

Abstract
The mechanisms that govern leukocyte transmigration through the endothelium are not yet fully defined. Junctional adhesion molecule (JAM) is a newly cloned member of the immunoglobulin superfamily which is selectively concentrated at tight junctions of endothelial and epithelial cells. A blocking monoclonal antibody (BV11 mAb) directed to JAM was able to inhibit monocyte transmigration through endothelial cells in in vitro and in vivo chemotaxis assays. In this study, we report that BV11 administration was able to attenuate cytokine-induced meningitis in mice. The intravenous injection of BV11 mAb significantly inhibited leukocyte accumulation in the cerebrospinal fluid and infiltration in the brain parenchyma. Blood–brain barrier permeability was also reduced by the mAb. We conclude that JAM may be a new target in limiting the inflammatory response that accompanies meningitis.
Keywords: endothelium, tight junction, meningitis, vascular permeability, blood–brain barrier
Maintenance of cerebral homeostasis requires a strict control of the extravascular traffic of cells and macromolecules from blood to the cerebrospinal fluid (CSF) and brain parenchyma. In physiological conditions, this task is well accomplished by both the epithelial lining that covers choroid plexus vessels, and the highly impermeable endothelium of cerebral capillaries which penetrates the brain parenchyma and constitutes the blood–brain barrier 1 2.
However, in acute inflammatory states such as bacterial meningitis, the processes that regulate blood–brain barrier functions are severely modified, thus bringing about diffused leukocyte invasion in CSF and brain parenchyma, which contributes to a severe pathological alteration of cerebral function 3 4 5 6. Inhibition of leukocyte recruitment 7 8 has been shown to reduce protein accumulation in the CSF and improve the survival rate in experimental models of meningitis.
The mechanisms that regulate leukocyte passage through endothelial cells and infiltration at inflammatory sites are still largely obscure. It is currently believed that leukocytes leave the circulation by first adhering to endothelial cells and then migrating through the interendothelial junctions 9 10. However, a few morphological studies reported that leukocytes may traverse endothelial cells through transcellular pathways 11.
Endothelial cell–cell junctions are complex structures formed by different adhesive molecules. At least two types of junctional adhesive structures have been described in the endothelium 12 13: the adherens junctions (AJs) formed by clusters of transmembrane proteins belonging to the cadherin family and the tight junctions (TJs). The transmembrane proteins at TJs are occludin 14 and a recently identified tetraspan protein family called claudins 15. These proteins codistribute with a complex network of cytoskeletal and signaling proteins inside the cells 16. These structures present common features between endothelial and epithelial cells.
Endothelial cells express several other adhesive proteins at intercellular contacts located outside AJs and TJs 5 13. Platelet endothelial cell adhesion molecule (PECAM) is one of the most relevant. It is a member of the immunoglobulin superfamily, promotes endothelial homotypic adhesion, and is clustered at intercellular contacts. Antibodies directed to PECAM are able to inhibit leukocyte transmigration in in vitro and in vivo systems 17 18 19.
We have recently identified and cloned a new junctional molecule in endothelial and epithelial cells called JAM (for junctional adhesion molecule [20]). JAM is a small transmembrane immunoglobulin-like molecule that codistributes with TJ components and promotes homotypic cell–cell adhesion 20. An mAb directed to JAM (BV11) was found to inhibit spontaneous and chemokine-induced chemotaxis of monocytes 20. JAM is highly expressed in cells that present well-organized TJs, such as the ependymal cells and the endothelium of the blood–brain barrier 20.
In this paper, we studied the effect of BV11 on experimental meningitis induced by intracerebroventricular (ICV) injection of inflammatory cytokines in mice 21. This model is characterized by a significant inflammatory cell influx and increase in blood–brain barrier permeability. The data reported show that JAM inhibition blocked monocyte and neutrophil infiltration into the CSF and brain parenchyma. In addition, albumin influx in CSF was strongly reduced by the JAM mAb. These data strongly suggest that functional blocking of JAM may provide a novel approach in limiting acute inflammatory reactions.
Materials and Methods
Reagents.
Reagents were purchased from the following sources: RPMI 1640, PBS, and FCS from GIBCO BRL; Immu-Mount from Shandon; OCT compound from Miles Labs; N-cetyl-N,N,N-trimethyl-ammonium bromide (TAB), Genziana violet, BSA, chloral hydrate, dianisidine dihydrochloride, erythrosine, FITC-conjugated bovine albumin (FITC-albumin), and heparin from Sigma Chemical Co.; human TNF-α from Genzyme Corp.; human IL-1β from Dompè; human monocyte chemotactic protein 3 (MCP-3) from Peprotech; Diff-Quik from Baxter Dade AG.
