cause slow folding. The PPOE perspective helps to illuminate these issues.
(1) It is often assumed that on-pathway intermediates must accumulate if they are stable relative to U, and are not present if no accumulation is seen. These views are built into multipathway models, as for lysozyme considered above. In fact, a kinetic intermediate will visibly populate only if it is more stable than all prior states and it is blocked by a barrier that is higher than all prior barriers (trough to peak). Experimentally, equilibrium native state HX has been able to detect folding intermediates under two-state folding conditions. They are stable, on-pathway, obligatory, and kinetically invisible. In the PPOE view, this is because on-pathway intermediates naturally fold forward more rapidly than the initial pathway step.
(2) The fact that on-pathway intermediates do not affect the measured folding rate does not make them unimportant for the folding process. In the PPOE model, normally occurring obligatory intermediates form after the initial inherently rate-limiting step, but they are kinetically silent only when all goes well. A corrupting error can cause an intermediate to accumulate and folding to be slowed. Slowing would not occur if the pathway did not obligately move through that particular intermediate. The initial step is rate-limiting and intermediates are invisible because the subsequent pathway goes forward efficiently. As for any kinetic pathway, the folding pathway is constructed by the obligatory intermediates that define it and would not progress efficiently without them, whether they occur invisibly after a rate-limiting nucleation step or not.
(3) The correlation between intermediate accumulation and slow folding has been widely noted, and a cause-and-effect relationship has often been drawn. Rather, in the PPOE analysis, intermediate accumulation and slowed folding occur together because both are caused by an inserted error-repair barrier. Uncorrupted intermediates promote rather than hinder folding, whether they are stable or not.
In summary, the absence of accumulation does not mean that intermediates are absent, or unstable, or unimportant, and the accumulation of off-pathway forms that are obstructive does not mean that uncorrupted on-pathway intermediates are obstructive. When these common misconceptions are put aside, available information can be seen to be wholly consistent with the PPOE model.
Summary
Spectroscopy-based and theory-based studies of kinetic protein folding are commonly interpreted in terms of multiple independent unrelated pathways, the IUP model. By contrast, detailed structural information depicts pathways that put predetermined units of the native protein into place in a sequential stabilization process, as in the PPOE model. Both models can quantitatively fit extensive data sets for the heterogeneous folding and unfolding of lysozyme at varied conditions and therefore seem able to fit the analogous behavior of other proteins in general. The specific rejection criteria developed by the Kiefhaber group eliminate any other minimal reaction schemes that are significantly different.
Fitting to the lysozyme data imposes different properties on the two different models, which makes possible a clear test against known information. The properties of the fitted IUP model are contrary to experimental results for many proteins. The properties of the fitted PPOE model are all seen experimentally. Theoretical simulations that exhibit many independent pathways seem to be pertinent to microscopic amino-acid-level behavior, at the level of individual foldon construction, rather than to macroscopic pathway behavior.
The PPOE model is based on clear experimental results that can be seen to derive from three physical principles, as follows.
(1) The units of protein folding are cooperative native-like structural elements (foldons) (Zimm and Bragg 1959; Lifson and Roig 1961; Krishna et al. 2003b).
(2) Prior structure templates the formation of complementary structure (sequential stabilization) (Watson and Crick 1953; Martin et al. 1996; DelMar et al. 2005; Uversky et al. 2005).
(3) Probabilistic misfolding errors, which are prevalent (Chi et al. 2003; Krishna et al. 2004; Yewdell 2005), can cause population fractions to block and the corrupted intermediates to accumulate (optional errors).
The first two principles dictate that proteins fold in macroscopic structural steps with the order of steps set by the way that foldon units are organized in the native protein. The third determines whether pathways appear to be kinetically two-state, three-state, or heterogeneous. The integration of these principles in the PPOE model unifies within a coherent mechanism a broad range of experimental observations concerning the properties of protein folding pathways and identifies their structural bases.
Materials and methods
The HEWL data analyzed here (Kiefhaber et al. 1997; Wildegger and Kiefhaber 1997; Bieri et al. 1999; Bieri and Kiefhaber 2001) were fit to the various kinetic schemes shown in Figure 2. Our analysis assumes, as in the prior analyses of Kiefhaber and coworkers, that unfolding amplitudes measured in the interrupted refolding experiments (fluorescence) represent concentrations of the individual species when corrected for signal strength.
