

Abstract
MHC class II (MHCII) molecules play a pivotal role in the induction and regulation of immune responses. The transcriptional coactivator class II transactivator (CIITA) controls MHCII expression. The CIITA gene is regulated by three independent promoters (pI, pIII, pIV). We have generated pIV knockout mice. These mice exhibit selective abrogation of interferon (IFN)-γ–induced MHCII expression on a wide variety of non-bone marrow–derived cells, including endothelia, epithelia, astrocytes, and fibroblasts. Constitutive MHCII expression on cortical thymic epithelial cells, and thus positive selection of CD4+ T cells, is also abolished. In contrast, constitutive and inducible MHCII expression is unaffected on professional antigen-presenting cells, including B cells, dendritic cells, and IFN-γ–activated cells of the macrophage lineage. pIV−/− mice have thus allowed precise definition of CIITA pIV usage in vivo. Moreover, they represent a unique animal model for studying the significance and contribution of MHCII-mediated antigen presentation by nonprofessional antigen-presenting cells in health and disease.
Keywords: knockout, promoter region, antigen-presenting cells, gene expression regulation, thymic selection
Introduction
MHC class II (MHCII) molecules play a pivotal role in the induction and regulation of the immune response. They are specialized for the presentation of peptides to the TCRs of CD4+ T cells. The engagement of MHCII–peptide complexes by the TCRs of CD4+ T cells is a pivotal interaction in the adaptive immune system because it regulates the development, activation, and survival of CD4+ T cells 1 2. During the course of these processes, the recognition of MHCII–peptide complexes can have several alternative consequences for the CD4+ T cells; it can trigger activation and proliferation, induce a state of anergy, or promote apoptosis. The precise outcome is determined by a variety of parameters, including the nature of the MHCII-positive cell and the density of the MHCII–peptide complexes displayed at its surface 2. Accordingly, the expression of MHCII molecules is tightly regulated in a cell-specific and quantitative fashion.
Basal or constitutive MHCII expression is the hallmark of three distinct types of bone marrow–derived cells, namely dendritic cells (DCs), B cells, and cells of the monocyte/macrophage lineage. These cells are collectively referred to as professional APCs. MHCII molecules are also expressed on cortical thymic epithelial cells (cTECs), where they mediate positive selection of CD4+ thymocytes 2. The majority of other cell types do not express basal levels of MHCII molecules but can generally be induced to express them in response to a number of stimuli, of which IFN-γ is by far the most potent and well known (for review see references 3–5). IFN-γ–induced MHCII expression has been described for a diverse set of non-bone marrow–derived cells in vitro and in vivo, including dermal fibroblasts, epithelial and endothelial cells in various organs, astrocytes, hepatocytes, and myocytes 3 4 5.
While the crucial functions of MHCII molecules on professional APCs and cTECs have been firmly established, the importance of IFN-γ–induced MHCII expression on non-bone marrow–derived cells is far from clear. Although numerous studies have confirmed that such nonprofessional APCs are capable of mediating MHCII-restricted antigen presentation in vitro, it remains unclear whether and how this ability actually participates in immune responses in vivo. This is an important question, because the induction of MHCII expression on non-bone marrow–derived cells is a widespread phenomenon that is intimately associated with many normal and pathological immune responses (for review see references 6 7 8 9 10 11 12). During the course of bacterial or viral infections, the induction of MHCII on macrophages and DCs helps to induce efficient immune responses directed against the pathogen. In addition, MHCII expression is upregulated in epithelia and endothelia, but the role of this is much less well understood. Ectopic expression of MHCII molecules on non-bone marrow–derived cells has also been described in the parenchyma of various organs in autoimmune diseases, such as autoimmune thyroiditis, multiple sclerosis, rheumatoid arthritis, inflammatory bowel disease, lupus nephritis, vitiligo, Sjögren's syndrome, and primary biliary cirrhosis. In mice and rats, systemic inflammatory stimuli such as the intravenous injection of IFN-γ results in a dramatic increase of MHCII throughout the body, including not only bone marrow–derived cells but also, for example, epithelial cells in the intestinal tract and kidney. MHCII expression has been observed on tumor cells from several neoplastic tissues. Finally, in organ transplantation, the expression of MHCII molecules on endothelial and epithelial cells in the transplant and in the host tissues is a hallmark of organ rejection.
In the majority of the above-mentioned situations, it has proved difficult to define the precise role of MHCII expression on nonprofessional APCs. This has been hampered by the lack of simple animal models in which MHCII expression on nonprofessional APCs can be uncoupled in vivo from that on professional bone marrow–derived APCs. Until now, such experiments relied essentially on the use of bone marrow transfer experiments in mice. However, elucidation of the molecular mechanisms controlling the transcription of MHCII genes has opened up new avenues for the manipulation of MHCII expression in vivo.
The class II transactivator (CIITA) 13 is a non–DNA-binding transcriptional coactivator that functions as a highly specific and essential transactivator of MHCII genes 3 5 14. It was first identified by virtue of the fact that it is defective in one form of the bare lymphocyte syndrome (BLS), a primary immunodeficiency disease resulting from a complete loss of constitutive and inducible MHCII expression in all cell types 5 15 16 17. The expression pattern of the gene encoding CIITA (called Mhc2ta in the mouse) is the primary determinant dictating the cell type specificity and induction of MHCII expression 3 5 14. A complex regulatory region containing at least three independent promoters (pI, pIII, and pIV) regulates expression of the Mhc2ta gene (Fig. 1) 18. In vitro studies with established cell lines and a limited number of primary cell types have indicated that pI and pIII drive expression in bone marrow–derived APCs such as DCs and B cells, while pIV is activated by IFN-γ in other cell types 18 19 20 21 22. No studies have addressed the question of which promoter directs CIITA expression in cTECs.
Figure 1.

Cell-specific and inducible MHCII expression is controlled by alternative usage of three promoters (pI, pIII, and pIV) of the Mhc2ta gene. pI and pIII are primarily active in DCs and B cells, respectively, while pIV is activated by IFN-γ. Usage of these promoters leads to the transcription (arrows) and splicing of alternative first exons (open boxes) to a shared second exon (shaded boxes). Incorporation of exons I and III leads to the synthesis of type I and type III CIITA mRNA encoding CIITA proteins having specific NH2-terminal extensions of 94 and 17 amino acids, respectively. In contrast, translation of type IV CIITA is initiated at the AUG found in the shared second exon. The three different forms of CIITA mRNA can be distinguished in RNase protection assay (rpa) with the indicated probes. CIITA activates transcription of MHCII genes by associating with proteins (RFX, X2BP, and NF-Y) that bind to MHCII promoters. ut, 5′ untranslated region.
To dissect the regulatory mechanisms controlling Mhc2ta expression in vivo, we have generated knockout mice in which pIV has been deleted. To our knowledge, this is the first description of a selective deletion of one promoter within a gene controlled by multiple promoters. Analysis of Mhc2ta and MHCII expression in pIV−/− mice demonstrates that the specificity of pIV is remarkably tight. Deletion of pIV selectively abrogates IFN-γ–induced MHCII expression in non-bone marrow–derived cells and constitutive MHCII expression in cTECs. In contrast, MHCII expression is unaffected in bone marrow–derived APCs, including DCs, B cells, and IFN-γ–activated cells of the macrophage lineage. These mice provide a unique and powerful tool to evaluate the significance and contribution of MHCII-mediated antigen presentation by nonprofessional APCs during the course of normal and pathological immune responses in vivo.
Materials and Methods
Generation of pIV−/− Mice.
Mapping and sequencing of the 5′ region of the Mhc2ta gene was reported previously 18. The targeting construct (see Fig. 2) was prepared in pBluescript (Stratagene) by standard PCR and recombinant DNA technology. It contains, in order: (i) a unique SfiI site to linearize the construct, (ii) a 5-kb fragment extending from the middle of the intron between exons I and III to the end of the intron between exons III and IV, (iii) an isolated loxP sequence, (iv) a 0.5-kb fragment containing pIV and its associated exon, (v) a loxP–flanked neomycin resistance gene (neo), and (vi) a 9-kb fragment situated in the intron downstream of exon IV. HindIII and BamHI sites were introduced to facilitate Southern blot analysis. The linearized targeting vector was transfected into E14.1 (129Sv) embryonic stem (ES) cells by electroporation, and G418-resistant clones were selected as described 23. Homologous recombinants were screened by PCR using a primer (5′-AGATTCAGTATGTTACACAAAGTAC-3′) situated upstream of the 5′ end of the targeting construct, and an internal primer (5′-TATGCTATACGAAGTTATAAGC-3′) lying at the 5′ loxP site. Positive clones were confirmed by Southern blotting using HindIII and BamHI digests in combination with a 3′ external probe and an internal probe (see Fig. 2). Two lines of mice carrying the pIV deletion were obtained as follows. (i) Several independent ES cell clones were injected into C57Bl/6 blastocysts. Chimeric mice were then crossed to a “deleter” strain expressing cre 24. Germline transmission was scored by coat color, and the deletion of pIV was confirmed by Southern blot. (ii) The ES cells were transfected with a cre expression vector (pOG231, originally obtained from S. O'Gorman (The Salk Institute, La Jolla, CA; reference 25), and subclones carrying the pIV deletion were then injected into C57Bl/6 blastocysts to obtain chimeric mice and germline transmission of the deleted locus. Blastocyst injections were performed at RCC (Itingen, Switzerland). Mice from the two independent lines were genotyped by PCR to distinguish between the wild-type (primers f 1+r1) and deleted (primers f 2+r2) alleles: f1, 5′-CCTAGGAGCCACGGAGCTG-3′; r1, 5′-TCCAGAGTCAGAGGTGGTC-3′; f 2, 5′-CAGACTATCCTGAAA-TGCC-3′; r2, 5′-CGAGATCTAGATATCGATAAGCTTG-3′. All experiments were performed with CIITA pIV−/− and pIV +/− littermates on a mixed Sv129–C57Bl/6 background. Animals were housed either under specific pathogen–free conditions at RCC or under standard conditions in a conventional mouse facility. The mice remain healthy in the conventional facility.
