

Abstract
To investigate the function of NF-κB RelA (p65), we generated mice deficient in this NF-κB family member by homologous recombination. Mice lacking RelA showed liver degeneration and died around embryonic day 14.5. To elucidate the role of RelA in lymphocyte development and function, we transplanted fetal liver cells of 13.5-day embryos from heterozygote matings into irradiated SCID mice. Within 4 weeks, both T and B cells had developed in the SCID mice receiving relA−/− fetal liver transplants, similar to the relA+/+ and +/− cases. T cells were found to mature to Thy-1+/TCRαβ+/CD3+/CD4+ or CD8+, while B cells had the ability to differentiate to IgM+/B220+ and to secrete immunoglobulins. However, the secretion of IgG1 and IgA was reduced in RelA-deficient B cells. Furthermore, both T and B cells lacking RelA showed marked reduction in proliferative responses to stimulation with Con A, anti-CD3, anti-CD3+anti-CD28, LPS, anti-IgM, and PMA+calcium ionophore. The results indicate that RelA plays a critical role in production of specific Ig isotypes and also in signal transduction pathways for lymphocyte proliferation.
The NF-κB family of transcription factors are conserved from flies to humans and regulate the expression of a wide variety of cellular and viral genes (reviewed in reference 1). Biochemical and molecular characteristics of NF-κB and their activation pathways have been extensively studied, especially in terms of immune and acute phase responses. The mammalian NF-κB proteins, RelA (p65), c-Rel, RelB, p50/p105, and p52/p100, share the “rel” homology domain which facilitates dimerization, nuclear translocation and DNA binding. Transactivation domains are also present at the COOH termini of RelA, c-Rel, and RelB. These NF-κB proteins form multiple interchangeable heterodimers and homodimers and their activity is regulated by binding of IκB inhibitory factors which determine the localization of NF-κB dimers, either in the cytoplasm or in the nucleus. Upon activation, NF-κB dimers dissociate from IκB and translocate to the nucleus and then bind to the κB sites in promoters and enhancers of NF-κB responsive genes, consequently activating their transcription.
The RelA/p50 heterodimer was the original NF-κB identified, as a transcription factor for Igκ light chain gene (2), and has the strongest transactivating capacity among NF-κB dimers as well as the most widely distributed κB binding activity (3). In the B cell lineage, RelA/p50 is the major NF-κB in pre-B cells, while c-Rel/p50 is predominant in mature B cells and RelB/p52 in plasmacytomas and LPS-activated B cells (4). This suggests that RelA/p50 plays an important role in certain steps of B cell development, although genes regulated by RelA/p50 have yet to be identified. In the T cell lineage, RelA/p50 has been reported to be critical for antigen activation (5) and cytokine production (3). Studies in vitro have suggested that RelA/ p50 regulates expression of the T cell receptor β chain gene (6), cytokine genes such as IL-2 (7), IL-6 (8), and TNFα (9), and the IL-2 receptor α chain gene (10). However, because of the presence of several related proteins and their pleiotropic effects, the specific roles of RelA in vivo remain to be elucidated. Studies on the functions of other NF-κB proteins have faced similar problems. To overcome this drawback, mutant mice lacking RelA (11), c-Rel (12), RelB (13), p50 (14), or IκBα (15) have been derived by homologous recombination to assess specific functions of individual NF-κB proteins. All except RelA-deficient mice demonstrate normal birth and development but with certain abnormalities in immune responses. In the case of RelA deficiency, however, embryonic mortality occurs, concomitant with liver degeneration (11), so that clarification of the function of this family member in the immune system has faced difficulties.
In the present study, we generated RelA-deficient mice with a targeting vector expected to yield a null mutation, different from the vector used in the previous study which would be expected to produce a truncated form of RelA (11). Our RelA-deficient mice also died during embryogenesis but in an attempt to explore the role of RelA in lymphoid development, we transplanted the fetal liver cells from relA−/− embryos into SCID mice and found that the RelA-deficient stem cells could then differentiate to mature T and B cells. To investigate the roles of RelA further, we examined RelA-deficient T and B cells for their functions and their proliferative responses to various stimuli.
Materials and Methods
Construction of the Targeting Vector.
The mouse relA gene was isolated from a C57BL/6 (B6)1 mouse genomic library using a mouse relA cDNA probe (codons 185-277, reference 16). The targeting vector was constructed in pBluescript as shown in Fig. 1. It contained 7 kb of the mouse relA gene including exons 1 to 6, PMC1-neo inserted into the first exon of relA at an NcoI site 3 bp downstream of the translation initiation codon, and the herpes simplex virus-thymidine kinase gene (HSV-tk) flanking at the 3′ end of the relA sequence. We expected that this targeting vector would generate a null mutant allele by homologous recombination.
Figure 1.

Structure of the relA targeting vector. The wild-type mouse relA allele is shown at the top. The targeting vector is in the middle and the predicted mutant allele is at the bottom. The area predicted to undergo homologous recombination is indicated by the dotted lines. Exons are indicated by closed boxes. The probes used for diagnostic DNA blot analysis are also indicated at the top. Restriction enzyme sites: H, HindIII; N, NcoI; P, PstI; Sm, SmaI; S, SphI.
Derivation of relA-deficient Mice.
20 μg DNA of the targeting vector was transfected into 5 × 107 CCE embryonic stem (ES) cells (kindly provided by Dr. Motoya Katsuki, Institute of Medical Science, University of Tokyo, Tokyo, Japan [17]) by electroporation (T-300; Biotechnologies & Experimental Research Inc., San Diego, CA). Transfected cells were cultured for 10 d in positive-negative selection medium (18) containing G418 (400 μg/ml; Sigma Chem. Co., St. Louis, MO) and Gancyclovir (5 μM, Demosine; F. Hoffmann-La Roche Ltd., Palo Alto, CA, provided by Tanabe Seiyaku Co. Ltd., Osaka, Japan). Growing colonies were tested for homologous recombination by DNA blot analysis using 5′ and 3′ flanking region probes. Eight clones with the targeted allele were obtained and injected into B6 blastocysts. The blastocysts were then transferred into the uterus of pseudopregnant Jcl: MCH(ICR) (MCH) mice. Three clones produced chimeric mice which transmitted a mutant allele to offspring by mating with B6 mice. Three relA mutant mouse strains, RKO-1, -2, and -3, were maintained by brother-sister mating of heterozygous mice in our animal facility. All three strains showed identical phenotypes and RKO-1 mice were mainly used in this study. B6 and MCH mice were purchased from Japan SLC Inc. (Hamamatsu, Japan) and Clea Japan Inc. (Tokyo, Japan), respectively.
