

Abstract
Insulin regulates blood glucose by promoting uptake by fat and muscle, and inhibiting production by liver. Insulin-stimulated glucose uptake is mediated by GLUT4, which translocates from an intracellular compartment to the plasma membrane. GLUT4 traffic and insulin secretion both rely on calcium-dependent, regulated exocytosis. Deletion of the voltage-gated potassium channel Kv1.3 results in constitutive expression of GLUT4 at the plasma membrane. Inhibition of channel activity stimulated GLUT4 translocation through a calcium dependent mechanism. The synaptotagmins (Syt) are calcium sensors for vesicular traffic, and Syt VII mediates lysosomal and secretory granule exocytosis. We asked if Syt VII regulates insulin secretion by pancreatic β cells, and GLUT4 translocation in insulin-sensitive tissues mouse model. Syt VII deletion (Syt VII −/−) results in glucose intolerance and a marked decrease in glucose-stimulated insulin secretion in vivo. Pancreatic islet cells isolated from Syt VII −/− cells secreted significantly less insulin than islets of littermate controls. Syt VII deletion disrupted GLUT4 traffic as evidenced by constitutive expression of GLUT4 present at the plasma membrane of fat and skeletal muscle cells and unresponsiveness to insulin. These data document a key role for Syt VII in peripheral glucose homeostasis through its action on both insulin secretion and GLUT4 traffic.
Keywords: Insulin, Glucose, Diabetes, Insulin resistance
Intracellular calcium plays a key role in glucose metabolism with respect to insulin secretion and glucose uptake. Insulin is the most important hormone for regulation of peripheral glucose metabolism. It is released into the circulation by pancreatic β cells in response to changes in blood glucose. The mechanism of release is well understood and initiated by glucose entry into the β cell through the glucose transporter GLUT2 [1]. Intracellular glucose is phosphorylated by the enzyme glucokinase and subsequently metabolized to generate ATP. The subsequent increase in ATP:ADP ratio inhibits the ATP-gated potassium channel (KATP), resulting in membrane depolarization, activation of voltage-gated calcium (Cav) channels, and in increased intracellular calcium [Ca]i. The rise in [Ca]i then stimulates the exocytosis of insulin containing vesicles.
Once in circulation, insulin binds to membrane receptors in liver to decrease glucose production, and in fat and skeletal muscle to increase glucose uptake. The transport of glucose into these cells is mediated by GLUT4. The transporter is stored in intracellular vesicles that are translocated to the plasma membrane after insulin-receptor activation. Insulin may also activate GLUT4 that is present on the cell surface [2]. While the molecular details are complex and some aspects are still poorly understood, it appears that [Ca]i plays an important role in the final step of vesicle fusion with the plasma membrane [3].
Members of the synaptotagmin family of membrane proteins emerged recently as strong candidates for Ca2+ sensors during exocytosis [4–8]. Synaptotagmins have long cytoplasmic regions containing two C2 domains, homologous to the regulatory C2-region of protein kinase C (PKC). Similar to what has been shown for PKC, synaptotagmin C2 domains mediate Ca2+ and phosphatidylserine (PS) binding activity, and are thought to promote exocytosis by facilitating the formation of SNARE complexes [9]. The brain-specific isoform Syt I is localized on neuronal synaptic vesicles, and extensive genetic and biochemical evidence indicates that it functions as a Ca2+ sensor for fast, synchronous neurotransmitter release [10–12]. The ubiquitous isoform Syt VII is localized on lysosomes of many cell types, and also on non-synaptic secretory granules of PC12 and pancreatic β cells, where it regulates Ca2+-triggered exocytosis [4–8,13]. Recent in vitro reconstitution experiments have directly demonstrated that both Syt I and Syt VII, in the absence of any other proteins, can confer Ca2+ sensitivity to SNARE-mediated membrane fusion [14,15].
The detection of Syt VII on pancreatic β cell granules, where it appears to regulate insulin secretion [5,13], prompted us to explore the role of Syt VII in glucose homeostasis in Syt VII−/− mice [16]. We find that Syt VII−/− mice are glucose intolerant, due to abnormalities not only in insulin secretion, but also in GLUT4 traffic.
Methods
Glucose tolerance test
Glucose and insulin tolerance measurements were carried out as described [17]. In brief, male 16–24-week-old Syt VII−/− mice [16] backcrossed into the C57BL/6 background were maintained according to National Institute of Health Guidelines, on standard rodent chow. Age-matched controls were given PBS IP. The experimental groups received glucose by IP injection and blood glucose levels were measured using a Glucometer (Bayer, West Haven, CT) at the indicated time intervals. Insulin levels were measured 30 min post-injection.
Glucose uptake in skeletal muscle and adipose tissue
Baseline and insulin-stimulated glucose uptake was measured in soleus muscle and epididymal fat, isolated from Syt VII−/− mice and control littermates, using the methods described by Slentz et al. [18]. The insulin concentration was 10 nM.