Figure 2.
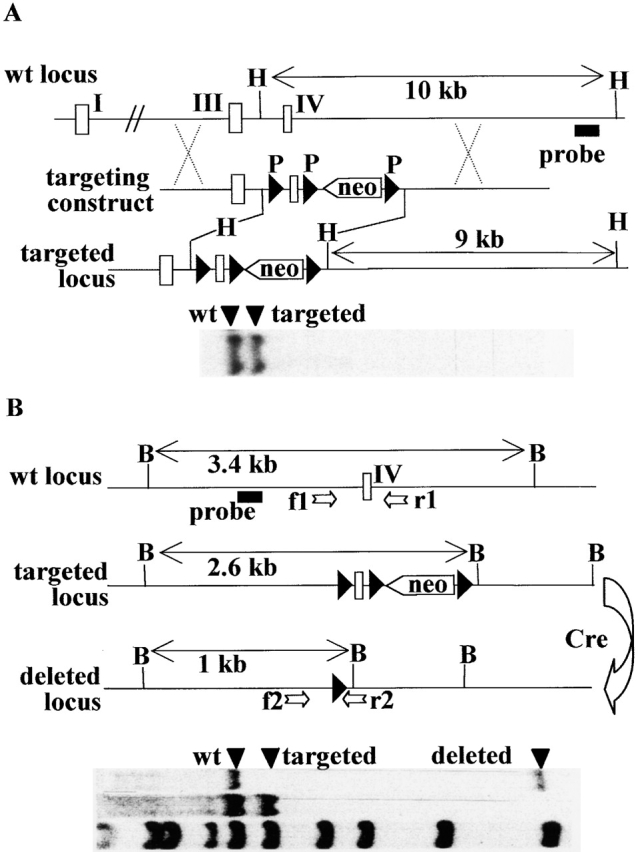

Generation of Mhc2ta pIV−/− mice. (A) The regulatory region of the wild type Mhc2ta locus, the targeting construct, and the targeted locus are depicted. Exons I, III, and IV are represented as open boxes, loxP sites (P) are shown as filled triangles, and the neo gene is indicated by an open arrow. HindIII sites (H) used for Southern blotting are indicated. A representative blot for a positive ES cell clone is shown below: the 3′ external probe (shown below the wild-type locus) hybridizes to a 10-kb fragment in the wild-type locus and to a 9-kb fragment in the targeted locus. (B) Cre-mediated deletion of the targeted locus. The wild-type, targeted, and deleted loci are depicted as in A. Arrows labeled f1, r1, f 2, and r2 represent PCR primers used for genotyping of mice (see C). BamHI sites (B) used for Southern blotting are indicated. A representative blot for heterozygous mice is shown below: the internal probe (shown below the wild-type locus) hybridizes to a 3.4-kb fragment in the wild-type locus, to a 2.6-kb fragment in the targeted locus, and to a 1-kb fragment in the deleted locus. λDNA digested with StyI was used as a molecular mass marker. (C) Genotyping of mice was done by PCR using the primer pair f1+r1 to amplify a 500-bp fragment specific for the wild-type locus and the primer pair f 2+r2 to amplify a 700-bp fragment diagnostic for the deleted locus. A representative experiment is shown for the offspring derived from a cross between two heterozygous pIV+/− mice. (D) Type IV CIITA mRNA is not induced in pIV−/− mice. MEF isolated from heterozygous (+/−) and homozygous (−/−) mice were cultivated in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of IFN-γ. The presence of type IV CIITA mRNA was assayed by RNase protection using the type IV specific probe (Fig. 1). A GAPDH probe was used as internal control. (E) Expression of types I and III CIITA mRNA is not affected by the deletion of pIV. Bone marrow–derived DCs generated from control littermates (+/−) and homozygous mutants (−/−) were analyzed by RNase protection using the type I–specific probe (top). Total spleen RNA from pIV+/− and pIV−/− animals was analyzed using the type III–specific probe (bottom). A TATA box binding protein (TBP) probe was used as internal control.
Four-Color Cytofluorimetric Analysis.
Ice-cooled single-cell suspensions were pretreated with Fc-block (anti-CD16/CD32; PharMingen) and then incubated with specific antibodies (PharMingen) directed against CD4 (RM4–5), CD8 (53–6.7), H-2Kb (AF6–88.5), I-Ab (AF6–120.1), CD11b (Mac1, M1/70), CD11c (N418, HL3), or B220 (CD45R, RA3–6B2). 104 (primary cultures) to 105 (suspensions from fresh organs) cells were analyzed on a FACSCalibur™ (Becton Dickinson).
Cell Preparation and Culture.
E14.1 ES cells from 129Sv mice were maintained as described 23. Mouse embryonic fibroblasts (MEFs) were isolated from 12.5-d-old fetuses. After the heads, livers, and internal organs were discarded, the remaining fetal tissues were minced, cells were resuspended in 0.05% trypsin and flushed through a 20-gauge needle, and DMEM was added to inactivate the trypsin. MEFs were stimulated for 24 h with 250 U/ml of recombinant mouse IFN-γ (Life Technologies). To isolate splenic and thymic DCs, the organs were digested with collagenase D (Boehringer Mannheim) before preparation of single-cell suspensions as described 26. Low-density cell fractions enriched in DCs (∼10%) were then recovered by centrifugation over an OptiPrep™ (Nycomed) density gradient 27. Myeloid and lymphoid DC subsets were distinguished on the basis of CD8 staining. DCs were generated from bone marrow precursors as described 28. Briefly, bone marrow cells were cultured for 12 d in RPMI containing 20 ng/ml murine GM-CSF (Peprotech) to drive DC differentiation. 250 U/ml of IFN-γ was added for the last 36 h, or maturation was induced with 10 μg/ml LPS (Sigma-Aldrich) for 36 h. DCs were identified by sorting for CD11c and excluding B220 cells. Peritoneal macrophages were harvested 4–6 d after intraperitoneal injection of 1 ml of 3% thioglycollate and were cultured for 72 h in DMEM in the presence or absence of 250 U/ml recombinant murine IFN-γ (Life Technologies). Primary brain cells were obtained from postnatal day 1 newborn mice. Brains were isolated, meninges were dissected out, the tissue was crushed between two frosted glass slides, and the single-cell suspension was cultured in DMEM/F12 (1:1; Life Technologies). 250 U/ml of recombinant mouse IFN-γ (Life Technologies) was added during the last 5 d of culture. After 12 d of culture, astrocytes and microglia were identified as CD11b − and CD11b + cells, respectively. Culture media were supplemented with 10% FCS, l-glutamine, β-mercaptoethanol, sodium pyruvate, and antibiotics. Single-cell suspensions from peripheral lymph nodes were prepared by crushing the tissue between two frosted glass slides. B cells were identified as B220 + cells.
Injection of Mice with IFN-γ.
Newborn mice were genotyped by PCR on tail DNA. pIV+/− and pIV −/− littermates were injected intraperitoneally at postnatal day 3 and 4 with 100,000 U of recombinant mouse IFN-γ (Life Technologies). The injected mice and age matched noninjected controls were snap frozen in OCT at postnatal day 5.
Immunohistochemistry.
8-μm sections from frozen adult organs and whole newborn mice were air dried, fixed in ice-cold acetone, blocked in PBS containing 0.6% H2O2, 5% goat serum, and 0.1% NaN3, and stained with digoxygenin-conjugated antibody directed against MHCII (M5/114; reference 29) and F4/80 (MCA497F; Serotec). Staining was revealed using antidigoxygenin peroxidase and 3-amino-9-ethyl carbazole (Sigma-Aldrich) to give a red precipitate. Sections were counterstained with methylene blue.
RNase Protection Analysis.
RNase protection assays were performed as described with 5–10 μg of RNA per sample 13. Probes specific for the different types of CIITA mRNA (Fig. 1) have been described 18 30. Quantification was performed by PhosphorImager analysis of the gels.
Antigen, Immunizations, and ELISA Techniques.
