

Abstract
Differentiation of dendritic cells (DCs) into particular subsets may act to shape innate and adaptive immune responses, but little is known about how this occurs during infections. Plasmacytoid dendritic cells (PDCs) are major producers of interferon (IFN)-α/β in response to many viruses. Here, the functions of these and other splenic DC subsets are further analyzed after in vivo infection with murine cytomegalovirus (MCMV). Viral challenge induced PDC maturation, their production of high levels of innate cytokines, and their ability to activate natural killer (NK) cells. The conditions also licensed PDCs to efficiently activate CD8 T cells in vitro. Non-plasmacytoid DCs induced T lymphocyte activation in vitro. As MCMV preferentially infected CD8α+ DCs, however, restricted access to antigens may limit plasmacytoid and CD11b+ DC contribution to CD8 T cell activation. IFN-α/β regulated multiple DC responses, limiting viral replication in all DC and IL-12 production especially in the CD11b+ subset but promoting PDC accumulation and CD8α+ DC maturation. Thus, during defense against a viral infection, PDCs appear specialized for initiation of innate, and as a result of their production of IFN-α/β, regulate other DCs for induction of adaptive immunity. Therefore, they may orchestrate the DC subsets to shape endogenous immune responses to viruses.
Keywords: plasmacytoid dendritic cell, interferon α/β, murine cytomegalovirus, antigen presentation, CD8 T lymphocyte
Introduction
Dendritic cells (DCs),* initially identified by their ability to initiate adaptive immune responses through antigen-specific activation of naive T cells (1), are now known to be also involved in innate immune responses through the secretion of antimicrobial cytokines such as IFN-α/β (2) or activation of other cellular effectors such as NK cells (3). They are heterogeneous populations. According to their frequency, DCs isolated from spleens of untreated, specific pathogen-free mice can be divided into three major subsets (4, 5): Ly6G/C−CD8α−CD11b+ (later referred to as CD11b+ DCs), Ly6G/C−CD8α+CD11b− (CD8α+ DCs), and Ly6G/C+CD8α+/−CD11b− plasmacytoid DCs (PDCs). The subsets can all be derived from a common B220+Ly6G/C−CD8α−CD11b+ precursor (6, 7), depending on the cytokine environment (5, 8), but different patterns of cytokine production by DC subsets may occur in response to particular microbes (4, 9) and function for antigen presentation can be assigned to different DC subsets depending on conditions of challenge (10–13). For example, both CD8α+ and CD11b+ DCs can be potent activators of naive T lymphocytes, but CD8α+ DCs have a unique ability to cross-present exogenous antigen in association with MHC class I molecules (11–13). By contrast, PDCs have been reported poor activators of CD4 T cells, even after stimulation in vitro (14–16). Thus, a picture is emerging of specialization of DC subsets in recognition of, and responses to, specific molecules expressed during microbial infections. In turn, the activation of these subsets may lead to specific pathways for differential regulation of downstream immune responses. However, knowledge on the respective roles of DC subsets in the course of microbial infections in vivo is scarce (9, 17–19), and extensive simultaneous analyses of the different functions of the major DC subsets in a given system remain to be done.
We have reported that splenic DCs are maintained as plasmacytoid, CD8α+ and CD11b+ subsets, and demonstrated the major contribution of PDCs to IFN-α/β and IL-12 production, during murine cytomegalovirus (MCMV) infection (9). However, the impact of in vivo viral infections on antigen presentation capability of PDCs and the relative consequences of particular DC functions on other DC subsets have not been evaluated previously. There is a high potential for PDCs to orchestrate both innate and acquired immune responses during certain viral infections, because the IFN-α/β cytokines produced by the subset can mediate multiple immunoregulatory functions (20). IFN-α/β regulate innate cytokine production during viral infections by promoting high levels of their own (9, 21) but limiting IL-12 production (9, 22). They also induce up-regulation of MHC class I molecules and regulate DC maturation in culture or in response to CFA or CpG in vivo (23–25).
The studies presented here were undertaken to examine the functional specialization of plasmacytoid, CD8α+ and CD11b+ DC subsets, and their regulation by IFN-α/β functions, during MCMV infection in vivo. The following questions were addressed. (a) What is the relative contribution of DC subsets to production of a panel of innate cytokines involved in defense and to NK cell activation? (b) Can PDCs activate naive T cells during viral infections? (c) Is MCMV replication within DCs part of the activation for cytokine production and/or antigen presentation? (d) Is PDC IFN-α/β production regulating DC responses during microbial infections in vivo? To examine these issues, DC subsets were isolated from the spleens of untreated or d1.5 MCMV-infected mice and analyzed for various parameters. The time point was chosen as it is relevant to functions involved in the activation of both innate and acquired immunity; day 1.5 corresponds to the peak of innate cytokine production (9, 26) and to the likely initiation of acquired immune responses (27–29). Globally, PDCs and other DC subsets mediated nonredundant, complementary functions. Even though they had acquired the ability to efficiently activate CD8 T cells in vitro, PDCs appeared specialized in the initiation of innate immune responses in vivo, and other DCs in the induction of T cell responses. However, multiple DC functions were regulated by PDC-derived IFN-α/β, in a specific way for each DC subset. Thus, the results suggest a coordinated specialization of DC subsets in the activation of different arms of the immune system with PDCs and their responses orchestrating the events during a viral infection in vivo.
Materials and Methods
Mice, Virus, and Treatments.
IFN-α/β receptor-deficient (IFN-α/βR−/−) 129 mice were bred under pathogen-free conditions in the animal care facility at Brown University. Specific pathogen-free wild-type 129 (129SvEv TacFBR) and C57BL/6 mice were purchased from Taconic Farms, as well as C57BL/6 RAG-2−/− mice transgenic for a TCR specific for the lymphocytic choriomeningitis virus (LCMV) epitope GP-33–41 (H2-Db), strain 4113-TcR P14 (30). Mice were 5 to 12 wk of age. Handling of mice and experimental procedures were conducted in accordance with institutional guidelines for animal care and use. Infections were initiated on d0 by intraperitoneal injection of 104 PFU of salivary gland-extracted MCMV Smith strain, WT, or clone RVG-102 (31) recombinant for the enhanced green fluorescence protein (GFP) under the promoter of the immediate early gene-1 (ie-1). In certain experiments, NK cells were depleted in vivo by intraperitoneal injection of anti-AGM1 antibody (Wako Pure Chemical Industries, Ltd.) 1 d before and 12 h after infection.
Preparation of Cell Suspensions and Subset Enrichment.
Spleens were digested by collagenase and cells suspensions prepared as described (9). DCs and NK cells were enriched using anti-CD11c or anti-DX5 magnetic beads and positive selection columns MS+ according to manufacturer's instructions (Miltenyi Biotec). Ly6G/C+ versus Ly6G/C−, CD11b+ versus CD11b−, or GFP+ versus GFP−, DC subsets were purified from enriched CD11c+ cells by fluorescence-activated cell sorting using a FACSVantage™ with a 70-μm nozzle (9). Similarly NK cells were further purified as DX5+CD3ɛ− cells. Purity and viability of sorted populations were analyzed immediately after sorting. CD8 T cells from P14 mice were prepared by negative selection using CD8 T cell enrichment kits, supplemented with CD11c-microbeads to ensure depletion of endogenous DC populations (Miltenyi Biotec).
Quantitation of Cytokines and Macrophage Inflammatory Protein 1α.
Supernatants from purified DC subsets or from cocultures of DCs with NK or T cells were harvested and tested in ELISA. Commercial kits were used for IL-12p40 and macrophage inflammatory protein (MIP)-1α (R&D Systems), as well as a custom antibody pair for IL-12p40 as described previously (26). Limits of detection were 20 pg/ml for IL-12p40 and <1.5 pg/ml for MIP-1α. The IFN-α, IFN-γ, IL-18, and TNF-α ELISA have been described previously (9, 32, 33). Limits of detection were 1,500 pg/ml for IFN-α, 5 pg/ml for IFN-γ, 30 pg/ml for IL-18, and <5 pg/ml for TNF-α. Cytokine titers were expressed as pg/ml or ng/ml for 106 cells for analyses in overnight DC cultures and as pg/ml or ng/ml for analyses in DC/NK or T cells cocultures.
