

Abstract
Recent data using MHC/peptide tetramers and dimers suggests that the T cell coreceptors, CD4 and CD8, although important for T cell activation, do not play a direct role in facilitating T cell receptor (TCR) binding to multivalent MHC/peptide ligands. Instead, a current model proposes that coreceptors are recruited only after a stable TCR–MHC/peptide complex has already formed and signaled. In contrast, we show using multimeric class I MHC/peptide ligands that CD8 plays a critical (in some cases obligatory) role in antigen-specific TCR binding. T cell activation, measured by calcium mobilization, was induced by multimeric but not monomeric ligands and also showed CD8 dependency. Our analysis using anti-CD8 antibodies revealed that binding to different epitopes of CD8 can either block or augment TCR–MHC/peptide interaction. These effects on TCR binding to high-affinity agonist ligands were even more pronounced when binding to multimeric low-affinity ligands, including TCR antagonists, was studied. Our data have important implications for the role of CD8 in TCR binding to MHC/peptide ligands and in T cell activation. In addition, our results argue against the view that multimeric MHC/peptide ligands bind directly and solely to the TCR; rather, our data highlight a pivotal contribution of CD8 for this association.
Keywords: MHC/peptide tetramers, T cell activation, cytotoxic T lymphocyte, calcium, flow cytometry
Introduction
The T cell coreceptors CD4 and CD8 are known to bind class II and class I molecules directly and to be critical for development and activation of most T cells 1 2 3. However, the function of these molecules in TCR binding to MHC/peptide ligands is unclear. A key role would be anticipated, as coengagement of the TCR and coreceptor greatly enhance T cell responses 1 4, and direct participation of the CD8 coreceptor in TCR–MHC/peptide ligand interactions has been suggested 5. However, these studies did not determine if the coreceptor functions to facilitate TCR engagement with ligand, enhance signal transduction, or both. Indeed, the model in which CD4 and CD8 assist in forming the TCR–MHC/peptide interaction has been repeatedly challenged. Xu and Littman suggested that efficient CD4 coreceptor function required binding of associated Lck with previously assembled TCR–MHC/peptide complexes after TCR activation 6. A similar role has been proposed for CD8 based on analogous data 7. Recent studies using multivalent peptide/class II MHC ligands have allowed more direct measurement of MHC/peptide binding to the TCR. These reports all reached the same, surprising conclusion: CD4 plays no role in forming a stable TCR interaction with multimeric ligands 8 9 10. Previous work showed that CD4 is important in interactions with agonist but not antagonist MHC/peptide ligands, concluding that a stable TCR interaction induced by agonist ligands was a prerequisite for recruitment of the coreceptor rather than the other way around 11 12.
Less had been reported on the role of CD8 in TCR–MHC/peptide binding. Yet recent work using surface plasmon resonance assays failed to detect any enhancement by CD8 of TCR binding to specific class I MHC/peptide ligands 13. Furthermore, CD8 binding to class I MHC molecules is known to be enhanced by activation of tyrosine kinases through the TCR 7 14 15. These data have lead to the conclusion that CD8, like CD4, might participate in T cell responses only after the TCR has already stably bound and been activated by its ligand 7 13. Finally, several groups have reported analysis of specific T cell populations using MHC/peptide multimers in the presence of anti-CD8 antibodies 16 17 18 19. This shows that multimer binding can occur in the presence of anti-CD8 antibody, although, importantly, the effect of antibody on CD8 function was not determined. Thus, a composite model from these studies is that both CD4 and CD8 are recruited into the TCR–peptide/MHC complex only after it has stably assembled and had an opportunity to participate in signal transduction 6 7 8 9 10 11 13 20 21 22.
Recent studies on the role of coreceptors in T cell activation, however, suggest that there may be key differences between the functional role of CD4 and CD8. Using soluble class I MHC/peptide complexes, it was shown that calcium mobilization could be induced by monomeric class I/peptide complexes, providing that CD8 was available 23. In the absence of CD8, dimeric MHC/peptide ligand was necessary and sufficient to induce such activation 24. A different pattern emerged from analysis of TCR–class II MHC interactions. Boniface et al. showed that CD4 was critical for very early activation events (the acidification response) induced by MHC/peptide multimers but that even high concentrations of MHC/peptide multimers were unable to induce a sustained calcium flux response 10. Furthermore, even in the presence of CD4, MHC/peptide monomers failed to induce any class II–restricted T cell response 10. These data further support the model in which CD4 has a critical role for class II–restricted T cell activation and mediates this effect after TCR encounter with multimeric MHC/peptide ligand. But the data from Delon et al. suggests that CD8 may play a different role, potentiating the response to rare and/or low-affinity ligands 23. It is difficult to compare these systems, however, as the approach used by Delon et al. did not determine if CD8 facilitated binding of the MHC/peptide complex to the TCR or whether the effect of CD8 could be attributed purely to enhanced signal transduction.
To address these issues, we studied the role of CD8 in TCR binding and activation using soluble multimeric MHC/peptide ligands. In contrast to the models discussed above, we demonstrate here that CD8 is critical in class I/peptide multimer binding to TCR in two well defined, class I–restricted TCR-transgenic systems. Indeed, in one of these systems (OT-I), the coreceptor was not only involved in but absolutely required for significant TCR binding to multimeric MHC/peptide ligands. Furthermore, we found that class I multimers, but not monomers, were capable of inducing rapid calcium mobilization and that this response is dependent on CD8 in both TCR-transgenic systems studied. Finally, we showed that different antibodies against CD8 had dramatically different results on multimer binding and T cell activation. Specifically, although most anti-CD8 antibodies blocked multimer binding to the T cell, one antibody enhanced specific multimer binding. This enhancement was especially dramatic in the case of multimers containing low-affinity TCR ligands, including TCR antagonists. Thus, our data is in contrast with that of other groups, who propose that TCR binding to antagonist ligands is coreceptor independent.
Materials and Methods
Mice and Cells.
6–12-wk-old OT-I, OT-I recombination activating gene (RAG)-1−/−, and 2C mice were generated and maintained under specific pathogen–free conditions. Major lymph nodes were harvested, and a single-cell suspension was generated as previously described 25. The majority of LN cells in OT-I RAG-1−/− mice are CD8+OT-I TCR+, and these cells were used without further purification. For most staining experiments and all activation experiments involving normal OT-I and 2C mice, the cells were passed over a “CD8 Cellect” column (Cytovax), which removes B cells and CD4 cells, thus enriching the population for CD8+ and/or CD4−8− T cells.
Peptides and MHC Multimers.
The peptides OVAp (SIINFEKL), SIYp (SIYRYYGL), G4 (SIIGFEKL), and E1 (EIINFEKL) have been described previously 26 27 28 and were synthesized by Research Genetics.
Production of Kb/β2m (microglobulin)/peptide multimers followed standard protocols 16 18. Plasmids encoding Kb molecules with the BirA recognition sequence at the COOH terminus and human β2m molecules (gifts of J. Altman, Emory University, Atlanta, GA and E. Pamer, Yale University, New Haven, CT, respectively) were transformed and overexpressed in Escherichia coli. The synthesized proteins were purified from inclusion bodies, denatured, and mixed with the appropriate Kb binding peptide, and the mixture was allowed to renature in suitable oxidoreductive conditions over 48 h. After biotinylation using BirA (Avidity), the monomeric MHC/peptide complexes were purified via fast protein liquid chromatography (FPLC) on a Superdex 200 column (Pharmacia Biotech, Inc.). The efficiency of biotinylation was assessed by sequential immunoprecipitation with avidin-conjugated beads (Pierce Chemical Co.), followed by anti-Kb antibody (Y3)-conjugated CNBr Sepharose beads (Sigma Chemical Co.), and was routinely estimated to be >80% efficient using this method. After concentration of the appropriate-sized fractions, the biotinylated Kb/peptide complexes were mixed with streptavidin (SA)–PE (Molecular Probes, Inc.) or SA–allophycocyanin (PharMingen) at a 4:1 molar ratio. Multimers which eluted in the predicted range of tetramers were purified via a second round of FPLC size exclusion. However, in some experiments the unpurified multimer preparation was used for staining with no significant difference in staining profiles. Multimers were tested over a range of doses for flow cytometry and were typically used at 10 μg/ml.