DNA and RNA Blot Analyses.
DNA and total RNA were isolated using the proteinase K/SDS and the guanidium thiocyanate/CsCl procedures, respectively (19, 20). DNA was digested by restriction enzymes and separated by agarose gel electrophoresis and then transferred to nitrocellulose filters (Schleicher & Schuell, Dassel, Germany). Total RNA was separated by 2.2 M formaldehyde agarose gel electrophoresis and transferred to nitrocellulose filters. RNA blots were analyzed by cDNA probes for relA, c-rel (codons 144-277, reference 21), p50 (codons 391-518, reference 22) and relB cDNA (codons 458-580, reference 23). DNA blots were analyzed with 5′ and 3′ flanking region probes (see Fig. 1).
Immunohistochemistry and Histology.
Whole embryos were fixed in buffered formalin and embedded in paraffin. Sagittal sections (5 μm) were reacted with rabbit anti-RelA specific antibody (no. sc372; Santa Cruz Biotechnology Inc., Santa Cruz, CA) and binding sites were visualized by an avidin-biotin enzyme complex (ABC) method (Vectastain Elite ABC kit; Vector Laboratories Inc., Burlingame, CA), and then counterstained in hematoxylin. For histological examination, the sections were stained also with hematoxylin-eosin (HE).
Transplantation of Fetal Liver Cells.
SCID mice between 5–8 wk age were irradiated (2.5 Gy; Hitachi MBR-1520R; Hitachi, Tokyo) and each injected intravenously with 3 × 106 fetal liver cells from ED13.5 relA+/+, +/−, or −/− embryos. 4 wk after the transplantation, these mice were used for experiments. SCID mice contain very few lymphocytes in the thymus, spleen and lymph nodes (24), due to a defect in the gene coding for DNAdependent protein kinase p350 (25). SCID mice were maintained in our breeding colony.
Serological Analysis.
Two-color analysis of cell surface antigens was performed with a FACScan® (Becton Dickinson and Co., Mountain View, CA). Thymocytes and spleen cells were stained with antibodies to H-2Kb (E121.46; Seikagaku Kogyo, Tokyo, Japan), Thy-1.2 (30-H12; Becton Dickinson and Co.), TCRαβ (H57-597; provided by Dr. R. T. Kubo, National Jewish Center for Immunology and Respiratory Medicine, Denver, CO [26]), CD3 (145-2C11; provided by Dr. J. A. Bluestone, The University of Chicago, IL [27]), CD4 (GK1.5; obtained from Dr. N. Shinohara, Mitsubishi Kasei Institute for Life Science, Machida, Japan, [28]), CD8 (53-6.7; obtained from Dr. N. Shinohara [29]), CD25 (IL-2Rα, 7D4; reference 30), CD44 (Pgp-1, NU5-50; Seikagaku Kogyo), B220 (RA3-6B2; PharMingen, San Diego, CA), and IgM (DAKO, Glostrup, Denmark).
ELISA for Measurement of Levels of Serum Ig.
For measurement of IgM, IgG1, IgG2a, IgG2b, IgA, and IgE, 96-well flat-bottom plates (Immuno Plate 430341; Nunc, Roskilde, Denmark) were pre-coated with affinity-purified goat anti–mouse Igκ (5 μg/ml, 100 μl/well; Bethyl Laboratories Inc., Montgomery, TX). For Igκ, plates were coated with affinity-purified rabbit anti-mouse IgG (5 μg/ml, 100 μl/well; Southern Biotechnology Associates, Inc., Birmingham, AL). Diluted serum samples and standard Ig were added and bound Ig were detected with horseradish peroxidase–labeled affinity-purified goat anti–mouse Ig isotype-specific antibodies or an anti-κ light chain-specific antibody (Southern Biotechnology). o-Phenylendiamine solution (0.04%; Sigma) was added to each well as a substrate. The optical density at 490 nm was measured with a microplate reader (model 3550; Bio-Rad, Hercules, CA).
IL-2 Bioassay.
3 × 105 spleen cells from transplanted mice were plated in 96-well plates (200 μl per well). Con A (2 μg/ml; Boehringer Mannheim GmbH, Mannheim, Germany), anti-CD28 (1 μg/ml; Caltag Laboratories, South San Francisco, CA), LPS (20 μg/ml; Sigma), anti-IgM (60 μg/ml; Capel Research Products, Durham, NC), and PMA (10 ng/ml; Sigma) plus calcium ionophore (100 ng/ml; Sigma) were added to the medium. For anti-CD3 antibody stimulation, plates were pre-coated with the antibody (10 μg/ml). After 18 h of culture, the supernatants were collected for the assay. To measure the levels of IL-2, serially diluted culture supernatants were added to IL-2–dependent NRB cells (5 × 103, reference 31) in 96-well plates. NRB cells were cultured for 44 h and their proliferation was quantitated using a CellTiter 96TM AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) and a microplate reader (model 3550; Bio-Rad). Recombinant human IL-2 (Takeda Chemical Industries, Osaka, Japan) was used as a standard.
Proliferation Assay.
Conditions for cell culture and stimulation were the same as for the IL-2 bioassay. After 48 h of culture, the proliferation response was measured by uptake of [3H]thymidine (New England Nuclear, Boston, MA) over a 16-h pulse. To determine the percentage of cells in apoptosis during the course of the proliferation assays, cells at 0, 24, 48, and 64 h after stimulation were stained by the TdT-mediated dUPT nick labeling (TUNEL) technique (32, 33), using an In Situ Cell Death Detection Kit (Boehringer Mannheim GmbH) and the results assessed by FACScan®.
Results and Discussion
Requirement of RelA for Embryonic Development of the Mouse.
