

Abstract
Mice with X-linked chronic granulomatous disease (CGD) generated by targeted disruption of the gp91phox subunit of the NADPH–oxidase complex (X-CGD mice) were examined for their response to respiratory challenge with Aspergillus fumigatus. This opportunistic fungal pathogen causes infection in CGD patients due to the deficient generation of neutrophil respiratory burst oxidants important for damaging A. fumigatus hyphae. Alveolar macrophages from X-CGD mice were found to kill A. fumigatus conidia in vitro as effectively as alveolar macrophages from wild-type mice. Pulmonary disease in X-CGD mice was observed after administration of doses ranging from 105 to 48 spores, none of which produced disease in wild-type mice. Higher doses produced a rapidly fatal bronchopneumonia in X-CGD mice, whereas progression of disease was slower at lower doses, with development of chronic inflammatory lesions. Marked differences were also observed in the response of X-CGD mice to the administration of sterilized Aspergillus hyphae into the lung. Within 24 hours of administration, X-CGD mice had significantly higher numbers of alveolar neutrophils and increased expression of the proinflammatory cytokines IL-1β and TNF-α relative to the responses seen in wild-type mice. By one week after administration, pulmonary inflammation was resolving in wild-type mice, whereas X-CGD mice developed chronic granulomatous lesions that persisted for at least six weeks. This is the first experimental evidence that chronic inflammation in CGD does not always result from persistent infection, and suggests that the clinical manifestations of this disorder reflect both impaired microbial killing as well as other abnormalities in the inflammatory response in the absence of a respiratory burst.
Chronic granulomatous disease (CGD)1 is an inherited disorder caused by defects in the phagocyte respiratory burst oxidase (NADPH oxidase) which generates superoxide (1), the precursor to hydrogen peroxide and other reactive oxygen derivatives with microbicidal activity. Hence, patients with CGD lack an essential host antimicrobial pathway, and develop recurrent and often severe bacterial and fungal infections. A second and distinctive feature of CGD is the frequent occurrence of inflammatory granulomas in lung, liver, skin, lymph nodes, or the lining of the gastrointestinal and genitourinary tracts (2–5). These chronic inflammatory lesions in some cases represent an attempt to contain an active site of infection, but can also be sterile, suggesting that their formation may be due to incomplete degradation of debris, impaired resolution of inflammation, or both (4, 6).
Aspergillus fumigatus is an opportunistic fungal pathogen that is an important cause of pulmonary and other infections in immunocompromised hosts, including patients with CGD. A. fumigatus exposure typically occurs via inhalation of conidia into the respiratory tract, where resistance to infection is twofold. Resident alveolar macrophages ingest and kill resting conidia, largely through nonoxidative mechanisms, while neutrophils use oxygen-dependent mechanisms to attack hyphae germinating from conidia that escape macrophage surveillance (7). The effectiveness of this system is evident from the observation that challenge with even large numbers of conidia fail to cause disease in immunocompetent animals (8). To cause disease, animals must be immunosuppressed either through the use of agents that impair the function of resident alveolar macrophages, such as glucocorticoids (7, 9), or those that produce profound granulocytopenia, such as cyclophosphamide or wholebody irradiation (7, 10).
The importance of the phagocyte respiratory burst in resistance to A. fumigatus has been established by the frequent occurrence of A. fumigatus infection in patients with CGD (4), which appears to result largely from a failure to kill hyphae by neutrophil oxidative mechanisms. Although purified cationic granule proteins isolated from human neutrophils are able to damage hyphae in a cell-free assay system, neutrophils from CGD patients do not cause significant damage in vitro (11), suggesting that under physiologic conditions, cellular nonoxidative mechanisms alone are insufficient to kill hyphae. Hydrogen peroxide, a product of the respiratory burst, has potent antihyphal activity in vitro (11), while neutrophil-mediated hyphal damage is strongly blocked by myeloperoxidase inhibitors, the hydrogen peroxide scavenger, catalase, and to a lesser extent, superoxide dismutase (12).
The role of oxidants in resistance to the conidial form of Aspergillus is probably less critical. Schaffner and others have reported that alveolar macrophages kill Aspergillus conidia effectively under anaerobic conditions, and that peripheral blood monocytes from patients with CGD kill conidia as effectively as monocytes from normal individuals (13). However, in a study suggesting that respiratory burst oxidants may play some role in the macrophage anticonidial activity, Washburn et al. reported that monocytes from CGD patients killed only about half as many conidia as normal monocytes within 2 h of phagocytosis. In addition, the anticonidial activity of normal monocytes was partially inhibited by catalase, suggesting that oxidative mechanisms contribute to macrophage anticonidial activity (14). No data have been published regarding the anticonidial potential of alveolar macrophages from CGD patients.
We have previously reported that X-CGD mice, generated by targeted disruption of the gp91phox subunit of the NADPH-oxidase, exhibit impaired clearance of Staphylococcus aureus and A. fumigatus, two common pathogens in human CGD (15). Respiratory challenge with A. fumigatus was associated with high rates of mortality in X-CGD mice, demonstrating a requirement for phagocyte-generated oxidants in murine host resistance to Aspergillus infection. The dose of conidia used in the previous study (3 × 106/mouse) is likely to far exceed normal levels of exposure (16, 17), probably contributing to the rapid progression of disease and early mortality observed.
In the present study, we have investigated the response to A. fumigatus at exposures that may be more representative of naturally occurring infection. Although alveolar macrophages from X-CGD mice were found to kill A. fumigatus conidia as effectively as normal macrophages, exposure to even very small numbers of conidia still resulted in bronchopneumonia, with persistence of viable A. fumigatus for at least 2–3 wk after respiratory challenge. Extensive areas of lung inflammation were often seen in X-CGD mice, with few detectable hyphae. Remarkably, administration of sterilized hyphal preparations also produced a chronic bronchopneumonia with a distinctive neutrophilic and granulomatous infiltrate in X-CGD mice but not wild-type mice. The early phase of the inflammatory response in X-CGD mice was associated with significantly higher numbers of alveolar neutrophils and increased levels of expression of IL-1β and TNF-α mRNA relative to wild-type mice. These data suggest that the absence of respiratory burst oxidants can result in altered progression of lung injury even to sterilized Aspergillus products, which may contribute to the development of chronic inflammatory lesions in CGD.
Materials and Methods
Animals.
X-CGD mice had previously been generated by targeted disruption of the gene encoding gp91phox, the large subunit of the NADPH respiratory burst oxidase complex (15). Phagocytes from these mice are unable to generate superoxide as measured by both the nitroblue tetrazolium test and cytochrome c reduction assays. Mice were housed in microisolator cages under specific pathogen-free conditions and fed autoclaved food and acidified water ad libitum. For experiments involving X-CGD mice generated by backcrossing carrier females with C57Bl/6J males for generations up to N7, wild-type littermates from the same backcross generation were used to minimize any potential bias contributed by differences in original 129-SV × C57Bl/6J genetic backgrounds. For experiments after generation N7, agematched wild-type C57Bl/6J mice were used. Mice studied in these experiments were from 6 to 10 wk old.