Flow Cytometric and Immunofluorescence Analyses.
Cell surface stainings were performed as described (9), using the following antibodies: CD11c-FITC, CD11c-PE, CD8α-APC, CD11b-FITC, CD11b-APC, Ly6G/C-FITC, Ly6G/C-APC, Ly6G/C-PE/Cy5, I-Ab-PE, H2-Kb-PE, CD40-PE, CD80-PE, CD86-PE, CD3ɛ-FITC, NK1.1-PE, and DX5-biotin with streptavidin-PE, purchased from BD Biosciences or eBioscience. Hyperimmune serum from MCMV-infected mice was used as a primary antibody and Cy5-conjugated affinity-purified F(ab)′2 fragment donkey anti–mouse IgG (Jackson ImmunoResearch Laboratories) as a secondary reagent for staining of viral membrane antigens. Freshly isolated cells were evaluated ex vivo for intracellular expression of IL-12 and IFN-γ as described (9), and of MCMV IE-1 protein using the Croma antibody (34) as a primary reagent and Cy5-conjugated donkey anti–mouse Ab as a secondary reagent. Samples were acquired using a FACSCalibur™ (Becton Dickinson), with the CELLQuest™ version 3.1 software package. Laser outputs were 15 mW at 488 and 635 nm wavelengths. For immunofluorescence studies of DC subset morphology and MHC class II expression, DCs were stained with CD11c-PE and Ly6G/C-FITC, purified by cell sorting in total CD11c+ cells or in Ly6G/C+ and Ly6G/C− subsets, restained in suspension with biotin-I-Ab followed by Texas Red-streptavidin and Ly6G/C-FITC. Specificity was assessed on total CD11c+ DCs using isotype control antibodies. Wright and Giemsa stainings were performed in parallel to ensure the lack of granulocyte contamination (unpublished data). Cytospins were done at various cell inputs. Sorted GFP+ and GFP− DCs isolated from d1.5 RVG102-infected 129 mice were also examined by immunofluorescence for intracellular localization of GFP. Slides were dried, fixed with 1% paraformaldehyde, washed, and mounted with anti-fading medium Vectashield (Vector Laboratories). Images were collected digitally with a single filter cube with a Spot, RT slider camera from Diagnostics, Inc. (MVI), and processed for publication with AdobePhotoshop.
RT-PCR for IL-15 mRNA.
Total RNA was extracted from ∼5 × 106 cells and retro-transcribed as described previously (9). 5 μl c-DNA was used as a template for PCR amplification using primers specific for IL-15 (35) synthesized by Operon. Amplifications were performed in a programmable thermal cycler (PTC-200; MJ Research) with 30 cycles and an annealing temperature of 54°C.
Real-time RT-PCR for Quantification of MCMV mRNA.
Total RNA was isolated using RNeasy (QIAGEN). To minimize contamination by viral genomic DNA, RNA was digested with DNase (RNase-Free DNase Set; QIAGEN). Spin columns (Zymogen) were used to concentrate samples. 100 ng total RNA was used for reverse transcription of three RVG102 mRNAs - GFP, the ie-1, and the MCMV late gene glycoprotein B (gB) - using gene-specific oligonucleotide primers and a commercially available kit (Omniscript; QIAGEN). Mock reactions omitting reverse transcriptase were run as negative controls. Quantification of cDNA was performed using a Light Cycler real-time PCR instrument (Roche Diagnostics) running Light Cycler software v3.5.3. Hybridization probe sets were designed using TM Utility v1.3, a freeware program from IT Biochem, and conjugated to dyes such as to produce a fluorescent signal upon hybridization to the accumulating PCR product (36). Hybridization of probes was detected with the Roche Fast Start Master DNA Hybridization Probes kit. 10% of the cDNA was used in the PCR reaction as template for mRNA quantification. Standards consisted of 10-fold dilutions of known concentrations of RVG102 genomic DNA (104 copies–10 copies). Fluorescence was recorded at the end of each annealing step. A single reaction consisted of 0.5 μM downstream primer, 1.0 μM upstream primer, 0.2 μM 3′FITC probe, 0.4 μM 5′LC Red 640 probe, 3 mM MgCl2, the kit master mixes at 1× concentration, and cDNA template in a total volume of 20 μl. Oligonucleotide primers and fluorochrome-labeled oligonucleotide probes were obtained from IT Biochem. The sequences as well as cycling parameters used in the reactions are given in Table I.
Table I.
Oligonucleotides and Cycling Parameters Used for Real-time RT PCR on MCMV Genes
| Gene | Primersa | Probes | Cyclesb |
|---|---|---|---|
| GFP | 5′TGCCGTCCTCGATGTTGTGG3′ | 5′LCRed640GCCACAACGTCTATATCATGGCCGACA3′ | 95°C 15 s |
| 5′AGCACGACTTCTTCAAGTCC3′ | 5′CCTGGGGCACAAGCTGGAGTACAACTACAAFITC3′ | 63°C 15 s | |
| 5′CCTTGATGCCGTTCTTCTGC3′ | 72°C 15 s | ||
| ie-1 | 5′TATCATGAGGTGTGCAATCT3′ | 5′LCRed640GGCCATCAAGCATGCTGCCAGGT3′ | 95°C 10 s |
| 5′TACAGGACAACAGAACGCTC3′ | 5′CCACGTGGGGAATGATAACAGCGACAFITC3′ | 60°C 15 s | |
| 5′CCTCGAGTCTGGAACCGAAA3′ | 72°C 10 s | ||
| gB | 5′GTAGTCCCGATACTCGTAGC3′ | 5′LCRed640GGAACCTTTCGGACGGAGAACTGTGAC3′ | 95°C 10 s |
| 5′AAGGACTACATCAACGATGCG3′ | 5′AGCTGGGCGAAAACAACGAGATTATGCTFITC3′ | 65°C 15 s | |
| 5′CACGACGAAGATCTTCCTGC3′ | 72°C 10 s |
The first sequence corresponds to the primer used in the reverse transcription, the two other sequences to the primers used in the real-time PCR.
Polymerase was first activated by heating for 10 min at 95°C.
NK Cell Activation Assay.
DC subsets were sorted from uninfected or d1.5 MCMV-infected 129 mice. 5 × 104 DCs were plated in coculture with 5 × 104 NK cells purified from the spleen of uninfected 129 mice. After 24 h, 100 μl of supernatant was harvested from each well for IFN-γ titration and replaced by 5 × 103 YAC cells labeled with 51Cr (PerkinElmer). In parallel, NK cells were sorted from uninfected or d1.5 MCMV-infected 129 mice, plated at 5 × 104/well, and used directly ex vivo for analysis of cytotoxic activity, or incubated for 24 h to measure spontaneous IFN-γ production. Wells with YAC cells alone were plated for evaluation of spontaneous and total releases. The chromium release assay was incubated for 6 h at 37°C, 5% CO2. Supernatant (50 μl) were transferred to a Lumaplate™, counted using the Topcount instrument (Packard Instrument Co./PerkinElmer), and the percentage specific release was calculated as described (37).
CD8 T Cell Antigen-specific Activation Assays.
DC subsets were sorted from uninfected or d1.5 MCMV-infected 129 mice, pulsed for 3 h at 37°C, 5% CO2 in a solution of the LCMV peptide GP33–41 (H-2Db), or with the LCMV peptide NP 396–404 (H-2Db) as a negative control. Unless specified, peptides were used at 10 μM. DCs were washed three times and resuspended in fresh medium. 5 × 104 viable naive GP33–41-specific CD8 T cells purified from the spleens of P14 mice were plated in each well with serial dilutions of the various DC suspensions. After 2 d of culture, 100 μl of supernatant was harvested from each well for IFN-γ titration, and cells were pulsed with 1 μCi of 3H-thymidine (PerkinElmer) in 100 μl of fresh medium for the last 18 h. Cells were collected and counted using the Packard Filtermate Universal Harvester and Topcount instruments (Packard Instrument Co.).
IFN-α ELISPOT Assays.