Antibodies and Flow Cytometry.
T cells (5 × 105) were stained in 50 μl of FACS buffer (PBS, 1% FCS, and 3 mM azide) or RP-10 with 3 mM azide at the temperatures and times indicated in the figures. Typically, MHC multimers (10 μg/ml) and anti-CD8 antibodies (50 μg/ml unless stated otherwise) were added simultaneously. In Fig. 4, OT-I T cells were stained with OVA/Kb multimers for 2 h at 4°C, washed twice, and reincubated with FACS buffer alone or with saturating concentrations of anti-CD8 FITC-conjugated antibodies. The following anti-CD8α antibodies were used: 3.168 (rat IgM; reference 29), 53.6.7 (rat IgG2a; references 30 and 31) (PharMingen), and CT-CD8a (rat IgG2a; Caltag Labs.). The anti-CD8β antibody used was 53.5.8 (rat IgG1; references 30 and 31; PharMingen). Cells were analyzed using a Becton Dickinson FACSCalibur™. For Ca2+ flux measurements, the T cells were loaded with Indo-1AM at 37°C as described previously 32, washed, and then incubated on ice with or without FITC-conjugated anti-CD8 antibodies for 30–60 min. After short centrifugation, the cells were resuspended into media at 37°C and analyzed immediately on a FACSVantage™ instrument. After establishing a baseline for 1 min, monomeric or multimeric MHC/peptide complexes (to 10 μg/ml) or free OVAp (to 200 nM) was added. As the molecular mass of OVA/Kb–SA–PE multimers is ∼400 kD, 10 μg/ml represents a molar concentration of ∼25 nM for the multimer and 100 nM for the OVAp molecules within the multimer. Thus, the concentration of free OVAp added in these experiments was double that of OVAp included in the multimer. The cells were quickly mixed and then analyzed by flow cytometry for an additional 7–15 min, as indicated. Data were analyzed using both CELLQuest™ (Becton Dickinson) and FlowJo (TreeStar) software.
Figure 4.

CD8 effect on multimer dissociation. (A) OVA/Kb multimers were allowed to bind to OT-I RAG−/− cells for 2 h at 4°C in the presence or absence of the anti-CD8 antibodies, as indicated. (B) In the same experiment, OT-I cells that were stained with OVA/Kb multimers in the absence of anti-CD8 antibody as in A were washed twice and then incubated for an additional 2 h at 4°C with or without anti-CD8 antibodies as indicated.
Results
We sought to determine if CD8 participates in binding of multivalent MHC/peptide ligands to class I–restricted TCRs. OT-I and 2C are TCR-transgenic mouse strains that bear receptors specific for the mouse class I molecule Kb complexed with OVAp and SIY, respectively 26 27 28. Using standard procedures, Kb/peptide multimeric complexes were synthesized bearing each of these peptides 16 18. As direct CD8 binding to Kb molecules has been reported 33, it was important to determine if interaction of these multimers with CD8 T cells was dependent on the specificity of the TCR. Fresh T cells were isolated from the lymph nodes of OT-I and 2C transgenic mice and stained with MHC/peptide multimers for 1 h at 37°C in tissue culture media containing azide to block T cell activation (conditions derived from Crawford et al. [9]). These binding assays revealed fine TCR specificity in multimer binding, such that the 2C T cells bound the Kb/SIY multimer but not the OVA/Kb multimer, whereas the reciprocal pattern of binding was observed for OT-I cells (Fig. 1 A). This correlates with reports of functional assays using these same receptors and ligands 25. Double staining under the same conditions with the anti-CD8α antibody 53.6.7 revealed that specific multimer staining was preserved in the presence of this antibody. It is important for subsequent experiments to note that there is a population of TCR expressing CD8− T cells in 2C animals (Fig. 1 B). These cells are known to express the clonotypic TCR at levels similar to the CD8+ 2C cells (34 35 36; our data not shown). Both CD8+ and CD8− 2C cells stain with the Kb/SIY multimer, although staining intensities differ for these two populations (Fig. 1 B).
Figure 1.

Binding of MHC/peptide multimers is dependent on the specificity of the TCR. (A) Lymph node cells from 2C and OT-I mice were depleted of B cells and CD4+ cells and stained for 1 h at 37°C with the indicated PE-conjugated multimers (Mult.). The mean fluorescence intensity (MFI) of 2C cells was 401 when stained with the SIY/Kb multimer versus 4 using the OVA/Kb multimer. MFI for OT-I cells was 5 for the SIY/Kb multimer and 326 with the OVA/ Kb multimer. (B) Staining as in A, except these cells were stained with the anti-CD8α antibody 53.6.7 in addition to the indicated multimer. Numbers represent the percentages of cells in respective quadrants.
We next tested a panel of antibodies that recognize distinct epitopes on the CD8α and -β chains to determine their effects on the TCR–MHC/peptide interaction. In stark contrast to the results using the 53.6.7 antibody, we found that saturating concentrations of two anti-CD8α antibodies, 3.168 (Fig. 2 A) and CT-CD8a (data not shown), and the anti-CD8β antibody 53.5.8 (data not shown), showed almost total blockade of Kb/OVA multimer binding to OT-I cells (Fig. 2 A and data not shown). Furthermore, we noted that 53.6.7 actually enhanced binding over that of the multimer without anti-CD8 (Fig. 2 A). To further quantitate the role of CD8 in multimer binding, a titration of the anti-CD8 antibodies was performed. The enhancing effect of 53.6.7 and blocking by the other CD8 antibodies was titratable and covered a similar dose range (Fig. 2 B). Importantly, no staining of OT-I cells with the noncognate Kb/SIY multimer was observed, regardless of the anti-CD8 antibody used (Fig. 2a and Fig. b). This indicates that the anti-CD8 antibodies did not change the fine specificity of multimer binding. Also, these results were not influenced by the dose of multimer used, as similar enhancement or blockade was observed using higher or lower doses of the OVA/Kb multimer (data not shown). Furthermore, similar profiles of enhancement or blockade were observed using unconjugated or allophycocyanin-conjugated anti-CD8 antibodies, arguing against some artifact of FACS compensation (data not shown).
Figure 2.


Variable effect of CD8 antibodies on MHC class I multimer binding OT-I LN T cells. LN cells from an OT-I RAG-1−/− animal were stained with the indicated PE-conjugated multimer (Mult.) at 37°C for 2 h, with or without simultaneous staining with saturating amounts of the indicated FITC-conjugated anti-CD8 antibodies. (A) Cells were stained with OVA/Kb multimer (top) or SIY/Kb multimer (bottom), with no anti-CD8 antibody (open histogram), 53.6.7 (dotted histogram), or 3.168 (filled histogram). The effect of the antibodies CT-CD8a or 53.5.8 was similar to that of 3.168 for both multimers, and data using these antibodies is omitted for clarity. (B) The MFI of staining with the indicated multimer in the presence of titrated anti-CD8 antibodies.