Animals.
CD1 male mice (25–30 g; Charles River) were housed with free access to food and water, under a 12-h light–dark cycle with constant temperature (21–23°C) and humidity (60 ± 5%). Procedures involving animals and their care were conducted in conformity with institutional guidelines that are in compliance with international laws and policies (European Economic Community Council Directive 86/609, OJ L 358,1; Dec. 12, 1987; National Institutes of Health Guide for the Care and Use of Laboratory Animals, NIH Publication No. 85-23, 1985).
Antibodies.
All the antibodies, unless otherwise specified, were rat anti–mouse mAbs and were used as sterile purified immunoglobulins. BV11 and BV12 mAbs were generated in our laboratory following a previously described procedure and characterized as described 20 22. BV12 recognizes a different JAM epitope than BV11 and is devoid of biological activity 20. Fab fragments of BV11 were prepared by standard procedures 23. Other antibodies used in this study were as follows: (a) HB-151, anti–HLA-DR5, used as isotype-matched control of BV11 (American Type Culture Collection); (b) MEC 7.46 to PECAM 22; (c) RB6-8C5 to granulocyte-differentiation antigen (CD15) was obtained from Dr. R.L. Coffman (DNAX, Palo Alto, CA). Endotoxin content of all the mAbs was tested by the Limulus assay with chromogenic detection (BioWhittaker). Tetramethyl rhodamine isothiocyanate (TRITC)-conjugated rabbit anti–rat IgG from Sigma Chemical Co. was used as secondary antibody in immunofluorescence microscopy.
Experimental Model of Meningitis.
A polyethylene cannula was permanently implanted in the right lateral ventricle (0.5 mm lateral and 1.0 mm posterior to bregma) 4 d before the onset of the experiment, as described elsewhere 24. At time 0, the animals were treated intravenously with mAbs at a standard dose of 100 μg/mouse in a final volume of 100 μl or with 100 μl of sterile, pyrogen-free saline. The dose of 100 μg/mouse for the mAbs was selected as the optimal dose to inhibit monocyte chemotaxis in vivo 20. After 10 min, mice were subjected to ICV treatment with a mixture of TNF-α (3 U/g) and IL-1β (1.25 U/g) dissolved in saline in a final volume of 2.5 μl (sham-injected animals received the same volume of saline). In a different set of experiments, animals were ICV-treated with 1 μg/mouse of MCP-3. After 6 h of treatment (except where otherwise specified), mice were anesthetized intraperitoneally with chloral hydrate (350 mg/kg), and 5–10 μl of CSF was drawn from the cisterna magna using a glass capillary with a tip of ∼300 μm 24. Careful surgery was conducted in order to avoid blood contamination of CSF. Mice were then decapitated, and brains were immediately cooled by liquid nitrogen and stored at −80°C until measurement of myeloperoxidase (MPO) activity or included in OCT compound, frozen in isopentane cooled by liquid nitrogen, and stored to −80°C, until preparation of brain tissue sections for immunofluorescence microscopy.
Quantitation of CSF and Total Blood Leukocytes.
CSF aliquots (2 μl) were immediately diluted in cool RPMI (containing 10% FCS) and kept in ice until cytospin centrifugation. Differential leukocyte counts of CSF samples were determined on Diff-Quik–fixed and erythrosine-stained cytospun slides by phase–contrast microscopy. Total blood leukocyte counts were determined by phase–contrast microscopy in heparin-anticoagulated blood diluted with Turk solution (1:20 [20]).
Immunofluorescence Microscopy.
Infiltrated leukocytes and vascular antigens were localized in brain tissue sections by immunofluorescence microscopy 20 24. Serial cryostat sections (20 μm) were cut horizontally from the brain, placed on glass coverslips, and dried overnight at room temperature 24. Tissue sections were fixed in cool methanol (5 min at −20°C), rinsed two times with PBS, incubated in PBS containing 2% of BSA (30 min at room temperature), and subsequently incubated for 1 h at room temperature with TRITC-conjugated anti–rat IgG and rinsed (three times, 5 min each) in PBS. Dried coverslips were then mounted in Immu-Mount, observed in a Zeiss Axiophot microscope, and images were recorded on Kodak TMax P3200 films.
Measurement of MPO Activity.
MPO activity was measured as described previously 25 26. In brief, the frozen tissues were weighed and homogenized in 20 volumes of 5 mM PBS, pH 6.0, at 4°C and then centrifuged at 30,000 g (30 min at 4°C). Supernatants were discarded, and pellets were extracted in 10 volumes of 0.5% TAB in 50 mM PBS, pH 6.0, at 25°C. Samples were frozen on dry ice (three freeze–thaw cycles) and then sonicated for 10 s at 25°C. Samples were subsequently incubated for 20 min at 4°C, centrifuged at 12,500 g for 15 min at 4°C, and MPO assay was performed as described 26 27. The absorbance change (ΔA) at 460 nm was measured over 2 min in the thermoregulated flow cell (25°C) of a Uvikon 860 spectrophotometer (Kontron Instruments). The data were derived using a kinetic computer program 26.