Mice heterozygous for a disrupted relA were normal and fertile, but no homozygous relA-deficient mice were born from heterozygote mating. Sequential DNA blot analysis and histological examination of embryos from timed matings of heterozygous mice were conducted. Until embryonic day (ED) 13.5, all embryos were apparently normal and homozygous mutants (−/−) were present in an expected ratio. On ED14.5, the relA−/− embryos were still present but some showed signs of abnormalities in their liver (Fig. 2 A). On ED15.5, a portion of the embryos became necrotic and were typed homozygous mutant (−/−), while normal embryos were all wild type (+/+) or heterozygous (+/−). RNA blot analysis of relA+/+ embryos from ED8.5 until birth as well as CCE ES cells showed the presence of relA transcripts, while no relA transcripts could be detected in relA−/− embryos at any stage (Fig. 2 B). Positive immunostaining with anti-RelA antibody correlated with the presence of RNA transcripts, showing that RelA proteins were present in almost all tissues of ED13.5 relA+/+ embryos, while no staining of relA−/− embryos (Fig. 2 C). Although a different vector construct was used in our study, the generated RelA-deficient mice showed the same phenotype as those of Beg et al. (11), indicating that RelA is essential for embryonic development of the mouse. In contrast to RelA-deficient mice, mice lacking other NF-κB proteins are known to develop normally at least until birth (12–14). The difference may simply reflect the fact that RelA is expressed ubiquitously from an early stage of development while the others are expressed in restricted tissues from a much later stage (34, 35). Identification of RelA responsive genes in developing embryos, especially in the liver, should open new avenues for elucidation of the function of NF-κB in embryonic development.
Figure 2.
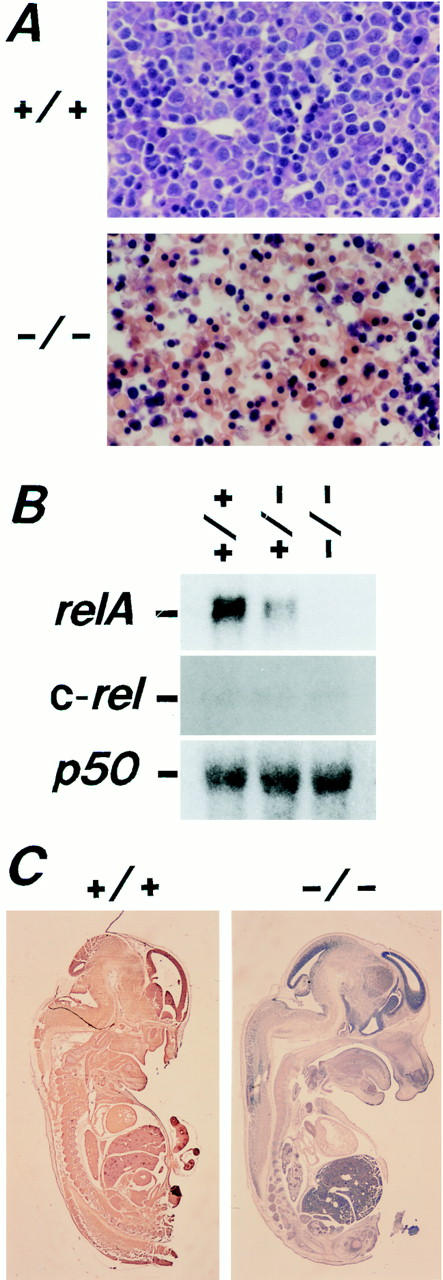
Liver degeneration and the absence of relA transcripts and RelA protein in relA−/− embryos. (A) Histological features of livers of ED14.5 relA+/+ and relA−/− embryos. In the liver of a relA+/+ embryo, hepatocytes with large cell size and light nuclear staining are mixed with the hematopoietic cells with dark nuclear staining. In the liver of relA−/− embryos, hepatocytes are disintegrated, while the hematopoietic cells are apparently normal. Magnification is 240-fold. (B) RNA blot analysis of ED13.5 relA+/+, +/−, and −/− embryos. 10 μg of total RNA were loaded per lane and analyzed with the relA, c-rel, relB, and p50 cDNA probes. relA transcripts were present in the relA+/+ and +/− embryos, but not in the relA−/− embryos. The relA+/+, +/− and −/− embryos expressed similar amounts of c-rel and p50 transcripts. No relB transcripts were detected in ED13.5 embryos (data not shown). (C) RelA protein in ED13.5 relA+/+ and relA−/− embryos. With rabbit antiRelA antibody and the ABC method, RelA protein is ubiquitously detected in the relA+/+ embryo, but is completely absent in the relA−/− embryo. Magnification is eightfold.
Transplantation of relA−/− Fetal Liver Cells.
During normal embryonic development, hematopoietic stem cells emerge in the fetal liver on ED9.5 (36). To test whether the RelA-deficient hematopoietic stem cells can develop in the fetal liver and also whether they can differentiate to mature lymphocytes, fetal liver cells of ED13.5 embryos were transplanted into irradiated SCID mice. Transplantation of fetal liver cells not only from relA+/+ and +/− but also from relA−/− embryos greatly increased the number of cells in the thymus, spleen and lymph nodes of SCID mice. The numbers of cells in spleen and lymph nodes of the mice transplanted with relA−/− fetal liver cells were similar to those receiving either relA+/+ or +/− fetal liver cells. The thymus from mice transplanted with relA−/− fetal liver cells contained fewer cells than those with wild-type or heterozygous fetal liver cells. The origin of the lymphocytes in transplanted mice was determined by testing the expression of H-2Kb antigen. The fetal liver cells were from crosses between 129 and B6, both of which express H-2KbDb, while the recipient SCID themselves express H-2KdDd. As shown in Fig. 3, >95% of lymphocytes of mice transplanted with fetal liver cells expressed H-2Kb, indicating that they were definitely of fetal liver origin. The donor origin of the lymphocytes in transplanted mice with fetal liver cells was further confirmed by the presence of disrupted relA genes and the absence of relA transcripts (Fig. 4). Thus, these results indicated that RelA-deficient hematopoietic stem cells can indeed develop in the fetal liver and also proliferate in SCID mice.
Figure 3.

The fetal liver origin of lymphocytes in transplanted SCID mice. By transplantation of the fetal liver from relA+/+, +/−, or −/− ED 13.5 embryos into irradiated SCID mice, the number of lymphocytes increased. Average numbers of the cells in the spleen, lymph nodes, and thymus in groups of three mice receiving transplants are indicated above the distribution plots. Staining in the presence and absence of mAb to H-2Kb is indicated by solid and open curves, respectively. Flow cytometric analysis of spleen cells, lymph node cells, and thymocytes showed the expression of H-2Kb, indicating the fetal liver origin.
Figure 4.