The IFN-α ELISPOT assays were adapted from IFN-γ ELISPOT described previously (38). In brief, Multiscreen–IP plates (Millipore) were coated overnight with 0.5 μg/well of the same primary antibody as used for the ELISA, washed, and blocked for 3 h with culture medium. Serial dilutions of enriched DCs from uninfected or d1.5 MCMV-infected 129 mice were then plated and incubated overnight at 37°C, 5% C02. Plates were washed and incubated with the same secondary antibody as in the ELISA, followed by biotinylated anti–rabbit antibody with minimal cross-reactivites between species (Jackson ImmunoResearch Laboratories) and extravidin-phosphatase alkaline (Sigma-Aldrich). Spots were developed and counted as described (38).
Statistical Analyses.
Statistical analyses were performed in Microsoft Excel 5.0 (Microsoft Corporation, Redmond, WA) using student's two-tailed t tests. Unless otherwise indicated, means ± SE are shown.
Results
Innate Cytokine/Chemokine Production and NK Cell Activation by DC Subsets.
The major producers of IFN-α/β and IL-12 during MCMV infections are CD11cdullLy6G/C+CD8α+/−CD11b− cells (9). To further characterize these cells, CD11c+ populations were isolated from spleens of d1.5 MCMV-infected mice, stained for cell surface markers, and examined microscopically. The CD11cdullLy6G/C+CD8α+/−CD11b− cells had plasmacytoid morphology and low expression of MHC class II as compared with CD11chighLy6G/C− DCs (Fig. 1) . Experiments were performed to determine if PDCs were induced to express other innate cytokines during MCMV infection. TNF-α, MIP-1α, IFN-γ, IL-15, and IL-18 were evaluated because these molecules also play critical roles early in the control of virus infection (32, 39, 40). In addition to IFN-α/β and IL-12, PDCs isolated from d1.5 MCMV-infected mice produced higher levels of TNF-α and MIP-1α than the other DC subsets (Fig. 2 A). They did not produce IFN-γ, which mostly originated from NK cells (Fig. 2 B) as reported previously (41). IL-15 mRNA expression was induced in total splenic leukocytes after MCMV infection. However, it segregated with non-DC populations upon sorting (Fig. 2 C). Similarly, IL-18 protein was induced in non-DC populations (unpublished data). Thus, PDCs are specialized in the production of a specific array of innate cytokines/chemokines in vivo during MCMV infection, and appear much more potent for this function than other DCs.
Figure 1.

Morphology and MHC class II expression by DC subsets. DCs were purified from d1.5 MCMV-infected 129 mice and stained for FITC-Ly6G/C and Texas-Red MHC class II (I-Ab). Cytospins were analyzed by fluorescent microcopy using a double red/green filter. Representative photographs of individual cells are shown. Scale bar is 10 μm. (A–C) CD11c+Ly6G/C+ purified cells. Most cells show a round morphology (A and B) and do not express detectable levels of MHC class II. Only few cells have dendrites (C). (D–F) CD11c+Ly6G/C− purified cells. All cells express high levels of MHC class II and most of them have a clear web of long dendrites (E and F). (G) One example of the few cells coexpressing Ly6G/C and MHC class II. (H and I) Photographs taken from total CD11c+ purified cells showing Ly6G/C+ MHC class II− plasmacytoid cells and Ly6G/C− MHC class II+ cells with developed dendrites in a single magnification field.
Figure 2.

Innate cytokine/chemokine production and NK cell activation by DC subsets. Mice were untreated or infected intraperitoneally for 1.5 d with 104 PFU MCMV. The ability of DC subsets to produce various soluble factors ex vivo and to rapidly activate NK cells in vitro was then evaluated. (A) Cytokine and MIP-1α titers in 24 h conditioned media from Ly6G/C+ (left) and Ly6G/C− (right) DC subsets isolated from the spleens of d1.5 MCMV-infected 129 mice. The top dot-plots show the populations after sorting. The purity was typically >95% for Ly6G/C+ and >99% for Ly6G/C− DCs. No or very low levels of cytokines were detected in DCs isolated from uninfected mice (unpublished data). The data are representative of at least three independent experiments. Similar results were obtained with DC subsets enriched from mice depleted of NK cells in vivo (unpublished data). (B) Flow cytometry analyses of IL-12 and IFN-γ expression ex vivo in Ly6G/C+ DCs (top panels) and in NK cells (bottom panels) from d1.5 MCMV-infected C57BL/6 mice. Similar results were obtained with cells from 129 mice, although with a lower production of IFN-γ (unpublished data). (C) Analyses of the expression of IL-15 mRNA by splenic leukocytes freshly isolated from d1.5 MCMV-infected or vehicle-treated 129 mice and fractionated for CD11c expression. IFN-α/β mRNA were detectable in all samples from infected mice (reference 9). (D) NK cell activation by DC subsets. Splenic NK cells were sorted from untreated 129 mice and cocultured for 24 h with Ly6G/C+ or Ly6G/C− DCs isolated from the spleen of untreated (d0) or d1.5 MCMV-infected 129 mice. As a positive control, NK cells were isolated from d1.5 MCMV-infected mice and tested directly ex vivo (gray bars). Cytotoxic activity against YAC cells (top panel) and IFN-γ production (bottom panel) were then evaluated as described in Materials and Methods. Cytotoxic responses in cocultures are shown after subtraction of the background measured in cultures of DCs or NK alone. No IFN-γ production was observed in cultures of DCs alone or NK cells isolated from uninfected mice alone. BLD, below level of detection.
The ability of DC subsets to rapidly activate NK cells in vitro was assessed. Ly6G/C+ and Ly6G/C− DCs were isolated from uninfected or d1.5 MCMV-infected mice and cocultivated with NK cells freshly isolated from uninfected mice. After 24 h, parameters of NK cell activation were measured (Fig. 2 D). Low levels of cytotoxicity were detectable in cultures of DCs alone from infected mice, even after in vivo depletion of NK cells (unpublished data). However, a clear induction of cytotoxic activity and IFN-γ production was detectable after coculture of NK cells with PDCs from infected mice, but not with PDCs from uninfected mice or with Ly6G/C− DCs. The levels of IFN-γ production and cytotoxic activity induced upon in vitro activation were within two- to fourfold of those of NK cells isolated at the peak time of in vivo activation, i.e., d1.5 after infection. No NK cell proliferation was detected in any of the conditions tested (unpublished data). Thus, in response to MCMV infection in vivo, PDCs specifically appear to acquire a potential for rapid activation of NK cell functions.
Antigen Presentation Potential.
The maturation status and antigen presentation potential of PDCs were compared with those of other DCs (Fig. 3) . Immature PDCs expressed far lower levels of MHC class II and costimulatory molecules than other DC subsets (Fig. 3 A). By contrast, their level of MHC class I expression was similar. After in vivo infection with MCMV, all DC subsets up-regulated MHC and costimulatory molecules. However, the expression of MHC class II and costimulatory molecules remained globally less on PDCs. Thus, PDCs mature in vivo in response to MCMV infection but may retain a lower potential for T cell priming than other DC subset.
Figure 3.