Isolation and Preparation of Aspergillus Inocula.
A clinical isolate of A. fumigatus (ATCC No. 90240; American Type Culture Collection, Rockville, MD) was cultured on Sabouraud dextrose agar (Becton Dickinson Labware, Lincoln Park, NJ) for 4 d at room temperature before use. Conidia were removed by flooding the culture dish with 5–10 ml of sterile PBS and gently scraping the mycelia with a sterile razor blade. Suspensions consisting mostly of single conidia were prepared by vortexing the mixture vigorously for 1 min followed by filtration through three-ply cotton gauze. Conidia were quantitated using a hemocytometer and diluted to the appropriate concentration in PBS. Quantitation was confirmed by plate culture of serial dilutions on Sabouraud dextrose agar. Conidia suspensions were always used on the day of harvest and kept on ice until needed. Viability as determined by quantitative culture was typically >95%.
Killing of A. fumigatus Conidia by Alveolar Macrophages.
For each experiment, alveolar macrophages were harvested from at least three wild-type and three X-CGD mice as previously described (18). Briefly, mice were killed by ketamine overdose and exsanguinated by inferior vena cava puncture. The thoracic cavity was opened and the trachea exposed by dissection. An 18-g angiocath (Becton Dickinson Vascular Access, Sandy, UT) was inserted in the trachea and the lungs were lavaged with 20 ml of prewarmed (37°C) PBS containing penicillin (50 U/ml; GIBCO BRL, Gaithersburg, MD) and streptomycin (50 μg/ml; GIBCO BRL) in 1 ml vol. Viability was always >95% and lavaged cells were >97% macrophages as determined by differential counts of Wright-Giemsa stained cytospins.
Assays for macrophage-mediated killing of A. fumigatus conidia were adapted from the method of Schaffner (13). Alveolar lavage cells were pooled, centrifuged at 600 g, washed once with PBS, and resuspended in RPMI 1640 containing 2 mM L-glutamine (GIBCO BRL), 50 U/ml penicillin, 50 μg/ml streptomycin, and 10% fetal bovine serum (Sigma Chemical Co., St. Louis, MO) at a concentration of 2.5 × 106 cells/ml. Freshly isolated conidia were suspended at a concentration of 12.5 × 106 conidia/ml in PBS and combined with an equal volume of macrophage cell suspension (1:5 macrophage/conidia). Macrophage/conidia mixtures were tumbled at ∼10 rpm for 45 min at 37°C, layered over 2 ml Histopaque-1119 (Sigma Chemical Co.), and centrifuged at 700 g for 20 min at 4°C. Interface cells were removed, washed once in PBS, and resuspended at 1 × 105 cells/ml. 1-ml aliquots were placed in polypropylene tubes (Falcon #2059; Becton Dickinson Labware) in triplicate and incubated for 0, 6, or 20 h at 37°C. Cells were lysed by addition of 9 ml distilled water containing 1% Triton X-100 (Sigma Chemical Co.) with vortexing for 3 min. The number of colony-forming units recovered was counted from serial dilutions of lysates plated on Sabouraud agar and incubated for 48 h at 37°C. Counted plates contained between 5 and 50 conidia. The number of viable conidia recovered at 6 h from control tubes containing no macrophages was not significantly different from the number of conidia recovered at 0 h.
Experimental Infection with A. fumigatus.
Mice were infected with A. fumigatus conidia as previously described (15). The trachea was exposed, a 24-g angiocath (Becton Dickinson Vascular Access) was inserted, and the inoculum was instilled in 35 μl of sterile saline containing 5% colloidal carbon (Eberhard Faber, Inc., Lewisburg, TN) to allow localization of the inoculum in each lung. The challenge dose was confirmed by both quantitative culture of homogenized lung tissue obtained from at least two wild-type mice 1–2 h after instillation and plate culture of the inoculum suspension. As prophylaxis against secondary bacterial infection, mice were given 1.25 mg ceftriaxone (Rocephin; Hoffman-La Roche, Nutley, NJ) immediately after infection, and again 24 h later, followed by oral tetracycline (5 mg/ml in drinking water) for the remainder of the experiment. Mice were examined daily and killed by halothane overdose when symptoms suggested advanced disease (lethargy, hunched posture, ruffled fur, and labored respirations) or at 2–3 wk. Lungs were removed and fixed in neutral buffered formalin for histologic examination of paraffin-embedded sections obtained from carbon-stained regions of lung. Sections were stained with hematoxylin and eosin for assessment of pathological changes or Grocott methamine silver for assessment of hyphae. Histologic sections were analyzed in regions containing colloidal carbon, which is a marker for regions receiving conidia. Mice were scored as positive for Aspergillus infection if they had pathological indications of disease, e.g., abscesses, extensive bronchopneumonia, or inflammatory nodules, on gross or histologic examination.
Administration of Sterile A. fumigatus Hyphal Cell Walls.
Mice were prepared as described above except that, instead of A. fumigatus conidia, 5 μg of sterile cell wall material isolated from A. fumigatus hyphae (see below) was administered in 35 μl of sterile saline containing 5% colloidal carbon. This dose was chosen after pilot experiments demonstrated that this dose produced a mild, selflimited, pneumonia with no mortality in wild-type mice. Higher doses from 50 to 500 μg caused severe acute pulmonary inflammation and occasional deaths in wild-type mice. Except where noted, at least five mice were used per experiment. Mice were given antibiotic prophylaxis as described above. Mice were killed at 24 h, 72 h, 1, 3, and 6 wk by halothane overdose, and lungs were prepared as described above for histologic examination. Peripheral blood counts were performed as described (19).
A. fumigatus hyphal cell walls were prepared from an overnight culture grown on Sabouraud dextrose agar at 37°C. The mycelia were scraped from a culture dish and suspended in 50 ml of PBS. The hyphae were washed five times in 50 ml of PBS, and then autoclaved to sterilize them. Sterilized hyphae were dried, frozen in liquid nitrogen, and ground to a fine powder with a mortar and pestle. The powder was washed three times in PBS, and then sonicated for 10 min at a setting of 100 with a 50% duty cycle (Vibra-cell; Sonics & Materials Inc., Danbury, CT). The resulting suspension was washed with PBS and centrifuged at 15,700 g to pellet. The pellet was lyophilized, weighed, and resuspended at a concentration of 3 mg/ml in double-distilled water. Sterile technique and sterile solutions were used throughout the procedure. Cultures of the final hyphal preparation for bacterial and fungal growth were negative.
Morphometric Quantitation of Lung Inflammatory Infiltrate.
The number of inflammatory cells (PMNs, large mononuclear cells, and small mononuclear cells) were counted in randomly selected alveoli containing colloidal carbon as previously described (20). Data were expressed as number of cells per 100 alveoli. Groups were compared using a two-tailed Student's t test for normally distributed data or a Mann-Whitney test for nonparametric data.
RNA Isolation and Northern Analysis.