NP (4-hydroxy-3-nitrophenyl acetyl; Cambridge Research Biochemicals) was conjugated to CGG (chicken gamma globulin; Sigma-Aldrich) and precipitated in alum (Sigma-Aldrich) as described elsewhere 31. Groups of adult (10–14 wk old) CIITA−/− ( 32), I-Aα2/2 ( 33), pIV−/−, and control wild-type mice were injected in the base of the tail with 25 μg of alum-precipitated NP-CGG. Plastic plates with 96 flat-bottomed wells were coated overnight at 4°C with 5 μg/ml of NP4–BSA to allow for the selective detection of high-affinity, NP-specific IgG antibodies in the tested sera. Plates were washed with PBS, 0.01% Tween-20 and then blocked with 1% BSA. After washing, dilutions of the test sera were added and incubated overnight at 4°C. Bound antibody was detected using biotinylated anti–mouse IgG (Amersham Pharmacia Biotech), followed by streptavidin-conjugated alkaline phosphatase (Roche Diagnostics). Plates were developed with p-nitrophenyl-phosphate (Sigma-Aldrich), and absorbance was read at 405 nm.
Results
Generation of CIITA Promoter IV Knockout Mice. To generate CIITA pIV knockout mice, we used a gene targeting strategy relying on the cre/loxP recombination system 34 35. Briefly, we generated a targeting construct designed to insert an isolated loxP site upstream of pIV and a loxP-flanked neo gene in the intron situated downstream of exon IV (Fig. 2 A). ES cell clones in which these mutations were integrated by homologous recombination were isolated by a PCR strategy (see Materials and Methods), and the presence of the mutated locus was confirmed by Southern blot experiments (Fig. 2 A). Mice carrying an 800-bp deletion removing pIV and the adjacent exon were then generated in parallel by two independent strategies. In the first strategy, the ES cells were used to generate mice carrying the targeted locus, and deletion of pIV, exon IV, and the neo gene was then achieved in vivo by crossing them with deleter mice expressing cre recombinase 24. Southern blotting and PCR approaches were used to follow transmission of the wild-type, targeted, and deleted loci (Fig. 2B and Fig. C). In the second strategy, the ES cells were transfected with a cre expression vector to generate the deletion in vitro. ES cells carrying the deleted locus were then used to generate mice. These two approaches led to the production of two independent lines of mice carrying exactly the same deletion of pIV and exon IV. All experimental results were identical in the two lines. The deleted allele is transmitted at a normal Mendelian frequency. Mice homozygous for the deleted allele are healthy and breed well in a conventional mouse facility.
To confirm that the deletion eliminates pIV-driven CIITA mRNA expression, we performed RNase protection assays with MEFs that had or had not been induced with IFN-γ (Fig. 2 D). A probe (Fig. 1) permitting CIITA type IV mRNA to be distinguished from transcripts derived from the other two promoters was used. No CIITA mRNA is detected in the absence of IFN-γ. In heterozygous pIV +/− MEFs, CIITA type IV mRNA is induced by treatment with IFN-γ. This induction is abolished in the homozygous pIV −/− MEFs. CIITA mRNA derived from pI and pIII is not detected in IFN-γ–treated MEFs from either pIV +/− or pIV −/− mice.
To determine whether the elimination of pIV affects expression of the remaining two promoters, we performed RNase protection assays with probes (Fig. 1) specific for types I and III CIITA mRNA. RNA prepared from cultures of bone marrow–derived DCs was used to analyze transcripts derived from pI, the preponderant type of CIITA mRNA in DCs 18 30. To analyze promoter III expression, we prepared RNA from total spleen. The majority of CIITA-positive cells in the spleen are B cells that express type III CIITA mRNA 18. The results show that the expression levels of types I and III CIITA mRNA in DCs and B cells are comparable in pIV+/− and pIV −/− mice (Fig. 2 E). The deletion of pIV does thus not have a significant effect on the expression of CIITA mRNA derived from the other two promoters.
Selective Loss of IFN-γ–induced MHCII Expression on Non-Bone Marrow–derived Cells.
CIITA is a key regulator of constitutive and inducible expression of MHCII genes. Therefore, to assess the consequences of the pIV deletion, we studied cell surface MHCII (I-A) expression in a variety of primary cell types isolated from pIV−/− mice. As pIV has previously been implicated in IFN-γ–induced MHCII expression in a variety of cell types in vitro 1819202122, we first concentrated on cells that activate MHCII expression upon exposure to this cytokine (Fig. 3).
Figure 3.
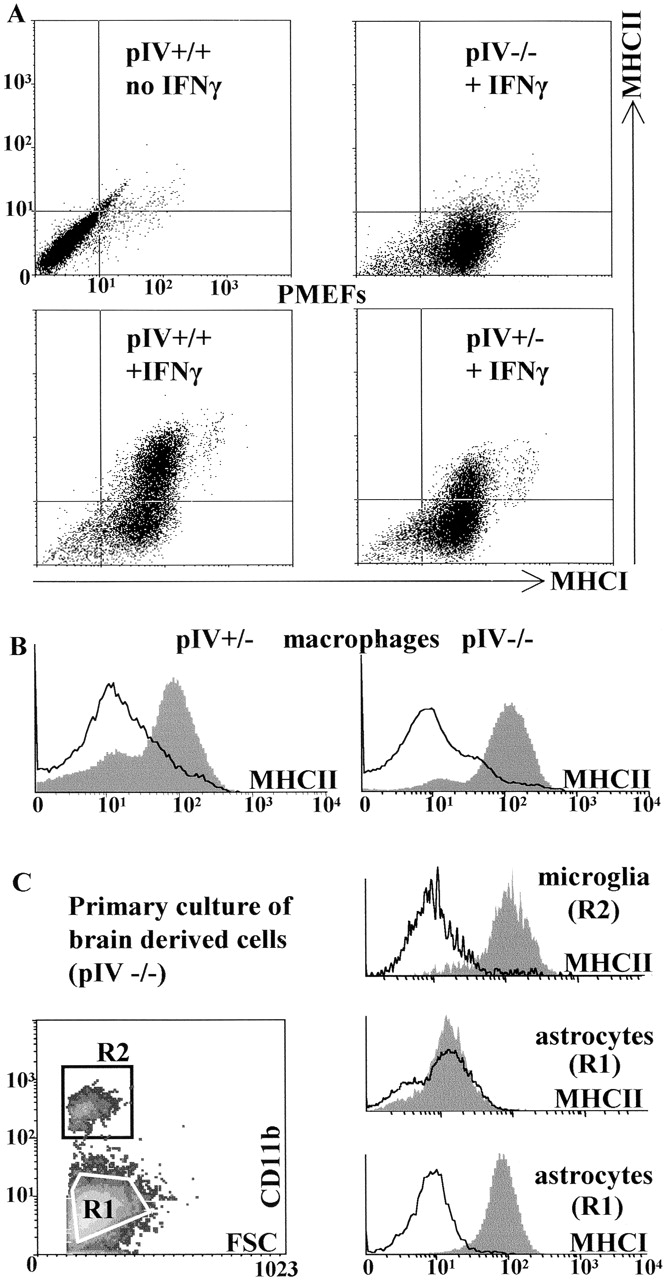
Selective loss of IFN-γ–induced MHCII expression on non-bone marrow–derived cells in pIV-deficient mice. (A) MEFs from pIV−/− fetuses exhibit a selective loss of IFN-γ–induced MHCII expression. MEFs isolated from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice were treated with IFN-γ and analyzed by two-color FACS® for the induction of MHCI and MHCII expression. (B) The deletion of pIV does not eliminate IFN-γ–induced MHCII expression in macrophages. Thioglycollate-elicited peritoneal macrophages were isolated from heterozygous (+/−) and homozygous (−/−) mice, cultured in the absence (open profile) or presence (filled profile) of IFN-γ, and then analyzed by FACS® for MHCII expression. (C) IFN-γ–induced MHCII expression is lost in astrocytes but retained in microglia from pIV-deficient mice. Brain-derived cells from pIV−/− mice were cultured in the absence (open profiles) or presence (solid profiles) of IFN-γ. CD11b+ microglial cells (R2, top right) and CD11b− astrocytes (R1, center right) were analyzed by FACS® for expression of MHCII. Activation of the astrocytes by IFN-γ was verified by examining the upregulation of MHCI expression (bottom right).
MEFs isolated from wild-type embryos do not express basal levels of I-A, but this expression is activated after exposure to IFN-γ (Fig. 3 A). In MEFs isolated from homozygous pIV−/− embryos, IFN-γ–induced I-A expression is completely eliminated (Fig. 3 A). In contrast to the effect on induction of I-A, the deletion of pIV has no repercussion on either the basal or induced levels of MHC class I (MHCI) expression (Fig. 3 A). Similar results have been obtained with dermal fibroblasts from adult mice (data not shown). It can thus be concluded that pIV is essential for IFN-γ–induced CIITA (and thus MHCII) expression in fibroblasts. This dependence is very strict, as a twofold reduction of I-A induction is already evident in heterozygous pIV +/− mice carrying only a single wild-type Mhc2ta allele (Fig. 3 A). The latter finding is consistent with previous experiments demonstrating that CIITA exerts a tight quantitative control over the level of MHCII expression in HeLa cells 36.
Surprisingly, the deletion of pIV has no effect on IFN-γ–induced I-A expression on peritoneal macrophages (Fig. 3 B). The level of I-A induced on pIV −/− macrophages is identical to that induced on pIV +/− macrophages (Fig. 3 B). Moreover, no difference in induction was observed between heterozygous pIV +/− and wild-type macrophages (data not shown). In contrast to the situation observed for fibroblasts, pIV is thus clearly not essential for IFN-γ–induced MHCII expression in macrophages.