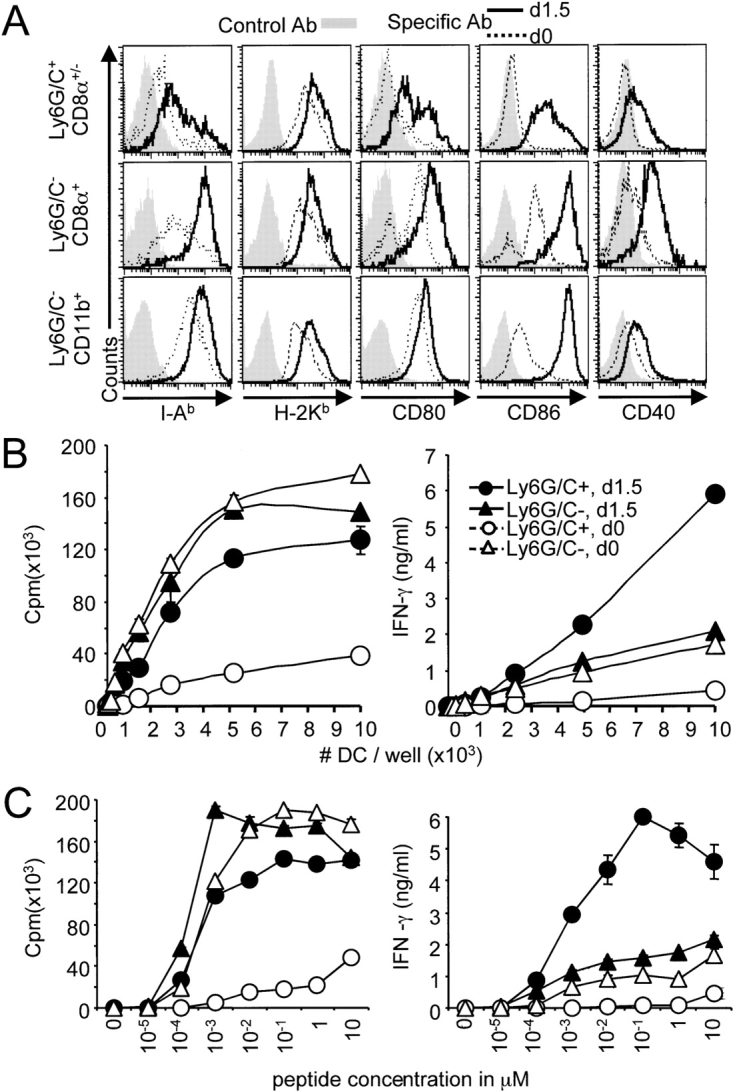
DC subset maturation and activation of T cells. (A) Expression of MHC and costimulatory molecules on DC subsets from uninfected (dotted line) and d1.5 MCMV-infected (bold line) 129 mice. Splenic DCs were enriched using CD11c microbeads and the expression of MHC class II I-Ab, class I H-2Kb, CD80, CD86, and CD40 molecules (x-axis) was analyzed on DC subsets (y-axis). Stainings with isotype control antibodies are shown as gray histograms. The data shown are representative of at least three independent experiments. (B) In vitro antigen-specific activation of CD8 T cells by DC subsets. Ly6G/C+ (•, ○) and Ly6G/C− (▴, ▵) DC subsets were sorted from uninfected (○, ▵) or d1.5 MCMV-infected (•, ▴) 129 mice, pulsed with the LCMV GP33–41 epitope, and cocultivated for 2 d with naive CD8 T cell expressing a transgenic TCR specific for this peptide. Proliferation (left panel) and IFN-γ production (right panel) were measured after 2 d. DCs pulsed with the LCMV NP396–404 epitope were used as a negative control, no activation of the CD8 T cells was detectable in the corresponding cultures (unpublished data). The data shown are representative of at least three independent experiments. (C) Peptide dose–response curve for the analysis of DC subset activation of CD8 T cells. Experiments were performed as above, except that DCs were incubated with serial dilutions of the GP33–41 peptide and plated at 10,000 cells/well. The data are representative of three independent experiments.
The ability of DCs to activate CD8 T cells was evaluated next. As both CD8α+ and CD11b+ DCs are known to have a high potential for antigen-specific priming of T lymphocytes in vitro when pulsed with peptides, these two subsets were analyzed together (i.e., Ly6G/C− DCs) for induction of CD8 T cell responses in vitro. Freshly isolated CD8 T cells expressing a transgenic TCR specific for the LCMV epitope GP-33–41 were cocultured with peptide-pulsed DC subsets (Fig. 3 B). Immature PDCs were far less efficient than their Ly6G/C- counterparts at priming CD8 T cells, both in terms of proliferation and IFN-γ production. However, after in vivo challenge with MCMV, PDCs became as potent as other DCs for antigen-specific activation of naive CD8 T cells, and induced higher levels of IFN-γ. The responses occurred only in the presence of an antigen-specific activation of the CD8 T cells, as they were not observed when DCs were pulsed with a negative control peptide (data not depicted). Peptide concentrations required to induce efficient activation of CD8 T cells by nonplasmacytoid DCs and mature PDCs were similar, with responses reaching a plateau at 1 nM (Fig. 3 C). Thus, immature PDCs are not very efficient at priming T cells. However, PDCs become very potent antigen-presenting cells for CD8 T lymphocytes in vivo in response to MCMV infection.
To evaluate the potential role of DC subsets for in vivo activation of antiviral CD8 T cells, their accessibility to viral antigens for presentation with MHC class I was evaluated during MCMV infection. To accomplish this, a recombinant virus RVG-102 expressing GFP under the ie-1 promoter (31) was used to track the DCs replicating MCMV or taking up apoptotic bodies from infected cells in vivo. A small proportion of DCs was clearly positive for GFP at d1.5 after infection (Fig. 4 A, top left dot plot). Flow cytometric analyses, using serum of hyper-immune mice for detecting overall membrane antigens, or the Croma antibody for specifically detecting the IE-1 protein, showed that most of the GFP+ DCs had high intensity expression of the MCMV antigens, whereas no specific staining was observed on GFP− DCs (unpublished data). To test for cells that might have been GFP− at the time of isolation because they were at very early stages of viral replication, sorted GFP− DCs (Fig. 4 A, bottom left dot plot) were reanalyzed for GFP expression after 24 h (bottom right dot plot) or 48 h (unpublished data) in culture. As compared with an initial fraction of 0.8–1% of GFP+ cells in total DCs, less than 0.03% of the negative DCs acquired GFP expression over a 48 h period of culture. Thus, at the time of ex vivo analysis, the populations at stages of productive virus replication preceding detectable GFP expression represented less than 4% of total infected DCs. The sensitivity of following GFP expression as an indication of infection was further demonstrated by ex vivo analysis of MCMV transcripts expression in total, GFP+ and GFP− DCs. These studies showed that most cells replicating MCMV score as GFP+ (Fig. 4 A, histograms) because GFP+ cells were highly enriched for mRNA expression of the ie-1 and gB MCMV genes as quantified by the sensitive real time RT-PCR analyses. Taken together, these results showed that analysis of GFP expression is a very sensitive assay for detecting infected cells in our system. Interestingly, most of the GFP+ cells appeared to be replicating the virus rather than taking up apoptotic bodies because 86% of the DCs expressed GFP throughout the cell as evaluated by fluorescent microscopy (Fig. 4 B, a–c). A punctuated pattern of expression was observed in only 14% of the cells (Fig. 4 B, d and e). Characterization of the DC subsets demonstrated that most of the GFP+ cells belonged to the CD8α+ population, whereas the number of GFP+ PDCs was very low (Fig. 4 C). Hence, although mature PDC can efficiently prime naive CD8 T cells in vitro, their contribution for this function during MCMV infection in vivo may be low as compared with CD8α+ DCs, due to a restricted access to viral antigen for presentation with MHC class I molecules.
Figure 4.

DC subset accessibility to MCMV antigens in vivo. (A) Analysis of the expression of MCMV transcripts in total, GFP+ and GFP− DCs. DCs were enriched from the spleen of 129 mice at 1.5 d after infection with the MCMV clone RVG-102, which express the GFP under the ie-1 promoter. DCs were then sorted in total, GFP+ and GFP− populations. The negative fraction was reanalyzed for GFP expression after 24 h in vitro culture, to allow expression of the marker in the eventual cells that were at very early stages of viral replication and scored negative at the time of isolation (bottom right dot plot). Number of events analyzed were 20,000 for total, 2,000 for GFP+, 770,000 for GFP−, and 650,000 for GFP− 24 h DCs. Percentages of GFP+ DCs are given above each dot plot. The data are from one representative experiment out of three. RNA was extracted from the GFP+, total and GFP− DC populations ex vivo, and transcripts for GFP, ie-1, and the glycoprotein-B (gB) were quantified by real-time RT-PCR (bar graphs). For the gB transcripts, values were 1,258, 57 and undetectable for triplicates cell pellets of GFP− DCs. The data are from one representative experiment of two. (B) Immunofluorescence analysis of GFP localization within positive DCs. Sorted GFP+ DCs were observed under a fluorescent microscope. No green signal was observed in the GFP− DCs under the same conditions (unpublished data). Scale bar is 10 μm. (C) Analysis of GFP expression in DC subsets. DCs were enriched from the spleen of 129 mice at 1.5 d after infection with RVG-102. The simultaneous expression of GFP (x-axis) and typical membrane markers (y-axis) was analyzed by flow cytometry within CD11c+ cells (left panel) or CD11c+Ly6G/C− cells (middle and right panels). The percentages of cells in the quadrants are given above each dot-plot. The data are representative of at least three independent experiments.