For a scheme involving n species, there will be (n − 1) nonzero macroscopic relaxation phases that are functions of the microscopic rate constants kij. Each kij, connecting species i and j, was assumed to depend on denaturant concentration D according to the usual relationship:
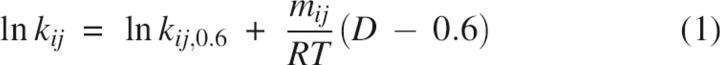 |
where k ij,0.6 are the reference values at 0.6 M GdmCl, the denaturant concentration where the interrupted folding experiments were done, and mij is the slope of the dependence on denaturant (D) of the term RT ln kij. The final fit values are in Figures 3, 5, 6, and 7.
For fitting the data to a given kinetic scheme, grid search (Bevington and Robinson 1994) and eigen methods were used. Eigenvalues of the rate matrix are the observed macroscopic relaxation rates. The right eigenvectors define the ratios of the corresponding amplitudes in the population kinetics. The exact amplitudes were calculated to match the starting condition (100% U) in kinetic experiments. The procedure used is as follows. Starting from an initial set of trial microscopic rate constants and m-values, the rate matrix was set up and solved for its eigen solutions using canned routines in the linear algebra package CLAPACK (Anderson et al. 1999; http://www.netlib.org/clapack/). The eigenvalues (macroscopic rates) as a function of denaturant concentration were used to fit the chevron data. The eigenvectors with pre-factors determined from the starting condition (100% U) were used to fit the population kinetics.
The ordinate values used in Figures 3, 6, and 7 (middle panels) represent concentrations calculated by the Kiefhaber group, based on their fit after normalizing fitted fluorescence to N = 1. The data in Figure 5 show the observed unfolding amplitudes. In order to globally refit the data, we had to recompute proportionality constants for the fluorescence signals of intermediate species (denoted as S in the Figures). S-values relative to the native signal normalized to 1 were determined using the standard linear regression relationship for calculated C(t) vs. measured At as in Equation 2:
 |
At is the measured fluorescence signal (unfolding amplitude) in interrupted folding experiments; C(t) is the fractional molecular concentration calculated from a given set of microscopic rate constants at the folding time t. S is not an independent parameter as evident from Equation 2. It is determined by the micro-rate constants to match the experimental data. S equal to 1 in Figures 3, 6, and 7 would represent exact agreement with the concentrations calculated by Kiefhaber et al.
Goodness of the chevron and kinetic fit was judged using the global reduced χ2 parameter defined as follows (Bevington and Robinson 1994):
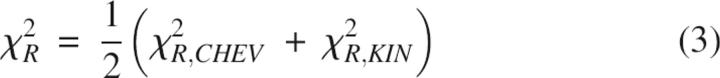 |
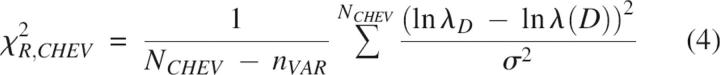 |
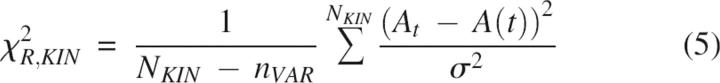 |
N CHEV and N KIN represent the number of chevron and kinetics data points; n VAR is the number of free parameters in a given scheme (for example in the T model, n VAR = 6 rate constants + 6 m-values = 12); λD and λ(D) are the experimentally measured and the calculated macroscopic relaxation rates at the denaturant concentration D; At and A(t), equal to S × C(t), are the measured and calculated fluorescence amplitudes at the folding time t in interrupted folding experiments. Standard deviations σ for ln λD and At were arbitrarily taken to be 2% of the full scale, namely, 0.2 and 0.02, respectively. The fitting procedure was repeated iteratively by varying each of the initial parameters in steps of ±10% to ±0.01% until the best fit (least χR 2) was obtained. Machine-generated random numbers were used to set the order in which the initial parameters were selected for step increments.
All computer programs were home written in ANSI C on the Microsoft Visual C++ 6 platform. The correctness of the programs was checked by comparing predicted kinetic curves with those obtained using the KINSIM (Barshop et al. 1983; http://www.biochem.wustl.edu/cflab/message.html) and DYNAFIT (Kuzmič 1996; http://www.biokin.com/dynafit) programs. The parameters constrained by the data, shown in boldface in Figures 3, 5, 6, and 7, were determined by starting from different sets of initial values in multiple runs and noting parameters that did and did not change.