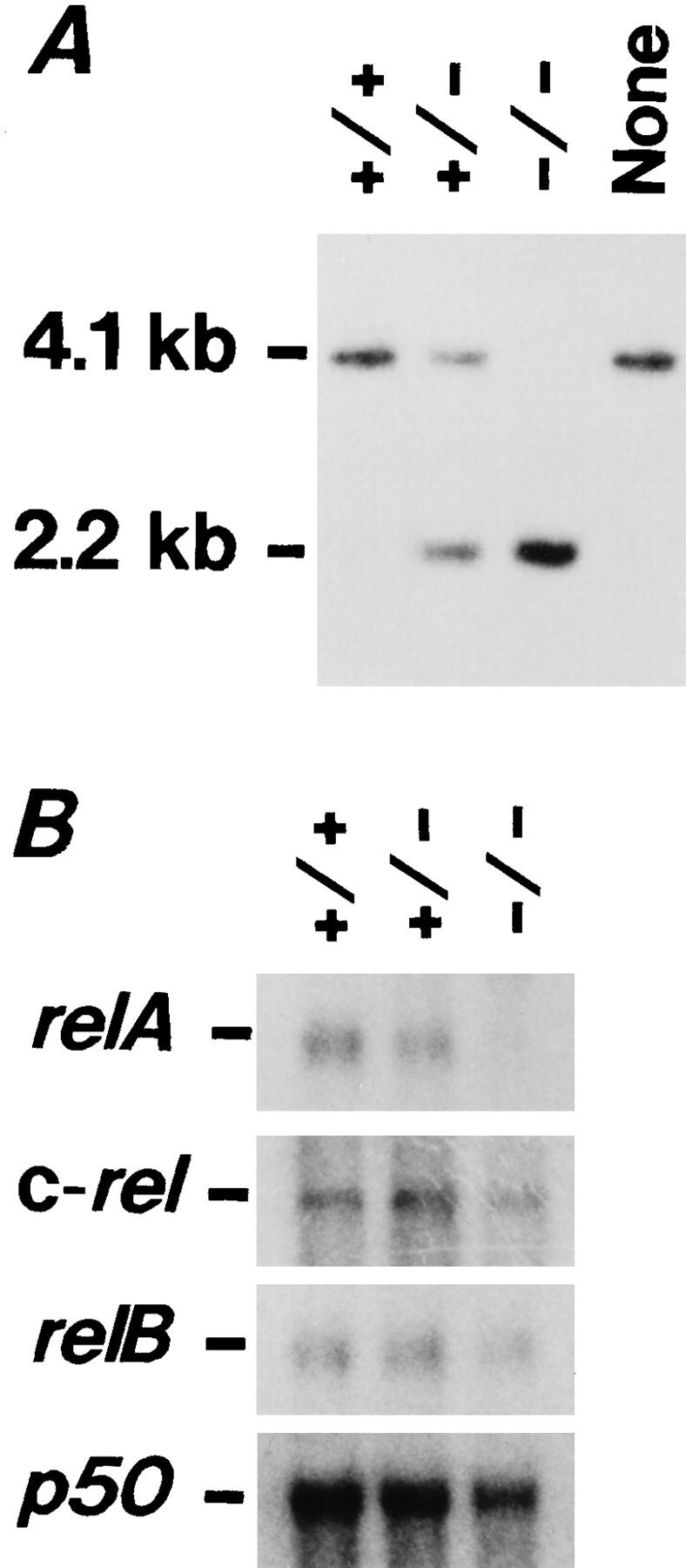
DNA and RNA blot analyses of the spleen cells of the transplanted SCID mice. (A) DNA blot analysis. DNA digested by PstI was analyzed with a 5′ franking probe (see Fig. 1); the wild-type relA allele yielding a 4.1-kb fragment, and the mutant allele a 2.2-kb fragment. (B) RNA blot analysis. Note the lack of relA transcripts in the spleen cells of mice transplanted with relA−/− fetal liver cells. The relA−/−, +/+, and +/− spleen cells express similar amounts of c-rel, relB, and p50 transcripts.
Development of T and B Cells in the Absence of RelA.
To test whether RelA-deficient stem cells can differentiate into T and B cells, the cell surface markers on lymphocytes of transplanted mice were examined. As shown in Fig. 5, the relA−/− hematopoietic stem cells differentiated to Thy1+/TCRαβ+ /CD3+/CD4+ or CD8+ T cells and IgM+/ B220+ B cells in the periphery, similarly to the relA+/+ and +/− stem cells. In addition, testing of the expression of activation markers of T cells, IL-2Rα (CD25) and CD44 (Pgp-1), revealed 6.6 and 2.5% of relA−/− spleen cells to be positive, respectively, with no significant differences from the relA+/+ or +/− cases (data not shown). The results with cells in lymph nodes were essentially identical to those of spleens (data not shown). The relA−/− thymuses with much fewer cells than those of relA+/+ and +/−, showed reduction of the CD4+/CD8+ immature population, which may have been caused by the absence of RelA. Altogether, these results suggested that RelA is not necessary for the maturation of lymphocytes or that the loss of RelA function can be compensated for by other members of the NF-κB family. In this regard, it was interesting to note that none of the mice deficient in any NF-κB subunit, whether RelA, c-Rel, RelB, or p50, showed abnormality in the development of T cells and B cells. There is a vast amount of evidence indicating the importance of NF-κB in lymphocyte development (for review see references 1, 3, 37). Thus, it is most likely that the development of T and B cells proceeds with certain combinations of NF-κB subunits and may not require the presence of specific NF-κB dimers.
Figure 5.