We considered that staining the cells at 37°C could allow effects such as CD8 and/or TCR capping and internalization to occur. Furthermore, it is known that even high concentrations of azide do not prevent ligand-triggered TCR internalization at 37°C (37, 38, and our unpublished observations). To control for these phenomena, we compared staining performed entirely at 4°C in the presence of 3 mM azide. These conditions prevent antibody-induced internalization and TCR downregulation (37, 38, and our unpublished observations). As shown in Fig. 3, multimer binding was slightly improved with staining at 37 versus 4°C, as expected from Crawford et al. 9. However, the effects of the anti-CD8 antibodies were identical under both conditions: the antibody 53.6.7 slightly enhanced multimer binding (Fig. 3), whereas antibodies 3.168 (Fig. 3), CT-CD8a, and 53.5.8 (not shown) all blocked multimer binding completely at both temperatures. Similar results were obtained when the cells were stained for longer times at these temperatures (data not shown). These data indicate that the CD8 effects observed are not dependent on T cell activation or TCR/CD8 internalization. However, given the potential complications of TCR and/or CD8 internalization and signaling at higher temperatures, subsequent staining was performed at 4°C in the presence of 3 mM azide.
Figure 3.

Effects of temperature on multimer staining and effects of anti-CD8 antibodies. OT-I RAG-1−/− LN cells were stained at 4°C (1 h, left panels) or 37°C (1 h, right panels) with the SIY/Kb multimer (shaded) or OVA/Kb multimer (open) without anti-CD8 antibody (A) or with CD8 antibody 53.6.7 (B) or 3.168 (C). The block with 3.168 causes the OVA/Kb multimer to overlap the histogram for the negative control, SIY/Kb multimer.
Using this system, we could also study the dynamic nature of MHC/peptide multimer binding. As we know that the half-life of the OT-I TCR–OVA/Kb complex is relatively short, 39 the binding of individual “heads” of the OVA/Kb multimer to the TCR is expected to be dynamic rather than static, such that each head of the multimer dissociates and reassociates with TCRs over time. If CD8 was involved in this process, then anti-CD8 antibodies might affect the stability of prebound MHC multimers. To test this, we compared the effects of anti-CD8 antibodies on multimer binding to OT-I cells under two conditions: (a) when antibodies and OVA/Kb multimers were added simultaneously (Fig. 4 A) or (b) when OVA/Kb multimers were allowed to bind first and the cells were subsequently incubated with anti-CD8 antibodies (Fig. 4 B). In the absence of anti-CD8 antibodies at either step, multimer staining appears quite stable, decreasing only slightly in the second incubation (compare Fig. 4A and Fig. B). As expected, multimer staining was greatly decreased when the blocking anti-CD8 antibodies (3.168 or CT-CD8a) were added during multimer binding (Fig. 4 A), but there was also significant loss of multimer when these antibodies were added only in the second incubation (Fig. 4 B). This displacement effect could be observed kinetically, in that multimer binding was not reduced to the same extent after only 45 min (rather than 2 h) of incubation with the blocking anti-CD8 antibodies (data not shown). It is also important to note that the blockade of multimer binding was more efficient when multimer and anti-CD8 antibodies were added simultaneously rather than sequentially (compare Fig. 4A and Fig. B). In contrast to these results, the “enhancing” anti-CD8 antibody (53.6.7) did not cause loss of prebound multimer but, on the contrary, appeared to stabilize multimer staining at the level observed at the beginning of the second incubation (compare Fig. 4A and Fig. B). Together, these data support the model of a dynamic nature of multimer binding to the TCR and suggest that CD8 participates in both the initial association of the TCR with MHC/peptide multimer and the stability of this interaction.
As mentioned previously, 2C TCR–transgenic mice are interesting in that they develop a natural population of T cells that are positive for the 2C TCR yet are CD8− 34. Such T cells are functional, as they can respond to TCR ligand, albeit only at high doses 35 36. These results suggest that 2C cells may be relatively coreceptor independent, and hence we were interested in what role CD8 might play in multimer binding to the 2C receptor. All 2C cells stained specifically with the Kb/SIY multimer in the absence of anti-CD8 antibody, but the profile was bi-modal, with a multimerhigh and a multimerlow population (Fig. 5 A). The percentages of multimerlow and multimerhigh populations correlate with the percentage of CD8− and CD8+ cells, respectively (data not shown), suggesting that CD8 plays a role in multimer binding to the 2C. We confirmed a role for CD8 using anti-CD8 antibodies. Staining with 3.168, CT-CD8a, or 53.5.8 caused disappearance of the Kb/SIY multimerhigh population, this group of cells becoming multimerlow. In contrast, 53.6.7 did not cause this change in multimer staining (Fig. 5 A) and, in some experiments, slightly increased the staining on the Kb/SIY multimerhigh population. As in the case of OT-I cells, the staining using nonspecific multimer was not significantly affected by use of any anti-CD8 antibody (Fig. 5 B).
Figure 5.
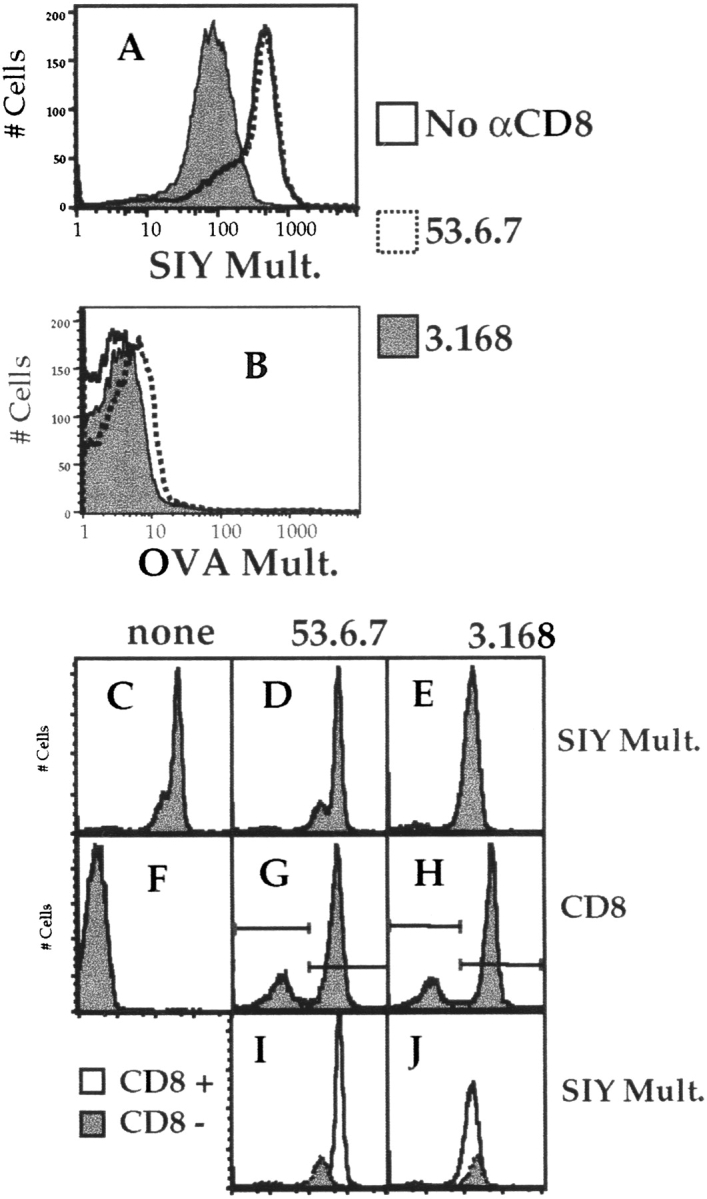
Role of CD8 in multimer binding to 2C cells. 2C LN cells were depleted of B cells/CD4+ T cells and stained for 2 h at 4°C with PE-conjugated SIY/Kb multimer (A) or OVA/Kb multimer (B), with no anti-CD8 antibody (open histogram), 53.6.7 (dotted histogram), or 3.168 (filled histogram). The effect of the antibodies CT-CD8a or 53.5.8 was similar to that of 3.168 for both multimers, and data using these antibodies is omitted for clarity. In a separate experiment, SIY/Kb multimer staining of 2C T cells was determined in the absence (C) or presence of the anti CD8 antibodies 53.6 (D) or 3.168 (E). F, G, and H show CD8/FITC staining for the cells in C, D, and E, respectively. The multimer staining of gated CD8− (shaded) and CD8+ (open) populations revealed with 53.6.7 (I) or 3.168 (J) is also shown.