Given the radically different effect of the pIV deletion on fibroblasts and peritoneal macrophages, we extended our analysis to two additional IFN-γ–inducible cell types, astrocytes and microglial cells. These two cell types are abundant in the central nervous system (CNS). Astrocytes are non-bone marrow–derived glial cells, while microglia are CNS-resident macrophages. Both cell types are known to respond to IFN-γ by upregulating expression of MHCII genes 37383940. We therefore studied IFN-γ–induced I-A expression on astrocytes and microglia in primary brain cell cultures (Fig. 3 C). As observed by others, I-A expression is induced by IFN-γ on both microglial cells and astrocytes from wild-type mice (data not shown). The deletion of pIV does not abolish IFN-γ–induced I-A expression on pIV −/− microglia. In contrast, IFN-γ–induced I-A expression is completely abrogated on the pIV −/− astrocytes. As observed for MEFs, the induction of MHCI expression is not affected in pIV −/− astrocytes (Fig. 3 C). Taken together, these results demonstrate that pIV is essential for IFN-γ–induced expression of CIITA and MHCII in non-bone marrow–derived cells such as fibroblasts and astrocytes but is dispensable in bone marrow–derived cells such as macrophages and microglia.
CIITA Promoter Usage in IFN-γ–activated Macrophages.
The finding that IFN-γ–induced MHCII expression is not affected in macrophages from pIV −/− mice was surprising because pIV has previously been shown to be activated by IFN-γ in primary cells and cell lines belonging to the monocyte/macrophage lineage 1819202122. We therefore examined CIITA promoter usage in IFN-γ–activated peritoneal macrophages from pIV −/− mice and littermate controls (Fig. 4). This analysis was done by RNase protection experiments using probes specific for transcripts derived from the three Mhc2ta promoters (Fig. 1). Under the culture conditions used, neither the control macrophages nor pIV−/− macrophages express basal levels of CIITA mRNA. In agreement with previous studies, the control macrophages express CIITA type IV mRNA after activation with IFN-γ. However, they also express high levels of CIITA type I mRNA and a trace amount of type III mRNA. In pIV −/− macrophages, IFN-γ–induced expression of CIITA type IV mRNA is completely abolished as expected. On the other hand, IFN-γ–induced expression of type I and type III mRNA is retained. Quantification has indicated that CIITA type I mRNA is the predominant form induced in both the control and pIV −/− macrophages (Fig. 4 D). This explains why cell surface MHCII expression in IFN-γ–activated macrophages is identical in pIV −/− and control macrophages (Fig. 3 B).
Figure 4.
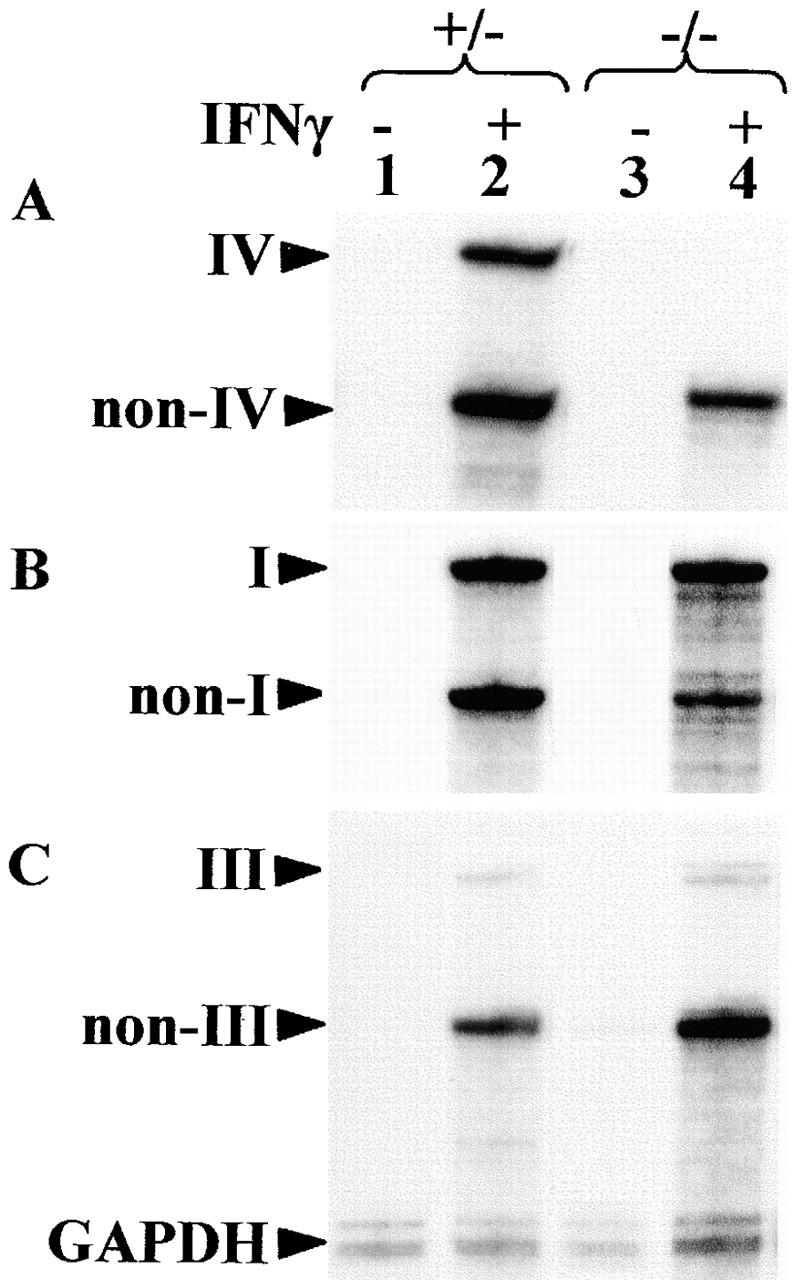

IFN-γ–induced activation of Mhc2ta expression is mediated largely by pI. Thioglycollate-elicited peritoneal macrophages from control (+/−) or homozygous mutant (−/−) mice were cultured in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of IFN-γ. Expression of CIITA was analyzed by RNase protection using probes specific for type IV (A), type I (B), and type III (C) mRNA. The band corresponding to the specific mRNA (I, III, or IV) and the band representing mRNA derived from the other two promoters (NonI, NonIII, and NonIV) are indicated. A probe for GAPDH mRNA was used as internal control. The percentages of total CIITA mRNA corresponding to types I, III, and IV were quantified by PhosphorImager analysis of RNase protection experiments similar to those shown in A–C (D).
pIV Is Not Required for MHCII Expression in Bone Marrow–derived Cells.
Previous in vitro studies had suggested that pI and pIII are active primarily in DCs and B cells, respectively 18 30 41. The strategy used to excise pIV was designed such that it should not interfere with the synthesis of CIITA mRNA derived from pI and pIII. The analysis of CIITA type I transcripts in DCs from bone marrow cultures and of CIITA type III transcripts in the spleen suggests that this is indeed the case (Fig. 2 E). To further confirm this finding, we studied the effect of the pIV deletion on I-A expression in various B cell and DC preparations (Fig. 5). B cells were isolated from the bone marrow, lymph nodes, and spleen. DC preparations included thymic DCs, bone marrow–derived DCs, bone marrow–derived DCs treated with IFN-γ or induced to maturate with LPS, and splenic DCs of the lymphoid (CD8+) and myeloid (CD8−) subsets. In all cases, the levels of I-A expression in cells from the pIV−/− mice were identical to those observed in cells from control littermates (Fig. 5). The function of pI and pIII in B cells and DC is thus independent of pIV.
Figure 5.