Role of Viral Infection and IFN-α/β on DC Subset Cytokine Production.
Until recently, IFN-α/β production during viral infection was believed to occur mainly in infected cells through triggering of intracellular receptors by viral components (42). However, it has been recently shown that IFN-α/β can also be induced in vitro in uninfected DCs through activation by glycosylated viral proteins (43) or double-stranded RNA (44). During ongoing viral infections in vivo, it is not known whether cytokine production is induced in single DCs as a result of viral replication inside the cell, of uptake of apoptotic bodies from dying infected cells, or of other signals delivered by neighboring infected cells. To address the relationship between expression of viral products and production of IFN-α/β, the frequencies of IFN-α spot-forming cells (SFCs) and of GFP+ cells were measured within DCs isolated from RVG-102–infected mice, by ELISPOT and flow cytometry, respectively (Fig. 5 A). After infection, the proportion of PDCs secreting IFN-α was consistently higher than those expressing GFP. These results were confirmed, and extended to IL-12 production, by comparing the levels of cytokines secreted by sorted GFP+ versus GFP− DCs (unpublished data). Moreover, when GFP and IL-12 expression were analyzed simultaneously in DC subsets isolated from d1.5 MCMV-infected mice, by flow cytometry, it was demonstrated that the two occurred in largely distinct populations of cells (Fig. 5 B). This suggests that, at the single cell level, viral replication or uptake of apoptotic bodies from infected cells is neither required nor sufficient for induction of either IFN-α/β or IL-12 production in DCs.
Figure 5.
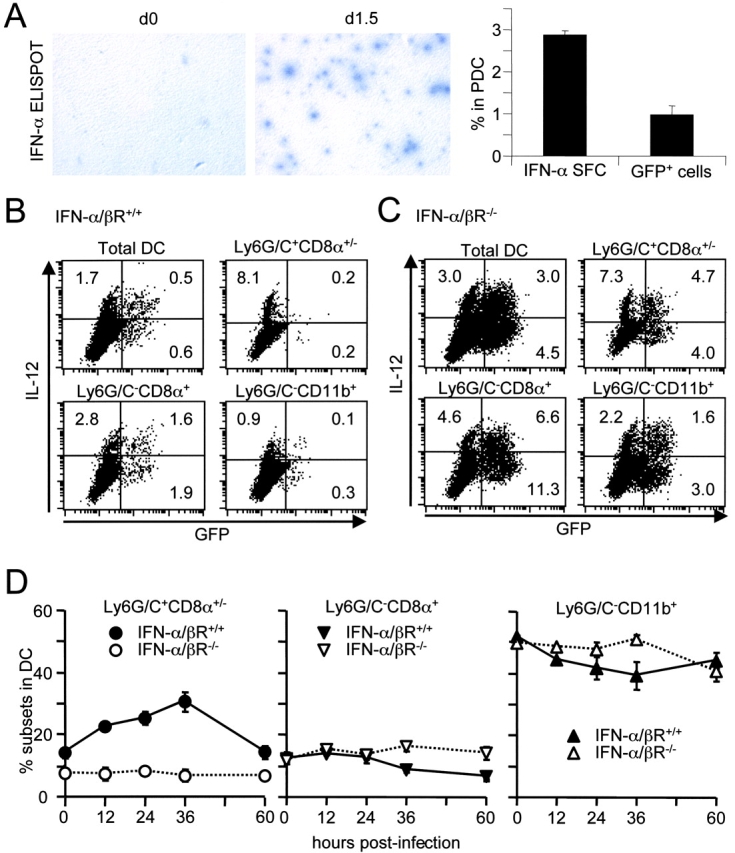
Cytokine production and expression of MCMV products in DC subsets. (A) Comparison of the frequencies of cells secreting IFN-α or expressing viral products in PDCs. The left panel shows photographs of IFN-α ELISPOT assays performed with DC populations from splenic leukocytes of 129 mice uninfected (d0) or at 1.5 d after infection with the MCMV clone RVG-102 (d1.5). The frequencies of IFN-α spot forming cells (SFC) versus GFP+ cells within PDCs are given on the right (average and standard deviations for three mice). (B) Simultaneous analysis of GFP (x-axis) and IL-12 (y-axis) expression in DC populations from splenic leukocytes of 129 IFN-α/βR+/+ at 1.5 d after infection with the MCMV clone RVG-102. The data are representative of at least three independent experiments. (C) Similar panel for IFN-α/βR−/− mice. The data are representative of two independent experiments. (D) Impact of IFN-α/β functions on DC subset accumulation in the spleen. IFN-α/βR+/+ (•, ▾, ▴) versus IFN-α/βR−/− (○, ▿, ▵) mice were infected with MCMV for various length of times and the frequency of subsets (Ly6G/C+: ○, •, CD8α+: ▿, ▾, and CD11b+: ▵, ▴) in splenic DCs was analyzed by flow cytometry. The data shown are representative of at least three independent experiments for the 36 h time point.
Mice lacking IFN-α/β functions produce more IL-12, especially in CD11b+ DCs, and this is associated with enhanced viral replication in the spleen (9). To determine whether the increase in IL-12 production by CD11b+ DCs was a result of enhanced exposure to viral products in those cells, we analyzed the relationship between GFP expression and IL-12 production in DC from RVG-102–infected IFN-α/βR−/− mice. In the absence of IFN-α/β functions, a significant increase in GFP expression was observed in all DC subsets (Fig. 5 C, Table II). An increase in the relative contribution of the CD11b+ DC subset to MCMV replication was consistently observed in the IFN-α/βR−/− mice at d1.5 and d2 after infection, where they accounted for 50% or more of the total GFP+ DCs versus less than 30% in the IFN-α/βR+/+ mice. However, in the absence of IFN-α/β functions, IL-12 production and GFP expression still occurred in largely distinct populations of cells (Fig. 5 C, Table II). Moreover, a significant increase in IL-12 production was observed in uninfected DCs, mostly in the CD11b+ subset but not in PDCs (Table II). These observations confirm that IFN-α/β inhibits IL-12 production preferentially in the CD11b+ DC subset, and show that this occurs in part independently of changes in expression of viral products within the cells.
Table II.
Impact of IFN-α/β Functions on MCMV Infection of, and IL-12 Production by, DC Subsets
| Percent GFP versus IL-12 expression in DC subsets
|
||||
|---|---|---|---|---|
| Mouse strain
|
||||
| DC subseta | Population | IFN-α/βR+/+
(n = 3)c |
IFN-α/βR−/−
(n = 4)c |
Pb |
| Ly6G/C+CD8α+/− | GFP+IL-12− | 0.26 ± 0.04 | 3.38 ± 0.40 | <10−4 |
| GFP+IL-12+ | 0.25 ± 0.05 | 5.16 ± 0.45 | <10−5 | |
| GFP−IL-12+ | 7.15 ± 1.81 | 7.95 ± 0.61 | NS | |
| Ly6G/C−CD8α+ | GFP+IL-12− | 2.63 ± 0.89 | 11.54 ± 0.83 | <10−4 |
| GFP+IL-12+ | 1.30 ± 0.26 | 8.17 ± 1.21 | <10−3 | |
| GFP−IL-12+ | 2.63 ± 0.53 | 4.50 ± 0.09 | <10−2 | |
| Ly6G/C−CD11b+ | GFP+IL-12− | 0.29 ± 0.09 | 2.42 ± 0.23 | <10−3 |
| GFP+IL-12+ | 0.08 ± 0.03 | 1.31 ± 0.23 | <10−3 | |
| GFP−IL-12+ | 0.79 ± 0.12 | 1.95 ± 0.21 | <10−3 | |
129 IFN-α/βR+/+ and IFN-α/βR−/− mice were infected for 1.5 d with MCMV RVG102 which encodes GFP under the promoter of the ie-1 gene. DCs were enriched from spleens and stained for membrane markers and intracellular IL-12 as described in Materials and Methods.
Statistical significance.
Mean ± standard deviation.