More directly, we analyzed Kb/SIY multimer binding on both the 2C CD8+ and CD8− populations, identified using either 53.6.7 or 3.168 (Fig. 5C–J). Multimer binding to the CD8+ population was higher than to the CD8− population when 53.6.7 was used to reveal CD8, whereas CD8+ T cells stained with 3.168-bound multimer no better than CD8− 2C cells (Fig. 5C–J). In this experiment, there was a slight enhancement of SIY/Kb multimer staining of the CD8+ population when 53.6.7 was used (compare Fig. 5C and Fig. D). These results are thus analogous to the influence of anti-CD8 antibodies on OT-I TCR binding, with the main difference between the systems being the degree of multimer binding in the absence/blockade of CD8: negligible in the case of the OT-I receptor, but merely reduced in the case of multimer binding to 2C TCR.
Reports from Madrenas et al. and Hampl et al. indicate that TCR binding to TCR antagonist ligands is coreceptor (CD4) independent 11 12. To characterize the role of CD8 in binding TCR antagonists in our system, we used Kb/peptide multimers containing altered peptide ligands with known affinity for the OT-I TCR. The variant peptide G4 is a weak agonist/antagonist for OT-I, whereas E1 is an antagonist 26 32 40. The OT-I affinity for these ligands is known and matches their biological function, with the rank order of affinity being OVA>G4>E1 (reference 39 and Alam, S.M., and N.R.J. Gascoigne, personal communication). Staining OT-I cells with these Kb/peptide multimeric complexes revealed that the order of affinity matches the intensity of staining, such that multimer complexes containing OVA stained more intensely than those containing G4 and the E1 multimer stained only slightly (but reproducibly) above the negative control level (Fig. 6). That the rank order of staining mirrors TCR affinity is consistent with data from Crawford et al. 9. We next tested the effect of anti-CD8 antibodies on binding of these multimers. Once again, the “blocking” anti-CD8 antibodies 3.168 (Fig. 6), CT-CD8a, and 53.5.8 (not shown) totally negated binding of all multimeric ligands. In contrast, the enhancing effect of 53.6.7 was observed for all of the specific OT-I ligands and was extremely marked for the low-affinity ligand E1/Kb, bringing staining with this multimer well above the level of the control (SIY/Kb; Fig. 6). These data demonstrate that binding of multimers containing low-affinity MHC/peptide ligands is still strongly influenced by CD8 participation. Furthermore, these data indicate that use of the “enhancing” 53.6.7 antibody can significantly augment staining of a multimer containing an antagonist ligand (E1/Kb).
Figure 6.

Role of CD8 binding of multimeric altered peptide ligands. OT-I cells were incubated at 4°C for 2 h with Kb multimers containing OVA, SIY, G4, or E1 peptides in the absence of anti-CD8 antibodies (A) or in the presence of saturating amounts of 53.6.7 (B) or 3.168 (C). In the case of 3.168, the histograms with all the multimers overlap that of the negative control SIY/Kb multimer. Results are representative of four separate experiments.
Data from Delon et al. 23 indicated that monomeric MHC/peptide ligands could stimulate a Ca2+ flux provided that CD8 was available, whereas data from Boniface et al. 10 indicated that even multimeric MHC class II/peptide ligands fail to induce a sustained Ca2+ response. Hence, it was possible that our experiments using multimer staining would not be predictive of the capacity of these ligands to activate CD8 T cells. We thus wished to explore the capacity of our class I MHC/peptide monomers and multimers to stimulate naive T cells and study the role of CD8 in such stimulation.
As an early activation event that can easily be studied in real time, we focused on induction of Ca2+ mobilization measured by flow cytometry. Use of fluorochrome-labeled multimers allowed us to study multimer binding in real time and correlate this with Ca2+ mobilization. As shown in Fig. 7, OT-I T cells bound specific multimers rapidly (within seconds), and this slightly preceded the initiation of a Ca2+ flux response. The vast majority of OT-I T cells (82–91% over three experiments) participated in robust Ca2+ mobilization under these conditions, and the level of intracellular Ca2+ did not return to baseline over the time course studied. These data therefore indicate that the Ca2+ flux response induced by cognate MHC/peptide multimers is synchronous and sustained. In contrast to these results, nonspecific multimers neither bound nor induced Ca2+ mobilization (Fig. 7), and neither did the OT-I TCR antagonist E1/Kb multimer (data not shown).
Figure 7.

Density plot showing real time analysis of multimer binding and Ca2+ flux response. Indo-1AM–loaded OT-I RAG-1−/− LN cells were analyzed by FACS® for 1 min, at which time OVA/Kb (left panels) or SIY/Kb (right panels) PE-conjugated multimers (10 μg/ml) were added. Analysis of multimer binding (A) and Ca2+ mobilization (as reflected by changes in the fluorescence of the Indo-1 dye; (B) was determined for the same population of cells. Analysis of this and two other experiments indicates that 83–91% of OT-I cells mobilized Ca2+ in response to the OVA/Kb multimer.
Because the OT-I T cells themselves express Kb, it was possible that OVA peptide was released from the multimeric complexes and presented via T cell–T cell interactions. As a control for this, we added double the concentration of free OVA peptide used in the multimer sample. This resulted in no Ca2+ mobilization (Fig. 8 A), indicating that MHC/peptide multimers were responsible for the activation event. In contrast to previous reports in another class I MHC–restricted system 23, we saw no activation of Ca2+ flux by monomeric OVA/Kb (Fig. 8 A). We went further to test whether we could induce OT-I T cell activation by multimerization of the monomeric ligands “on-the-fly” by addition of SA–PE into the sample and following the response for a further 8 min. This approach showed a slight but noticeable rise in intracellular Ca2+ consistent with a presumably inefficient assembly of OVA/Kb multimers and subsequent OT-I activation (Fig. 8 A). The inefficiency of this response is to be expected, as multimerization takes several hours in our standard protocol, making the weak Ca2+ flux observed even more significant.
Figure 8.