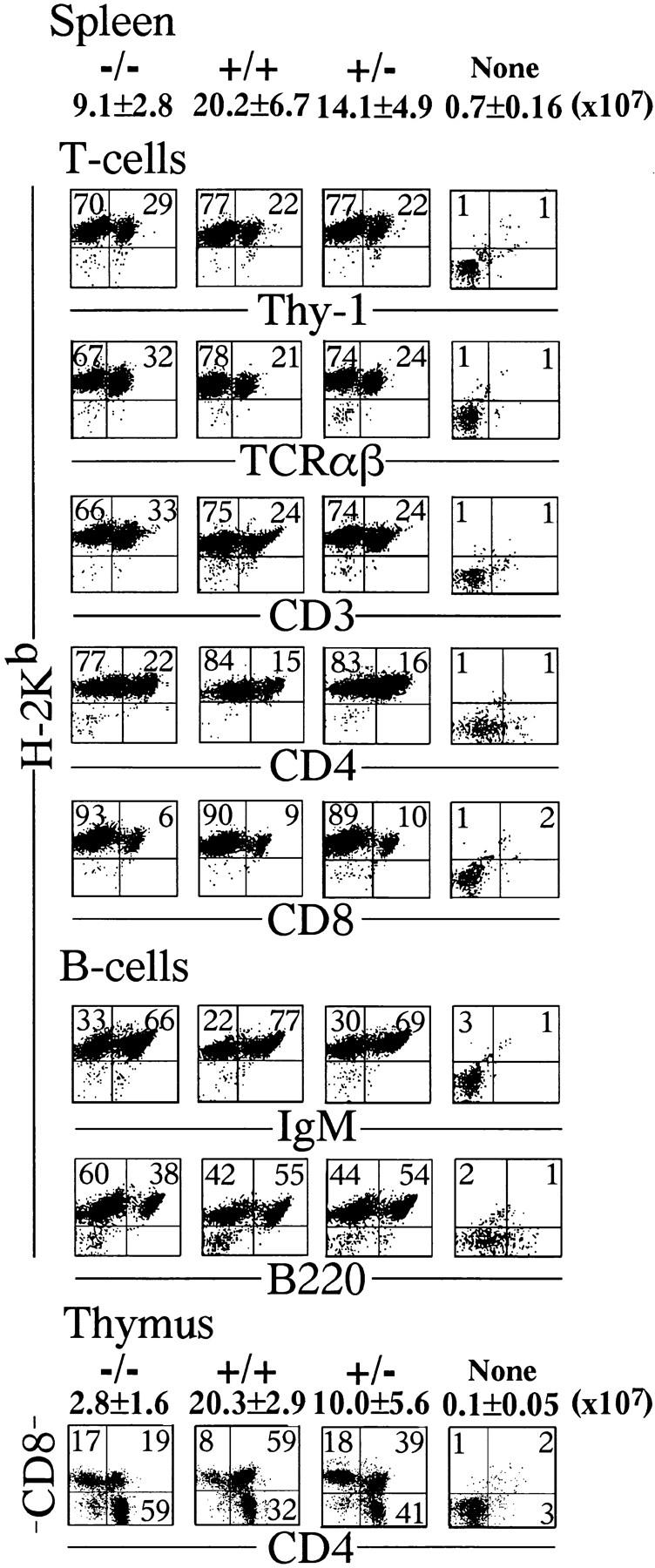
Surface antigen profiles of spleen cells and thymocytes of SCID mice transplanted with ED13.5 fetal liver cells. Cells were examined by two-color staining using various combinations of antibodies. The expression of H-2Kb by spleen cells confirms their fetal liver origin. As for T cell markers, Thy-1, TCRαβ, CD3, CD4, and CD8 were examined and as for B cell makers, IgM and B220 were examined. Even relA−/− fetal liver cells develop normally to mature T and B cells in the transplanted SCID mice.
Reduced Production of IgG1 and IgA by relA−/− B Cells.
As the RelA-deficient B and T cells matured normally, the role of RelA in lymphocyte function was examined. To assess B cell function, the levels of serum Ig isotypes in SCID mice transplanted with fetal liver cells were measured (Fig. 6). The results showed that relA−/− B cells were capable of secreting Ig as well as switching classes of Ig isotypes. The levels of total Ig and individual classes of IgM, IgG2a, IgG2b, IgE, and Igκ produced by relA−/− B cells were comparable to those with relA+/+ or +/− B cells. Although RelA was originally identified, together with p50, as an enhancer binding protein for an Igκ chain gene, surprisingly the absence of RelA had no effect on the levels of Igκ production. The RelA-deficient B cells, however, produced 10-fold and 100-fold less IgG1 and IgA, respectively, than the control B cells. Reduced production of certain Ig classes has been also reported in mice deficient in p50 (14) and c-Rel (12): IgG1, IgA, and IgE in the former and IgG1 and IgG2a in the latter. Thus, each NF-κB member is critically involved in the production of certain Ig isotypes, presumably by regulating the transcription of Ig genes directly and/or acting on various cytokine genes which ultimately control Ig class switching and production. In this regard, it is interesting to note that IgA reduction has also been reported in IL-6-deficient mice (38) and that the expression of IL-6 is controlled by RelA/p50 heterodimers (8).
Figure 6.

The serum Ig levels in SCID mice transplanted with fetal liver cells. 4 wk after the transplantation, mice with relA−/− fetal livers were able to secrete Ig in their sera, with the total amount being comparable to those with relA+/+ or relA+/− cells, but the IgA and IgG1 levels were found to be significantly lower. SCID mice secrete hardly detectable levels of any Ig isotypes. Open, striped, and closed circles correspond to mice transplanted with relA+/+, +/−, and −/− fetal liver cells, respectively.
IL-2 and IL-2Rα in relA−/− T Cells.
In T cells, the RelA/p50 heterodimer has been reported to be a potent transcription factor for the IL-2 gene after stimulation by various agents (39). The relA−/− spleen cells, however, produced similar levels of IL-2 to relA+/+ or +/− spleen cells after stimulation with Con A, anti-CD3, anti-CD3+ anti-CD28, or PMA+calcium ionophore (Table 1). The results were in contrast to those for c-Rel–deficient mice which showed ∼50-fold reduction in IL-2 production after stimulation by anti-CD3, and anti-CD3+anti-CD28 and threefold reduction by the PMA+calcium ionophore. Thus, it was strongly suggested that the main component of the NF-κB transcription factor for the IL-2 gene in vivo is c-Rel rather than RelA.
Table 1.
Levels of IL-2 Production by Spleen Cells from SCID Mice Transplanted with Fetal Liver Cells in Response to Various Stimuli
| relA genotype of fetal liver donor* | Stimuli‡ | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Con A | Anti-CD3 | Anti-CD28 | Anti-CD3 + Anti-CD28 | PMA + calcium ionophore | None | |||||||
| IL-2 levels | ||||||||||||
| U/ml § | ||||||||||||
| +/+ | 167 ± 16.2‖ | 63.7 ± 10.3 | 5.7 ± 0.84 | 446 ± 19.8 | 361 ± 69.8 | 4.5 ± 1.42 | ||||||
| +/− | 146 ± 18.5 | 42.8 ± 5.27 | 5.2 ± 0.82 | 448 ± 10.9 | 312 ± 26.1 | 2.0 ± 0.05 | ||||||
| −/− | 118 ± 16.8 | 60.3 ± 8.11 | 6.5 ± 1.08 | 402 ± 88.0 | 276 ± 42.3 | 2.4 ± 0.32 | ||||||
| None | 1.5 ± 0.66 | 3.4 ± 1.03 | 2.3 ± 0.62 | 5.2 ± 1.84 | 6.5 ± 1.32 | 1.4 ± 0.32 | ||||||
3 × 106 fetal liver cells from relA +/+, +/−, or −/− embryos were transplanted into irradiated SCID mice.