Lungs from untreated wild-type and X-CGD mice were removed and frozen immediately in liquid nitrogen then stored at −70°C. Lungs from mice given sterile A. fumigatus hyphae were removed 24 h, 72 h, or 1 wk after treatment and stored similarly. Lung tissue was ground to a powder with a mortar and pestle chilled with liquid nitrogen, dissolved in Solution D (4 M guanidine thiocyanate, 25 mM sodium citrate, 0.5% N-lauryl sarcosine, and 0.1 M 2-mercaptoethanol; all from Sigma Chemical Co.) and total RNA extracted as described (21). 10 μg of RNA was run on a 1% agarose/formaldehyde gel and transferred to Magnacharge nylon membranes (Micron Seps Inc., Westboro, MA) according to the manufacturer's instructions. Radiolabeled cDNA probes were prepared by random priming (Prime-a-Gene kit; Promega Corp., Madison, WI) according to the manufacturer's instructions. The murine IL-1β cDNA was provided by Dr. Patrick Gray (Genentech, Inc., South San Francisco, CA), the TGFβ1 cDNA by Dr. Rik Derynck (Genentech, Inc.), and the JE (murine monocyte chemotactic protein-1), KC (murine homologue of the human groα gene), and TNF-α cDNAs were from the American Type Culture Collection. To correct for differences in loading, the Northern blots were probed for β-actin. Differences in expression were quantitated by densitometry (GS-700 Densitometer; Bio Rad Labs., Hercules, CA) and were normalized to β-actin expression.
Results
Killing of A. fumigatus Conidia by Alveolar Macrophages from X-CGD Mice.
To evaluate the role of phagocyte-derived oxidants in resistance to the conidial form of A. fumigatus, we compared the ability of alveolar macrophages from wild-type and X-CGD mice to kill conidia in vitro. No differences were observed in the phagocytic potential of X-CGD versus wild-type macrophages as measured by the phagocytic index, i.e., the ratio of ingested conidia to macrophages, or the percentage of macrophages phagocytosing at least one conidia (Table 1). 6 h after phagocytosis, the majority of ingested conidia in both wild-type and X-CGD macrophages had been killed, with a similar percentage of conidia killed in cultures containing wild-type macrophages (86 ± 18%) as were killed in cultures containing macrophages from X-CGD mice (92 ± 10%). By 20 h of incubation, both wild-type and X-CGD mice had effected more than a log reduction in viable conidia. However, we are unable to draw any firm conclusion regarding macrophage killing at this time point since control cultures containing no macrophages also show a similar loss in viable conidia, presumably through germination and subsequent adherence and aggregation of hyphae.
Table 1.
Anticonidial and Phagocytic Potential of Wild-type and X-CGD Alveolar Macrophages
| Experimental group | Phagocytic index | Percent phagocytosis | Percent of conidia killed | |||
|---|---|---|---|---|---|---|
| Wild type | 2.2 ± 0.3 | 61 ± 2 | 86 ± 18 | |||
| X-CGD | 2.1 ± 0.4 | 55 ±13 | 92 ± 10 |
Alveolar macrophages were allowed to ingest conidia for 45 min at 37°C (1:5 macrophage to conidia ratio), and then incubated an additional 6 h at 37°C. Percent killing of ingested conidia was then determined by lysing macrophages and plating in at least triplicate cultures on Sabaroud agar. Phagocytic index (the ratio of ingested conidia to macrophages) and percent phagocytosis (the percentage of macrophages ingesting at least one conidia) were determined by counting at least 200 cells in Wright-Giemsa stained cytospins. Data are expressed as mean ± SD of two independent experiments using alveolar macrophages pooled from three to five mice per experiment.
Experimental Infection with A. fumigatus.
Wild-type and X-CGD mice were challenged with A. fumigatus conidia at doses ranging from 1 × 105 to 48 per animal, as administered by intratracheal instillation. No mortality was observed in wild-type mice, even at the highest dose tested. In contrast, pulmonary disease occurred in all X-CGD mice after Aspergillus exposure, with 24 of the 28 mice dead or moribund within 11–21 d due to progressive lung disease (Fig. 1). X-CGD mice given 1 × 105 conidia developed a rapidly progressing and uniformly fatal bronchopneumonia with all mice dead or moribund by the fourth day after infection, similar to what was previously observed with exposure to 3 × 106 conidia (15). Administration of lower doses resulted in a longer asymptomatic period and survival of a small number of mice (Fig. 1).
Figure 1.

Survival curve of X-CGD mice receiving from 1 × 105 to 48 A. fumigatus conidia. Mice were examined daily and the percent of mice surviving in each treatment group was plotted versus time from intratracheal administration of the indicated dose of conidia (day 0). ○, 1 × 105 conidia/mouse (n = 5); ▿, 7 × 103 conidia/mouse (n = 5); ▵, 1 × 103 conidia/mouse (n = 8); □, 540 conidia/mouse (n = 5); ⋄, 48 conidia/mouse (n = 5).
Fungal cultures of lung homogenates obtained at autopsy showed persistence of viable colony-forming units (cfu) of A. fumigatus in 27 out of 28 X-CGD mice after respiratory challenge (Table 2). Only one X-CGD mouse, who was studied at 21 d after exposure to 48 spores, did not have fungus cultured from lung tissue, although both abscesses and chronic inflammatory lesions with rare hyphal forms visible by silver stain were seen in lung sections prepared from this mouse (not shown). In contrast, very few, if any, viable A. fumigatus cfu were cultured from lungs obtained from wild-type mice, even after 3–4 d of challenge with 1 × 105 conidia (Table 2).
Table 2.
Culture of A. fumigatus from Lung Tissue after Intratracheal Administration of Conidia to X-CGD and Wild-type Mice*
| Conidia dose | Duration of study (days after conidia challenge) | Wild-type mice | X-CGD mice | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean cfu‡/lung | Range | Mean cfu‡/lung | Range | |||||||
| 100,000 | 3–4 | 35 | 0–128 (n = 6) | 22,000 | 2,000–40,000 (n = 4) | |||||
| 7,000 | 3–9 | Not done | 8,500 | 400–16,000 (n = 5) | ||||||
| 1,000 | 7–11 | 100 | 0–200 (n = 2) | 3,800 | 200–12,000 (n = 8) | |||||
| 540 | 8–11 | 0 | n/a (n = 3) | 870 | 60–4,000 (n = 5) | |||||
| 48 | 4–21 | Not done | 225 | 0–680 (n = 5) | ||||||
Lungs were homogenized in 1 ml PBS per lung and serial dilutions were plated on Sabouraud's media in triplicate. Colonies were counted from plates containing 10–50 colonies.
cfu detected by plate culture.
In agreement with previous studies (8, 15), the histologic appearance of lungs from wild-type mice challenged intratracheally with 1 × 105 conidia and killed 3 d after infection, appeared either normal or showed only a mild peribronchial pneumonia (data not shown). No hyphae were visible in silver-stained sections (data not shown).