Immunocytochemistry
Primary cultures of epididymal fat, isolated from Syt VII mice and control littermates under sterile conditions, were established as described by Cabrero et al. [19]. Cells were treated with either PBS, 100 nM insulin, for 30 min at 37 μC, and fixed with 3% paraformaldehyde in PBS for 3 min at room temperature. GLUT4 expression was detected in adipocytes by immunofluorescence (anti-GLUT4 antibody, Cell Signaling Technology, Beverly, MA). The Texas red-conjugated secondary antibody was obtained from Vector lab (Burlingame, CA). Samples were examined by confocal fluorescence microscopy after labeling.
Colocalization of GLUT4 and Syt VII was carried out in primary cultures of epididymal fat by immunofluorescence using an anti-GLUT4 antibody (Cell Signaling Technology, Beverly, MA) and a rabbit anti-Syt VII polyclonal antibody [16].
Western blot analysis
Homogenates were prepared from white fat of Syt VII−/− mice. Protein (10 μg) was resolved by 10% SDS–PAGE and transferred to a nitrocellulose membrane, which was probed with an anti-Syt VII polyclonal antibody (1:200). Immunoreactive protein bands were visualized by enhanced chemiluminescence (Perkin–Elmer, Boston, MA).
Isolated islet insulin secretion
Islet isolation
The detailed methodologies employed to assess insulin output from collagenase-isolated islets have been previously described [20–22]. All animals were treated in a manner that complied with the NIH Guidelines for the Care and Use of Laboratory Animals. The animals were fed ad lib. After Nembutal (pentobarbital sodium, 50 mg/kg; Abbott, North Chicago, IL)-induced anesthesia, islets were isolated by collagenase digestion and handpicked, using a glass loop pipette, under a stereomicroscope into Krebs–Ringer bicarbonate (KRB) medium supplemented with 3 mM glucose. They were free of exocrine contamination. At the time of islet isolation the body weights of wild-type controls averaged 29.6 ± 13.2 g (n = 6). The knockout body weights averaged 25.3 ± 1.9 g (n = 6).
Perifusion studies
Islets were loaded onto nylon filters (Sefar America Inc., Kansas City, MO) and perifused in a KRB buffer at a flow rate of 1 ml/min for 30 min in the presence of 3 mM glucose to establish basal and stable insulin secretory rates as described [21]. After this 30-min stabilization period, they were then perifused with 20 mM glucose. Perifusate solutions were gassed with 95% O2/5% CO2 and maintained at 37 μC. Insulin released into the medium was measured by radioimmunoassay [23].
Reagents
Hank’s solution was used for the islet isolation. The perifusion medium consisted of 115 mM NaCl, 5 mM KCl, 2.2 mM CaCl2, 1 mM MgCl2, 24 mM NaHCO3, and 0.17 g/dl bovine serum albumin. The 125I-labeled insulin for the insulin assay was purchased from Perkin-Elmer Life Sciences (Boston, MA). Bovine serum albumin (RIA grade), glucose, carbachol, and the salts used to make the Hank’s solution and perifusion medium were purchased from Sigma (St. Louis, MO). Rat insulin standard (lot #615-ZS-157) was the generous gift of Dr. Gerald Gold, Eli Lilly Co. (Indianapolis, IN). Collagenase (Type P) was obtained from Roche Diagnostics (Indianapolis, IN).
Statistical analysis
Statistical significance was determined using Student’s t test for unpaired data or analysis of variance in conjunction with the Newman–Keuls test for unpaired data. A p value ≤ 0.05 was taken as significant. Values presented in the figures and results represent means ± SEs of at least three observations.
Results
Abnormal glucose metabolism in Syt VII −/− mice
Syt VII is expressed in the pancreas [5,13] and colocalizes with insulin containing granules [5]. Syt VII overexpression in the insulin-secreting β cell line RINm5F was associated with increased carbachol-induced insulin secretion, suggesting a possible role for Syt VII in the regulation of insulin secretion [5]. The physiological relevance of these in vitro findings was examined in the Syt VII−/− mouse model [16]. At the end of a 4-h fasting period, blood glucose was significantly higher in Syt VII−/− compared to WT mice (154 ± 8 vs 123 ± 9 mg/dl, n = 4, p < 0.05). The difference in fasting blood glucose was not accounted for by body weight (29.00 ± 2.61 g for Syt VII−/−, n = 4, and 29.02 ± 0.18 g for WT, n = 4). Peripheral glucose metabolism was examined in more detail using a glucose tolerance test. As shown in Fig. 1A, Syt VII−/− mice were significantly more hyperglycemic than WT at 90 and 120 min, indicating a defect in peripheral glucose metabolism.
Fig. 1.