3 × 105 spleen cells were stimulated as described in the Materials and Methods.
The levels of IL-2 were measured by bioassay using NRB cells (see the Materials and Methods).
Mean ± SD. The results obtained from three mice were averaged.
RelA has been reported to be involved also in the upregulation of IL-2Rα expression with stimulation by various agents (40). Before stimulation, the levels of IL-2Rα on relA−/−, +/+, or +/− lymphocytes were similar to one another as mentioned above. With stimulation by PMA+ calcium ionophore or Con A, the levels of IL-2Rα expression on relA−/− T cells increased and did not significantly differ from relA+/+ or relA+/− T cells (data not shown). The c-Rel-deficient T cells also showed no reduction in the basal or induced expression of IL-2Rα (41). These observations suggest the following two possibilities: (a) neither RelA nor c-Rel is required for the expression of IL-2Rα, or (b) both can participate in the control of IL-2Rα expression and one works in the absence of the other. Identification of binding subunits to the κB motif of IL-2Rα in the absence of RelA or c-Rel and derivation of mice lacking both RelA and c-Rel should sort out these possibilities.
Impaired Proliferative Response of relA−/− Lymphocytes.
To further analyze the role of RelA in lymphocyte activation, spleen cells from mice transplanted with relA−/− fetal liver cells were stimulated with various agents. With both T cell specific stimuli, Con A, anti-CD3 and anti-CD3+ anti-CD28, and B cell–specific stimuli, LPS and anti-IgM, relA−/− spleen cells showed a much lower [3H]thymidine uptake than relA+/+ or +/− spleen cells (Table 2). To test whether this low response of relA−/− spleen cells is due to reduced cellular proliferation or to increased apoptotic cell death, the percentages of cells in apoptosis during the course of proliferation assays were determined. As shown in Fig. 7, the percentages and the actual numbers of apoptotic cells with relA−/− were not significantly different from the relA+/− case. Although the number of viable cells may not be as indicative as [3H]thymidine uptake because only a small component of the spleen cells can proliferate in response to certain mitogenic stimuli, relA−/− yielded constantly fewer viable cells than relA+/−. These results indicate that RelA-deficient lymphocytes indeed have an impaired proliferative response to various mitogens. As the production of IL-2 and the expression of IL-2Rα were normal in RelA-deficient T cells, the results suggested that RelA is also involved in yet unidentified critical steps of proliferative responses. Furthermore, RelA-deficient lymphocytes exhibited impaired responses to various stimuli whose signals are transduced by distinctive pathways (42, 43). Thus, RelA may be involved in each single pathway or in a critical merging step downstream of these different pathways. Identification of RelA responsive genes involved in proliferation should reveal the role of RelA in these responses. T and B cells of c-Rel–deficient mice have also been found to demonstrate a defective proliferation response to various stimuli, generally with severe reduction (12). These results indicate that RelA and c-Rel are essential for certain steps of proliferation and that they cannot compensate for each other. It is interesting to note that relA−/− lymphocytes showed an impaired proliferative response to PMA+calcium ionophore in this study while c-rel−/− lymphocytes respond normally to this agent (12). Presumably, the involvement of RelA in proliferative responses is thus wider. Furthermore, relA−/− embryonic fibroblasts also showed reduced proliferation after PMA+ calcium ionophore stimulation, down to 30% of the levels of their relA+/+ or +/− counterparts (data not shown). As expression of relA is not restricted to lymphocytes, in contrast to that of c-rel (44), this also suggests a role in a wider range of biological processes.
Table 2.
In Vitro Proliferation of Spleen Cells from SCID Mice Transplanted with Fetal Liver Cells in Response to Various Mitogenic Stimuli
| relA genotype of fetal liver donor* | Stimuli* | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Con A | Anti-CD3 | Anti-CD3 + Anti-CD28 | LPS | Anti-IgM | PMA + calcium ionophore | None | ||||||||
| [3H]Thymidine uptake | ||||||||||||||
| cpm | ||||||||||||||
| +/+ | 19,328 ± 3,771§ | 46,465 ± 4,898 | 94,775 ± 2,612 | 18,336 ± 2,924 | 15,571 ± 2,003 | 73,837 ± 2,261 | 305 ± 73 | |||||||
| (n = 6)‡ | (n = 4) | (n = 3) | (n = 6) | (n = 4) | (n = 2) | (n = 6) | ||||||||
| +/− | 23,031 ± 3,491 | 49,169 ± 6,557 | 92,642 ± 5,692 | 20,076 ± 7,856 | 18,560 ± 4,059 | 85,702 ± 6,130 | 505 ± 146 | |||||||
| (n = 9) | (n = 7) | (n = 6) | (n = 9) | (n = 7) | (n = 5) | (n = 9) | ||||||||
| −/− | 3,621 ±960 | 18,343 ± 3,030 | 18,865 ± 4,575 | 4,717 ± 1,026 | 5,879 ± 1,076 | 20,863 ± 6,150 | 403 ± 168 | |||||||
| (n = 9) | (n = 7) | (n = 6) | (n = 9) | (n = 7) | (n = 5) | (n = 9) | ||||||||
| None | 381 ± 145 | 911 ± 352 | 860 ± 215 | 807 ± 285 | 689 ± 285 | 1,707 ± 734 | 298 ± 75 | |||||||
| (n = 7) | (n = 5) | (n = 4) | (n = 7) | (n = 5) | (n = 3) | (n = 7) | ||||||||
Figure 7.