In Vivo Permeability Assay.
Sham- or cytokine-treated mice (with or without mAb pretreatment) were injected intravenously with FITC-albumin (1.5 mg/mouse) 1 h before time of killing. CSF was collected as usual, and 4 μl was immediately diluted in 100 μl of ice-cold phosphate buffer and kept at 4°C. Samples were then placed in a 96-well microtiter plate, and fluorescence content was measured in a fluorimeter (Cytofluor 2300 Fluorescence Measurement System; Millipore Co.) at 492-nm absorbance and 520-nm emission wavelengths, respectively.
Results
Vascular JAM staining was observed in brain tissue sections of sham- or cytokine-treated animals that had received mAb BV11 in vivo. BV11 staining was evident in different sized blood vessels that penetrate the brain parenchyma (Fig. 1 a) and in those of choroid plexus (Fig. 1 b). No specific staining with the secondary antibody was observed when the animals were treated with saline; when the mice were treated with BV12 or when brain slices were treated with BV11 in vitro, we obtained a similar immunofluorescence pattern as that reported in Fig. 1 (not shown). No major difference in JAM staining was observed in cytokine- or sham-treated animals. These data confirm previous work 20 showing that, despite the complex organization of the TJs in the brain microvasculature, JAM is located more superficially and can be available to mAb staining.
Figure 1.
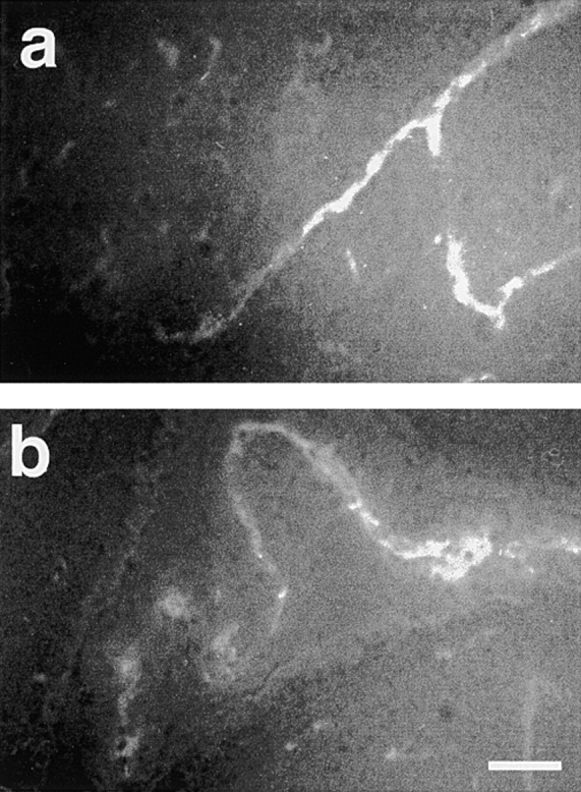
Binding of anti-JAM antibodies to brain vessels. Mice were treated intravenously with BV11 mAb. mAb binding was evidenced by TRIC-conjugated anti–rat IgG. BV11 was able to bind vessels in the brain parenchyma (a) and in the choroid plexus (b).
Mice ICV-treated with TNF-α (3 U/g body wt) and IL-1β (1.25 U/g body wt) showed a time-dependent CSF recruitment of both neutrophils and monocytes (Fig. 2 A, panel a). Maximal neutrophil recruitment at 6 h was almost 90% of the total leukocytes. Monocyte infiltration did not exceed 14% of the total recruited leukocytes. A comparable time course of leukocyte recruitment, measured as MPO activity, was observed in brain extracts of cytokine-treated mice (Fig. 2 A, panel b). After 6 h of treatment, cytokines induced a 3.7-fold increase of MPO activity compared with sham-treated animals. As reported in Fig. 2 B, panel a, the CSF of sham-treated animals was virtually devoid of circulating cells. In contrast, CSF of cytokine-treated mice contained a large number of recruited leukocytes (Fig. 2 B, panel b). This response was markedly reduced by intravenous administration of BV11 mAb (100 μg/mouse; Fig. 2 B, panel c).
Figure 2.