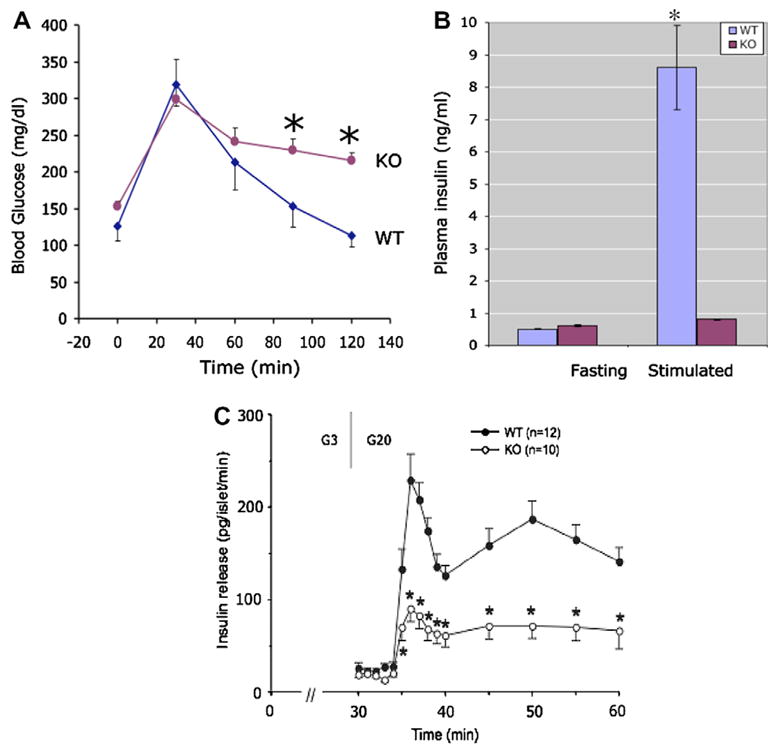
Decreased insulin secretion in Syt VII−/− mice. (A) Abnormal glucose tolerance test in Syt VII−/−: WT (wild-type littermate, n = 4) compared to KO (Syt VII−/−, n = 4). Blood glucose measured at indicated times following intraperitoneal injection of glucose (2 g/kg body weight). Stars indicate p values less than 0.05. (B) Decreased glucose-stimulated insulin secretion in vivo in Syt VII−/−: WT (wild-type littermate, n = 4) compared to KO (Syt VII−/−, n = 4). Animals were subjected to a 4 h fast and plasma insulin measured at before and 30 min following intraperitoneal injection of glucose (2 g/ kg body weight). A star indicates a p value less than 0.05. (C) Insulin secretory responses of islets isolated from wild-type and Syt VII knockouts: Groups of 14–18 islets were isolated from wild type (closed circles) or Syt VII knockout mice and perifused for 30 min with 3 mM glucose (G3) to establish stable, basal rates of insulin release. They were then perifused for 30 min with 20 mM glucose (G20), onset indicated by the first vertical line. Mean values ± SEMs of at least six perifusions are shown. This figure has not been corrected for the dead space in the perifusion apparatus, 2.5 ml or approximately 2.5 min with a flow rate of 1 ml/min. A star indicates a p value less than 0.05.
Defect in insulin secretion in Syt VII−/− mice
The mechanism underlying the observed abnormality in glucose metabolism could entail a defect in insulin secretion, resistance to the action of insulin, or a combination of both. To differentiate between these possibilities, plasma insulin was measured in the fasting state and 30 min following glucose administration. Wild-type mice increased insulin levels 10-fold in response to a glucose load (Fig. 1B). In contrast, glucose administration failed to significantly increase plasma insulin in Syt VII−/− mice (Fig. 1B), suggesting that these mice have a defect in glucose-stimulated, Syt VII-dependent insulin secretion, consistent with previous in vitro findings [5,13]. To confirm the results of the in vivo results and to further examine the process of insulin release in Syt VII−/− mice, insulin secretion was studied in isolated, perifused islets cells.
Islets were isolated from control and KO animals and perifused to establish the dynamics of insulin secretion. As shown in Fig. 1C, islets isolated from control animals exhibited a brisk secretory response when stimulated with 20 mM glucose. In contrast, islets isolated from Syt VII knockout animals released significantly less insulin at virtually all time points after the onset of stimulation.