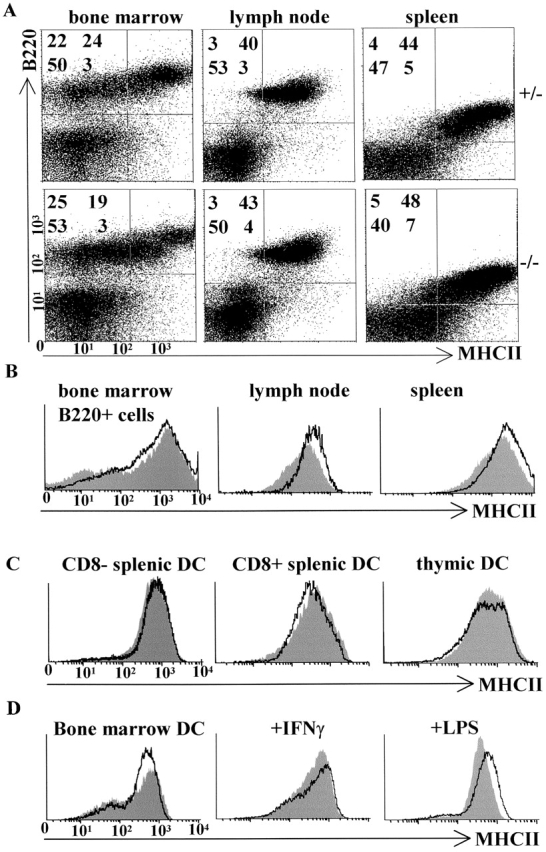
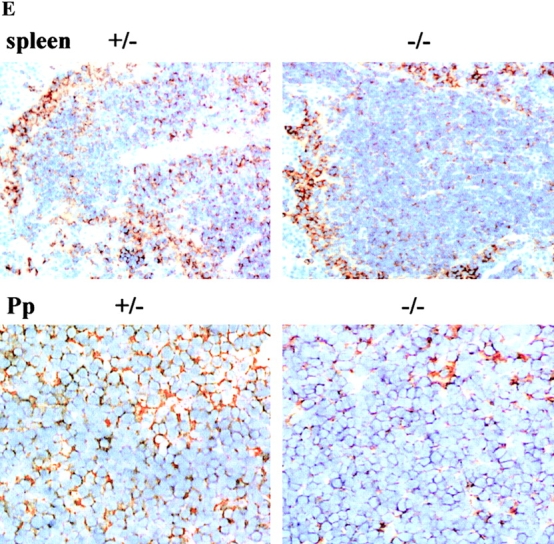
Deletion of Mhc2ta pIV does not affect constitutive MHCII expression in bone marrow–derived cells and secondary lymphoid tissues. (A) Cells isolated from the bone marrow, lymph nodes, and spleens of pIV−/− mice and pIV+/− littermate controls were analyzed by two-color FACS® for the expression of MHCII and the B cell marker B220. The percentage of cells in the four quadrants is indicated for each panel. (B) FACS® analysis of MHCII expression on ex vivo B cells isolated from the bone marrow, lymph nodes, and spleens of pIV−/− mice (solid profiles) and control littermates (open profiles). (C) MHCII expression on ex vivo DCs isolated from pIV−/− mice (solid profiles) and control pIV+/− littermates (open profiles) was examined by four-color FACS® analysis. Cells studied were thymic (CD11c+B220−) DCs and splenic DCs of the myeloid (CD11c+B220−CD8−) and lymphoid (CD11c+B220−CD8+) subsets. (D) MHCII expression was analyzed by FACS® on CD11c+ bone marrow–derived DCs from pIV−/− mice (solid profiles) and control littermates (open profiles). The DCs were analyzed as such, after induction with IFN-γ, or after treatment with LPS to induce maturation. (E) Sections of the spleen and Peyer's patches from adult pIV−/− mice and control pIV+/− littermates were stained for MHCII expression (brown color).
Further confirmation that the deletion of pIV does not affect MHCII expression on bone marrow–derived cells was obtained by staining tissue sections with antibodies against I-A (Fig. 5 E and Fig. 6). Various lymphoid and nonlymphoid tissues were examined. The overall architecture of the spleen in pIV−/− mice is normal, as assessed by the examination of sections stained with hematoxylin and eosin and sections stained for distribution of the DC marker CD11c to distinguish between the T and B cell zones (data not shown). The pattern of I-A expression in the splenic white pulp (B cells and DCs) and on marginal zone macrophages of pIV −/− mice is identical to that observed in control mice (Fig. 5 E). Similarly, pIV −/− and control mice exhibit very similar I-A expression patterns in Peyer's patches (Fig. 5 E) and lymph nodes (data not shown). Finally, I-A expression in the pIV−/− mice is also normal on scattered cells in various tissues, including the brain, lungs, liver, and intestine (Fig. 6). These MHCII-positive cells are detected both before and after administration of IFN-γ (Fig. 6). Staining of adjacent sections with cell lineage markers (B220 for B cells, CD11c for DCs, and F4/80 for macrophages) has indicated that these MHCII-positive cells are primarily tissue macrophages. Colocalization of staining for I-A and the macrophage marker F4/80 is shown for the brain, lung, and intestine in Fig. 6. Taken together these results demonstrate that pIV of the Mhc2ta gene is not required for MHCII expression in B cells, DCs, and macrophages.
Figure 6.
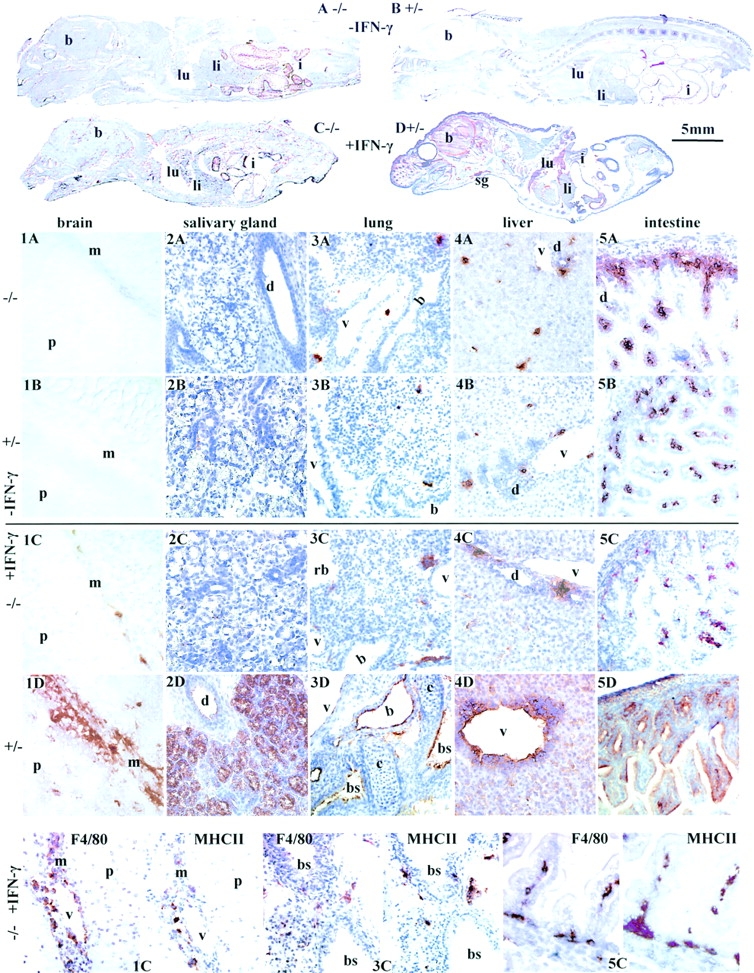
Deletion of Mhc2ta pIV results in a selective loss of IFN-γ–induced MHCII expression on non-bone marrow–derived cells. Sections of newborn mice were stained for MHCII expression (brown color). (Top) Whole body sections of newborn pIV−/− mice (A and C) and control pIV+/− littermates (B and D) that had been injected (C and D) or had not been injected (A and B) with IFN-γ. Positions of the brain (b), lungs (lu), liver (li), intestines (i), and salivary glands (sg) are indicated. (1A–5D) Higher magnifications of representative regions of organs from the mice shown in A–D. Brain sections (1A–1D): m, meninges; p, parenchyma. Submandibular salivary gland sections (2A–2D): d, secretory duct. Lung sections (3A–3D): v, pulmonary veins; b, bronchioles; rb, respiratory bronchiole; bs, bronchus. Liver sections (4A–4D): v, central vein; d, bile duct. Sections through the intestines (5A–5D) show the visceral peritoneum, crypts, and villi. (Bottom) Adjacent sections from the brain (1C), lung (3C), and intestine (5C) of IFN-γ–injected pIV−/− mice were stained for expression of MHCII and a macrophage-specific marker (F4/80).
Loss of IFN-γ–induced MHCII Expression on Non-Bone Marrow–derived Cells In Vivo.
Widespread induction of MHCII expression can be obtained in vivo by the injection of IFN-γ into mice and rats 42434445. We used this approach to determine whether our observation that IFN-γ–induced MHCII expression is abrogated in pIV −/− fibroblasts and astrocytes can be extended to other non-bone marrow–derived cells in vivo. Newborn mice were injected intraperitoneally with IFN-γ on postnatal days 3 and 4 and were then killed on day 5. Whole embryo cryosections were stained for I-A expression. Fig. 6 shows whole body sections and details of representative regions of the brain, salivary gland, lung, liver, and intestines from control (pIV +/− ) and knockout (pIV −/− ) mice that had or had not been injected with IFN-γ. After administration of IFN-γ, a strong induction of I-A expression is observed in the control pIV+/− mice on a variety of non-bone marrow–derived cell types throughout the body. I-A expression in the control mice is, for instance, induced on meninges, epithelial cells lining the acini and main duct of the submandibular salivary gland, epithelial cells lining the airways in the lower respiratory tract, epithelial cells lining the bile ducts in the liver (data not shown), epithelial cells lining the villi and glands in the crypts of the intestine, many vascular endothelial cells, such as those of the veins in the lungs and liver, and hepatocytes (Fig. 6, panels 1D–5D). Other cell types include parietal and visceral peritoneum, parietal and visceral pleura, and ependymal and plexus epithelial cells in the brain and skin fibroblasts, mostly around hair follicles (data not shown). In all of the cell types mentioned above, the IFN-γ–induced I-A expression is eliminated in the pIV knockout mice (Fig. 6, panels 1C–5C). These results thus confirm that pIV is essential for IFN-γ–induced MHCII expression in a wide variety of non-bone marrow–derived cells in vivo.
I-A expression in pIV−/− mice is retained on scattered cells in organs such as the brain, spinal cord (data not shown), lung, and liver (Fig. 6). The distribution and localization of these cells suggests that they are tissue macrophages. This has been confirmed by staining serial sections for expression of I-A and the macrophage marker F4/80 (Fig. 6, bottom panels). Therefore, as observed for microglia and peritoneal macrophages in vitro (Fig. 3), the deletion of pIV does not abolish MHCII expression by cells of the macrophage lineage in vivo.