Acknowledgments
We thank Thomas Kiefhaber for kindly providing the lysozyme folding data. For a critical reading of the manuscript and helpful comments, we thank Robert Baldwin, Yawen Bai, Ken Dill, Neville Kallenbach, Thomas Kiefhaber, Leland Mayne, Leslie Milk, Hillary Nelson, George Rose, John Skinner, Tobin Sosnick, and our laboratory members. This work was supported by NIH research grant GM 031847.
Footnotes
Reprint requests to: Mallela M.G. Krishna, Johnson Research Foundation, Department of Biochemistry and Biophysics, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6059, USA; e-mail: kmallela@mail.med.upenn.edu; fax: (215) 898-2415.
Abbreviations: HEWL, hen egg white lysozyme; Cyt c, cytochrome c; HX, hydrogen exchange; GdmCl, guanidinium chloride; IUP, independent unrelated pathways model; PPOE, predetermined pathway–optional error model; U, I, and N, unfolded, intermediate, and native states; Iix, kinetically blocked intermediate.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062655907.
References
- Anderson E., Bai, Z., Bischof, C., Blackford, S., Demmel, J., Dongarra, J., Du Croz, J., Greenbaum, A., Hammarling, S., and McKenney, A., et al. 1999. LAPACK users’ guide. Society for Industrial and Applied Mathematics, Philadelphia.
- Bai Y.. 1999. Kinetic evidence for an on-pathway intermediate in the folding of cytochrome c . Proc. Natl. Acad. Sci. 96: 477–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Sosnick, T.R., Mayne, L., and Englander, S.W. 1995. Protein folding intermediates: Native-state hydrogen exchange. Science 269: 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin R.L.. 1995. The nature of protein folding pathways: The classical versus the new view. J. Biomol. NMR 5: 103–109. [DOI] [PubMed] [Google Scholar]
- Barshop B.A., Wrenn, R.F., and Frieden, C. 1983. Analysis of numerical methods for computer simulation of kinetic processes: Development of KINSIM—A flexible, portable system. Anal. Biochem. 130: 134–145. [DOI] [PubMed] [Google Scholar]
- Bevington P.R. and Robinson, D.K. 1994. Data reduction and error analysis for the physical sciences. McGraw-Hill, New York.
- Bhuyan A.K. and Udgaonkar, J.B. 2001. Folding of horse cytochrome c in the reduced state. J. Mol. Biol. 312: 1135–1160. [DOI] [PubMed] [Google Scholar]
- Bieri O. and Kiefhaber, T. 2001. Origin of apparent fast and non-exponential kinetics of lysozyme folding measured in pulsed hydrogen exchange measurements. J. Mol. Biol. 310: 919–935. [DOI] [PubMed] [Google Scholar]
- Bieri O., Wildegger, G., Bachmann, A., Wagner, C., and Kiefhaber, T. 1999. A salt-induced kinetic intermediate is on a new parallel pathway of lysozyme folding. Biochemistry 38: 12460–12470. [DOI] [PubMed] [Google Scholar]
- Bilsel O. and Matthews, C.R. 2000. Barriers in protein folding reactions. Adv. Protein Chem. 53: 153–207. [DOI] [PubMed] [Google Scholar]
- Bilsel O., Zitzewitz, J.A., Bowers, K.E., and Matthews, C.R. 1999. Folding mechanism of the α-subunit of tryptophan synthase, an α/β barrel protein: Global analysis highlights the interconversion of multiple native, intermediate, and unfolded forms through parallel channels. Biochemistry 38: 1018–1029. [DOI] [PubMed] [Google Scholar]
- Bollen Y.J.M., Sánchez, I.E., and van Mierlo, C.P.M. 2004. Formation of on- and off-pathway intermediates in the folding kinetics of Azotobacter vinelandii apoflavodoxin. Biochemistry 43: 10475–10489. [DOI] [PubMed] [Google Scholar]
- Brooks C.L.. 1998. Simulations of protein folding and unfolding. Curr. Opin. Struct. Biol. 8: 222–226. [DOI] [PubMed] [Google Scholar]
- Bryngelson J.D., Onuchic, J.N., Socci, N.D., and Wolynes, P.G. 1995. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins 21: 167–195. [DOI] [PubMed] [Google Scholar]
- Capaldi A.P., Kleanthous, C., and Radford, S.E. 2002. Im7 folding mechanism: Misfolding on a path to the native state. Nat. Struct. Biol. 9: 209–216. [DOI] [PubMed] [Google Scholar]
- Cecconi C., Shank, E.A., Bustamante, C., and Marqusee, S. 2005. Direct observation of the three-state folding of a single protein molecule. Science 309: 2057–2060. [DOI] [PubMed] [Google Scholar]