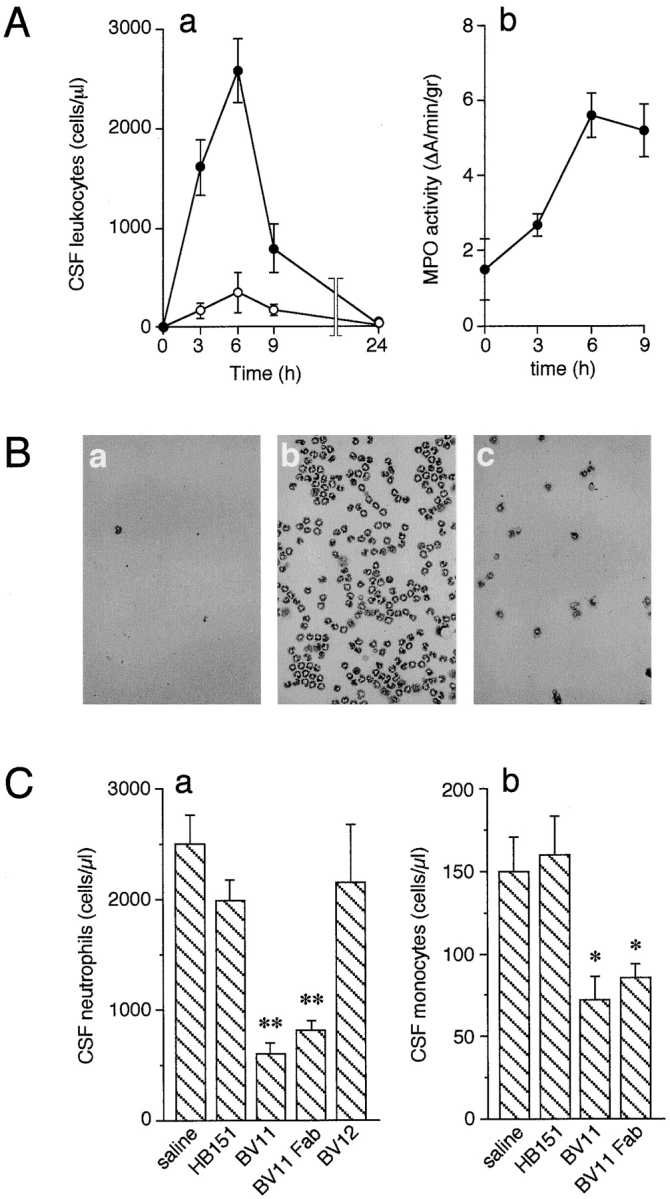
(A) Time course of leukocyte accumulation in the CSF (a) or brain parenchyma (b) after ICV injection of IL-1β and TNF-α. (a) The inflammatory cytokines caused a marked increase of CSF neutrophils (•) and monocytes (○). Data are expressed as percentage of total cell number in CSF counted after cytokine administration. The number of cells in sham-treated animals was essentially undetectable (see also B, panel a). Data are means ± SD of not less than five animals for each point from a typical experiment out of six performed. (b) MPO accumulation in the brain, as a parameter of leukocyte infiltration, was enhanced in a time-dependent manner. Data are means ± SD of at least five animals for each point from a typical experiment out of five performed. (B) Representative CSF samples collected at time 0 (a) and 6 h after ICV injection of cytokines (b, c). BV11 administration (c) strongly reduced leukocyte accumulation. (C) Administration of BV11 inhibited neutrophil and monocyte accumulation in the CSF after ICV cytokine injection. (a) BV11 and BV11 Fab significantly reduced neutrophil number in the CSF at 6 h after IL-1β and TNF-α administration. mAbs BV12, directed to JAM, and HB-151, as an isotype-matched control, were ineffective. (b) Monocyte accumulation after IL-1β and TNF-α was significantly reduced by BV11 and BV11 Fab but not by HB-151. Data are means ± SD of at least five animals from a typical experiment out of six performed. *P < 0.05; **P < 0.01 by analysis of variance and Dunnet test compared with saline treatment.
mAb BV11 was able to reduce neutrophil accumulation in the CSF by ∼76%. This activity was retained by its Fab fragment, whereas an isotype-matched control, mAb HB-151, was ineffective (Fig. 2 C, panel a). mAb BV12, which is able to bind JAM to a different epitope than BV11, did not show a significant inhibitory activity (Fig. 2 C, panel a). Monocyte recruitment was also reduced by ∼50% by BV11 or its Fab fragments, whereas HB-151 was ineffective (Fig. 2 C, panel b). At a longer time, 9 h after treatment, inhibition was still apparent, even if slightly lower, reaching 52% for neutrophils (not shown). The inhibitory effect of BV11 was observed also when mice were injected ICV with MCP-3 (1 μg/mouse 28). Monocytes and neutrophils were efficiently recruited by MCP-3 ICV, and their number was significantly reduced in mice pretreated with BV11, but not with HB-151 and BV12 (Fig. 3, a and b).