Gross and histologic abnormalities after A. fumigatus respiratory challenge were observed in the lungs of all X-CGD mice at all doses of conidia studied. X-CGD mice that died or were moribund within the first week after Aspergillus challenge had similar pathology. Histologic findings, which included extensive peribronchiolar and alveolar inflammation consisting of neutrophils, necrotic debris, and edema, were similar to that previously described for X-CGD mice challenged with 3 × 106 conidia (not shown) (15). In X-CGD mice that survived beyond 1 wk, chronic inflammatory changes became more evident. Areas of almost complete neutrophil consolidation surrounded by a more diffuse mononuclear cell infiltrate and edema were a common finding (Fig. 2 a), as was the formation of wellcircumscribed abscesses (not shown). Intact hyphae, as visualized by silver staining, were detected in all X-CGD mice challenged with Aspergillus. Regions with an extensive neutrophil infiltrate typically contained numerous hyphae (Fig. 2 b), while areas of chronic inflammation had few hyphae (not shown). However, occasional regions with numerous focal collections of neutrophils did not contain detectable hyphae (Fig. 2 c). In X-CGD mice who survived for more than 2 wk after Aspergillus challenge, chronic inflammatory lesions were typically distributed in a nodular pattern and were comprised largely of mononuclear cells (Fig. 2 d) along with other cell types frequently associated with granulomatous inflammation including large, highly vacuolated macrophages (foam cells; not shown) and giant cells (Fig. 2 e). Neutrophils were often present in the centers of these nodules (Fig. 2 e), and occasionally, necrotic debris (not shown).
Figure 2.
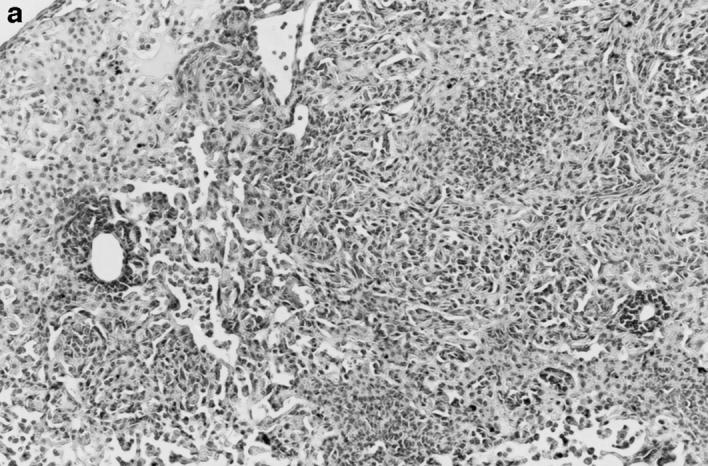


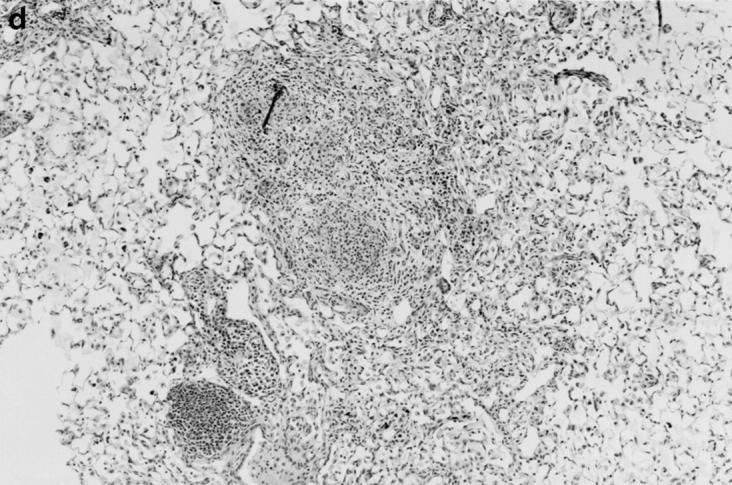

Pulmonary disease in X-CGD mice after administration of A. fumigatus conidia. (a) Lung tissue obtained 11 d after challenge with 540 A. fumigatus conidia and stained with hematoxylin and eosin. Note areas of neutrophil inflammation surrounded by mononuclear cells (original magnification of 100). (b) Grocott methamine stain of lung from X-CGD mouse with neutrophil inflammation; note branching filamentous hyphae (original magnification of 100). (c) Grocott methamine silver staining of the same tissue shown in a with no visible hyphae (original magnification of 100). (d) Lung tissue obtained 21 d after challenge with 48 conidia and stained with hematoxylin and eosin. Low power view of a representative inflammatory lesion showing nodular region of inflammation and destruction of underlying lung tissue (original magnification of 100). (e) Higher power view of chronic inflammatory lesion consisting of neutrophils, epithelioid macrophages, and giant cells (original magnification of 400).
Granulomatous Inflammation in X-CGD Mice Elicited by Sterile A. fumigatus Hyphae.
The observation of intense acute and chronic inflammation in X-CGD mice with no or small numbers of hyphae (Fig. 2, a and c) led us to speculate that the defect in phagocyte oxidant production was associated with a prolonged inflammatory response that was independent of active fungal infection. To separate the effects of persistent infection in X-CGD mice from the inflammatory response to Aspergillus, we administered sterile hyphal fragments to wild-type and X-CGD mice.
Striking differences in the appearance of the alveolar inflammatory infiltrate in wild-type versus X-CGD mice were evident even during the first 72 h after instillation of hyphae. At 24 h, an intense neutrophil alveolar infiltrate was seen in X-CGD mice, with foci of neutrophils often found in tight clusters around an eosinophilic center, with many cells assuming a spindle-shaped orientation towards the center of the inflammatory focus (Fig. 3 a). In contrast, in wild-type mice, the predominantly neutrophilic inflammatory infiltrate was diffusely scattered among alveoli without this focal pattern (Fig. 3 b). By 72 h, increased numbers of large mononuclear cells, debris, and a fibrinous exudate were seen in wild-type lung samples, again in a diffuse distribution (not shown). In X-CGD lung, focal accumulations of neutrophils were still numerous and were often surrounded by mononuclear cells (not shown).
Figure 3.


Lung inflammation in wild-type mice and X-CGD mice 24 h after challenge with sterile A. fumigatus hyphae. Lung tissue was obtained 24 h after intratracheal administration of 5 μg of sterile hyphae and staining of representative sections with hematoxylin and eosin. (a) X-CGD lung with pneumonia and focal centers of neutrophil accumulation (original magnification of 400). (b) Lung from a wild-type mouse with comparatively mild, diffuse neutrophil infiltrate. (original magnification of 400).
Morphometric analysis of the inflammatory cell infiltrate elicited by intratracheal administration of sterile hyphal fragments was also performed to obtain a quantitative measure of the differences between wild-type and X-CGD mice. At both 24 and 72 h, the neutrophils were the predominant cell type in both X-CGD and wild-type alveoli (Fig. 4). However, X-CGD mice had almost five times the amount of neutrophils as wild-type controls. The number of mononuclear cells was also significantly higher (almost double) in X-CGD mice compared to wild-type mice 72 h after administration of hyphae (Fig. 4). No differences in the peripheral leukocyte counts were observed between wild-type and X-CGD mice at either of these time points (data not shown).
Figure 4.