- Chaffotte A.F., Guillou, Y., and Goldberg, M.E. 1992. Kinetic resolution of peptide bond and side chain far-UV circular dichroism during the folding of hen egg white lysozyme. Biochemistry 31: 9694–9703. [DOI] [PubMed] [Google Scholar]
- Chamberlain A.K. and Marqusee, S. 2000. Comparison of equilibrium and kinetic approaches for determining protein folding mechanisms. Adv. Protein Chem. 53: 283–328. [DOI] [PubMed] [Google Scholar]
- Chamberlain A.K., Handel, T.M., and Marqusee, S. 1996. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat. Struct. Biol. 3: 782–787. [DOI] [PubMed] [Google Scholar]
- Chamberlain A.K., Fischer, K.F., Reardon, D., Handel, T.M., and Marqusee, A.S. 1999. Folding of an isolated ribonuclease H core fragment. Protein Sci. 8: 2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi E.Y., Krishnan, S., Randolph, T.W., and Carpenter, J.F. 2003. Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 20: 1325–1336. [DOI] [PubMed] [Google Scholar]
- Chu R., Pei, W., Takei, J., and Bai, Y. 2002. Relationship between the native-state hydrogen exchange and folding pathways of a four-helix bundle protein. Biochemistry 41: 7998–8003. [DOI] [PubMed] [Google Scholar]
- Colón W., Elöve, G.A., Wakem, P., Sherman, F., and Roder, H. 1996. Side chain packing of the N- and C- terminal helices play a critical role in the kinetics of cytochrome c folding. Biochemistry 35: 5538–5549. [DOI] [PubMed] [Google Scholar]
- DelMar C., Greenbaum, E.A., Mayne, L., Englander, S.W., and Woods Jr, V.L. 2005. Structure and properties of α-synuclein and other amyloids determined at the amino acid level. Proc. Natl. Acad. Sci. 102: 15477–15482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill K.A. and Chan, H.S. 1997. From Levinthal to pathways to funnels. Nat. Struct. Biol. 4: 10–19. [DOI] [PubMed] [Google Scholar]
- Dobson C.M., Evans, P.A., and Radford, S.E. 1994. Understanding how proteins fold: The lysozyme story so far. Trends Biochem. Sci. 19: 31–37. [DOI] [PubMed] [Google Scholar]
- Dobson C.M., Šali, A., and Karplus, M. 1998. Protein folding: A perspective from theory and experiment. Angew. Chem. Int. Ed. Engl. 37: 868–893. [DOI] [PubMed] [Google Scholar]
- Elöve G.A., Bhuyan, A.K., and Roder, H. 1994. Kinetic mechanism of cytochrome c folding: Involvement of the heme and its ligands. Biochemistry 33: 6925–6935. [DOI] [PubMed] [Google Scholar]
- Englander S.W.. 2000. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 29: 213–238. [DOI] [PubMed] [Google Scholar]
- Englander S.W., Sosnick, T.R., Mayne, L.C., Shtilerman, M., Qi, P.X., and Bai, Y. 1998. Fast and slow folding in cytochrome c . Acc. Chem. Res. 31: 737–744. [Google Scholar]
- Eyles S.J., Radford, S.E., Robinson, C.V., and Dobson, C.M. 1994. Kinetic consequences of the removal of a disulfide bridge on the folding of hen lysozyme. Biochemistry 33: 13038–13048. [DOI] [PubMed] [Google Scholar]
- Feng H., Takei, J., Lipsitz, R., Tjandra, N., and Bai, Y. 2003. Specific non-native hydrophobic interactions in a hidden folding intermediate: Implications for protein folding. Biochemistry 42: 12461–12465. [DOI] [PubMed] [Google Scholar]
- Feng H., Zhou, Z., and Bai, Y. 2005. A protein folding pathway with multiple folding intermediates at atomic resolution. Proc. Natl. Acad. Sci. 102: 5026–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A.R.. 1995. Optimization of rates of protein folding: The nucleation-condensation mechanism and its implications. Proc. Natl. Acad. Sci. 92: 10869–10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes E.J. and Wand, A.J. 1998a. Local dynamics and stability of apocytochrome b562 examined by hydrogen exchange. Biochemistry 37: 3687–3698. [DOI] [PubMed] [Google Scholar]
- Fuentes E.J. and Wand, A.J. 1998b. Local stability and dynamics of apocytochrome b562 examined by the dependence of hydrogen exchange on hydrostatic pressure. Biochemistry 37: 9877–9883. [DOI] [PubMed] [Google Scholar]
- Gladwin S.T. and Evans, P.A. 1996. Structure of very early protein folding intermediates: New insights through a variant of hydrogen exchange labelling. Fold. Des. 1: 407–417. [DOI] [PubMed] [Google Scholar]