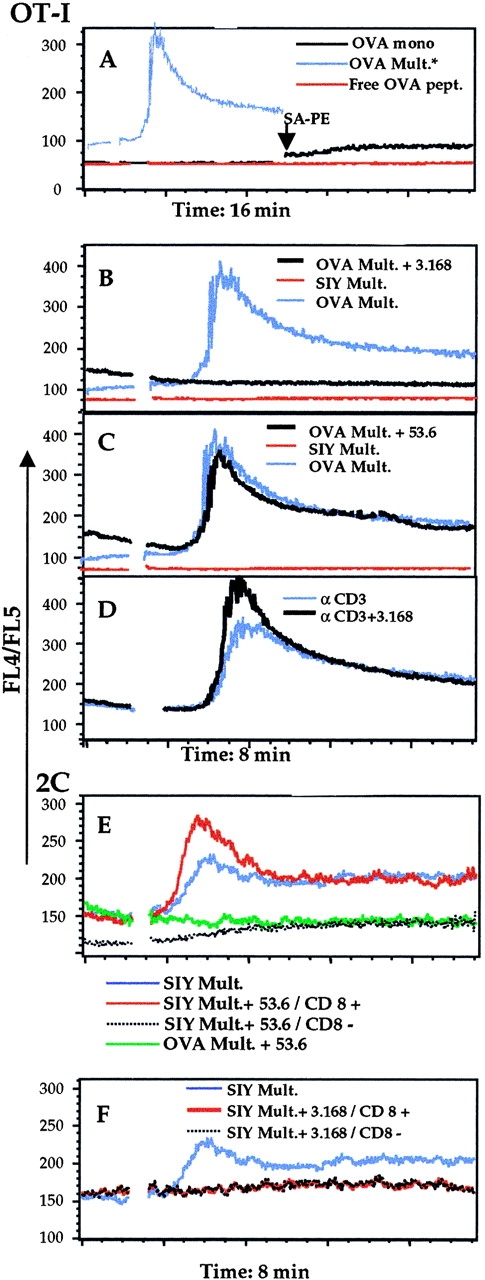
Ca2+ flux response in OT-I or 2C T cells induced by MHC/peptide monomers and multimers. (A) Ca2+ mobilization in OT-I RAG-1−/− LN cells treated with OVA/Kb multimers (blue line), free OVA peptide (red line), or OVA/Kb monomeric complexes (black line). In the case of the monomers, SA–PE was added at 8 min (arrow) and the sample reanalyzed for a further ∼8 min. The OVA/Kb multimer trace is taken from an 8-min time course and overlaid here as a representation of a responding population. (B and C) OT-I cells were pretreated with either 3.168 (B) or 53.6.7 (C) antibodies (black lines) before stimulation with OVA/Kb multimers or were exposed to SIY/Kb multimers (red lines) or OVA/Kb multimers (blue lines) in the absence of anti-CD8 antibodies. (D) In parallel, OT-I cells were stimulated by addition of anti-CD3 antibody 500.A2 (αCD3 antibody) plus cross-linking goat anti–mouse Ig, with (black line) or without (blue line) pretreatment with 3.168. 2C cells were stimulated with SIY/Kb or OVA/Kb multimers in the presence or absence of anti-CD8 antibodies 53.6.7 (E) or 3.168 (F). Response to the SIY/Kb multimer in the absence of any CD8 antibodies is shown in both panels (blue line). In E, the CD8+ (red line) and CD8− (black line) populations are delineated by 53.6.7. Exposure to OVA/Kb multimers in the presence of 53.6.7 (green line) served as a negative control in this response. In F, the CD8+ (black line) and CD8− (red line) populations are delineated by 3.168. All traces indicate the median of the responding population.
Further analysis of activation with multimer complexes investigated the role of CD8. Again, the OVA/Kb multimer induced a strong, sustained Ca2+ flux in OT-I cells that was not induced by the control (SIY/Kb) multimer (Fig. 8B and Fig. C). This flux was similar in magnitude and duration to that induced by cross-linked anti-CD3 antibody 500.A2 (Fig. 8 D). To study the role of CD8 in this process, we pretreated the cells with anti-CD8 antibody, either 3.168 or 53.6.7. Fluorochrome-labeled anti-CD8 antibodies and multimers were used so that staining of the responding T cell populations could be studied during the experiment. Activation of the OT-I cells was unaffected by prestaining with the 53.6.7 antibody, whereas activation was completely blocked using 3.168 (Fig. 8B and Fig. C). It was possible that pretreatment with 3.168 was inducing more than simple blockade of multimer binding. This was a special concern, as anti-CD8–treated cells frequently showed slight elevations in basal levels of intracellular Ca2+ not observed with unstained control populations (Fig. 8). However, pretreatment of OT-I cells with the “blocking” 3.168 antibody had no effect on the response to anti-CD3 (Fig. 8 D), which argues against a general inhibitory effect of the anti-CD8 antibody.
Similar experiments were used for 2C cells. We took advantage of the naturally occurring population of CD8− 2C cells. Anti-CD3 antibody cross-linking was able to induce Ca2+ flux in both populations of CD8+ and CD8− 2C cells (data not shown) and, as for OT-I cells, activation of 2C cells was observed only using the specific multimer (in this case SIY/Kb; Fig. 8 E). Intriguingly, from preliminary experiments it was evident that multimer induced activation failed to stimulate all of the 2C cells. To determine if there was a difference in the capacity of CD8+ and CD8− populations to respond to SIY/Kb multimers, we used the 53.6.7 antibody to separate these subsets. The CD8− population failed to respond to SIY/Kb multimer stimulation, whereas the CD8+ population responded well (Fig. 8 E). In the presence of 3.168, however, neither the CD8− nor CD8+ populations responded (Fig. 8 F). Over the entire time course, there was a slight rise in intracellular Ca2+ in the CD8− population (Fig. 8E and Fig. F), and we are investigating the possibility that the response of CD8− cells is kinetically delayed. In any case, the effect of CD8 on T cell activation by multimeric ligands mirrored the staining profile, with the important exception that, although CD8− 2C cells can bind well to the SIY/Kb multimer in the staining protocol, they do not respond efficiently to it by Ca2+ flux.
We also studied a later activation parameter, the upregulation of CD69, which was induced after 3 h by agonist ligands (OVA/Kb in the case of OT-I and SIY/Kb for 2C cells) but not by noncognate or nonagonistic multimeric ligands (data not shown). These data are in keeping with the results of the Ca2+ flux experiments and suggest that stimulation with cognate MHC/peptide multimers is capable of inducing new gene transcription.
Discussion
The production of synthetic MHC/peptide multimers has caused a revolution in T cell biology, allowing detection of antigen-specific T cell populations by using increased avidity to compensate for the very low affinity of TCRs for MHC/peptide ligands 16 41. Although there have been numerous papers published showing the capacity of multimers to bind antigen-specific TCRs, there has been comparatively little analysis of the role of the coreceptors in binding and T cell activation by these multimeric ligands.
A few reports using multimeric class II MHC/peptide ligands to analyze the minimal requirements for TCR binding and T cell activation reached the unanimous conclusion that the CD4 coreceptor is critical for activation of proximal signal transduction events but not required at all for TCR binding to either dimeric or multimeric MHC/peptide ligands 8 9 10. Our data using class I MHC/peptide multimers differ from these results in several key ways. First, we observe that TCR binding to cognate class I multimers is highly CD8 dependent, as shown by blockade with anti-CD8 antibodies and analysis of CD8− class I–restricted T cells. The degree of CD8 dependence differed between the two TCR systems studied, but in both cases the multimer interaction was profoundly influenced by CD8 participation. Second, in contrast to the data of Boniface et al. 10, we found that Ca2+ flux was efficiently induced by low doses of class I/peptide multimers. Moreover, the size of the responding population and uniformly raised levels of intracellular Ca2+ (Fig. 7) suggest that this is a sustained Ca2+ flux response, rather than the transient “partial agonist–like” response observed by Boniface et al. 10. However, using the flow cytometric approach it is difficult to determine the extent of Ca2+ oscillations in individual cells, so accurate resolution of this question will require single-cell analysis. Our functional assay data also differs from that of Delon et al. and Abastado et al., who showed that monomeric class I MHC/peptide complexes induced sustained Ca2+ flux, provided that CD8 was accessible, whereas dimeric MHC/peptide complexes could activate even in the absence of CD8 23 24. In contrast, we found that MHC/peptide monomers fail to activate Ca2+ flux and that the activation induced by multimeric MHC/peptide complexes is still highly CD8 dependent. We also showed that generation of multimers from monomer ligands during the time course of the Ca2+ flux experiment could induce OT-I T cell activation, albeit inefficiently. In comparing our results with those of Delon et al. 23, it is important to note that they studied primed CTLs, whereas we describe responses of naive T cells. Differences between the minimal activation requirements for naive versus effector cells are under investigation.