Morphometric analysis of lung sections 24 and 72 h after challenge with sterile A. fumigatus hyphae. Lung tissue was obtained either 24 or 72 h after intratracheal administration of 5 μg of sterile hyphae. 100 randomly selected alveoli containing carbon were scored for numbers of neutrophils, large mononuclear cells, and small mononuclear cells. Data are expressed as mean ± SD. Open bars, wild-type mice; filled bars, X-CGD mice. *, significantly different from wild type (P <0.0005, t test); ‡, significantly different from wild type (P <0.05, t test); §, significantly different from wild type (P <0.005, t test).
One week after administration of 5 μg of sterile hyphal fragments, 11 out of 13 wild-type mice still had evidence of a mild, diffuse alveolar pneumonia, consisting of neutrophils and mononuclear cells (not shown). In two wildtype mice, a more severe chronic inflammatory response developed, characterized by extensive infiltration of alveoli with large mononuclear cells and a fibrinous exudate (not shown). In contrast, at 1 wk, 8 out of 8 X-CGD mice were found to have a distinctive granuloma-like inflammatory infiltrate consisting of neutrophil microabscesses surrounded by large mononuclear cells with an epithelioid morphology (not shown). By 21 d, the contrast between wild-type and X-CGD mice was even more striking. The pneumonia in 3 out of 3 wild-type mice had almost entirely resolved (Fig. 5 a), whereas 3 out of 3 X-CGD mice still had numerous inflammatory nodules (Fig. 5 b) which often become confluent (Fig. 5 c). These foci continued to show a characteristic granuloma-like structure, with a central area of neutrophils surrounded by epithelioid mononuclear cells (Fig. 5 d). Lungs obtained from 3 of 3 X-CGD mice 6 wk after administration of sterile hyphal fragments showed persistence of these inflammatory lesions. No evidence of hyphal growth was evident in Grocott methamine silver–stained sections (not shown), and cultures of lungs from X-CGD mice killed at 1 wk showed no growth of fungi and grew similar numbers of bacteria to that observed in wild-type mice (<1 × 104 cfu/lung).
Figure 5.
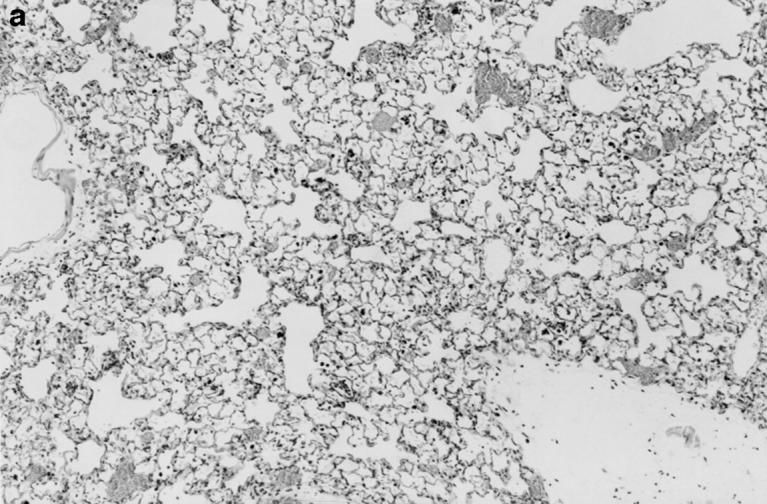



Inflammatory response in wild-type and X-CGD mice lungs 21 d after challenge with sterile A. fumigatus hyphae. Lung tissue was obtained 21 d after intratracheal administration of 5 μg of sterile hyphae and stained with hematoxylin and eosin. (a) Representative section of lung from a wild-type mouse. Inflammation has largely resolved, although some residual edema and peribronchiolar inflammation remain (original magnification of 100). (b) Lung from X-CGD mouse with scattered nodular inflammatory foci (original magnification of 100). (c) Lung from X-CGD mouse with almost complete consolidation and destruction of lung parenchyma (original magnification of 100). (d) Higher power of area in 6 b, showing central neutrophil microabscess and surrounding epithelioid cells (original magnification of 400).
Expression of Cytokines in Lungs after Intratracheal Administration of Sterile A. fumigatus Hyphae.
To determine if the differences in the inflammatory response to sterilized A. fumigatus hyphae between X-CGD and wild-type mice observed by histologic examination were associated with differences in cytokine expression, we examined Northern blots of total lung RNA from X-CGD and wild-type mice using probes for several proinflammatory mediators, as well as TGFβ1, a potent antiinflammatory cytokine (22).
Of the cytokines examined, IL-1β, TNF-α, JE, and KC showed very low, if any, levels of expression in untreated mice, with no significant differences observed between X-CGD and wild-type mice (Figs. 6 and 7). TGFβ1 expression was detectable in untreated mice at similar levels in wild type and X-CGD (Figs. 6 and 7). The most striking differences in mRNA expression after instillation of hyphae were observed with the proinflammatory cytokines IL-1β and TNF-α. Although markedly increased in both X-CGD and wild-type mice by 24 h after instillation, the expression of IL-1β and TNF-α in X-CGD mice was nearly 5- and 8.5-fold, respectively, higher than levels seen in wild-type animals (Fig. 7, a and b). By 72 h, expression of both cytokine mRNAs had dropped nearly to baseline levels in wild-type mice, while expression in X-CGD mice was even higher than at 24 h and remained persistently elevated for at least a week (Fig. 7, a and b). In addition, we also examined expression of two chemokines, KC and JE. KC, the murine groα homologue, binds to the murine orphan IL-8 receptor and is a potent neutrophil chemoattractant and activator (23), while JE, the murine homologue of monocyte chemoattractant protein-1 has similar activity for monocytes (24). Expression of KC and JE peaked at 24 h in both wild-type and X-CGD mice, and then declined (Fig. 6). Expression of both KC (Fig. 7 c) and JE (data not shown) were significantly higher at 24 h in X-CGD mice relative to wild-type controls, and KC remained significantly elevated in X-CGD versus wild-type mice at 72 h and 1 wk (three- to fivefold, respectively). TGFβ1 was significantly elevated in X-CGD mice relative to wild-type controls at 24 h, but not at later time points (Fig. 7 d).
Figure 6.

Northern analysis of total lung RNA in wild-type and X-CGD mice receiving sterile A. fumigatus hyphae. Lung tissue was obtained from wild-type (WT) and X-CGD (CGD) mice which were untreated (Control) or given 5 μg of sterile hyphal fragments by intratracheal administration. Mice given hyphal fragments were killed either at 24 h, 72 h, or 1 wk after instillation and the lungs removed and total RNA isolated as described in Materials and Methods. Blots were sequentially probed with the indicated cDNAs.
Figure 7.

Densitometric analysis of cytokine expression in wild-type and X-CGD mice receiving A. fumigatus hyphae. Autoradiographs were scanned and optical density calculated after background subtraction. Optical density was expressed as arbitrary density units and then normalized to β-actin expression. Data expressed as mean ± SD. (a) IL1β; (b) TNF-α; (c) KC; (d) TGFβ1. Open bars, wild-type mice; filled bars, X-CGD mice. *, significantly different from wild type (P <0.05, Mann-Whitney U test); ‡, significantly different from wild type (P <0.05, t test); §, significantly different from wild type (P <0.0005, t test); ∥, significantly different from wild type (P <0.005, t test).