- Goldberg M.E. and Guillou, Y. 1994. Native disulfide bonds greatly accelerate secondary structure formation in the folding of lysozyme. Protein Sci. 3: 883–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang L., Bédard, S., Krishna, M.M.G., Lin, Y., and Englander, S.W. 2002. Cytochrome c folding pathway: Kinetic native-state hydrogen exchange. Proc. Natl. Acad. Sci. 99: 12173–12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki L.S., Evans, P.A., Dobson, C.M., and Radford, S.E. 1994. Tertiary interactions in the folding pathway of hen lysozyme: Kinetic studies using fluorescent probes. Biochemistry 33: 5212–5220. [DOI] [PubMed] [Google Scholar]
- Jacob J., Krantz, B., Dothager, R.S., Thiyagarajan, P., and Sosnick, T.R. 2004. Early collapse is not an obligate step in protein folding. J. Mol. Biol. 338: 369–382. [DOI] [PubMed] [Google Scholar]
- Kamagata K., Arai, M., and Kuwajima, K. 2004. Unification of the folding mechanisms of non-two-state and two-state proteins. J. Mol. Biol. 339: 951–965. [DOI] [PubMed] [Google Scholar]
- Kiefhaber T.. 1995. Kinetic traps in lysozyme folding. Proc. Natl. Acad. Sci. 92: 9029–9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefhaber T., Grunert, H.P., Hahn, U., and Schmid, F.X. 1992. Folding of RNase T1 is decelerated by a specific tertiary contact in a folding intermediate. Proteins 12: 171–179. [DOI] [PubMed] [Google Scholar]
- Kiefhaber T., Bachmann, A., Wildegger, G., and Wagner, C. 1997. Direct measurement of nucleation and growth rates in lysozyme folding. Biochemistry 36: 5108–5112. [DOI] [PubMed] [Google Scholar]
- Kim P.S. and Baldwin, R.L. 1982. Specific intermediates in the folding reactions of small proteins and the mechanism of protein folding. Annu. Rev. Biochem. 51: 459–489. [DOI] [PubMed] [Google Scholar]
- Kim P.S. and Baldwin, R.L. 1990. Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem. 59: 631–660. [DOI] [PubMed] [Google Scholar]
- Klein-Seetharaman J., Oikawa, M., Grimshaw, S.B., Wirmer, J., Duchardt, E., Ueda, T., Imoto, T., Smith, L.J., Dobson, C.M., and Schwalbe, H. 2002. Long-range interactions within a nonnative protein. Science 295: 1719–1722. [DOI] [PubMed] [Google Scholar]
- Korzhnev D.M., Salvatella, X., Vendruscolo, M., Nardo, A.A.D., Davidson, A.R., Dobson, C.M., and Kay, L.E. 2004. Low-populated folding intermediates of Fyn SH3 characterized by relaxation dispersion NMR. Nature 430: 586–590. [DOI] [PubMed] [Google Scholar]
- Krishna M.M.G. and Englander, S.W. 2005. The N-terminal to C-terminal motif in protein folding and function. Proc. Natl. Acad. Sci. 102: 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna M.M.G., Lin, Y., Mayne, L., and Englander, S.W. 2003a. Intimate view of a kinetic protein folding intermediate: Residue-resolved structure, interactions, stability, folding and unfolding rates, homogeneity. J. Mol. Biol. 334: 501–513. [DOI] [PubMed] [Google Scholar]
- Krishna M.M.G., Lin, Y., Rumbley, J.N., and Englander, S.W. 2003b. Cooperative omega loops in cytochrome c: Role in folding and function. J. Mol. Biol. 331: 29–36. [DOI] [PubMed] [Google Scholar]
- Krishna M.M.G., Lin, Y., and Englander, S.W. 2004. Protein misfolding: Optional barriers, misfolded intermediates, and pathway heterogeneity. J. Mol. Biol. 343: 1095–1109. [DOI] [PubMed] [Google Scholar]
- Krishna M.M.G., Maity, H., Rumbley, J.N., Lin, Y., and Englander, S.W. 2006. Order of steps in the cytochrome c folding pathway: Evidence for a sequential stabilization mechanism. J. Mol. Biol. 359: 1411–1420. [DOI] [PubMed] [Google Scholar]
- Kulakarni S.K., Ashcroft, A.E., Carey, M., Masselos, D., Robinson, C.V., and Radford, S.E. 2006. A near-native state on the slow refolding pathway of hen lysozyme. Protein Sci. 8: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmič P.. 1996. Program DYNAFIT for the analysis of enzyme kinetic data: Application to HIV proteinase. Anal. Biochem. 237: 260–273. [DOI] [PubMed] [Google Scholar]
- Levinthal C.. 1968. Are there pathways for protein folding? J. Chim. Phys. 65: 44–45. [Google Scholar]
- Levinthal C.. 1969. How to fold graciously. Mossbauer Spectroscopy in Biological Systems Proceedings, University of Illinois Bulletin, University of Illinois Press, Urbana.