The profound effect of CD8 on OVA/Kb multimer binding to OT-I was initially unexpected, as the current literature suggests that multimeric MHC/peptide ligands bind efficiently to the TCR alone and because the affinity of the OT-I receptor is in the same range as class II MHC–restricted TCRs, which evidently do not require coreceptor participation for binding 8 9 10 41 42. We were concerned that anti-CD8 antibody binding may indirectly influence accessibility to the TCR, for example through some consequence of T cell activation or by induction of TCR internalization. This is unlikely given the consistency in results when parameters such as Ig isotype, temperature, and duration of staining and the presence or absence of azide were varied. Furthermore, expression of TCRs as assessed by CD3 staining was unaffected by exposure to anti-CD8 antibodies (data not shown), arguing against modulation of the TCR in these experiments. Perhaps most convincing are the parallel results obtained using the 2C system, in which multimer staining was not lost after anti-CD8 blockade but merely reduced to approximately the same level as naturally occurring CD8− 2C cells. Furthermore, the similarity of our data to that of Luescher et al. 5, who showed CD8 dependence for TCR binding to monomeric MHC/peptide ligands, suggests that our results are not an artifact of using multimeric TCR ligands. Instead, we are left with the idea that the inherent affinity of the OT-I TCR is insufficient to allow even multimeric ligand binding if CD8 participation is blocked. Further work will be required to determine how this obligate role for CD8 for OT-I TCR engagement by multimers is mediated. An interesting result in this context was the capacity of blocking anti-CD8 antibodies to induce loss of prebound MHC/peptide multimers (Fig. 4). This suggests that the MHC/peptide–TCR interaction is dynamic in nature and that “stable” multimer binding reflects a series of TCR–ligand release and rebinding, which is influenced by CD8. In keeping with this, the “enhancing” antibody 53.6.72 appeared to stabilize prebound MHC/peptide multimer (Fig. 4), raising the possibility that binding by this antibody lengthens the half-life of the MHC/peptide–TCR (–CD8) interaction. Thus, we show that cumulative multimer binding is enhanced by CD8 but can be reversed by sustained blockade of certain CD8 combining sites. On the other hand, recent measurements of cell surface–associated 2C TCR binding to the SIY/Kb complex indicate that the affinity of this interaction is extremely high 43. Thus, the interaction of this TCR with the SIY/Kb ligand may inherently be less coreceptor dependent.
We also demonstrated that CD8 played a critical role in OT-I TCR binding to multimers containing low-affinity, altered peptide ligands. In keeping with the predictions of Crawford et al. 9, we saw a ranking of multimer binding consistent with multimer affinity for the OT-I TCR. However, in our case this was again strongly influenced by manipulation of the coreceptor. Most striking is the effect of anti-CD8 on binding to the E1/Kb, which we previously showed was a low-affinity TCR antagonist 26 39 40. Binding to this multimeric ligand is detectable but weak on normal OT-I cells but is strongly enhanced by the 53.6 antibody, whereas it is completely blocked by the other anti-CD8 antibodies tested. Interestingly, previous work had indicated that high levels of CD8 expression could convert this ligand into a weak agonist 40. Our data is in contrast, however, with the conclusion made by Madrenas et al. and Hampl et al., who proposed that the CD4 coreceptor plays no role in binding to TCR antagonists 11 12. Aside from potential differences between the roles of CD4 and CD8 in antagonist recognition, it is interesting to note that there are also large differences in the ratio of TCR affinity for agonists versus antagonists in these systems. Thus, TCR antagonists bind with only three- to fivefold lower affinity than agonists in the OT-I system 39, but the difference in the 2B4 system used by Lyons et al. was 10–50-fold 44. Thus, differences in the involvement of the coreceptor in encounter with antagonists may relate to the core TCR affinity for these ligands. This raises the concern, however, that coreceptor participation in antagonist recognition may vary depending on the affinity of the particular TCR and hence may not be generalizable.
We conclude that TCR interactions and activation by multimeric MHC/peptide ligands on the surfaces of living CD8+ cells typically involve CD8. These conclusions are similar to those presented by Luescher et al. 5 using a Kd-restricted T hybridoma system in which TCR binding to MHC/peptide monomers could be detected. Thus, data from three different TCR systems involving two MHC class I alleles were strikingly similar and imply that this role of CD8 for TCR–MHC/peptide interactions can be generalized, at least in the mouse system. An intriguing outcome of these studies was the diverse effect of different antibodies to CD8. The CD8α antibody 53.6.7 enhanced TCR association by cognate (but not noncognate) MHC/peptide ligands, whereas the CD8α antibodies CT-CD8a and 3.168 and the CD8β antibody 53.5.8 all dramatically blocked TCR binding and, correspondingly, T cell activation. It is not clear why some CD8 antibodies augment TCR–MHC/peptide binding and others block it. Presumably, 53.6.7 favors encounter between CD8 and the TCR or CD8 and class I, whereas the other antibodies block these interactions. Incidentally, our results explain why the role of CD8 in multimer binding had not been appreciated until this report: previous studies in the mouse exclusively used the 53.6.7 antibody to stain for CD8α 17 18 19, which would be expected to augment rather than block multimer binding. It is also of interest that the CD8β antibody tested shows efficient blockade of multimer binding. CD8β expression is known to enhance T cell responses 45 46 and development 47 48 49, but it is unclear whether this chain plays a direct role in MHC class I binding or T cell signaling 2 3 50. Interestingly, 53.5.8 does not appear to occlude the CD8α chain, as determined by antibody binding competition 51, and has only a mild effect on CD8 binding to immobilized class I molecules 5, implying a minimal role in CD8-mediated adhesion. Thus, it is tempting to speculate that CD8β blockade may affect the TCR–CD8 rather than the CD8–MHC interaction. Such a role for CD8β is supported by experiments indicating that CD8β is more efficient than CD8α at association with the TCR 52. Also in support of our observations is a report describing partial blockade by 53.5.8 of multimer binding to polyclonal antigen–specific T cells 53.
The results observed in the 2C system deserve special attention. Although SIY/Kb binding was clearly not entirely dependent on CD8, overt activation of these cells by multimer ligands did require CD8. This matches well with reports that anti-CD8–treated 2C cells and/or CD8− 2C cells fail to respond to physiological densities of MHC/peptide antigen expressed on APCs 35, although this CD8 requirement could be overcome by very high antigen density 36. Thus, our multimeric system appears to mirror the response of 2C to physiological levels of antigen expressed by APCs. Interestingly, our data also correlate with findings of Garcia et al. 33, who used surface plasmon resonance to show a role for CD8 in enhancing TCR–MHC/peptide interactions, including the 2C receptor. These data were reinterpreted by Wyer et al. 13, who suggested that CD8 multimers contributed to the evident enhancement of TCR binding observed by Garcia et al. As we deliberately used multimeric MHC/peptide ligands in this work, it may not be surprising that we observed a similar enhancing role for CD8 in our system.