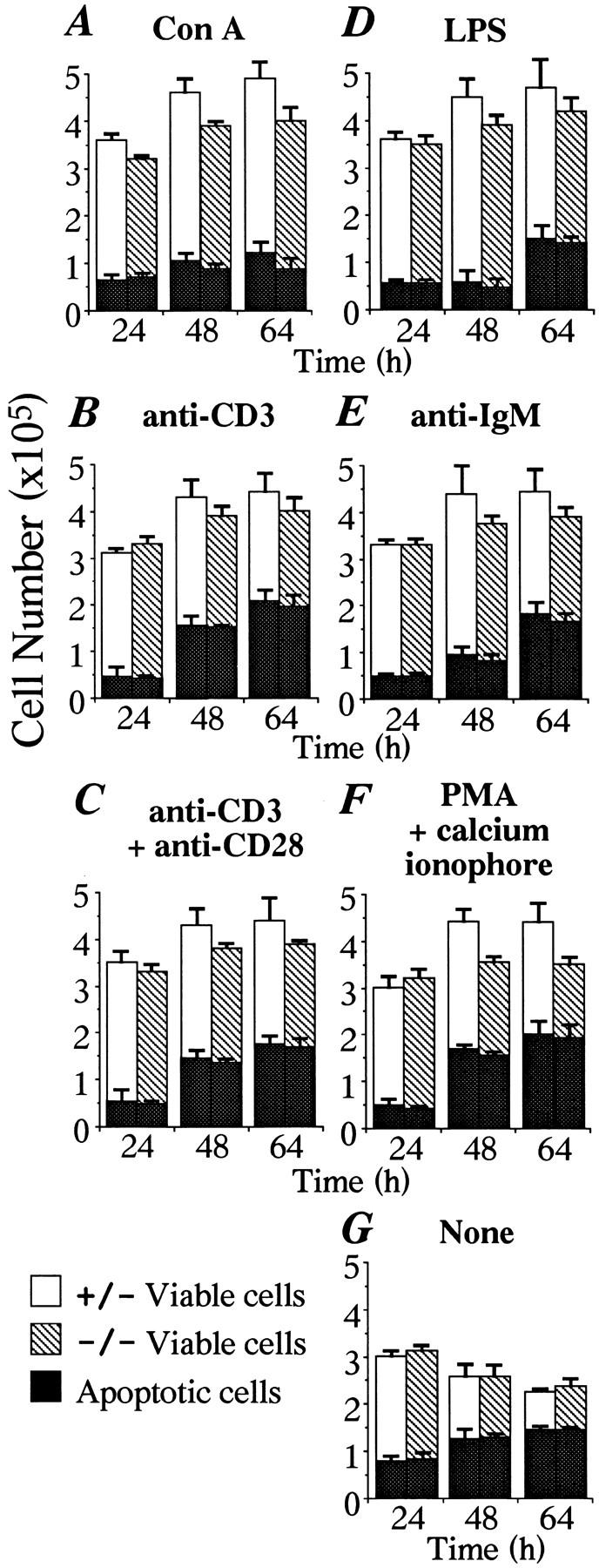
Cells in apoptosis during the course of proliferation assays. Spleen cells from mice transplanted with relA+/− or −/− fetal liver cells were stimulated with various mitogens as in Table 2. The percentages of apoptotic cells were determined using the TUNEL technique with a FACScan® (32, 33). The numbers of viable cells were determined by the trypan blue exclusion test. At the beginning of stimulation, no cells were in apoptosis. The averages and SD values from three independent experiments are shown in the figure.
In conclusion, transplantation of fetal liver cells into SCID mice in the present investigation allowed light to be cast on a number of the functions of RelA in the immune system, despite the fact that the relA−/− genotype is lethal for embryos. Further study with in vitro and in vivo immunization by T-dependent and -independent antigens should facilitate clarification of RelA roles. In addition, since lymphocytes can be rescued from dying embryos by transplantation of fetal liver cells as described here, mice lacking multiple NF-κB proteins, such as RelA and c-Rel, or RelA and RelB, should now be testable for their actions exerted in concert.
Acknowledgments
We thank Drs. Motoya Katsuki, Hitoshi Niwa, and Kunio Tsujimura for their valuable suggestions. We also thank Mineko Izawa, Yasue Matsudaira, Hitomi Nishiwaki, Hiromi Tamaki, and Satoshi Ozeki for their excellent technical assistance. We are grateful to Dr. Malcolm A. Moore for his editorial assistance.
This work was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas and a Grantin-Aid for General Scientific Research from the Ministry of Education, Science, Sports and Culture, Japan. This work was also supported in part by grants from the Naito and Imanaga Foundations.
Footnotes
1 Abbreviations used in this paper: ABC, avidin-biotin enzyme complex; B6, C57BL/6; ED, embryonic day; ES, embryonic stem; HE, hematoxylineosin; HSV-tk, herpes simplex virus-thymidine kinase; MCH, Jcl: MCH(ICR); TUNEL, TdT-mediated dUTP nick end labeling.
References
- 1.Baldwin AS., Jr The NF-κB and IκB proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 2.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. [PubMed] [Google Scholar]
- 3.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 4.Liou H-C, Sha WC, Scott ML, Baltimore D. Sequential induction of NF-κB/Rel family protein during B-cell terminal differentiation. Mol Cell Biol. 1994;14:5349–5359. doi: 10.1128/mcb.14.8.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verweij CL, Geerts M, Aarden LA. Activation of interleukin-2 gene transcription via T-cell surface molecule CD28 is mediated through an NF-κB-like response element. J Biol Chem. 1991;266:14179–14182. [PubMed] [Google Scholar]
- 6.Jamieson C, Mauxion F, Sen R. Identification of a functional NF-κB binding site in the murine T cell receptor β2 locus. J Exp Med. 1989;170:1737–1743. doi: 10.1084/jem.170.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh P, Tan T-H, Rice NR, Sica A, Young HA. The interleukin 2 CD28-responsive complex contains at least three members of the NF-κB family: c-Rel, p50, and p65. Proc Natl Acad Sci USA. 1993;90:1696–1700. doi: 10.1073/pnas.90.5.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-κB transcription factor. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four κB-like motifs and of constitutive and inducible forms of NF-κB. Mol Cell Biol. 1990;10:1498–1506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamieson C, McCaffrey PG, Rao A, Sen R. Physiologic activation of T cells via the T cell receptor induces NF-κB. J Immunol. 1991;147:416–420. [PubMed] [Google Scholar]
- 11.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature (Lond) 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 12.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-relproto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 13.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck R-P, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/ Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 14.Sha WC, Liou H-C, Toumanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 15.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 16.Nolan GP, Ghosh S, Liou H-C, Tempst P, Baltimore D. DNA binding and IκB inhibition of the cloned p65 subunit of NF-κB, a rel-related polypeptide. Cell. 1991;64:961–969. doi: 10.1016/0092-8674(91)90320-x. [DOI] [PubMed] [Google Scholar]
- 17.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature (Lond) 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 18.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2in mouse embryoderived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature (Lond) 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 19.Blin N, Stafford DW. A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res. 1976;3:2303–2308. doi: 10.1093/nar/3.9.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 21.Bull P, Morley KL, Hoekstra MF, Hunter T, Verma IM. The mouse c-relprotein has an N-terminal regulatory domain and a C-terminal transcriptional transactivation domain. Mol Cell Biol. 1990;10:5473–5485. doi: 10.1128/mcb.10.10.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh S, Gifford AM, Riviere LR, Tempst P, Nolan GP, Baltimore D. Cloning of the p50 DNA binding subunit of NF-κB: homology to rel and dorsal. . Cell. 1990;62:1019–1029. doi: 10.1016/0092-8674(90)90276-k. [DOI] [PubMed] [Google Scholar]