- Lifson S. and Roig, A. 1961. On the theory of the helix-coil transition in polypeptides. J. Chem. Phys. 34: 1963–1974. [Google Scholar]
- Maity H., Maity, M., and Englander, S.W. 2004. How cytochrome c folds, and why: Submolecular foldon units and their stepwise sequential stabilization. J. Mol. Biol. 343: 223–233. [DOI] [PubMed] [Google Scholar]
- Maity H., Maity, M., Krishna, M.M.G., Mayne, L., and Englander, S.W. 2005. Protein folding: The stepwise assembly of foldon units. Proc. Natl. Acad. Sci. 102: 4741–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E.L., Rens-Domiano, S., Schatz, P.J., and Hamm, H.E. 1996. Potent peptide analogues of a G protein receptor-binding region obtained with a combinatorial library. J. Biol. Chem. 271: 361–366. [DOI] [PubMed] [Google Scholar]
- Matagne A., Radford, S.E., and Dobson, C.M. 1997. Fast and slow tracks in lysozyme folding: Insight into the role of domains in the folding process. J. Mol. Biol. 267: 1068–1074. [DOI] [PubMed] [Google Scholar]
- Matagne A., Chung, E.W., Ball, L.J., Radford, S.E., Robinson, C.V., and Dobson, C.M. 1998. The origin of the α-domain intermediate in the folding of hen lysozyme. J. Mol. Biol. 277: 997–1005. [DOI] [PubMed] [Google Scholar]
- Milne J.S., Xu, Y., Mayne, L.C., and Englander, S.W. 1999. Experimental study of the protein folding landscape: Unfolding reactions in cytochrome c . J. Mol. Biol. 290: 811–822. [DOI] [PubMed] [Google Scholar]
- Miranker A., Robinson, C.V., Radford, S.E., Aplin, R.T., and Dobson, C.M. 1993. Detection of transient protein folding populations by mass spectrometry. Science 262: 896–900. [DOI] [PubMed] [Google Scholar]
- Muñoz V., Lopez, E.M., Jager, M., and Serrano, L. 1994. Kinetic characterization of the chemotactic protein from Escherichia coli, CheY. Kinetic analysis of the inverse hydrophobic effect. Biochemistry 33: 5858–5866. [DOI] [PubMed] [Google Scholar]
- Nall B.T.. 1994. Proline isomerization as a rate-limiting step. In Mechanisms of protein folding (ed. R.H. Pain), pp. 80–103. Oxford, UK.