Discussion
In this study, we have investigated the host response to A. fumigatus in mice that lack phagocyte respiratory burst oxidase activity due to targeted disruption in the X-linked gene for gp91phox. A. fumigatus is an opportunistic fungal pathogen that accounts for ∼15% of infections that occur in patients with CGD, typically causing what can be life-threatening pneumonias (25). Previous work in our laboratory has demonstrated that X-CGD mice develop an invasive, necrotizing bronchopneumonia after respiratory challenge with large numbers of A. fumigatus conidia (15). We now show that exposure to as few as 50 conidia can establish pulmonary infection in X-CGD mice. Furthermore, administration of sterilized Aspergillus hyphae produce chronic granulomatous inflammation in X-CGD mice, indicating that viable organisms are not a prerequisite for the development of chronic inflammatory lesions in CGD.
That even a small number of A. fumigatus conidia appear to be sufficient to cause infection in X-CGD mice illustrates the importance of respiratory burst oxidants in the host defense against this organism. We found that alveolar macrophages obtained from X-CGD mice ingested and killed conidia as efficiently as wild-type alveolar macrophages, consistent with previous studies showing that macrophage anticonidial activity is due to nonoxidative processes (13, 14). However, although X-CGD alveolar macrophages may function effectively as the first line of defense, it is likely that some conidia resist intracellular killing or are not ingested before germination. Phagocytosis of conidia in vitro by wild-type macrophages has been shown to be rapid and efficient both in the presence and absence of opsonins, but some manage to evade phagocytosis (11, 26), perhaps due to the release of soluble antiphagocytic substances from conidia (27). Hyphae germinating from conidia that escape macrophage-mediated killing would be efficiently killed by neutrophils in wild-type mice by oxidative mechanisms (11, 12) that are deficient in CGD. We found that viable fungus persisted for days to weeks in X-CGD mice, even after exposure to small numbers of conidia, and intact hyphae were frequently seen by silver-staining of lung tissue. As has been pointed out by others, precise quantitation of the load of viable Aspergillus is difficult since it has no reproducible infective unit (28). Individual colonies on plate culture can arise from either conidia, small pieces of hyphae, or mycelial clumps.
After exposure to small numbers of conidia, X-CGD mice survived for up to 21 d, which may simply represent a slower progression of infection or may reflect some degree of nonoxidative antihyphal activity, as suggested by the presence of abscesses containing few, or no, hyphae. One report has described a compensatory enhancement of nonoxidative antihyphal activity in monocytes from human CGD patients (29). This activity was found in fractions enriched for cytoplasmic granules, did not require exogenous peroxide or halide, and was inhibited by polyanions, suggesting granule-associated cationic proteins as potential mediators of this activity. Abscess formation may also help to slow the invasion of surrounding tissue by walling off growing hyphae.
It is important to note that differences in nonoxidative antimicrobial activity have been noted between human and murine phagocytes, so that results from these murine studies cannot necessarily be extrapolated to patients. For example, murine neutrophils lack defensins (30), a class of cationic antimicrobial peptides, but appear to have a higher capacity to generate nitric oxide (31). Nevertheless, our data suggest that exposure even to low doses of A. fumigatus spores may pose a serious risk of infection in X-CGD patients.
Although bacterial and fungal infections are an important component of the clinical syndrome associated with CGD, complications arising from granulomatous inflammation can also cause considerable morbidity (4). It has long been postulated that the granulomatous manifestations of this disorder reflect an intrinsic defect in the resolution of inflammatory responses (4). However, a requisite role of the respiratory burst in inflammation outside of its function in microbial killing has yet to be clearly defined. The production of superoxide results in an increase in pH within the phagocytic vacuole that may be important for optimizing the digestive activity of granule proteins released into this compartment (6, 32). Abnormal vacuolar pH regulation in CGD phagocytes could hence lead to defective digestion of ingested microbial products or debris and a prolongation of the inflammatory process. Respiratory burst oxidants have also been implicated in the inactivation of chemoattractant factors (33, 34) and in the inactivation of proteinase inhibitors and activation of latent metalloproteinases (35). It has also been noted that oxygen radicals satisfy many of the requirements for second messengers (36, 37), and that some transcription factors appear to be regulated by their redox state (38), suggesting that oxidants may have direct effects on gene expression. For example, oxidant stress may upregulate interleukin-8 expression (39).
Abnormal inflammatory responses in response to injury have previously been described in both murine and human CGD. In the initial response to intraperitoneal injection of sterile thioglycollate, both gp91phox −/− (X-CGD) and p47phox −/− (autosomal CGD) mice exhibit an initial increase in the steady-state number of peritoneal exudate neutrophils that is ∼twofold higher than that observed for wild-type mice (15, 40). In X-CGD mice, the percentage of exudate neutrophils declines with kinetics that are comparable to those observed in wild-type mice, but absolute neutrophil numbers remain persistently elevated over levels seen in exudate obtained from wild-type controls (15). Similarly, in human X-CGD patients, the percentage of neutrophils measured with the Rebuck skin window technique is abnormally elevated from 8 to at least 24 h after abrasion (41). Persistent pulmonary inflammation has been reported in a CGD patient after infection with A. fumigatus. In this case, amphotericin treatment of biopsy proven A. fumigatus produced an initial clinical response. Pulmonary function subsequently deteriorated, and a second lung biopsy showed evidence of a sterile interstitial pneumonitis, which was successfully treated with corticosteroids (42). However, chronic inflammatory changes have not been observed in the lungs of X-CGD mice surviving a single episode of either grampositive or gram-negative pneumonia (Dinauer, M., M. Gifford, and C. Doerschuk, manuscript in preparation). These studies suggest that inflammatory responses to only some inciting stimuli may be abnormal in CGD.
In the present study, we found that pulmonary administration of sterilized A. fumigatus hyphae produced an altered inflammatory response in X-CGD mice relative to wildtype controls. This is the first experimental evidence that the absence of phagocyte respiratory burst oxidants can be associated with the development of chronic inflammatory lesions even without an inciting bacterial infection. Our studies also suggest that the underlying mechanisms responsible for the pathologic response to sterilized hyphae in X-CGD mice include aberrant production of proinflammatory cytokines. In wild-type mice, sterilized hyphae produced a self-limited pneumonia, with development of a neutrophil-rich alveolar infiltrate and increased expression of IL-1β, TNF-α, and the chemokine KC, a neutrophil chemoattractant (43) during the first few days of the response. In X-CGD mice, these early responses were markedly exaggerated, and expression of IL-1β, TNF-α, and KC remained elevated one week after administration of sterilized hyphae. At one week after hyphae administration, X-CGD mice had developed a granulomatous, neutrophilcontaining alveolar infiltrate that persisted for at least 6 wk. Expression of TGFβ1, a potent antiinflammatory cytokine (22), was similar in both wild-type and X-CGD mice, suggesting that the defect in the inflammatory response is not due to an inability to generate appropriate antiinflammatory responses. However, the net balance of cytokine expression in the lungs of X-CGD mice in response to sterilized hyphae appears to be tipped towards the proinflammatory side of the equation, which could promote a continued inflammatory response rather than its resolution.