Syt VII regulates GLUT4 traffic in adipocytes
Insulin tolerance studies revealed no significant differences between Syt VII−/− and WT mice in their overall response to an insulin challenge (Fig. 2A). Since skeletal muscle accounts for more than 80% of peripheral glucose uptake in vivo, we did not expect to find any in vitro differences in the insulin responsiveness of skeletal muscle of WT and Syt VII−/− mice. Fig. 2B confirms that skeletal muscle isolated from Syt VII−/− mice responds as well as WT muscle to insulin, and increases glucose uptake to a similar degree. In contrast, fat cells isolated from Syt VII−/− mice failed to increase glucose uptake upon stimulation by insulin (Fig. 2C), suggesting a significant degree of insulin resistance in the adipose tissue of Syt VII−/− mice. These results prompted us to examine the effect of Syt VII deletion on GLUT4 traffic in adipose tissue. In WT fat cells, in the basal state, GLUT4 is largely confined to an intracellular vesicular compartment (Fig. 3A, left panel). The addition of insulin causes GLUT4 to move from the intracellular pool to the plasma membrane. In marked contrast to WT cells, Syt VII−/− cells not only constitutively express GLUT4 at the plasma membrane (Fig. 3A, right panel), but also fail to increase plasma membrane associated GLUT4 in the presence of insulin. Western blot studies also demonstrated that Syt VII deletion did not alter overall GLUT4 content (Fig. 3B). Syt VII is expressed in GLUT4 containing vesicles (Fig. 4A and C), as is Kv1.3 (Fig. 4B and C), a voltage-gated ion channel known to regulate GLUT4 traffic through a calcium-dependent mechanism.
Fig. 2.
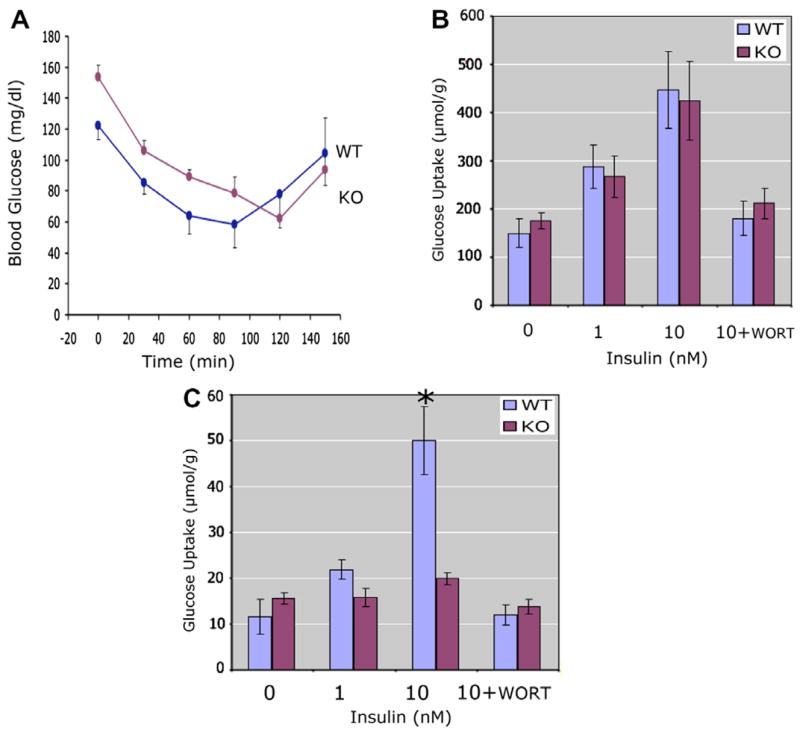
Insulin resistance in Syt VII−/− adipocytes. (A) Insulin tolerance test: WT (wild-type littermate, n = 4) compared to KO (Syt VII−/−, n = 4). Blood glucose measured at indicated times following intraperitoneal injection of insulin (2.55 μg/kg body weight). (B) Insulin-stimulated glucose uptake in skeletal muscle: skeletal muscle isolated from WT (wild-type littermate, n = 4) compared to KO (Syt VII−/−, n = 4). WORT is wortmanin (100 nM). (C) Insulin-stimulated glucose uptake in adipocytes: Adipose tissue isolated from WT (wild-type littermate, n = 4) compared to KO (Syt VII−/−, n = 4). WORT is wortmanin (100 nM). A star indicates a p value less than 0.05.
Fig. 3.