- Nishimura C., Dyson, H.J., and Wright, P.E. 2006. Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J. Mol. Biol. 355: 139–156. [DOI] [PubMed] [Google Scholar]
- Oas T.G. and Kim, P.S. 1988. A peptide model of a protein folding intermediate. Nature 336: 42–48. [DOI] [PubMed] [Google Scholar]
- Plaxco K.W., Simons, K.T., and Baker, D. 1998. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277: 985–994. [DOI] [PubMed] [Google Scholar]
- Plaxco K.W., Millett, I.S., Segel, D.J., Doniach, S., and Baker, D. 1999. Chain collapse can occur concomitantly with the rate-limiting step in protein folding. Nat. Struct. Biol. 6: 554–556. [DOI] [PubMed] [Google Scholar]
- Plaxco K.W., Simons, K.T., Ruczinski, I., and Baker, D. 2000. Topology, stability, sequence, and length: Defining the determinants of two-state protein folding kinetics. Biochemistry 39: 11177–11183. [DOI] [PubMed] [Google Scholar]
- Plotkin S.S. and Onuchic, J.N. 2002a. Understanding protein folding with energy landscape theory. Part I: Basic concepts. Q. Rev. Biophys. 35: 111–167. [DOI] [PubMed] [Google Scholar]
- Plotkin S.S. and Onuchic, J.N. 2002b. Understanding protein folding with energy landscape theory. Part II: Quantitative concepts. Q. Rev. Biophys. 35: 205–286. [DOI] [PubMed] [Google Scholar]
- Ptitsyn O.B.. 1995. Molten globule and protein folding. Adv. Protein Chem. 47: 83–229. [DOI] [PubMed] [Google Scholar]
- Radford S.E., Dobson, C.M., and Evans, P.A. 1992. The folding of hen lysozyme involves partially structured intermediates and multiple pathways. Nature 358: 302–307. [DOI] [PubMed] [Google Scholar]
- Raschke T.M. and Marqusee, S. 1997. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nat. Struct. Biol. 4: 298–304. [DOI] [PubMed] [Google Scholar]
- Religa T.L., Markson, J.S., Mayor, U., Freund, S.M.V., and Fersht, A.R. 2005. Solution structure of a protein denatured state and folding intermediate. Nature 437: 1053–1056. [DOI] [PubMed] [Google Scholar]
- Roder H., Elöve, G.A., and Englander, S.W. 1988. Structural characterization of folding intermediates in cytochrome c by H-exchange labeling and proton NMR. Nature 335: 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojsajjakul T., Wintrode, P., Vadrevu, R., Matthews, C.R., and Smith, D.L. 2004. Multi-state unfolding of the α subunit of tryptophan synthase, a TIM barrel protein: Insights into the secondary structure of the stable equilibrium intermediates by hydrogen exchange mass spectrometry. J. Mol. Biol. 341: 241–253. [DOI] [PubMed] [Google Scholar]
- Rothwarf D.M. and Scheraga, H.A. 1996. Role of non-native aromatic and hydrophobic interactions in the folding of egg white lysozyme. Biochemistry 35: 13797–13807. [DOI] [PubMed] [Google Scholar]
- Roux P., Delepierre, M., Goldberg, M.E., and Chaffotte, A.-F. 1997. Kinetics of secondary structure recovery during the refolding of reduced hen egg white lysozyme. Proc. Natl. Acad. Sci. 272: 24843–24849. [DOI] [PubMed] [Google Scholar]
- Sánchez I.E. and Kiefhaber, T. 2003a. Evidence for sequential barriers and obligatory intermediates in apparent two-state protein folding. J. Mol. Biol. 325: 367–376. [DOI] [PubMed] [Google Scholar]
- Sánchez I.E. and Kiefhaber, T. 2003b. Hammond behavior versus ground state effects in protein folding: Evidence for narrow free energy barriers and residual structure in unfolded states. J. Mol. Biol. 327: 867–884. [DOI] [PubMed] [Google Scholar]
- Schmid F.X.. 1983. Mechanism of folding of ribonuclease A. Slow refolding is a sequential reaction via structural intermediates. Biochemistry 22: 4690–4696. [DOI] [PubMed] [Google Scholar]
- Segel D.J., Bachmann, A., Hofrichter, J., Hodgson, K.O., Doniach, S., and Kiefhaber, T. 1999. Characterization of transient intermediates in lysozyme folding with time-resolved small-angle X-ray scattering. J. Mol. Biol. 288: 489–499. [DOI] [PubMed] [Google Scholar]
- Silow M. and Oliveberg, M. 1997. Transient aggregates in protein folding are easily mistaken for folding intermediates. Proc. Natl. Acad. Sci. 94: 6084–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman J.A. and Harbury, P.B. 2002. The equilibrium unfolding pathway of a (β/α)8 barrel. J. Mol. Biol. 324: 1031–1040. [DOI] [PubMed] [Google Scholar]