Figure 3.

Administration of BV11 inhibited neutrophil and monocyte accumulation in the CSF after ICV injection of MCP-3. BV11 significantly reduced monocyte (a) and neutrophil (b) number in the CSF at 6 h after MCP-3 administration, whereas mAbs HB-151 and BV12 were ineffective. Data are means ± SD of at least five animals from a typical experiment out of three performed. *P < 0.01 by analysis of variance and Dunnet test compared with saline treatment.
In all of the experiments reported above, the number of circulating leukocytes was never substantially altered by infusion of any type of mAb in cytokine- or sham-treated mice. Blood samples were taken by puncture of the left heart ventricle. In a typical experiment, in animals treated ICV with IL-1β and TNF-α for 6 h, circulating leukocytes were as follows: 9.1 ± 2.2 × 106/ml in saline; 8.3 ± 1.3 × 106 in HB-151; 10.1 ± 2 × 106 in BV11; and 10.5 ± 1.8 × 106 in BV12-treated animals. In comparison, in animals treated ICV with saline for 6 h, circulating leukocytes were as follows: 8.6 ± 1.2 × 106/ml in saline; 8.8 ± 1.1 × 106 in HB-151; 9.9 ± 1.9 × 106 in BV11; and 9.5 ± 1.2 × 106 in BV12-treated animals.
In addition, we were unable to detect increases in neutrophil trapping in lungs, spleen, liver, and kidneys by immunofluorescence analysis of CD15-stained tissue sections in animals treated with any of the mAbs used in this study.
As further control, we measured MPO activity (see below) in lungs of mice treated for 6 h with saline, BV11, BV12, HB-151 (100 μg/mouse), or BV11 Fab fragments (200 μg/mouse). The values, expressed as ΔA/min/g, were not significantly different: 34.6 ± 0.3 in saline; 27.6 ± 0.2 in HB-151; 27.1 ± 0.7 in BV12; 29.6 ± 0.6 in BV11; and 26.8 ± 0.7 in BV11 Fab–treated animals.
Neutrophil infiltration of the brain was also evaluated by histological means. Brain tissue sections of sham-treated mice showed essentially no staining of neutrophils with the anti-CD15 mAb RB6-8C5 (Fig. 4 A, panels a and d). In cytokine-treated mice, infiltrated neutrophils could be recognized in the surrounding areas of cerebral blood vessels (Fig. 4 A, panel b) and in submeningeal spaces (Fig. 4 A, panel e). Pretreatment of the mice with mAb BV11 (Fig. 4 A, panels c and f) but not with HB-151 (not shown) visibly reduced neutrophil extravasation.
Figure 4.

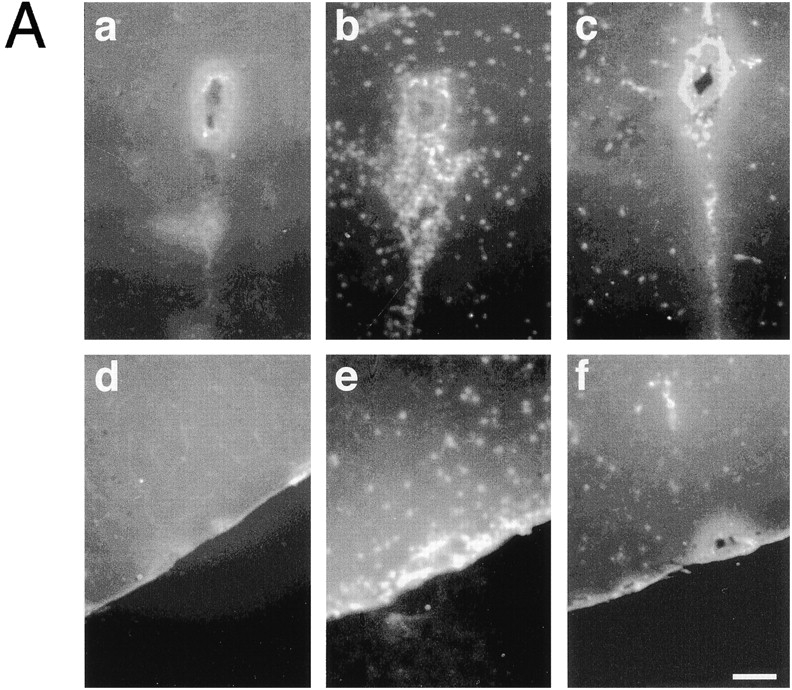
(A) Neutrophil infiltration in the brain detected by immunofluorescence analysis of areas surrounding blood vessels (a–c) or submeningeal space (d–f). Sham-treated animals showed essentially no staining with an anti-CD15 mAb (a and d). When mice were treated with IL-1β and TNF-α for 6 h, a marked increase in neutrophil infiltration was observed (b and e). This phenomenon was inhibited by BV11 administration (c and f). (B) MPO activity in brain extracts. ICV administration of IL-1β and TNF-α increased MPO activity in the brain (compare sham with saline). BV11 significantly reduced MPO accumulation, whereas HB-151 and BV12 were ineffective. Data are means ± SD of five animals per group of a typical experiment out of four performed. *P < 0.01 versus saline treatment by analysis of variance and Dunnet test.