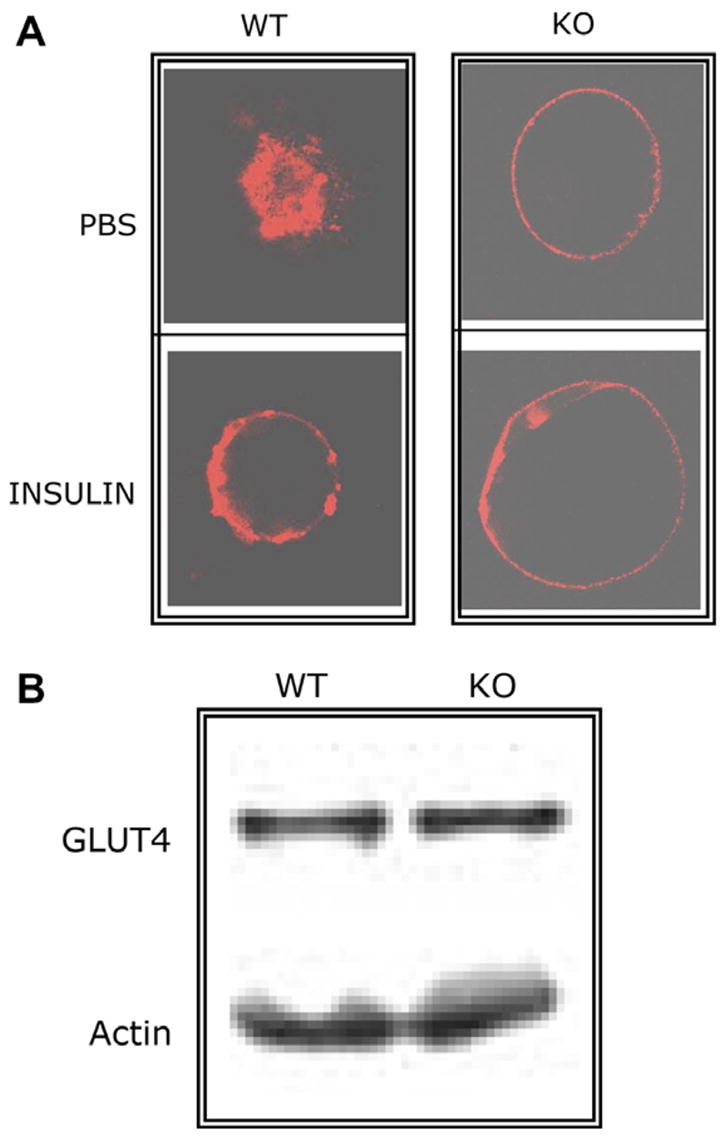
Dysregulated GLUT4 traffic in Syt VII−/− mice. (A) GLUT4 traffic in WT and Syt VII−/− fat cells: GLUT4 distribution examined by confocal microscopy in adipocytes, isolated from wild-type littermate (WT, n = 3) and Syt VII−/−, (KO, n = 3), and maintained in primary culture. PBS is phosphate buffered saline. Insulin dose is 100 nM. Representative experiment shown. (B) Total GLUT4 content unaltered by Syt VII deletion: Unfractionated adipocyte proteins separated by SDS gel electrophoresis and total GLUT4 content measured by Western blotting. WT is wild-type littermate, and KO is Syt VII−/−. Representative experiment shown.
Fig. 4.
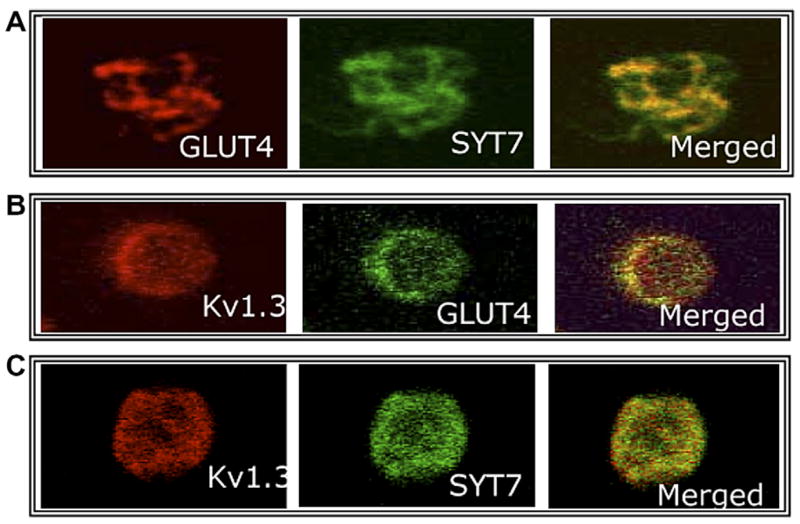
Colocalization of GLUT4, Syt VII, and Kv1.3. Unstimulated adipocytes isolated from wild-type mice, maintained in primary culture, and examined by confocal microscopy. Colocalization of (A) GLUT4 and Syt VII, (B) Kv1.3 and GLUT4, (C) Kv1.3 and Syt VII.
Discussion
Synaptotagmins belong to a family of transmembrane proteins that coordinate changes in intracellular calcium related to membrane fusion events [24]. There are 16 vertebrate synaptotagmins. Syt 1 is predominantly expressed in neural tissue and serves as a calcium sensor critical for rapid neurotransmitter release. In contrast, Syt VII is expressed in a wide variety of tissues, including kidney, heart, and pancreas. In the pancreas, Syt VII is found on both lysosomes and insulin-secreting vesicles [5].