A prolonged period of neutrophil recruitment and inflammation could also be indicative of the relative persistence of A. fumigatus cell wall material as an inflammatory stimulus in X-CGD mice compared with thioglycollate, localized skin injury, or bacteria. Chitin and glucans are major structural components found in Aspergillus hyphae (44) and have also been shown to potentiate macrophage activation (45, 46). Mechanisms which eliminate fungal cell wall components in vivo are not well understood, but may be dependent on appropriate pH regulation of the phagocytic vacuole or still undefined processes that require an intact respiratory burst. However, it is not clear how a delayed digestion of fungal material in X-CGD could explain the marked increase in inflammatory cell numbers and cytokine expression observed during the early phases of the response in X-CGD mice.
A final comment relates to the chronic pulmonary lesions elicited in X-CGD mice by respiratory exposure to A. fumigatus, which are a mixture of neutrophilic and granulomatous inflammation. Granulomas have classically been loosely grouped into two major types, immune or hypersensitivity granulomas which are characterized by accumulation of epithelioid cells, lymphocytes, and plasma cells in response to a persistent antigen that induces a delayed-type hypersensitivity response, and foreign body granulomas which typically lack lymphocytes and are formed in response to nonimmunologically active, but persistent agents (47). The chronic lesions seen in X-CGD mice, although resembling granulomas in certain aspects such as persistence and the accumulation of epithelioid macrophages, are atypical in that they frequently include foci of neutrophils and debris, and are hence perhaps better described as granulomatous microabscesses. Pathology similar to that observed in X-CGD mice has previously been described in patients with CGD, particularly in association with fungal infections (2, 5).
In summary, our data demonstrate that infection of X-CGD mice with A. fumigatus can occur even with administration of as few as 50 conidia via the respiratory route, consistent with the high frequency of A. fumigatus infection seen in CGD patients. Administration of either low doses of conidia or sterile fungal cell walls to X-CGD mice produced chronic lung inflammation, often with granulomatous features. An understanding of how sterilized hyphal fragments produce chronic inflammatory lesions in the X-CGD mouse model may shed light on why chronic inflammation is a frequent complication of human CGD and the role played by respiratory burst oxidants outside of its function in host defense. Future studies will focus on identifying fungal mediators of chronic inflammation, clearance of fungal debris, and further investigation on the role of proinflammatory and antiinflammatory cytokines in response to pulmonary injury in X-CGD mice. This model of Aspergillus infection and chronic inflammation should also provide an excellent means to test the efficacy of various antifungal, antiinflammatory, and immunomodulatory strategies in treating CGD.
Footnotes
The authors would like to thank Attilio Orazi and Karla John for preparation of histology sections and Jim Cumming for assistance in mouse colony maintenance.
This work was supported by National Institutes of Health grant R01 HL52565 and a Biomedical Research grant from the Arthritis Foundation.
1 Abbreviations used in this paper: cfu, colony-forming units; CGD, chronic granulomatous disease.
References
- 1.Dinauer MC. The respiratory burst oxidase and the molecular genetics of chronic granulomatous disease. Crit Rev Clin Lab Sci. 1993;30:329–369. doi: 10.3109/10408369309082591. [DOI] [PubMed] [Google Scholar]
- 2.Carson MJ, Chadwick DL, Brubaker CA, Cleland RS, Landing BH. Thirteen boys with progressive septic granulomatosis. Pediatrics. 1965;35:405–412. [PubMed] [Google Scholar]
- 3.Johnston RB, Jr, McMurry J. Chronic familial granulomatosis. Am J Dis Child. 1967;114:370–378. doi: 10.1001/archpedi.1967.02090250068002. [DOI] [PubMed] [Google Scholar]
- 4.Gallin JI, Buescher ES, Seligmann BE, Nath J, Gaither T, Katz P. Recent advances in chronic granulomatous disease. Ann Intern Med. 1983;99:657–674. doi: 10.7326/0003-4819-99-5-657. [DOI] [PubMed] [Google Scholar]
- 5.Moskaluk CA, Pogrebniak HW, Pass HI, Gallin JI, Travis WD. Surgical pathology of the lung in chronic granulomatous disease. Am J Clin Pathol. 1994;102:684–691. doi: 10.1093/ajcp/102.5.684. [DOI] [PubMed] [Google Scholar]
- 6.Segal A. The NADPH oxidase and chronic granulomatous disease. Mol Med Today. 1996;2:129–135. doi: 10.1016/1357-4310(96)88723-5. [DOI] [PubMed] [Google Scholar]
- 7.Schaffner A, Douglas H, Braude A. Selective protection against conidia by mononuclear and against mycelia by polymorphonuclear phagocytes in resistance to aspergillus. J Clin Invest. 1982;69:617–631. doi: 10.1172/JCI110489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon DM, Polack A, Walsh TJ. Fungus dosedependent primary pulmonary aspergillosis. Infect Immun. 1989;57:1452–1456. doi: 10.1128/iai.57.5.1452-1456.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaffner A. Therapeutic concentrations of glucocorticoids suppress the antimicrobial activity of human macrophages without impairing their responsiveness to gamma interferon. J Clin Invest. 1985;76:1755–1764. doi: 10.1172/JCI112166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sidranksy HEV, Beede H. Experimental pulmonary aspergillosis. Arch Pathol. 1965;79:299–309. [PubMed] [Google Scholar]
- 11.Diamond RD, Clark RA. Damage to Aspergillus fumigatus and Rhizopus oryzaehyphae by oxidative and nonoxidative microbicidal products of human neutrophils in vitro. Infect Immun. 1982;38:487–495. doi: 10.1128/iai.38.2.487-495.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diamond, R.D., R. Krzesicki, B. Epstein, and J. Wellington. 1978. Damage to hyphal forms of fungi by human leukocytes in vitro. A possible host defense mechanism in Aspergillosis and Mucormycosis. <JNL>Am. J. Pathol. 91:313–328. [PMC free article] [PubMed]
- 13.Schaffner A, Douglas H, Braude AI, Davis CE. Killing of aspergillus spores depends on the anatomical source of the macrophage. Infect Immun. 1983;42:1109–1115. doi: 10.1128/iai.42.3.1109-1115.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Washburn RG, Gallin JI, Bennett JE. Oxidative killing of Aspergillus fumigatusproceeds by parallel myeloperoxidase-dependent and -independent pathways. Infect Immun. 1987;55:2088–2092. doi: 10.1128/iai.55.9.2088-2092.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollock JD, Williams DA, Gifford MAC, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 16.Arnow PM, Sadigh M, Costas C, Weil D, Chudy R. Endemic and epidemic aspergillosis associated with in-hospital replication of Aspergillusorganisms. J Infect Dis. 1991;164:998–1002. doi: 10.1093/infdis/164.5.998. [DOI] [PubMed] [Google Scholar]