To quantify leukocyte infiltration, we measured MPO activity in brain extracts. The basal MPO activity was increased by about three times in animals treated with TNF/IL-1 and was reduced in mice pretreated with mAb BV11 but not with HB-151 (Fig. 4 B).
As another marker of acute inflammatory reaction in the brain, we tested the effect of mAb BV11 on the extravasation of FITC-albumin. Cytokine treatment induced a 3.4-fold increase of FITC-albumin extravasation in CSF (Fig. 5). This enhancement of permeability was significantly reduced in mice pretreated with mAb BV11 but not with the control mAb, HB-151.
Figure 5.

Cytokine (IL-1β and TNF-α) treatment increased FITC-albumin accumulation in the CSF (compare sham with saline). BV11 administration significantly reduced this parameter, whereas HB-151 was ineffective. Data are means ± SD of seven animals of a typical experiment out of three performed. *P < 0.01 by analysis of variance and Dunnet test compared with saline treatment.
Discussion
As shown previously by others 29, the direct injection of TNF-α and IL-1β ICV induces acute inflammatory reactions in the brain parenchyma largely reminiscent of bacterial meningitis.
Reduction of inflammation in meningitis can decrease mortality 3 7 30 31. Inhibition of leukocyte adhesion to endothelial cells by β2 integrin–blocking mAbs in bacterial meningitis reduces CSF protein accumulation 7 8 and improves the survival rate. Furthermore, in mice deficient in endothelial P- and E-selectins, leukocyte and protein influx in the CSF upon inflammatory cytokine ICV injection was virtually abolished 21.
In this paper, we extend these observations by showing that inhibition of a novel leukocyte ligand, JAM, which regulates leukocyte transmigration 20, is effective in reducing inflammatory reactions in experimental meningitis. Intravenous pretreatment of mice with the anti-JAM mAb BV11 strongly reduced leukocyte extravasation and albumin permeability in the CSF and brain parenchyma promoted by ICV administration of cytokines.
The effect of BV11 was specific and not influenced by leukocyte Fc receptor, since BV11 Fab fragments retained the activity and an isotype-matched control mAb was ineffective. In addition, BV12, an mAb directed to JAM but recognizing a different epitope 20, was devoid of activity. Although in previous work 20 we observed that JAM was important in controlling monocyte extravasation, in the present model we confirm and extend this observation to neutrophils, which constitute the majority of the cellular exudate.
The effect of JAM appears to be specific for leukocyte transmigration, since BV11 does not inhibit monocyte or neutrophil adhesion to endothelial cells 20. In addition, BV11 also reduced the effect of MCP-3 on CSF accumulation of both cells types. MCP-3 does not induce endothelial inflammatory reactions like IL-1β/TNF-α, and therefore these data are in favor of a specific effect of the mAb on leukocyte recruitment.
Despite reduction in albumin influx in CSF, it is unlikely that BV11 acts on endothelial permeability directly since it did not modify basal or IL-1/TNF–increased endothelial permeability in vitro in the absence of leukocytes (20; and data not shown). A more likely hypothesis is that for its localization at intercellular junctions, JAM binds leukocytes and directs their migration through the junctions. Since leukocyte transmigration through cytokine-activated endothelial cells may lead to an increase in permeability 32 33, JAM inhibition would cause an indirect protection of blood–brain barrier breakdown.
A similar mechanism has been indicated for PECAM 19, and it is possible that these two proteins constitute a new class of agents which collaborate in promoting cell movement through endothelial cell junctions. JAM is located at TJs at the most apical domain of the intercellular cleft 20, whereas PECAM is found at a more basal site of the junctions 34. JAM may be necessary for the first interaction of circulating cells, which would then move along the cleft.
An important issue is JAM counterreceptor in monocytes and neutrophils. Monocytes and neutrophils obtained from the peritoneal fluid of thioglycollate-injected mice 20 or neutrophils from femoral bone marrow bind very poorly mAbs BV11 and BV12. Therefore, it is conceivable that these cells recognize JAM through a heterophilic receptor and that this interaction is inhibited by BV11.