A number of physiological signals including glucose, amino acids, and acetylcholine can stimulate insulin secretion by the pancreatic β cell. The cytoskeletal network plays a key role in the process of insulin secretion by forming a bridge between the endoplasmic reticulum, the Golgi, and the plasma membrane. Under basal conditions, a small fraction of the cytoplasmic pool of insulin-secreting vesicles is capable of fusing with the plasma membrane. This group of vesicles is called the ready releasable pool (RRP), and mediates the first phase of insulin release, which occurs within 3–5 min of an appropriate physiological stimulus [1]. The second phase of insulin release depends on further priming of the cytoplasmic pool and insulin synthesis and processing. The specificity of vesicle fusion relies on the recognition of the vesicle SNARE VAMP-2 by the target SNARE SNAP 25 [25]. A rise in intracellular calcium precedes the fusion of insulin vesicles with the plasma membrane.
In vitro studies suggest that Syt VII mediates calcium-induced insulin secretion in pancreatic β cells. Syt VII is expressed on insulin vesicles of the rat pancreatic β cell line RINm5F. Overexpression of Syt VII resulted in a significant increase in carbachol and calcium-induced insulin secretion [5]. Regulation of exocytosis by Syt VII involves specific SNARE interactions. During exocytosis-mediated repair, Syt VII interacts with the lysosomal vSNARE VAMP7 and the plasma membrane target SNAREs Syntaxin 4 and SNAP23 [26]. Here we have shown that Syt VII−/− mice are diabetic, as evidenced by an increase in fasting blood sugar and an abnormal glucose tolerance test. The defect in peripheral glucose metabolism is partly explained by a significant decrease in insulin release. Blood insulin levels remain low after glucose infusion in vivo, and isolated perifused pancreatic islets secrete significantly less insulin than controls. Both phases of insulin release are affected in Syt VII−/− mice. These findings provide strong support for the hypothesis that Syt VII plays a key role in mediating insulin secretion through the fusion of insulin vesicles with the plasma membrane. However, that insulin secretion persists at a reduced level suggests that additional, possibly compensatory, mechanisms mediate insulin secretion.
Syt VII−/− mice develop an inflammatory autoimmune disease [16] and it could be argued that the defect in insulin secretion is secondary to an immune-mediated destruction on the endocrine pancreas, similar to what occurs in juvenile-onset (Type 1) diabetes mellitus. This is an unlikely explanation for two major reasons. First, the autoimmune state observed in Syt VII−/− mice appears to be limited to skin and skeletal muscle, perhaps because, as the authors suggest, a defect in plasma membrane repair is most critical in tissues exposed to significant mechanical stress. While the inflammatory reaction and fibrosis was easily detected in skin and skeletal muscle, brain, liver, kidney, heart, spleen, and pancreas were normal [16]. Second, when maximally stimulated using glucose and either forskolin or carbachol, Syt VII−/− β cells released insulin as well as control cells, indicating that there is no significant immune destruction of β cells in Syt VII−/− mice.
We also tested, using insulin tolerance tests, whether insulin resistance played a role in the diabetic state observed in Syt VII−/− mice, and were unable to detect any significant differences when compared to control mice. The interpretation of the data is complicated by the fact that skeletal muscle accounts for more than 80% of peripheral glucose uptake, and that Syt VII−/− mice have an autoimmune myositis [16]. Adipose tissue increases peripheral glucose uptake in response to insulin and mediates about 20% of the observed response. Our in vitro data indicate that glucose uptake in skeletal muscle was not significantly different from control, confirming the in vivo insulin tolerance test. In contrast, Syt VII−/− adipocytes exhibited significant insulin resistance.
Two major pathways mediate glucose uptake in insulin-sensitive tissues [27–29]. One involves the binding of insulin to its plasma membrane receptor (a receptor tyrosine kinase, IR). IR autophosphorylation initiates intracellular signaling events, ultimately resulting in the translocation of the glucose carrier GLUT4 to the plasma membrane and to increased glucose uptake. The PI3K and CAP/Cbl pathways are critical to the action of insulin on glucose transport. The other pathway for glucose uptake is clearly seen during muscular contraction, where GLUT4 translocates to the plasma membrane and increases cell glucose uptake in the absence of insulin. Contraction-mediated GLUT4 translocation is clearly Ca2+ dependent, and recent studies also indicate a key role of calcium in insulin-dependent traffic of GLUT4 [3]. GLUT4 traffic is abnormally regulated in Syt VII−/− adipocytes. Under basal conditions, in the absence of insulin GLUT4 is distributed in a manner consistent with the plasma membrane in Syt VII−/− adipocytes. Furthermore, the addition of insulin fails to noticeably alter GLUT4 traffic. This is in marked contrast with control adipocytes, in which GLUT4 is confined to intracellular compartments at baseline and traffics to the plasma membrane in the presence of insulin. These data suggest that Syt VII participates in the regulation of GLUT4 traffic, perhaps by sensing changes in intra-cellular calcium.