Lastly, a technical consequence of our studies is that the use of CD8 antibodies in flow cytometric analysis can drastically influence TCR binding to MHC/peptide ligands. Moreover, our data using low-affinity TCR ligands together with the enhancing anti-CD8 antibody 53.6.7 indicates that significant multimer staining may be seen using ligands that fail to induce a functional response. It is not clear what influence the commonly used anti–human CD8 antibodies will exert, but the effects documented here raise a cautionary note.
Acknowledgments
We thank John Altman and Eric Pamer for plasmids and protocols, Kris Hogquist, Marc Jenkins, and Matt Mescher for critical review of the manuscript, and members of the Jameson and Hogquist labs for helpful discussions.
This work was supported in part by grants from the National Institutes of Health (AI38903) and the American Cancer Society (JFRA-639 and RPG-99-264) to S.C. Jameson. M.A. Daniels is supported by a National Institutes of Health Immunology Training Grant (T32 AI-07313).
Footnotes
Abbreviations used in this paper: RAG, recombination activating gene; SA, streptavidin.
Address correspondence to Stephen C. Jameson, Center for Immunology and Dept. of Lab Medicine and Pathology, University of Minnesota Medical School, Box 334 FUMC, 420 Delaware St. SE, Minneapolis, MN 55455. Phone: 612-625-1496; Fax: 612-625-2199; E-mail: james024@tc.umn.edu
References
- Janeway C.A., Jr. The T cell receptor as a multicomponent signalling machineCD4/CD8 coreceptors and CD45 in T cell activation. Annu. Rev. Immunol. 1992;10:645–674. doi: 10.1146/annurev.iy.10.040192.003241. [DOI] [PubMed] [Google Scholar]
- Miceli M.C., Parnes J.R. Role of CD4 and CD8 in T cell activation and differentiation. Adv. Immunol. 1993;53:59–122. doi: 10.1016/s0065-2776(08)60498-8. [DOI] [PubMed] [Google Scholar]
- Zamoyska R. CD4 and CD8modulators of T-cell receptor recognition of antigen and of immune responses? Curr. Opin. Immunol. 1998;10:82–87. doi: 10.1016/s0952-7915(98)80036-8. [DOI] [PubMed] [Google Scholar]
- Eichmann K., Boyce N.W., Schmidt-Ullrich R., Jonsson J.I. Distinct functions of CD8(CD4) are utilized at different stages of T-lymphocyte differentiation. Immunol. Rev. 1989;109:39–75. doi: 10.1111/j.1600-065x.1989.tb00019.x. [DOI] [PubMed] [Google Scholar]
- Luescher I.F., Vivier E., Layer A., Mahiou J., Godeau F., Malissen B., Romero P. CD8 modulation of T-cell antigen receptor-ligand interactions on living cytotoxic T lymphocytes. Nature. 1995;373:353–356. doi: 10.1038/373353a0. [DOI] [PubMed] [Google Scholar]
- Xu H., Littman D.R. A kinase-independent function of Lck in potentiating antigen-specific T cell activation. Cell. 1993;74:633–643. doi: 10.1016/0092-8674(93)90511-n. [DOI] [PubMed] [Google Scholar]
- Thome M., Germain V., DiSanto J.P., Acuto O. The p56lck SH2 domain mediates recruitment of CD8/p56lck to the activated T cell receptor/CD3/zeta complex. Eur. J. Immunol. 1996;26:2093–2100. doi: 10.1002/eji.1830260920. [DOI] [PubMed] [Google Scholar]
- Hamad A.R., O'Herrin S.M., Lebowitz M.S., Srikrishnan A., Bieler J., Schneck J., Pardoll D. Potent T cell activation with dimeric peptide–major histocompatibility complex class II ligandthe role of CD4 coreceptor. J. Exp. Med. 1998;188:1633–1640. doi: 10.1084/jem.188.9.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford F., Kozono H., White J., Marrack P., Kappler J. Detection of antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity. 1998;8:675–682. doi: 10.1016/s1074-7613(00)80572-5. [DOI] [PubMed] [Google Scholar]
- Boniface J.J., Rabinowitz J.D., Wulfing C., Hampl J., Reich Z., Altman J.D., Kantor R.M., Beeson C., McConnell H.M., Davis M.M. Initiation of signal transduction through the T cell receptor requires the multivalent engagement of peptide/MHC ligands. Immunity. 1998;9:459–466. doi: 10.1016/s1074-7613(00)80629-9. [DOI] [PubMed] [Google Scholar]
- Madrenas J., Chau L.A., Smith J., Bluestone J.A., Germain R.N. The efficiency of CD4 recruitment to ligand-engaged TCR controls the agonist/partial agonist properties of peptide–MHC molecule ligands. J. Exp. Med. 1997;185:219–229. doi: 10.1084/jem.185.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampl J., Chien Y.-H., Davis M.M. CD4 augments the response of a T cell to agonist but not to antagonist ligands. Immunity. 1997;7:379–385. doi: 10.1016/s1074-7613(00)80359-3. [DOI] [PubMed] [Google Scholar]
- Wyer J.R., Willcox B.E., Gao G.F., Gerth U.C., Davis S.J., Bell J.I., van der Merwe P.A., Jakobsen B.K. T cell receptor and coreceptor CD8 αα bind peptide-MHC independently and with distinct kinetics. Immunity. 1999;10:219–225. doi: 10.1016/s1074-7613(00)80022-9. [DOI] [PubMed] [Google Scholar]
- O'Rourke A.M., Rogers J., Mescher M.F. Activated CD8 binding to class I protein mediated by the T-cell receptor results in signalling. Nature. 1990;346:187–189. doi: 10.1038/346187a0. [DOI] [PubMed] [Google Scholar]
- O'Rourke A.M., Mescher M.F. Cytotoxic T-lymphocyte activation involves a cascade of signalling and adhesion events. Nature. 1992;358:253–255. doi: 10.1038/358253a0. [DOI] [PubMed] [Google Scholar]
- Altman J.D., Moss P.A.H., Goulder P.J.R., Barouch D.H., McHeyzer-Williams M.G., Bell J.I., McMichael A.J., Davis M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- Murali-Krishna K., Altman J.D., Suresh M., Sourdive D.J., Zajac A.J., Miller J.D., Slansky J., Ahmed R. Counting antigen-specific CD8 T cellsa reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- Busch D.H., Pilip I.M., Vijh S., Pamer E.G. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 1998;8:353–362. doi: 10.1016/s1074-7613(00)80540-3. [DOI] [PubMed] [Google Scholar]
- Flynn K.J., Belz G.T., Altman J.D., Ahmed R., Woodland D.L., Doherty P.C. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 1998;8:683–691. doi: 10.1016/s1074-7613(00)80573-7. [DOI] [PubMed] [Google Scholar]
- Killeen N., Littman D.R. Helper T-cell development in the absence of CD4-p56lck association. Nature. 1993;364:729–732. doi: 10.1038/364729a0. [DOI] [PubMed] [Google Scholar]
- O'Rourke A.M., Ybarrondo B., Mescher M.F. CD8 and antigen-specific T cell adhesion cascades. Semin. Immunol. 1993;5:263–270. doi: 10.1006/smim.1993.1030. [DOI] [PubMed] [Google Scholar]
- Madrenas J., Germain R.N. Variant TCR ligandsnew insights into the molecular basis of antigen-dependent signal transduction and T-cell activation. Semin. Immunol. 1996;8:83–101. doi: 10.1006/smim.1996.0011. [DOI] [PubMed] [Google Scholar]
- Delon J., Gregoire C., Malissen B., Darche S., Lemaitre F., Kourilsky P., Abastado J.P., Trautmann A. CD8 expression allows T cell signaling by monomeric peptide-MHC complexes. Immunity. 1998;9:467–473. doi: 10.1016/s1074-7613(00)80630-5. [DOI] [PubMed] [Google Scholar]