We had previously reported that mice lacking IFN-α/β functions produced less of these cytokines (9). Here, the impact of IFN-α/β functions on the kinetics of DC subset accumulation in the spleen during MCMV infection was examined (Fig. 5 D). In contrast to other DC subsets, PDCs frequency increased within the DC population of the spleen to reach a peak around 36 h after MCMV challenge, at the time of maximal IFN-α/β production, in IFN-α/βR+/+ mice. This increase was completely abrogated in IFN-α/βR−/− mice. Thus, IFN-α/β functions are necessary for the accumulation of PDCs, the major IFN-α/β producers, in the spleen during MCMV infection, and this likely contributes to the amplification of the cytokines.
Role of IFN-α/β on Maturation of DC Subsets.
The ex vivo expression of MHC and costimulatory molecules by DC subsets was next compared between IFN-α/βR+/+ and IFN-α/βR−/− mice at d1.5 MCMV infection (Fig. 6 A). No major changes were observed for MHC class II expression in the absence of IFN-α/β functions. However, the up-regulation of MHC class I and CD86 was reduced on all DC subsets, but to different extents. The maturation of the CD8α+ DC subpopulation was especially dependent upon intact IFN-α/β responses, as the up-regulation of MHC class I, CD80, and CD86 was almost completely abrogated, and that of CD40 reduced. In contrast, the maturation of the CD11b+ DCs was much less affected. Finally, the injection of biologically active recombinant IFN-α in the absence of viral infection did not induce detectable maturation of the DCs (unpublished data). The differential regulation of DC subset maturation by IFN-α/β was confirmed at the functional level, as twice as many CD11b− DCs were required to induce equivalent levels of CD8 T cell antigen-specific proliferation in IFN-α/βR−/− mice as compared with their IFN-α/βR+/+ counterparts (Fig. 6 B). The levels of IFN-γ secreted by the CD8 T cells were also decreased when using CD11b− DCs from IFN-α/βR−/− mice as antigen-presenting cells. Thus, even during a viral infection where there is ongoing replication of a pathogen and production of a complex array of innate cytokines, intact IFN-α/β functions are required to promote the maturation of DCs, especially of the CD8α+ subset.
Figure 6.

Impact of IFN-α/β functions on DC subset maturation. (A) Expression of MHC and costimulatory molecules on DC subsets from untreated (dotted line) or d1.5 MCMV-infected IFN-α/βR+/+ (bold line) versus IFN-α/βR−/− (gray histogram) mice. (B) In vitro activation of antigen-specific proliferation of CD8 T cells by CD11b− (⋄, ♦) and CD11b+ (▵, ▴) DC subsets isolated from d1.5 MCMV-infected IFN-α/βR+/+ (plain line) versus IFN-α/βR−/− (dotted line) mice. Bar graphs show IFN-γ production by the CD8 T cells under the same culture conditions. Experiments were performed as described in the legend of Fig. 3 B.
Discussion
We have established the critical contribution of mouse PDCs to IFN-α/β production during MCMV infection (9). Here the functions and regulation of the three major splenic DC subsets (i.e., PDCs, CD8α+, and CD11b+) are further characterized at d1.5 after MCMV infection. PDCs from MCMV-infected mice secreted high levels of innate cytokines and induced rapid activation of NK cells ex vivo. PDCs from untreated mice had only weak antigen presentation capabilities. MCMV infection in vivo induced PDC maturation, licensing them to efficiently activate CD8 T cells in vitro. Non-plasmacytoid DCs did not produce IFN-α/β, secreted only low levels of the other innate cytokines, and did not induce detectable NK cell activation upon short coculture. However, they were very efficient at activating naive CD8 T cells in vitro. Expression of MCMV antigens seemed largely restricted to CD8α+ DCs, suggesting that the contribution of other DC subsets for in vivo activation of antiviral CD8 T cells may be low. Thus, PDCs were specialized in the activation of innate immune responses early during MCMV infection, whereas other DCs were likely to initiate acquired immune responses. However, PDC-derived IFN-α/β regulated multiple DC functions during MCMV infection in vivo, with different effects on DC subsets. In particular, the cytokines were necessary for maturation of CD8α+ DCs. Altogether, the results suggest that PDCs are a cornerstone for induction of both innate and adaptive immune responses to MCMV infection in vivo, through production of various cytokines, direct activation of NK cells, and indirect regulation of T cell functions by IFN-α/β–mediated effects on other DCs.
Different mechanisms are in place to induce cytokine production by cells from the innate immune system in response to microbial challenges. Cells can produce cytokines as a consequence of recognition of molecular patterns shared within a class of microbes, through pattern-recognition receptors (PRRs; reference 45). Alternatively, responses can be triggered by host products generated during inflammation, such as heat shock proteins (hsp; reference 46). Both cytoplasmic and membrane-bound PRRs have been involved in IFN-α/β induction during in vitro viral challenges (42–44). All these mechanisms of danger recognition are relevant to DC physiology, because DCs are susceptible to infection by many viruses, can engulf apoptotic bodies from dying cells, and express a variety of membrane-bound PRRs or hsp ligands. Here, the relationship between expression of viral products and production of cytokines in DC subsets was examined during MCMV infection. A virus recombinant for the GFP under the ie-1 promoter was used to track DCs that were productively infected by MCMV in vivo or had engulfed whole apoptotic bodies from neighboring infected cells. GFP and cytokine expression occurred in largely distinct populations of DCs. As the results show that uptake of apoptotic bodies from infected cells or productive viral infection within DCs was neither required nor sufficient for induction of cytokine production, some of the other pathways must contribute to the induction of cytokine production by DCs during MCMV infection. These likely include recognition of viral components through Toll-like receptors (TLRs; reference 45) or other receptors. Although beyond the scope of this report, ongoing studies in our laboratory are aimed at defining these.
Several investigators have shown the occurrence of an auto-amplification pathway regulating the transcription of interferon genes during viral infections in vitro (47). Our group has demonstrated that intact IFN-α/β functions were required for high level productions of IFN-α/β during LCMV (21) and MCMV infections in vivo (9). As IFN-α/β have been shown to promote the survival of PDCs in vitro both in the mouse (16) and in the human (48), other mechanisms in addition to transcriptional regulation of the interferon genes could be involved in the positive feedback loop for production of the cytokines in vivo during viral infections. Indeed, IFN-α/β functions were required for accumulation of PDCs in the spleen at the time of local peak production of innate cytokines. Thus, one of the mechanisms by which IFN-α/β enhance their own production during MCMV infection is the promotion of PDC accumulation.
Both in mice and in humans, in vitro studies have demonstrated that DCs can activate NK cells through membrane contacts and that this may be at least partially independent of IFN-α/β and IL-12 production (for a review, see reference 3). DCs have also been shown to activate NK-dependent antitumoral responses in vivo (49). However, the roles of the different DC subsets for this function have not been reported. Here, only DCs isolated from d1.5 MCMV-infected mice were able to activate NK cytotoxicity and IFN-γ production in 24 h of coculture, and this function segregated with PDCs. Preliminary results suggested that IFN-α/β functions and IL-12 production were required for optimization of NK cell responses. Whether other cytokines or cell–cell contacts are involved still needs to be investigated. Whatever the case, these results demonstrate that PDCs are more efficient than other DC subsets at activating NK cells early during MCMV infection.
In consistency with the observations reported in vitro with resting human PDCs (48, 50), mouse PDCs isolated from uninfected mice were relatively poor at antigen presentation. Responses to MCMV infection induced PDCs to become very potent antigen-presenting cells for priming CD8 T cells in vitro, with induction of higher levels of IFN-γ production than other DCs. In vivo, expression of MCMV antigens seemed largely restricted to CD8α+ DCs. These cells have also been shown to have the unique ability to cross-present exogenous antigen in association with MHC class I (11–13). Therefore, CD8α+ but not CD11b+ or PDCs are likely to play a major role in the direct activation of MCMV-specific CD8 T cells in vivo. However, PDCs may be necessary for optimal induction of CD8 T cell responses during MCMV infection, through IL-12 or IFN-α/β–mediated effects on the T lymphocytes themselves or on other DC subsets. Indeed, it is shown here that IFN-α/β functions are required for CD8α+ DC maturation in vivo during MCMV infection, and we have previously reported that IFN-α/β promote CD8 T cell IFN-γ production in vivo during viral infections (33, 51, 52).