- 23.Ryseck R-P, Bull P, Takamiya M, Bours V, Siebenlist U, Dobrzanski P, Bravo R. RelB, a new Rel family transcription activator that can interact with p50-NF-κB. Mol Cell Biol. 1992;12:674–684. doi: 10.1128/mcb.12.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature (Lond) 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 25.Kirchgessner CU, Patil CK, Evans JW, Cuomo CA, Fried LM, Carter T, Oettinger MA, Brown JM. DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science (Wash DC) 1995;267:1178–1183. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- 26.Kubo RT, Born W, Kappler JW, Marrack P, Pigeon M. Characterization of a monoclonal antibody which detects all murine αβ T cell receptors. J Immunol. 1989;142:2736–2742. [Google Scholar]
- 27.Leo O, Foo M, Sachs DH, Samelson LE, Bluestone JA. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci USA. 1987;84:1374–1378. doi: 10.1073/pnas.84.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dialynas DP, Quan ZS, Wall KA, Pierres A, Quintans J, Loken MR, Pierres M, Fitch FW. Characterization of the murine T cell surface molecule, designated L3T4, identified by monoclonal antibody GK1.5: similarity of L3T4 to the human Leu-3/T4 molecule. J Immunol. 1983;131:2445–2551. [PubMed] [Google Scholar]
- 29.Ledbetter J, Herzenberg L. Xenogenic monoclonal antibodies to mouse lymphoid differentiation antigen. Immunol Rev. 1979;47:63–90. doi: 10.1111/j.1600-065x.1979.tb00289.x. [DOI] [PubMed] [Google Scholar]
- 30.Malek TR, Robb RJ, Shvach EM. Identification and initial characterization of a rat monoclonal antibody reactive with the murine interleukin 2 receptor-ligand complex. Proc Natl Acad Sci USA. 1983;80:5694–5698. doi: 10.1073/pnas.80.18.5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsutake T, Sasaki T, Ogino T, Kaku M, Nakayama E. Prevention of mortality by in vivodepletion of αβ T cells in murine lethal listeriosis and involvement of γδ T cells in bacterial elimination. Immunobiol. 1995;193:71–83. doi: 10.1016/s0171-2985(11)80156-7. [DOI] [PubMed] [Google Scholar]
- 32.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sgonic R, Boeck G, Dietrich H, Grhber J, Recheis H, Wick G. Simultaneous determination of cell surface antigens and apoptosis. Trends Genet. 1994;10:41–42. doi: 10.1016/0168-9525(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 34.Carrasco D, Weih F, Bravo R. Developmental expression of the mouse c-relproto-oncogene in hematopoietic organs. Development. 1994;120:2991–3004. doi: 10.1242/dev.120.10.2991. [DOI] [PubMed] [Google Scholar]
- 35.Carrasco D, Ryseck R-P, Bravo R. Expression of relBtranscripts during lymphoid organ development: specific expression in dendritic antigen-presenting cells. Development. 1993;118:1221–1231. doi: 10.1242/dev.118.4.1221. [DOI] [PubMed] [Google Scholar]
- 36.Johnson GR, Jones RO. Differentiation of the mammalian hepatic primordium in vitro. I. Morphogenesis and the onset of hematopoiesis. J Embryol Exp Morphol. 1973;30:83–96. [PubMed] [Google Scholar]
- 37.Clevers HC, Grosschedl R. Transcriptional control of lymphoid development: lessons from gene targeting. Immunol Today. 1996;17:336–343. doi: 10.1016/0167-5699(96)10019-0. [DOI] [PubMed] [Google Scholar]
- 38.Ramsay AJ, Husband AJ, Ramshaw IA, Bao S, Matthaei KI, Koehler G, Kopf M. The role of interleukin-6 in mucosal IgA antibody responses in vivo. . Science (Wash DC) 1994;264:561–563. doi: 10.1126/science.8160012. [DOI] [PubMed] [Google Scholar]
- 39.Lai J, Horvath G, Subleski J, Bruder J, Ghosh P, Tan T. RelA is a potent transcriptional activator of the CD28 response element within the interleukin 2 promoter. Mol Cell Biol. 1995;15:4260–4271. doi: 10.1128/mcb.15.8.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Algarte M, Lecine P, Costello R, Plet A, Olive D, Imbert J. In vivoregulation of interleukin-2 receptor α gene transcription by the coordinated binding of constitutive and inducible factors in human primary T cells. EMBO (Eur Mol Biol Organ) J. 1995;20:5060–5072. doi: 10.1002/j.1460-2075.1995.tb00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerondakis S, Strasser A, Metcalf D, Grigoriadis G, Scheerlinck J-PY, Grumont RJ. Rel-deficient T cells exhibit defects in production of interleukin 3 and granulocyte-macrophage colony stimulating factor. Proc Natl Acad Sci USA. 1996;93:3405–3409. doi: 10.1073/pnas.93.8.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geppert TD, Davis LS, Gur H, Wacholtz MC, Lipsky PE. Accessory cell signals involved in T-cell activation. Immunol Rev. 1990;117:5–66. doi: 10.1111/j.1600-065x.1990.tb00566.x. [DOI] [PubMed] [Google Scholar]
- 43.Gold MR, Defranco AL. Biochemistry of B lymphocyte activation. Adv Immunol. 1994;55:221–295. doi: 10.1016/s0065-2776(08)60511-8. [DOI] [PubMed] [Google Scholar]
- 44.Grumont RJ, Gerondakis S. The murine c-relproto-oncogene encodes two mRNAs the expression of which is modulated by lymphoid stimuli. Oncogene Res. 1990;5:245–254. [PubMed] [Google Scholar]