- Abastado J.P., Lone Y.C., Casrouge A., Boulot G., Kourilsky P. Dimerization of soluble major histocompatibility complex–peptide complexes is sufficient for activation of T cell hybridoma and induction of unresponsiveness. J. Exp. Med. 1995;182:439–447. doi: 10.1084/jem.182.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels M.A., Schober S.L., Hogquist K.A., Jameson S.C. Cutting edgea test of the dominant negative signal model for TCR antagonism. J. Immunol. 1999;162:3761–3764. [PubMed] [Google Scholar]
- Hogquist K.A., Jameson S.C., Heath W.R., Howard J.L., Bevan M.J., Carbone F.R. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Sha W.C., Nelson C.A., Newberry R.D., Kranz D.M., Russell J.H., Loh D.Y. Selective expression of an antigen receptor on CD8-bearing T lymphocytes in transgenic mice. Nature. 1988;335:271–274. doi: 10.1038/335271a0. [DOI] [PubMed] [Google Scholar]
- Udaka K., Wiesmuller K.H., Kienle S., Jung G., Walden P. Self-MHC-restricted peptides recognized by an alloreactive T lymphocyte clone. J. Immunol. 1996;157:670–678. [PubMed] [Google Scholar]
- Sarmiento M., Glasebrook A.L., Fitch F.W. IgG or IgM monoclonal antibodies reactive with different determinants on the molecular complex bearing Lyt 2 antigen block T cell-mediated cytolysis in the absence of complement. J. Immunol. 1980;125:2665–2762. [PubMed] [Google Scholar]
- Ledbetter J.A., Herzenberg L.A. Xenogeneic monoclonal antibodies to mouse lymphoid differentiation antigens. Immunol. Rev. 1979;47:63–90. doi: 10.1111/j.1600-065x.1979.tb00289.x. [DOI] [PubMed] [Google Scholar]
- Ledbetter J.A., Rouse R.V., Micklem H.S., Herzenberg L.A. T cell subsets defined by expression of Lyt-1,2,3 and Thy-1 antigens. Two-parameter immunofluorescence and cytotoxicity analysis with monoclonal antibodies modifies current views. J. Exp. Med. 1980;152:280–295. doi: 10.1084/jem.152.2.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson S.C., Carbone F.R., Bevan M.J. Clone-specific T cell receptor antagonists of major histocompatibility complex class I–restricted cytotoxic T cells. J. Exp. Med. 1993;177:1541–1550. doi: 10.1084/jem.177.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia K.C., Scott C.A., Brunmark A., Carbone F.R., Peterson P.A., Wilson I.A., Teyton L. CD8 enhances formation of stable T-cell receptor/MHC class I molecule complexes. Nature. 1996;384:577–581. doi: 10.1038/384577a0. [DOI] [PubMed] [Google Scholar]
- Russell J.H., Meleedy-Rey P., McCulley D.E., Sha W.C., Nelson C.A., Loh D.Y. Evidence for CD8-independent T cell maturation in transgenic mice. J. Immunol. 1990;144:3318–3325. [PubMed] [Google Scholar]
- Cai Z., Sprent J. Resting and activated T cells display different requirements for CD8 molecules. J. Exp. Med. 1994;179:2005–2015. doi: 10.1084/jem.179.6.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z., Sprent J. Influence of antigen dose and costimulation on the primary response of CD8+ T cells in vitro. J. Exp. Med. 1996;183:2247–2257. doi: 10.1084/jem.183.5.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z., Kishimoto H., Brunmark A., Jackson M.R., Peterson P.A., Sprent J. Requirements for peptide-induced T cell receptor downregulation on naive CD8+ T cells. J. Exp. Med. 1997;185:641–651. doi: 10.1084/jem.185.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valitutti S., Muller S., Dessing M., Lanzavecchia A. Signal extinction and T cell repolarization in T helper cell-antigen-presenting cell conjugates. Eur. J. Immunol. 1996;26:2012–2016. doi: 10.1002/eji.1830260907. [DOI] [PubMed] [Google Scholar]
- Alam S.M., Travers P.J., Wung J.L., Nasholds W., Redpath S., Jameson S.C., Gascoigne N.R.J. T-cell receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- Jameson S.C., Hogquist K.A., Bevan M.J. Specificity and flexibility in thymic selection. Nature. 1994;369:750–752. doi: 10.1038/369750a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMichael A.J., O'Callaghan C.A. A new look at T cells. J. Exp. Med. 1998;187:1367–1371. doi: 10.1084/jem.187.9.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M.M., Boniface J.J., Reich Z., Lyons D., Hampl J., Arden B., Chien Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- Sykulev Y., Vugmeyster Y., Brunmark A., Ploegh H.L., Eisen H.N. Peptide antagonism and T cell receptor interactions with peptide-MHC complexes. Immunity. 1998;9:475–483. doi: 10.1016/s1074-7613(00)80631-7. [DOI] [PubMed] [Google Scholar]
- Lyons D.S., Lieberman S.A., Hampl J., Boniface J.J., Chien Y., Berg L.J., Davis M.M. A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- Wheeler C.J., von Hoegen P., Parnes J.R. An immunological role for the CD8 beta-chain. Nature. 1992;357:247–249. doi: 10.1038/357247a0. [DOI] [PubMed] [Google Scholar]
- Renard V., Romero P., Vivier E., Malissen B., Luescher I.F. CD8β increases CD8 coreceptor function and participation in TCR–ligand binding. J. Exp. Med. 1996;184:2439–2444. doi: 10.1084/jem.184.6.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K., Negishi I., Kuida K., Louie M.C., Kanagawa O., Nakauchi H., Loh D.Y. Requirement for CD8β chain in positive selection of CD8-lineage T cells. Science. 1994;263:1131–1133. doi: 10.1126/science.8108731. [DOI] [PubMed] [Google Scholar]
- Crooks M.E., Littman D.R. Disruption of T lymphocyte positive and negative selection in mice lacking the CD8β chain. Immunity. 1994;1:277–285. doi: 10.1016/1074-7613(94)90079-5. [DOI] [PubMed] [Google Scholar]
- Itano A., Cado D., Chan F.K., Robey E. A role for the cytoplasmic tail of the beta chain of CD8 in thymic selection. Immunity. 1994;1:287–290. doi: 10.1016/1074-7613(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Zamoyska R. The CD8 coreceptor revisitedone chain good, two chains better. Immunity. 1994;1:243–246. doi: 10.1016/1074-7613(94)90075-2. [DOI] [PubMed] [Google Scholar]
- Eichmann K., Ehrfeld A., Falk I., Goebel H., Kupsch J., Reimann A., Zgaga-Griesz A., Saizawa K.M., Yachelini P., Tomonari K. Affinity enhancement and transmembrane signaling are associated with distinct epitopes on the CD8 alpha beta heterodimer. J. Immunol. 1991;147:2075–2081. [PubMed] [Google Scholar]
- Kwan Lim G.E., McNeill L., Whitley K., Becker D.L., Zamoyska R. Co-capping studies reveal CD8/TCR interactions after capping CD8 beta polypeptides and intracellular associations of CD8 with p56(lck) Eur. J. Immunol. 1998;28:745–754. doi: 10.1002/(SICI)1521-4141(199802)28:02<745::AID-IMMU745>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Busch D.H., Pamer E.G. T cell affinity maturation by selective expansion during infection. J. Exp. Med. 1999;189:701–710. doi: 10.1084/jem.189.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]