- 17.Beaumont F, Kauffman HF, Sluiter HJ, De Vries K. Environmental aerobiological studies in allergic bronchopulmonary aspergillosis. Allergy (Cph) 1984;39:183–193. doi: 10.1111/j.1398-9995.1984.tb02623.x. [DOI] [PubMed] [Google Scholar]
- 18.Wilkes DS, Heidler KM, Bowen LK, Quinlan WM, Doyle NA, Cummings OW, Doerschuk CM. Allogeneic bronchoalveolar lavage cells induce the histology of acute lung allograft rejection, and deposition of IgG2a in recipient murine lungs. J Immunol. 1995;155:2775–2783. [PubMed] [Google Scholar]
- 19.Pollock J, Williams D, Gifford M, Li L, Du X, Fisherman J, Orkin S, Doerschuk C, Dinauer M. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocytes superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 20.Doerschuk CM, Winn RK, Coxson HO, Harlan JM. CD18-dependent and -independent mechanisms of neutrophil emigration in the pulmonary and systemic microcirculation of rabbits. J Immunol. 1990;144:2327–2333. [PubMed] [Google Scholar]
- 21.Kubo H, Morgenstern D, Quinlan W, Ward P, Dinauer M, Doerschuk C. Preservation of complement-induced lung injury in mice with deficiency of NADPH oxidase. J Clin Invest. 1996;97:2680–2684. doi: 10.1172/JCI118718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ulich TR, Songmei Y, Guo K, Yi ES, Remick D, del Castillo J. Intratracheal injection of endotoxin and cytokines. II. Interleukin-6 and transforming growth factor beta inhibit acute inflammation. Am J Pathol. 1991;138:1097–1101. [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI. Chemokine binding and activities mediated by the mouse IL-8 receptor. J Immunol. 1995;155:2158–2164. [PubMed] [Google Scholar]
- 24.Yoshimura T, Yukhi N, Moore SK, Appella E, Lerman MI, Leonard EJ. Human monocyte chemoattractant protein-1 (MCP-1). Full length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE . FEBS Lett. 1989;244:487–493. doi: 10.1016/0014-5793(89)80590-3. [DOI] [PubMed] [Google Scholar]
- 25.Curnutte, J. 1992. Disorders of granulocyte function and granulopoiesis. In Hematology of Infancy and Childhood, 4th ed. N. Oski, editor, W.B. Saunders Company, Philadelphia. 904–937.
- 26.Lehrer RI, Jan RG. Interaction of Aspergillus fumigatuswith human leukocytes and serum. Infect Immun. 1970;1:345–350. doi: 10.1128/iai.1.4.345-350.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robertson MD, Seaton A, Milne LJR, Raeburn JR. Resistance of spores of Aspergillus fumigatusto ingestion by phagocytic cells. Thorax. 1987;42:466–472. doi: 10.1136/thx.42.6.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lehmann P, White L. Chitin assay used to demonstrate renal localization and cortisone-enhanced growth of Aspergillus fumigatusmycelium in mice. Infect Immun. 1975;12:987–992. doi: 10.1128/iai.12.5.987-992.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diamond RD, Huber E, Haudenschild CC. Mechanisms of destruction of Aspergillus fumigatushyphae mediated by human monocytes. J Infect Dis. 1983;147:474–483. doi: 10.1093/infdis/147.3.474. [DOI] [PubMed] [Google Scholar]
- 30.Eisenhauer PB, Lehrer RI. Mouse neutrophils lack defensins. Infect Immun. 1992;60:3446–3447. doi: 10.1128/iai.60.8.3446-3447.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denis M. Human monocytes/macrophages: NO or no NO. J Leukocyte Biol. 1994;55:682–684. doi: 10.1002/jlb.55.5.682. [DOI] [PubMed] [Google Scholar]
- 32.Segal A, Geisow M, Garcia R, Harper A, Miller R. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature (Lond) 1981;290:406–409. doi: 10.1038/290406a0. [DOI] [PubMed] [Google Scholar]
- 33.Clark RA, Klebanoff S. Chemotactic factor inactivation by the myeloperoxidase-hydrogen peroxide-halide system. J Clin Invest. 1979;64:913–920. doi: 10.1172/JCI109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clark RA, Szot S, Venkatasubramanian K, Schiffman E. Chemotactic factor inactivation by myeloperoxidase-mediated oxidation of methionine. J Immunol. 1980;124:2020–2026. [PubMed] [Google Scholar]
- 35.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–375. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 36.Schreck R, Rieber P, Baeurle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO (Eur Mol Biol Organ) J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brumell JH, Burkhardt AL, Bolen JB, Grinstein S. Endogenous reactive oxygen intermediates activate tyrosine kinases in human neutrophils. J Biol Chem. 1996;271:1455–1461. doi: 10.1074/jbc.271.3.1455. [DOI] [PubMed] [Google Scholar]
- 38.Staal FJT, Roederer M, Herzenberg LA, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa β and transcription of human immunodeficiency virus. Proc Natl Acad Sci USA. 1990;87:9943–9947. doi: 10.1073/pnas.87.24.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeForge LE, Preston AM, Takeuchi E, Kenney J, Boxer LA, Remick DG. Regulation of interleukin-8 gene expression by oxidant stress. J Biol Chem. 1993;268:25568–25576. [PubMed] [Google Scholar]
- 40.Jackson SH, Gallin JI, Holland SM. The p47phox knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gallin JI, Buescher ES. Abnormal regulation of inflammatory skin responses in male patients with chronic granulomatous disease. Inflammation. 1983;7:227–232. doi: 10.1007/BF00917259. [DOI] [PubMed] [Google Scholar]
- 42.Quie PG, Belani KK. Corticosteroids for chronic granulomatous disease. J Pediatr. 1987;111:393–394. doi: 10.1016/s0022-3476(87)80460-2. [DOI] [PubMed] [Google Scholar]
- 43.Bozic CR, Kolakowski LF, Gerard NP, Garcia-Rodriguez C, von Uexkull-Gulenband C, Conklyn MJ, Breslow R, Showell HJ, Gerard C. Expression and biologic characterization of the murine chemokine KC. J Immunol. 1995;154:6048–6057. [PubMed] [Google Scholar]
- 44.Ruiz-Herrera J. Chemical components of the cell wall of Aspergillus species. Arch Biochem Biophys. 1967;122:118–125. doi: 10.1016/0003-9861(67)90130-0. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki K, Okawa Y, Hashimoto K, Suzuki S, Suzuki M. Protecting effect of chitin and chitosan in experimentally induced murine candidiasis. Microbiol Immunol. 1984;28:903–912. doi: 10.1111/j.1348-0421.1984.tb00746.x. [DOI] [PubMed] [Google Scholar]
- 46.Sherwood ER, Williams DL, McNamee RB, Jones EL, Browder IW, Di Luzio NR. In vitro tumoricidal activity of resting and glucan-activated Kupffer cells. J Leukocyte Biol. 1987;42:69–75. doi: 10.1002/jlb.42.1.69. [DOI] [PubMed] [Google Scholar]
- 47.Boros DL. Granulomatous inflammations. Prog Allergy. 1978;24:183–267. doi: 10.1159/000401230. [DOI] [PubMed] [Google Scholar]