Loss of MHCII Expression on cTECs.
To examine the effect of the deletion of pIV on the expression of MHCII in the thymus, we stained thymic sections with antibodies directed against I-A (Fig. 7 A). In wild-type thymi, the cortex exhibits a fine reticular pattern of I-A expression characteristic of cTECs. The more diffuse staining seen in the medulla is attributable to DCs, B cells, and macrophages. Unexpectedly, pIV−/− thymi exhibit a selective loss of the reticular I-A staining on cTECs (Fig. 7 A). Only a residual patchy staining remains in the cortex. In contrast to the cortex, the diffuse staining on bone marrow–derived cells in the medulla is unchanged (Fig. 7 A). A very similar pattern of MHCII expression has been described in MHCII-deficient mice repopulated with wild-type bone marrow cells 46.
Figure 7.


Deletion of Mhc2ta pIV results in the loss of MHCII expression on cTECs and leads to impaired positive selection of CD4+ T cells. (A) Thymic sections from pIV−/− mice and pIV+/− littermate controls were stained (brown color) for expression of MHCII, the DC marker CD11c, or the macrophage marker F4/80. Regions corresponding to the cortex (c) and medulla (m) are indicated. (B) T cell populations in the thymus, lymph nodes, and spleen of pIV−/− mice and pIV+/− littermate controls were analyzed by FACS® for expression of CD8 and CD4. The percentages of single-positive CD4, single-positive CD8, and double-positive cells are indicated. (C) The ability to generate a primary T cell–dependent antibody response was examined in pIV−/− mice, in pIV+/− littermates, and in negative controls (CIITA−/− and I-Aα2/− mice). High-affinity IgG titers produced after immunization with NP-CGG were measured at the indicated dilutions of the sera.
To identify the cell type that retains residual MHCII expression in the thymic cortex of pIV−/− mice, we stained serial sections with cell lineage–specific antibodies. The patchy MHCII staining in the cortex is matched by the macrophage-specific (anti-F4/80) staining that we performed on adjacent sections (Fig. 7 A). This is consistent with the fact that macrophages from pIV −/− mice retain MHCII expression (Fig. 3, Fig. 4, and Fig. 6). In contrast to the expression of F4/80, cells expressing CD11c (DCs) are restricted to the medulla and cortico-medullary junction (Fig. 7 A), which is in accordance with previous results obtained on other mouse strains 47. Staining with an antikeratin antibody is consistent with a conserved architecture of the thymic stroma (data not shown).
To determine whether MHCII expression could be restored by IFN-γ in pIV −/− cTECs, we examined I-A expression in the thymi of newborn mice that had received high doses of this cytokine. The MHCII staining pattern observed in the thymic cortex of IFN-γ–injected pIV−/− mice is similar to that seen in noninjected pIV −/− mice (data not shown). Therefore, as observed for other extrahematopoietic cells, pIV is essential for CIITA expression in cTECs, and its absence cannot be compensated for by IFN-γ–mediated activation of pI or pIII.
Impaired Positive Selection of CD41 T Cells and T Cell–dependent Immune Responses.
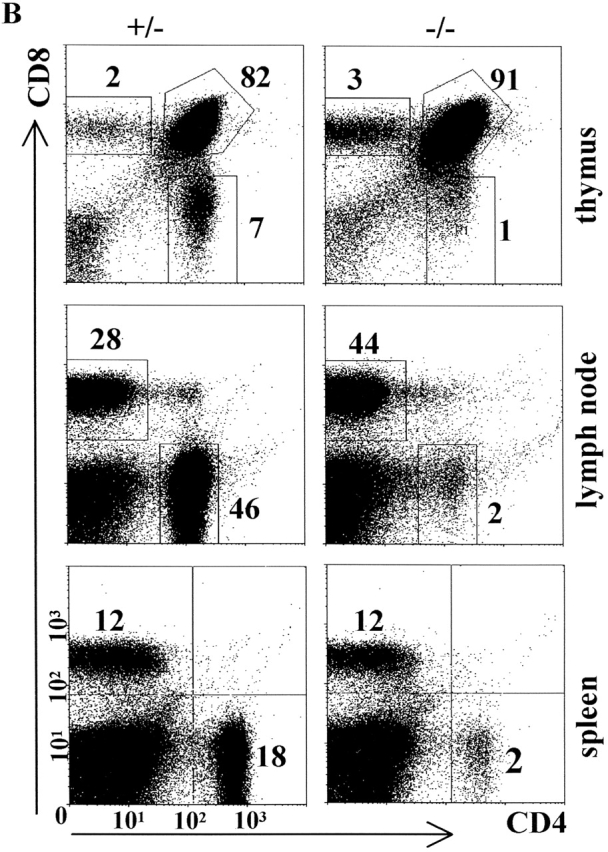
The selective absence of MHCII on cTECs in pIV−/− mice has a profound impact on positive selection of CD4 + T cells. Percentages of CD4+ T cells are reduced 7–10-fold in the thymus and up to 20-fold in peripheral lymphoid organs of pIV−/− mice (Fig. 7 B). Similar numbers were found in older pIV −/− mice, indicating that the presence of MHCII-positive APCs in peripheral organs is not sufficient in itself to lead with time to the accumulation of significant numbers of MHCII-restricted CD4+ T cells (data not shown).
To document the functional consequences of the impairment of positive selection, we examined the ability of the pIV−/− mice to generate a T cell–dependent immune response (Fig. 7 C). pIV −/− and control mice were immunized with NP-CGG, and primary antibody responses at day 15 were measured. One-half of the pIV −/− mice produced low levels of high-affinity IgG during the primary response, whereas no response was detected in negative control mice (I-Aα−/− and CIITA −/− mice) (Fig. 7 C, left panel). Titration curves indicate that the IgG titers produced by the responding pIV−/− mice are at least 20-fold lower than the titers obtained in control littermates (Fig. 7 C, right panel). All but one of the pIV−/− mice produced detectable but reduced levels of high-affinity IgG after a second antigen challenge (data not shown). Taken together, these results indicate that the residual CD4 + T cells in pIV−/− mice can sustain a T cell–dependent immune response, albeit with a strongly reduced efficiency.
Discussion
The realization that CIITA is a key regulatory factor for transcription of MHCII genes has kindled a great deal of interest in the molecular mechanisms regulating its expression 3 5 14. A large and complex regulatory region controls expression of the Mhc2ta gene. In mice, this region contains three independent promoters (pI, pIII, and pIV) spread out over 12 kb of DNA 18. Until now, the specificity of these promoters has been addressed almost exclusively in vitro. We therefore opted for an in vivo approach relying on the generation of Mhc2ta promoter knockout mice. A mouse lacking pIV of the Mhc2ta gene has been constructed. The analysis of this mouse has demonstrated that pIV is essential for constitutive MHCII expression in cTECs and IFN-γ–induced MHCII expression in non-bone marrow–derived cells, whereas it is dispensable for constitutive and inducible MHCII expression in DCs, B cells, and cells of the macrophage lineage. This selective loss of MHCII expression on non-bone marrow–derived cells represents a novel and unusual phenotype. It contrasts sharply with the phenotype of mice carrying null mutations of the Mhc2ta gene, which exhibit an almost complete loss of both constitutive and IFN-γ–induced MHCII expression in all tissues and cell types 324849.
To generate the pIV knockout mice, we used a strategy relying on the cre/loxP recombination system 34 35. The choice of the cre/loxP system was motivated by two advantages. First, it permits excision of the neo gene and its associated regulatory elements, thereby ensuring that activity of the remaining Mhc2ta promoters (pI and pIII) and the splicing pattern of precursor RNAs is not perturbed. Second, the cre/loxP system permits the generation of conditional knockout mice in which pIV can be excised in a temporally or spatially restricted manner 34 35. To achieve this, we are in the process of generating mice in which pIV and its associated exon are left intact but flanked by loxP sites. In these mice, pIV-dependent MHCII expression can be abrogated at specific points in time and/or in defined cell types by the introduction of inducible and/or tissue-specific cre transgenes.
The Mhc2ta pIV knockout mouse exhibits a selective loss of IFN-γ–induced MHCII expression on non-bone marrow–derived cells. This is evident for a variety of cell types exposed to IFN-γ in vitro or in vivo, including fibroblasts, astrocytes, hepatocytes, and endothelial and epithelial cells in various organs (Table ). In these cell types, IFN-γ–induced MHCII expression exhibits a strict dependence on pIV, and the remaining two promoters cannot compensate for its absence. This is consistent with previous RNase protection experiments and reporter gene assays, suggesting that pIV is the main Mhc2ta promoter that is activated by IFN-γ in vitro in cells such as rodent astrocytes 21 22 30, human melanoma cells 18, and mouse melanoma cells (data not shown) (Table ). However, it is at odds with experiments showing that the human pIII promoter can be activated by IFN-γ, albeit with relatively low efficiency, in PP2 fibroblasts, human umbilical vein endothelial cells (HUVECs), and certain cell lines (Table ; references 18, 20, and 50). This discrepancy may reflect a species-specific difference. The sequences implicated in IFN-γ induction of the human pIII promoter lie far upstream in a region that is not conserved between the human and mouse genes. An alternative explanation may be that reporter gene assays do not always reproduce faithfully the activity of the endogenous promoters. Experiments performed with the choriocarcinoma cell line Jar provide a good illustration of this point: pIV-driven reporter gene constructs can be activated by IFN-γ in Jar cells (Table ), but hypermethylation of the endogenous promoter renders it refractory to this induction 51.