In conclusion, the data reported here support the concept that JAM could constitute a novel target for modulating leukocyte extravasation at sites of inflammation. Therefore, manipulation of the molecular organization of endothelial junctions may be an effective approach to control vascular permeability and leukocyte traffic.
Acknowledgments
This study was supported by Human Frontiers Science Programme (grant RG0006/1997M), the Italian Association for Cancer Research, the European Community (projects Biomed BMH4-CT950875, BMH4-CT960669, BMH4-CT983380, and BMO4-CT0337), and the Italian National Research Council (CNR 97.01299.PF49). P. Fruscella is the recipient of a fellowship of Fondazione Valenti.
References
- Rowland L.P., Fink M.E., Rubin L. Cerebrospinal fluidblood-brain barrier, brain edema, and hydrocephalus. In: Kandel E.R., Schwartz J., Jessel T.M., editors. Principles of Neural Science. Elsevier Science Inc; New York: 1991. pp. 1050–1060. [Google Scholar]
- Perry V.H., Anthony D.C., Bolton S.J., Brown H.C. The blood-brain barrier and the inflammatory response. Mol. Med. Today. 1997;3:335–341. doi: 10.1016/S1357-4310(97)01077-0. [DOI] [PubMed] [Google Scholar]
- Quagliarello V.J., Scheld W.M. New perspectives on bacterial meningitis Clin. Infect. Dis. 17 1993. 603 608quiz 609–610 [DOI] [PubMed] [Google Scholar]
- Quagliarello V.J., Long W.J., Scheld W.M. Morphologic alterations of the blood–brain barrier with experimental meningitis in the rat. Temporal sequence and role of encapsulation. J. Clin. Invest. 1986;77:1084–1095. doi: 10.1172/JCI112407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharief M.K., Ciardi M., Thompson E.J. Blood-brain barrier damage in patients with bacterial meningitisassociation with tumor necrosis factor-alpha but not interleukin-1 beta. J. Infect. Dis. 1992;166:350–358. doi: 10.1093/infdis/166.2.350. [DOI] [PubMed] [Google Scholar]
- Smith A.L. Neurologic sequelae of meningitis. N. Engl. J. Med. 1988;319:1012–1014. doi: 10.1056/NEJM198810133191510. [DOI] [PubMed] [Google Scholar]
- Tuomanen E.I., Saukkonen K., Sande S., Cioffe C., Wright S.D. Reduction of inflammation, tissue damage, and mortality in bacterial meningitis in rabbits treated with monoclonal antibodies against adhesion-promoting receptors of leukocytes. J. Exp. Med. 1989;170:959–969. doi: 10.1084/jem.170.3.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Llorens X., Jafari H.S., Severien C., Parras F., Olsen K.D., Hansen E.J., Singer I.I., McCracken G.H., Jr. Enhanced attenuation of meningeal inflammation and brain edema by concomitant administration of anti-CD18 monoclonal antibodies and dexamethasone in experimental Haemophilus meningitis . J. Clin. Invest. 1991;88:2003–2011. doi: 10.1172/JCI115527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof B.A., Dunon D. Leukocyte migration and adhesion. Adv. Immunol. 1995;58:345–416. doi: 10.1016/s0065-2776(08)60623-9. [DOI] [PubMed] [Google Scholar]
- Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigrationthe multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Feng D., Nagy J.A., Pyne K., Dvorak H.F., Dvorak A.M. Neutrophils emigrate from venules by a transendothelial cell pathway in response to FMLP. J. Exp. Med. 1998;187:903–915. doi: 10.1084/jem.187.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani M.G., Dejana E. Interendothelial junctionsstructure, signalling and functional roles. Curr. Opin. Cell Biol. 1997;9:674–682. doi: 10.1016/s0955-0674(97)80121-4. [DOI] [PubMed] [Google Scholar]
- Dejana E. Endothelial adherens junctionsimplications in the control of vascular permeability and angiogenesis. J. Clin. Invest. 1997;100:S7–S10. [PubMed] [Google Scholar]
- Furuse M., Hirase T., Itoh M., Nagafuchi A., Yonemura S., Tsukita S. Occludina novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K., Furuse M., Fujimoto K., Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc. Natl. Acad. Sci. USA. 1999;96:511–516. doi: 10.1073/pnas.96.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J.M., Balda M.S., Fanning A.S. The structure and regulation of tight junctions. Curr. Opin. Cell Biol. 1993;5:772–776. doi: 10.1016/0955-0674(93)90024-k. [DOI] [PubMed] [Google Scholar]