Altogether, the results presented here suggest a model of DC subset specialization and DC cross-talk during MCMV infection, with a central role for IFN-α/β functions in regulating both innate and acquired immune responses (Fig. 7) . Early in response to MCMV challenge, PDCs are activated to produce IFN-α/β. The cytokines can elicit mechanisms directly mediating antiviral effects (arrow 1). They have also a number of important immunoregulatory functions. In particular, they induce NK cell cytotoxicity (arrow 2). Simultaneous PDC production of IL-12 is inducing NK cell IFN-γ responses. The PDC-derived IFN-α/β is also controlling the accumulation, maturation and cytokine production of DC subsets. IFN-α/β promote increased proportions of PDCs in the spleen, and this is likely to contribute to the amplification loop for secretion of the cytokines (arrow 3). PDC-produced IFN-α/β are also generally required for maturation of DCs, especially the CD8α+ subset (arrow 4). As this subset harbor viral products, it is likely to initiate acquired CD8 T cell responses by presentation of MCMV-derived epitopes in association with the MHC class I molecules. Thus, IFN-α/β induction of DC maturation may indirectly contribute to the activation of CD8 T cells. In addition, IFN-α/β are enhancing the maturation of, and inhibiting IL-12 production by, CD11b+ DCs (arrow 5). Because CD11b+ DCs have been reported particularly efficient for priming CD4 T cells in other systems (12), IFN-α/β–mediated regulation of this DC subset may be in place to shape CD4 T cell responses during viral challenges. Moreover, PDCs can participate in the activation of anti-MCMV CD8 T cell responses through mechanisms in addition to those downstream of IFN-α/β–induced CD8α+ DC maturation (arrow 6). In particular, if armed with antigen, they may directly support antigen-specific activation of the CD8 T cells. In addition, their production of cytokines may enhance IFN-γ production by CD8 T cells. Hence, during MCMV infection in vivo, PDCs are specialized in the activation of innate immune responses but, as a result of their maturation and production of IFN-α/β, they may also play a central role in the orchestration of global DC functions and downstream adaptive immune responses.
Figure 7.
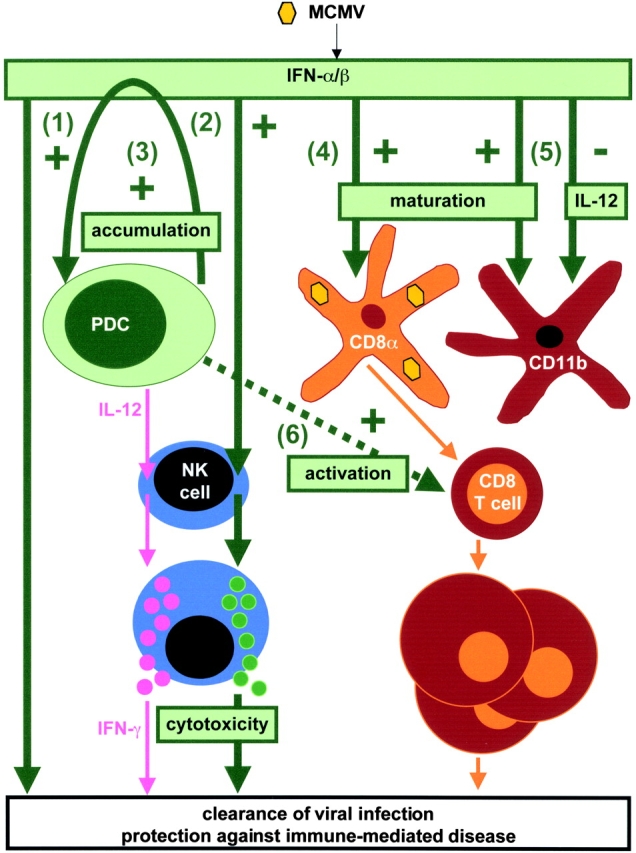
Model for DC subset functional specialization and cross-talk in vivo during MCMV infection. See Discussion for Figure comments.
Acknowledgments
The authors wish to thank Drs. Giorgio Trinchieri, Francine Brière, Carine Asselin-Paturel (Schering-Plough, Dardilly, France), Khuong Nguyen, Laurent Brossay (Brown University), and Eric Vivier (Center For Immunology at Marseille-Luminy, France) for insightful discussions. We are also grateful to Dr. H.W. Virgin for providing us with the Croma hybridoma (Washington University School of Medicine, St. Louis, MO), and to Sara Spangenberger and Dr. Paul McMillan for assistance with the cell sorting experiments.
This work was supported by National Institutes of Health grants CA41268 and CA70076. M. Dalod is a fellow of the Cancer Research Institute. R. Salomon is a recipient of National Institutes of Health grant F31GM20760.
Marc Dalod's present address is Centre d'Immunologie de Marseille-Luminy, INSERM-CNRS-Univ. Méditerranée, F-13288 Marseille Cedex 09, France.
Footnotes
Abbreviations used in this paper: DC, dendritic cell; GFP, green fluorescence protein; ie-1, immediate early gene-1; LCMV, lymphocytic choriomeningitis virus; MCMV, murine cytomegalovirus; MIP, macrophage inflammatory protein; PDC, plasmacytoid DC; PRR, pattern-recognition receptor.
References
- 1.Steinman, R.M. 1991. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 9:271–296. [DOI] [PubMed] [Google Scholar]
- 2.Colonna, M., A. Krug, and M. Cella. 2002. Interferon-producing cells: on the front line in immune responses against pathogens. Curr. Opin. Immunol. 14:373–379. [DOI] [PubMed] [Google Scholar]
- 3.Zitvogel, L. 2002. Dendritic and natural killer cells cooperate in the control/switch of innate immunity. J. Exp. Med. 195:F9–F14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 5.Ardavin, C., G. Martinez del Hoyo, P. Martin, F. Anjuere, C.F. Arias, A.R. Marin, S. Ruiz, V. Parrillas, and H. Hernandez. 2001. Origin and differentiation of dendritic cells. Trends Immunol. 22:691–700. [DOI] [PubMed] [Google Scholar]
- 6.Bruno, L., T. Seidl, and A. Lanzavecchia. 2001. Mouse pre-immunocytes as non-proliferating multipotent precursors of macrophages, interferon-producing cells, CD8alpha(+) and CD8alpha(-) dendritic cells. Eur. J. Immunol. 31:3403–3412. [DOI] [PubMed] [Google Scholar]
- 7.Martinez del Hoyo, G., P. Martin, H.H. Vargas, S. Ruiz, C.F. Arias, and C. Ardavin. 2002. Characterization of a common precursor population for dendritic cells. Nature. 415:1043–1047. [DOI] [PubMed] [Google Scholar]
- 8.Gilliet, M., A. Boonstra, C. Paturel, S. Antonenko, X.L. Xu, G. Trinchieri, A. O'Garra, and Y.J. Liu. 2002. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3-ligand and granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 195:953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalod, M., T.P. Salazar-Mather, L. Malmgaard, C. Lewis, C. Asselin-Paturel, F. Briere, G. Trinchieri, and C.A. Biron. 2002. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 195:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moron, G., P. Rueda, I. Casal, and C. Leclerc. 2002. CD8alpha(−) CD11b(+) dendritic cells present exogenous virus-like particles to CD8(+) T cells and subsequently express CD8alpha and CD205 molecules. J. Exp. Med. 195:1233–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.den Haan, J.M., S.M. Lehar, and M.J. Bevan. 2000. CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pooley, J.L., W.R. Heath, and K. Shortman. 2001. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 166:5327–5330. [DOI] [PubMed] [Google Scholar]
- 13.Iyoda, T., S. Shimoyama, K. Liu, Y. Omatsu, Y. Akiyama, Y. Maeda, K. Takahara, R.M. Steinman, and K. Inaba. 2002. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195:1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakano, H., M. Yanagita, and M.D. Gunn. 2001. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 194:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bjorck, P. 2001. Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte-macrophage colony-stimulating factor-treated mice. Blood. 98:3520–3526. [DOI] [PubMed] [Google Scholar]
- 16.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, and G. Trinchieri. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 17.Wick, M.J. 2002. The role of dendritic cells during Salmonella infection. Curr. Opin. Immunol. 14:437–443. [DOI] [PubMed] [Google Scholar]
- 18.Scott, P., and C.A. Hunter. 2002. Dendritic cells and immunity to leishmaniasis and toxoplasmosis. Curr. Opin. Immunol. 14:466–470. [DOI] [PubMed] [Google Scholar]
- 19.Jiao, X., R. Lo-Man, P. Guermonprez, L. Fiette, E. Deriaud, S. Burgaud, B. Gicquel, N. Winter, and C. Leclerc. 2002. Dendritic cells are host cells for mycobacteria in vivo that trigger innate and acquired immunity. J. Immunol. 168:1294–1301. [DOI] [PubMed] [Google Scholar]