Table 1.
CIITA Promoter Activity in IFN-γ–treated Cells
| Approach | Cell type | pI | pIII | pIV |
|---|---|---|---|---|
| I. | Endothelium | − | − | +++ |
| Bronchial epithelium | − | − | +++ | |
| Pleural epithelium | − | − | +++ | |
| Peritoneal epithelium | − | − | +++ | |
| Intestinal epithelium | − | − | +++ | |
| Glands, intestinal tract | − | − | +++ | |
| Bile duct epithelium | − | − | +++ | |
| Skin fibroblasts | − | − | +++ | |
| MEFs | − | − | +++ | |
| Astrocytes | − | − | +++ | |
| II. | MEFs | − | − | +++ |
| Astrocytes | − | − | +++ | |
| Macrophages | +++ | +− | + | |
| Microglia | ++ | ND | + | |
| THP1 monocytes | − | − | +++ | |
| PP2 fibroblasts | ND | + | +++ | |
| HUVECs | ND | + | +++ | |
| Melanoma Me67.1, 67.8 | − | − | +++ | |
| HeLa cells | ND | − | +++ | |
| B16 murine melanoma | − | − | +++ | |
| Jar choriocarcinoma cells | ND | − | +++ | |
| U373 glioblastoma cells | ND | + | ++ | |
| 2fTGH fibrosarcoma cells | ND | + | +++ | |
| P19 embryonal carcinoma | ND | + | +++ | |
| RAW 264.7 macrophages | ND | + | +++ |
Approach I: Absence of MHCII on pIV−/− cells (FACS® or histology). Approach II: RNase protection or reporter gene assays.
The deletion of pIV does not affect IFN-γ–induced CIITA and MHCII expression in peritoneal macrophages and microglia in vitro. Induction of MHCII expression on tissue macrophages in the lung, CNS, intestine, liver, and thymus is also retained. Thus, although induction of pIV activity is indeed observed in cells of the macrophage lineage, this is not an absolute requirement for upregulation of MHCII expression. This was rather unexpected, because pIV was until now believed to be the major promoter responsible for IFN-γ–induced expression in macrophages 18 21. We show here that enhanced MHCII expression in IFN-γ–activated mouse macrophages is in fact ensured mainly by pI and to only a minor extent by pIII and pIV (Fig. 4 and Table ). This is consistent with our previous finding that transcripts derived from pI are detected in IFN-γ–activated microglia 30. Surprisingly, IFN-γ–responsive sequences have not been identified in the vicinity of pI. This raises the possibility that IFN-γ–activated expression of pI is mediated by a distant, as yet unidentified enhancer region. Alternatively, it is possible that IFN-γ does not affect pI directly. Instead, enhanced activity of pI could be an indirect consequence of macrophage activation. The answer to this question will have to await further dissection of the regulatory mechanisms controlling pI.
Examination of MHCII expression in the thymi of pIV−/− mice revealed an unsuspected function of pIV. cTECs in pIV−/− mice are devoid of MHCII molecules, and positive selection of CD4+ T cells is consequently impaired. The activation of pIV in cTECs must occur via mechanisms that are distinct from those mediating IFN-γ–induced expression in other cell types. Indeed, although cTECs can upregulate MHCII expression in response to IFN-γ in vitro 52, these cells generally express MHCII molecules in a constitutive manner in vivo and when maintained in culture as three-dimensional aggregates 53. This constitutive MHCII expression by cTECs is independent of the IFN-γ stimulatory pathway 54. Moreover, mice deficient for IFN-γ or its receptor do not exhibit impaired positive selection of CD4+ T cells and have normal CD4+ T cell counts in the thymus and periphery 54 55.
The fact that CD4+ T cells do not accumulate in MHCII-positive peripheral lymphoid organs of the pIV−/− mice indicates that the defect in positive selection is severe. The extent of CD4+ T cell depletion in pIV−/− mice is currently being analyzed in greater detail. Preliminary results indicate that the percentages of CD4+ T cells is not significantly higher than in I-Aα2/− or CIITA−/− mice. Nevertheless, inefficient T cell–dependent immune responses can be elicited in pIV−/− mice, indicating that the residual CD4+ T cell compartment is competent for providing help to MHCII-positive B cells. That this was not observed previously in the classical CIITA knockout mice can be explained by the fact that the latter lacked MHCII-positive APCs and B cells.
The induction of MHCII expression on non-bone marrow–derived cells is widespread in many normal and pathological CD4+ T cell–mediated immune responses, including those called into play during the course of autoimmune, allergic, infectious, and neoplastic diseases, as well as graft rejection. However, the contribution of this ectopic MHCII expression on nonprofessional APCs is not well defined in many of these situations. The pIV-deficient mouse constitutes a novel means to address this question because the deletion of pIV results in a highly selective loss of MHCII-mediated antigen presentation by nonprofessional APCs. In particular, we hope that the pIV knockout mouse will be useful for a number of mouse models of CD4+ T cell–mediated autoimmune diseases in which ectopic MHCII expression may represent a crucial feature. The following models represent good examples. TGF-β1–deficient mice develop a fatal systemic CD4+ T cell–mediated autoimmune disease. In this model, MHCII expression is induced widely on nonlymphoid cells of target tissues before the appearance of other signs of inflammation 56 57. Similarly, induction of MHCII expression in the CNS is a key aspect of experimental autoimmune encephalitis, a widely accepted animal model for multiple sclerosis 58 59. Induction of MHCII expression in the kidney has been implicated in models of experimental glomerulonephritis 60. Finally, mice expressing IFN-γ transgenes driven by tissue-specific promoters suffer from various inflammatory or autoimmune syndromes characterized by ectopic expression of MHCII molecules 61 62 63 64 65. For the pIV−/− mice to be useful in such models, it will be necessary to restore positive selection by one of the following approaches. First, positive selection could be restored by transplanting a wild-type thymus. Second, the conditional cre/loxP-mediated knockout strategy could be used to delete pIV in adult mice after the mature T cell repertoire has been established. Finally, positive selection could be restored by introducing a CIITA transgene controlled by a promoter (K14) driving expression in thymic epithelial cells.
pIV−/− mice represent the first dissection by gene targeting techniques of a complex regulatory region consisting of multiple promoters. The precise and highly selective effect of the loss of pIV on MHCII expression suggests that the regulatory region controlling the Mhc2ta gene is readily amenable to genetic manipulations in vivo without resulting in unwanted disturbances. For instance, our results emphasize the fact that pI and pIII are functionally independent of pIV. The different Mhc2ta promoters thus do not appear to be subjected to a significant amount of cross-talk, and they do not affect each other's activity. This paves the way for further targeting experiments designed to define the specificity of the Mhc2ta promoters in vivo. In addition to furthering our understanding of the molecular mechanisms controlling expression of CIITA and MHCII genes, these experiments will also generate mice exhibiting novel patterns of compartmentalized MHCII expression. We anticipate that such mice will be valuable for dissecting the respective roles of MHCII-mediated antigen presentation by different professional and nonprofessional APCs in normal and pathological immune responses.
Acknowledgments
We are grateful to Bernard Mach for providing the environment in which this work was initiated. We thank Annick Mühlethaler-Mottet and L. Otten for the isolation and mapping of genomic clones and for plasmids used to make RNase protection probes. We appreciate the help of Giovanna Badic, Jeannine Bamat, and Estelle Säuberli with immunohistology. We are grateful to K. Rajewsky and his colleagues for providing the E14.1 ES cells and the “deleter” cre mice, and to M. Friedly, J. Zákány, and D. Duboule for their help in establishing the ES cell culture.
Jean-Marc Waldburger was supported by an M.D./Ph.D. fellowship from the Roche Research Foundation and by a fellowship generously provided by Professor A.F. Junod (Hopital Cantonal, Geneva). Work in the laboratory of W. Reith was supported by the Swiss National Science Foundation, the Gabriella Giorgi-Cavaglieri Foundation, the Ernst and Lucie Schmidheiny Foundation, and the Swiss Multiple Sclerosis Foundation. Work in the laboratory of H. Acha-Orbea was supported by the Swiss National Science Foundation and the Gabriella Giorgi-Cavaglieri Foundation. Work in the laboratory of A. Fontana was supported by the Swiss National Science Foundation and the Swiss Multiple Sclerosis Foundation. The collaboration between the laboratories in Geneva and Zurich was supported by the Swiss National Science program, NCCR (Neural Plasticity and Repair).
Footnotes
H. Acha-Orbea and W. Reith contributed equally to this work.
Abbreviations used in this paper: CIITA, class II transactivator; CNS, central nervous system; cTECs, cortical thymic epithelial cells; DCs, dendritic cells; ES, embryonic stem; MEFs, mouse embryonic fibroblasts; MHCI, MHC class I; MHCII, MHC class II.