- DeLisser H.M., Newman P.J., Albelda S.M. Molecular and functional aspects of PECAM-1/CD31. Immunol. Today. 1994;15:490–495. doi: 10.1016/0167-5699(94)90195-3. [DOI] [PubMed] [Google Scholar]
- Newman P.J. The biology of PECAM-1. J. Clin. Invest. 1997;99:3–8. doi: 10.1172/JCI119129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W.A., Weigl S.A., Deng X., Phillips D.M. PECAM-1 is required for transendothelial migration of leukocytes. J. Exp. Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Padura I., Lostaglio S., Schneemann M., Williams L., Romano M., Fruscella P., Panzeri C., Stoppacciaro A., Ruco L., Villa A. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T., Frenette P.S., Hynes R.O., Wagner D.D., Mayadas T.N. Cytokine-induced meningitis is dramatically attenuated in mice deficient in endothelial selectins. J. Clin. Invest. 1996;97:2485–2490. doi: 10.1172/JCI118695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchi A., Garlanda C., Lampugnani M.G., Resnati M., Matteucci C., Stoppacciaro A., Schnurch H., Risau W., Ruco L., Mantovani A., Dejana E. Monoclonal antibodies specific for endothelial cells of mouse blood vessels. Their application in the identification of adult and embryonic endothelium. Eur. J. Cell Biol. 1994;63:247–254. [PubMed] [Google Scholar]
- Parham P. Preparation and purification of active fragments from mouse monoclonal antibodies. In: Weir D.M., editor. Cellular Immunology., Vol. 1. 4th ed. Blackwell Science, Inc.; Malden, MA: 1986. [Google Scholar]
- De Simoni M.G., Sironi M., De Luigi A., Manfridi A., Ghezzi P. Intracerebroventricular injection of interleukin 1 induces high circulating levels of interleukin 6. J. Exp. Med. 1990;171:1773–1778. doi: 10.1084/jem.171.5.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki Y., Matsuo Y., Zagorski J., Matsuura N., Onodera H.I., Kogure K. New therapeutic possibility of blocking cytokine-induced neutrophil chemoattractant on transient ischemic brain damage in rats. Brain Res. 1997;759:103–111. doi: 10.1016/s0006-8993(97)00251-5. [DOI] [PubMed] [Google Scholar]
- Goldblum S.M., Wu K.M., Jay M. Lung myeloperoxidase as a measure of pulmonary leukostasis in rabbits. J. Appl. Physiol. 1985;59:1978–1985. doi: 10.1152/jappl.1985.59.6.1978. [DOI] [PubMed] [Google Scholar]
- Bradley P.P., Priebat D.A., Christensen R.D., Rothstein G. Measurement of cutaneous inflammationestimation of neutrophil content with an enzyme marker. J. Investig. Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- Fioretti F., Fradelizi D., Stoppacciaro A., Ramponi S., Ruco L., Minty A., Sozzani S., Garlanda C., Vecchi A., Mantovani A. Reduced tumorigenicity and augmented leukocyte infiltration after chemotactic protein-3 (MCP-3) gene transferperivascular accumulation of dendritic cells in peritumoral tissue and neutrophil recruitment within the tumor. J. Immunol. 1998;161:342–346. [PubMed] [Google Scholar]
- Saukkonen K., Sande S., Cioffe C., Wolpe S., Sherry B., Tuomanen E. The role of cytokines in the generation of inflammation and tissue damage in experimental gram-positive meningitis. J. Exp. Med. 1990;171:439–448. doi: 10.1084/jem.171.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuomanen E. Adjunctive therapy of experimental meningitisagents other than steroids. In: Schonfeld H., Helwig H., editors. Bacterial Meningitis. Antibiotic Chemotherapy. S. Karger AG; Basel: 1992. pp. 184–191. [DOI] [PubMed] [Google Scholar]
- Tuomanen E. Modulation of inflammation in bacterial meningitis. Isr. J. Med. Sci. 1994;30:339–341. [PubMed] [Google Scholar]
- Del Maschio A., Zanetti A., Corada M., Rival I., Ruco L., Lampugnani M.G., Dejana E. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J. Cell Biol. 1996;135:497–510. doi: 10.1083/jcb.135.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton S.J., Anthony D.C., Perry V.H. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience. 1998;86:1245–1257. doi: 10.1016/s0306-4522(98)00058-x. [DOI] [PubMed] [Google Scholar]
- Ayalon O., Sabanai H., Lampugnani M.G., Dejana E., Geiger B. Spatial and temporal relationships between cadherins and PECAM-1 in cell–cell junctions of human endothelial cells. J. Cell Biol. 1994;126:247–258. doi: 10.1083/jcb.126.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]