- Sosnick T.R., Mayne, L., Hiller, R., and Englander, S.W. 1994. The barriers in protein folding. Nat. Struct. Biol. 1: 149–156. [DOI] [PubMed] [Google Scholar]
- Sosnick T.R., Mayne, L., and Englander, S.W. 1996. Molecular collapse: The rate-limiting step in two-state cytochrome c folding. Proteins 24: 413–426. [DOI] [PubMed] [Google Scholar]
- Staley J.P. and Kim, P.S. 1990. Role of a subdomain in the folding of bovine pancreatic trypsin inhibitor. Nature 344: 685–688. [DOI] [PubMed] [Google Scholar]
- Uversky V.N., Oldfield, C.J., and Dunker, A.K. 2005. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18: 343–384. [DOI] [PubMed] [Google Scholar]
- Walkenhorst W.F., Green, S.M., and Roder, H. 1997. Kinetic evidence for folding and unfolding intermediates in staphylococcal nuclease. Biochemistry 36: 5795–5805. [DOI] [PubMed] [Google Scholar]
- Wallace L.A. and Matthews, C.R. 2002. Sequential vs. parallel protein-folding mechanisms: Experimental tests for complex folding reactions. Biophys. Chem. 101–102: 113–131. [DOI] [PubMed] [Google Scholar]
- Watson J.D. and Crick, F.H.C. 1953. Molecular structure of nucleic acids. A structure for deoxyribose nucleic acid. Nature 171: 737–738. [DOI] [PubMed] [Google Scholar]
- Weinkam P., Zong, C., and Wolynes, P.G. 2005. A funneled energy landscape for cytochrome c directly predicts the sequential folding route inferred from hydrogen exchange experiments. Proc. Natl. Acad. Sci. 102: 12401–12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman J.S. and Kim, P.S. 1995. A kinetic explanation for the rearrangement pathway of BPTI folding. Nat. Struct. Biol. 2: 1123–1130. [DOI] [PubMed] [Google Scholar]
- Wetlaufer D.B.. 1973. Nucleation, rapid folding, and globular intrachain regions in proteins. Proc. Natl. Acad. Sci. 70: 697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildegger G. and Kiefhaber, T. 1997. Three-state model for lysozyme folding: Triangular folding mechanism with an energetically trapped intermediate. J. Mol. Biol. 270: 294–304. [DOI] [PubMed] [Google Scholar]
- Wintrode P.L., Rojsajjakul, T., Vadrevu, R., Matthews, C.R., and Smith, D.L. 2005. An obligatory intermediate controls the folding of the α-subunit of tryptophan synthase, a TIM barrel protein. J. Mol. Biol. 347: 911–919. [DOI] [PubMed] [Google Scholar]
- Wolynes P.G., Onuchic, J.N., and Thirumalai, D. 1995. Navigating the folding routes. Science 267: 1619–1620. [DOI] [PubMed] [Google Scholar]
- Wu Y. and Matthews, C.R. 2002. Parallel channels and rate-limiting steps in complex protein folding reactions: Prolyl isomerization and the α subunit of Trp synthase, a TIM barrel protein. J. Mol. Biol. 323: 309–325. [DOI] [PubMed] [Google Scholar]
- Wu Y. and Matthews, C.R. 2003. Proline replacements and the simplification of the complex, parallel channel folding mechanism for the α subunit of Trp synthase, a TIM barrel protein. J. Mol. Biol. 330: 1131–1144. [DOI] [PubMed] [Google Scholar]
- Wu L.C., Laub, P.B., Elöve, G.A., Carey, J., and Roder, H. 1993. A noncovalent peptide complex as a model for an early folding intermediate of cytochrome c. Biochemistry 32: 10271–10276. [DOI] [PubMed] [Google Scholar]
- Xu Y., Mayne, L., and Englander, S.W. 1998. Evidence for an unfolding and refolding pathway in cytochrome c . Nat. Struct. Biol. 5: 774–778. [DOI] [PubMed] [Google Scholar]
- Yan S., Kennedy, S.D., and Koide, S. 2002. Thermodynamic and kinetic exploration of the energy landscape of Borrelia budgdorferi OspA by native-state hydrogen exchange. J. Mol. Biol. 323: 363–375. [DOI] [PubMed] [Google Scholar]
- Yan S., Gawlak, G., Smith, J., Silver, L., Koide, A., and Koide, S. 2004. Conformational heterogeneity of an equilibrium folding intermediate quantified and mapped by scanning mutagenesis. J. Mol. Biol. 338: 811–825. [DOI] [PubMed] [Google Scholar]
- Yewdell J.W.. 2005. Serendipity strikes twice: The discovery and rediscovery of defective ribosomal products (DRiPS). Cell. Mol. Biol. 51: 635–641. [PubMed] [Google Scholar]
- Zimm G.H. and Bragg, J.K. 1959. Theory of the phase transition between helix and random coil in polypeptide chains. J. Chem. Phys. 31: 526–535. [Google Scholar]
- Zwanzig R., Szabo, A., and Bagchi, B. 1992. Levinthal's paradox. Proc. Natl. Acad. Sci. 89: 20–22. [DOI] [PMC free article] [PubMed] [Google Scholar]