An alternative explanation for the observation that Syt VII deletion results in constitutive expression of GLUT4 at the plasma membrane relates to the possible requirement of Syt VII for endocytosis. All synaptotagmins bind to the clathrin adapter AP2 [30] and in the case of Syt 1, the AP2 binding site does not appear to be an internalization signal, but rather functions as a regulator of endocytosis [31]. Syt VII has been specifically implicated in endocytosis [32] and its internalization signals utilize two independent pathways that are masked by intramolecular inhibitions. Steady state levels of GLUT4 at the plasma membrane are determined by the rates of endocytosis and exocytosis. Syt VII deletion could slow the rate of endocytosis and cause an increase in plasma membrane associated GLUT4.
The voltage-gated potassium channel Kv1.3 plays a key role in the regulation of peripheral glucose metabolism. Channel inhibition in adipocytes leads to membrane depolarization, calcium release from intracellular stores, increased intracellular calcium, and ultimately to GLUT4 translocation to the plasma membrane [33]. Channel deletion results in constitutive expression of GLUT4 at the plasma membrane. Since Kv1.3 modulates intracellular calcium, Syt VII senses intracellular calcium, and are both expressed on GLUT4 vesicles, we speculate that Kv1.3 and Syt VII are components of the machinery that regulates calcium-dependent GUT4 traffic.
In summary, Syt VII modulates peripheral glucose homeostasis through its action on both insulin secretion and GLUT4 traffic. Syt VII deletion in mice leads to a diabetic state characterized by decreased insulin secretion by the pancreatic β cell, constitutive expression of GLUT4 at the plasma membrane, and decreased responsiveness to insulin in adipocytes.
Footnotes
References
- 1.Doyle ME, Egan JM. Pharmacological agents that directly modulate insulin secretion. Pharmacol Rev. 2003;55:105–131. doi: 10.1124/pr.55.1.7. [DOI] [PubMed] [Google Scholar]
- 2.Michelle Furtado L, Poon V, Klip A. GLUT4 activation: thoughts on possible mechanisms. Acta Physiol Scand. 2003;178:287–296. doi: 10.1046/j.1365-201X.2003.01160.x. [DOI] [PubMed] [Google Scholar]
- 3.Whitehead JP, Molero JC, Clark S, Martin S, Meneilly G, James DE. The role of Ca2+ in insulin-stimulated glucose transport in 3T3-L1 cells. J Biol Chem. 2001;276:27816–27824. doi: 10.1074/jbc.M011590200. [DOI] [PubMed] [Google Scholar]
- 4.Fukuda M, Kanno E, Satoh M, Saegusa C, Yamamoto A. Synaptotagmin VII is targeted to dense-core vesicles and regulates their Ca2+-dependent exocytosis in PC12 cells. J Biol Chem. 2004;279:52677–52684. doi: 10.1074/jbc.M409241200. [DOI] [PubMed] [Google Scholar]
- 5.Gao Z, Reavey-Cantwell J, Young RA, Jegier P, Wolf BA. Synaptotagmin III/VII isoforms mediate Ca2+-induced insulin secretion in pancreatic islet beta-cells. J Biol Chem. 2000;275:36079–36085. doi: 10.1074/jbc.M004284200. [DOI] [PubMed] [Google Scholar]
- 6.Martinez I, Chakrabarti S, Hellevik T, Morehead J, Fowler K, Andrews NW. Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol. 2000;148:1141–1149. doi: 10.1083/jcb.148.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 8.Wang P, Chicka MC, Bhalla A, Richards DA, Chapman ER. Synaptotagmin VII is targeted to secretory organelles in PC12 cells, where it functions as a high-affnity calcium sensor. Mol Cell Biol. 2005;25:8693–8702. doi: 10.1128/MCB.25.19.8693-8702.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman ER. Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat Rev Mol Cell Biol. 2002;3:498–508. doi: 10.1038/nrm855. [DOI] [PubMed] [Google Scholar]
- 10.Bai J, Chapman ER. The C2 domains of synaptotagmin—partners in exocytosis. Trends Biochem Sci. 2004;29:143–151. doi: 10.1016/j.tibs.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Koh TW, Bellen HJ. Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci. 2003;26:413–422. doi: 10.1016/S0166-2236(03)00195-4. [DOI] [PubMed] [Google Scholar]
- 12.Yoshihara M, Adolfsen B, Littleton JT. Is synaptotagmin the calcium sensor? Curr Opin Neurobiol. 2003;13:315–323. doi: 10.1016/s0959-4388(03)00063-1. [DOI] [PubMed] [Google Scholar]
- 13.Gut A, Kiraly CE, Fukuda M, Mikoshiba K, Wollheim CB, Lang J. Expression and localisation of synaptotagmin isoforms in endocrine beta-cells: their function in insulin exocytosis. J Cell Sci. 2001;114:1709–1716. doi: 10.1242/jcs.114.9.1709. [DOI] [PubMed] [Google Scholar]