- 20.Biron, C.A. 2001. Interferons α and β as immune regulators - a new look. Immunity. 14:661–664. [DOI] [PubMed] [Google Scholar]
- 21.Malmgaard, L., T.P. Salazar-Mather, C.A. Lewis, and C.A. Biron. 2002. Promotion of alpha/beta interferon induction during in vivo viral infection through alpha/beta interferon receptor/STAT1 system-dependent and -independent pathways. J. Virol. 76:4520–4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cousens, L.P., J.S. Orange, H.C. Su, and C.A. Biron. 1997. Interferon-alpha/beta inhibition of interleukin 12 and interferon-gamma production in vitro and endogenously during viral infection. Proc. Natl. Acad. Sci. USA. 94:634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montoya, M., G. Schiavoni, F. Mattei, I. Gresser, F. Belardelli, P. Borrow, and D.F. Tough. 2002. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood. 99:3263–3271. [DOI] [PubMed] [Google Scholar]
- 24.Le Bon, A., G. Schiavoni, G. D'Agostino, I. Gresser, F. Belardelli, and D.F. Tough. 2001. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 14:461–470. [DOI] [PubMed] [Google Scholar]
- 25.Cho, H.J., T. Hayashi, S.K. Datta, K. Takabayashi, J.H. Van Uden, A. Horner, M. Corr, and E. Raz. 2002. IFN-alphabeta promote priming of antigen-specific CD8(+) and CD4(+) T lymphocytes by immunostimulatory DNA-based vaccines. J. Immunol. 168:4907–4913. [DOI] [PubMed] [Google Scholar]
- 26.Ruzek, M.C., A.H. Miller, S.M. Opal, B.D. Pearce, and C.A. Biron. 1997. Characterization of early cytokine responses and an interleukin (IL)-6-dependent pathway of endogenous glucocorticoid induction during murine cytomegalovirus infection. J. Exp. Med. 185:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salazar-Mather, T.P., R. Ishikawa, and C.A. Biron. 1996. NK cell trafficking and cytokine expression in splenic compartments after IFN induction and viral infection. J. Immunol. 157:3054–3064. [PubMed] [Google Scholar]
- 28.Norbury, C.C., D. Malide, J.S. Gibbs, J.R. Bennink, and J.W. Yewdell. 2002. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat. Immunol. 3:265–271. [DOI] [PubMed] [Google Scholar]
- 29.Stoll, S., J. Delon, T.M. Brotz, and R.N. Germain. 2002. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science. 296:1873–1876. [DOI] [PubMed] [Google Scholar]
- 30.Pircher, H., K. Burki, R. Lang, H. Hengartner, and R.M. Zinkernagel. 1989. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 342:559–561. [DOI] [PubMed] [Google Scholar]
- 31.Henry, S.C., K. Schmader, T.T. Brown, S.E. Miller, D.N. Howell, G.G. Daley, and J.D. Hamilton. 2000. Enhanced green fluorescent protein as a marker for localizing murine cytomegalovirus in acute and latent infection. J. Virol. Methods. 89:61–73. [DOI] [PubMed] [Google Scholar]
- 32.Orange, J.S., and C.A. Biron. 1996. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J. Immunol. 156:4746–4756. [PubMed] [Google Scholar]
- 33.Pien, G.C., K.B. Nguyen, L. Malmgaard, A.R. Satoskar, and C.A. Biron. 2002. A unique mechanism for innate cytokine promotion of T cell responses to viral infections. J. Immunol. 169:5827–5837. [DOI] [PubMed] [Google Scholar]
- 34.Presti, R.M., D.L. Popkin, M. Connick, S. Paetzold, and H.W. Virgin IV. 2001. Novel cell type-specific antiviral mechanism of interferon gamma action in macrophages. J. Exp. Med. 193:483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang, X., S. Sun, I. Hwang, D.F. Tough, and J. Sprent. 1998. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 8:591–599. [DOI] [PubMed] [Google Scholar]
- 36.Tyagi, S., and F.R. Kramer. 1996. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 14:303–308. [DOI] [PubMed] [Google Scholar]
- 37.Robbins, S.H., K.B. Nguyen, N. Takahashi, T. Mikayama, C.A. Biron, and L. Brossay. 2002. Cutting edge: inhibitory functions of the killer cell lectin-like receptor G1 molecule during the activation of mouse NK cells. J. Immunol. 168:2585–2589. [DOI] [PubMed] [Google Scholar]
- 38.Dalod, M., M. Dupuis, J.C. Deschemin, D. Sicard, D. Salmon, J.F. Delfraissy, A. Venet, M. Sinet, and J.G. Guillet. 1999. Broad, intense anti-human immunodeficiency virus (HIV) ex vivo CD8(+) responses in HIV type 1-infected patients: Comparison with anti-Epstein-Barr virus responses and changes during antiretroviral therapy. J. Virol. 73:7108–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen, K.B., T.P. Salazar-Mather, M. Dalod, J.B. VanDeusen, X.-Q. Wei, F.Y. Liew, M.A. Caliguiri, J.E. Durbin, and C.A. Biron. 2002. Coordinated and distinct roles for IFN-α/β-, IL-12-, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 169:4279–4287. [DOI] [PubMed] [Google Scholar]
- 40.Salazar-Mather, T.P., J.S. Orange, and C.A. Biron. 1998. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1alpha (MIP-1alpha)-dependent pathways. J. Exp. Med. 187:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orange, J.S., B. Wang, C. Terhorst, and C.A. Biron. 1995. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J. Exp. Med. 182:1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mogensen, T.H., and S.R. Paludan. 2001. Molecular pathways in virus-induced cytokine production. Microbiol. Mol. Biol. Rev. 65:131–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milone, M.C., and P. Fitzgerald-Bocarsly. 1998. The mannose receptor mediates induction of IFN-alpha in peripheral blood dendritic cells by enveloped RNA and DNA viruses. J. Immunol. 161:2391–2399. [PubMed] [Google Scholar]
- 44.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 45.Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135–145. [DOI] [PubMed] [Google Scholar]
- 46.Singh-Jasuja, H., H.U. Scherer, N. Hilf, D. Arnold-Schild, H.G. Rammensee, R.E. Toes, and H. Schild. 2000. The heat shock protein gp96 induces maturation of dendritic cells and down-regulation of its receptor. Eur. J. Immunol. 30:2211–2215. [DOI] [PubMed] [Google Scholar]
- 47.Taniguchi, T., and A. Takaoka. 2002. The interferon-alpha/beta system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr. Opin. Immunol. 14:111–116. [DOI] [PubMed] [Google Scholar]
- 48.Kadowaki, N., S. Antonenko, J.Y. Lau, and Y.J. Liu. 2000. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J. Exp. Med. 192:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernandez, N.C., A. Lozier, C. Flament, P. Ricciardi-Castagnoli, D. Bellet, M. Suter, M. Perricaudet, T. Tursz, E. Maraskovsky, and L. Zitvogel. 1999. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat. Med. 5:405–411. [DOI] [PubMed] [Google Scholar]
- 50.Cella, M., F. Facchetti, A. Lanzavecchia, and M. Colonna. 2000. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat. Immunol. 1:305–310. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen, K.B., W.T. Watford, R. Salomon, S.R. Hofmann, G.C. Pien, A. Morinobu, M. Gadina, J.J. O'Shea, and C.A. Biron. 2002. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science. 297:2063–2066. [DOI] [PubMed] [Google Scholar]
- 52.Cousens, L.P., R. Peterson, S. Hsu, A. Dorner, J.D. Altman, R. Ahmed, and C.A. Biron. 1999. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J. Exp. Med. 189:1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]