- 14.Bhalla A, Tucker WC, Chapman ER. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell. 2005;16:4755–4764. doi: 10.1091/mbc.E05-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tucker WC, Weber T, Chapman ER. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 2004;304:435–438. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- 16.Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS, Andrews NW. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol. 2003;162:543–549. doi: 10.1083/jcb.200305131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J, Wang P, Li Y, Li G, Kaczmarek LK, Wu Y, Koni PA, Flavell RA, Desir GV. The voltage-gated potassium channel Kv1.3 regulates peripheral insulin sensitivity. Proc Natl Acad Sci USA. 2004;101:3112–3117. doi: 10.1073/pnas.0308450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slentz CA, Gulve EA, Rodnick KJ, Henriksen EJ, Youn JH, Holloszy JO. Glucose transporters and maximal transport are increased in endurance-trained rat soleus. J Appl Physiol. 1992;73:486–492. doi: 10.1152/jappl.1992.73.2.486. [DOI] [PubMed] [Google Scholar]
- 19.Cabrero A, Alegret M, Sanchez RM, Adzet T, Laguna JC, Vazquez M. Bezafibrate reduces mRNA levels of adipocyte markers and increases fatty acid oxidation in primary culture of adipocytes. Diabetes. 2001;50:1883–1890. doi: 10.2337/diabetes.50.8.1883. [DOI] [PubMed] [Google Scholar]
- 20.Zawalich WS, Diaz VA, Zawalich KC. Role of phosphoinositide metabolism in induction of memory in isolated perifused rat islets. Am J Physiol. 1988;254:E609–E616. doi: 10.1152/ajpendo.1988.254.5.E609. [DOI] [PubMed] [Google Scholar]
- 21.Zawalich WS, Zawalich KC, Kelley GG. Regulation of insulin release by phospholipase C activation in mouse islets: differential effects of glucose and neurohumoral stimulation. Endocrinology. 1995;136:4903–4909. doi: 10.1210/endo.136.11.7588223. [DOI] [PubMed] [Google Scholar]
- 22.Zawalich WS, Zawalich KC, Rasmussen H. Cholinergic agonists prime the beta-cell to glucose stimulation. Endocrinology. 1989;125:2400–2406. doi: 10.1210/endo-125-5-2400. [DOI] [PubMed] [Google Scholar]
- 23.Albano JD, Ekins RP, Maritz G, Turner RC. A sensitive, precise radioimmunoassay of serum insulin relying on charcoal separation of bound and free hormone moieties. Acta Endocrinol (Copenh) 1972;70:487–509. doi: 10.1530/acta.0.0700487. [DOI] [PubMed] [Google Scholar]
- 24.Andrews NW, Chakrabarti S. There’s more to life than neurotransmission: the regulation of exocytosis by synaptotagmin VII. Trends Cell Biol. 2005;15:626–631. doi: 10.1016/j.tcb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Wheeler MB, Sheu L, Ghai M, Bouquillon A, Grondin G, Weller U, Beaudoin AR, Bennett MK, Trimble WS, Gaisano HY. Characterization of SNARE protein expression in beta cell lines and pancreatic islets. Endocrinology. 1996;137:1340–1348. doi: 10.1210/endo.137.4.8625909. [DOI] [PubMed] [Google Scholar]
- 26.Rao SK, Huynh C, Proux-Gillardeaux V, Galli T, Andrews NW. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J Biol Chem. 2004;279:20471–20479. doi: 10.1074/jbc.M400798200. [DOI] [PubMed] [Google Scholar]
- 27.Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106:165–169. doi: 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol. 2002;12:65–71. doi: 10.1016/s0962-8924(01)02207-3. [DOI] [PubMed] [Google Scholar]
- 29.Watson RT, Pessin JE. Intracellular organization of insulin signaling and GLUT4 translocation. Recent Prog Horm Res. 2001;56:175–193. doi: 10.1210/rp.56.1.175. [DOI] [PubMed] [Google Scholar]
- 30.Jarousse N, Wilson JD, Arac D, Rizo J, Kelly RB. Endocytosis of synaptotagmin 1 is mediated by a novel, tryptophan-containing motif. Traffic. 2003;4:468–478. doi: 10.1034/j.1600-0854.2003.00101.x. [DOI] [PubMed] [Google Scholar]
- 31.Jarousse N, Kelly RB. The AP2 binding site of synaptotagmin 1 is not an internalization signal but a regulator of endocytosis. J Cell Biol. 2001;154:857–866. doi: 10.1083/jcb.200103040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dasgupta S, Kelly RB. Internalization signals in synaptotagmin VII utilizing two independent pathways are masked by intramolecular inhibitions. J Cell Sci. 2003;116:1327–1337. doi: 10.1242/jcs.00290. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Wang P, Xu J, Desir GV. Voltage-gated potassium channel Kv1.3 regulates GLUT4 trafficking to the plasma membrane via a Ca2+-dependent mechanism. Am J Physiol Cell Physiol. 2006;90:C345–C351. doi: 10.1152/ajpcell.00091.2005. [DOI] [PubMed] [Google Scholar]