References
- Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu. Rev. Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- Viret C., Janeway C.A. MHC and T cell development. Rev. Immunogenet. 1999;1:91–104. [PubMed] [Google Scholar]
- Harton J.A., Ting J.P. Class II transactivatormastering the art of major histocompatibility complex expression. Mol. Cell Biol. 2000;20:6185–6194. doi: 10.1128/mcb.20.17.6185-6194.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boss J.M. Regulation of transcription of MHC class II genes. Curr. Opin. Immunol. 1997;9:107–113. doi: 10.1016/s0952-7915(97)80166-5. [DOI] [PubMed] [Google Scholar]
- Reith W., Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu. Rev. Immunol. 2001;19:331–373. doi: 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- Sims T.N., Halloran P.F. MHC class II regulation in vivo in the mouse kidney. Microbes Infect. 1999;1:903–912. doi: 10.1016/s1286-4579(99)00227-0. [DOI] [PubMed] [Google Scholar]
- Bland P. MHC class II expression by the gut epithelium. Immunol. Today. 1988;9:174–178. doi: 10.1016/0167-5699(88)91293-5. [DOI] [PubMed] [Google Scholar]
- Marelli-Berg F.M., Lechler R.I. Antigen presentation by parenchymal cellsa route to peripheral tolerance? Immunol. Rev. 1999;172:297–314. doi: 10.1111/j.1600-065x.1999.tb01374.x. [DOI] [PubMed] [Google Scholar]
- Touraine J.L., Betuel H., Pouteil Noble C., Royo C. HLA class II antigensstructure, function, and expression in immunodeficiencies, autoimmune diseases, and allograft rejection. Adv. Nephrol. 1989;18:325–334. [PubMed] [Google Scholar]
- Davies T.F. The complex role of epithelial cell MHC class II antigen expression in autoimmune endocrine disease. Autoimmunity. 1990;8:87–89. doi: 10.3109/08916939008995725. [DOI] [PubMed] [Google Scholar]
- Guardiola J., Maffei A. Control of MHC class II gene expression in autoimmune infections and neoplastic diseases. Crit. Rev. Immunol. 1993;13:247–268. [PubMed] [Google Scholar]
- Bottazzo G.F., Todd I., Mirakian R., Belfiore A., Pujol-Borrell R. Organ-specific autoimmunitya 1986 overview. Immunol. Rev. 1986;94:137–169. doi: 10.1111/j.1600-065X.1986.tb01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steimle V., Otten L.A., Zufferey M., Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency. Cell. 1993;75:135–146. [PubMed] [Google Scholar]
- Waldburger J.M., Masternak K., Muhlethaler-Mottet A., Villard J., Peretti M., Landmann S., Reith W. Lessons from the bare lymphocyte syndromemolecular mechanisms regulating MHC class II expression. Immunol. Rev. 2000;178:148–165. doi: 10.1034/j.1600-065x.2000.17813.x. [DOI] [PubMed] [Google Scholar]
- Klein C., Lisowska Grospierre B., LeDeist F., Fischer A., Griscelli C. Major histocompatibility complex class II deficiencyclinical manifestations, immunologic features, and outcome. J. Pediatr. 1993;123:921–928. doi: 10.1016/s0022-3476(05)80388-9. [DOI] [PubMed] [Google Scholar]
- Masternak K., Muhlethaler-Mottet A., Villard J., Peretti M., Reith W. Molecular genetics of the bare lymphocyte syndrome. Rev. Immunogenet. 2000;2:267–282. [PubMed] [Google Scholar]
- Elhasid R., Etzioni A. Major histocompatibility complex class II deficiencya clinical review. Blood Rev. 1996;10:242–248. doi: 10.1016/s0268-960x(96)90008-9. [DOI] [PubMed] [Google Scholar]
- Muhlethaler-Mottet A., Otten L.A., Steimle V., Mach B. Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J. 1997;16:2851–2860. doi: 10.1093/emboj/16.10.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlethaler-Mottet A., Di Berardino W., Otten L.A., Mach B. Activation of the MHC class II transactivator CIITA by interferon-γ requires cooperative interaction between Stat1 and USF-1. Immunity. 1998;8:157–166. doi: 10.1016/s1074-7613(00)80468-9. [DOI] [PubMed] [Google Scholar]
- Piskurich J.F., Linhoff M.W., Wang Y., Ting J.P. Two distinct gamma interferon-inducible promoters of the major histocompatibility complex class II transactivator gene are differentially regulated by STAT1, interferon regulatory factor 1, and transforming growth factor beta. Mol. Cell Biol. 1999;19:431–440. doi: 10.1128/mcb.19.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikcevich K.M., Piskurich J.F., Hellendall R.P., Wang Y., Ting J.P. Differential selectivity of CIITA promoter activation by IFN-gamma and IRF-1 in astrocytes and macrophagesCIITA promoter activation is not affected by TNF-alpha. J. Neuroimmunol. 1999;99:195–204. doi: 10.1016/s0165-5728(99)00117-4. [DOI] [PubMed] [Google Scholar]
- Dong Y., Rohn W.M., Benveniste E.N. IFN-gamma regulation of the type IV class II transactivator promoter in astrocytes. J. Immunol. 1999;162:4731–4739. [PubMed] [Google Scholar]
- Torres R.M., Kühn R. Laboratory protocols for conditional gene targeting 1997. Oxford University Press; Oxford, UK: pp. 167 [Google Scholar]
- Schwenk F., Baron U., Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon P., Oberholzer J., Occhiodoro T., Morel P., Lou J., Trono D. Reversible immortalization of human primary cells by lentivector-mediated transfer of specific genes. Mol. Ther. 2000;2:404–414. doi: 10.1006/mthe.2000.0141. [DOI] [PubMed] [Google Scholar]
- Crowley M., Inaba K., Witmer-Pack M., Steinman R.M. The cell surface of mouse dendritic cellsFACS analyses of dendritic cells from different tissues including thymus. Cell Immunol. 1989;118:108–125. doi: 10.1016/0008-8749(89)90361-4. [DOI] [PubMed] [Google Scholar]
- Ruedl C., Rieser C., Bock G., Wick G., Wolf H. Phenotypic and functional characterization of CD11c+ dendritic cell population in mouse Peyer's patches. Eur. J. Immunol. 1996;26:1801–1806. doi: 10.1002/eji.1830260821. [DOI] [PubMed] [Google Scholar]
- Lutz M.B., Kukutsch N., Ogilvie A.L., Rossner S., Koch F., Romani N., Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A., Dorf M.E., Springer T.A. A shared alloantigenic determinant on Ia antigens encoded by the I-A and I-E subregionsevidence for I region gene duplication. J. Immunol. 1981;127:2488–2495. [PubMed] [Google Scholar]
- Suter T., Malipiero U., Otten L., Ludewig B., Muelethaler-Mottet A., Mach B., Reith W., Fontana A. Dendritic cells and differential usage of the MHC class II transactivator promoters in the central nervous system in experimental autoimmune encephalitis. Eur. J. Immunol. 2000;30:794–802. doi: 10.1002/1521-4141(200003)30:3<794::AID-IMMU794>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Nossal G.J., Karvelas M. Soluble antigen abrogates the appearance of anti-protein IgG1-forming cell precursors during primary immunization. Proc. Natl. Acad. Sci. USA. 1990;87:1615–1619. doi: 10.1073/pnas.87.4.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.H., Guerder S., Hong S.C., van Ewijk W., Flavell R.A. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity. 1996;4:167–178. doi: 10.1016/s1074-7613(00)80681-0. [DOI] [PubMed] [Google Scholar]
- Kontgen F., Suss G., Stewart C., Steinmetz M., Bluethmann H. Targeted disruption of the MHC class II Aa gene in C57BL/6 mice. Int. Immunol. 1993;5:957–964. doi: 10.1093/intimm/5.8.957. [DOI] [PubMed] [Google Scholar]
- Rajewsky K., Gu H., Kuhn R., Betz U.A., Muller W., Roes J., Schwenk F. Conditional gene targeting. J. Clin. Invest. 1996;98:600–603. doi: 10.1172/JCI118828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R., Schwenk F. Advances in gene targeting methods. Curr. Opin. Immunol. 1997;9:183–188. doi: 10.1016/s0952-7915(97)80133-1. [DOI] [PubMed] [Google Scholar]
- Otten L.A., Steimle V., Bontron S., Mach B. Quantitative control of MHC class II expression by the transactivator CIITA. Eur. J. Immunol. 1998;28:473–478. doi: 10.1002/(SICI)1521-4141(199802)28:02<473::AID-IMMU473>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Wong G.H., Bartlett P.F., Clark-Lewis I., Battye F., Schrader J.W. Inducible expression of H-2 and Ia antigens on brain cells. Nature. 1984;310:688–691. doi: 10.1038/310688a0. [DOI] [PubMed] [Google Scholar]
- Frei K., Siepl C., Groscurth P., Bodmer S., Schwerdel C., Fontana A. Antigen presentation and tumor cytotoxicity by interferon-gamma-treated microglial cells. Eur. J. Immunol. 1987;17:1271–1278. doi: 10.1002/eji.1830170909. [DOI] [PubMed] [Google Scholar]
- Benveniste E.N., Sparacio S.M., Bethea J.R. Tumor necrosis factor-alpha enhances interferon-gamma-mediated class II antigen expression on astrocytes. J. Neuroimmunol. 1989;25:209–219. doi: 10.1016/0165-5728(89)90139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierz W., Endler B., Reske K., Wekerle H., Fontana A. Astrocytes as antigen-presenting cells. I. Induction of Ia antigen expression on astrocytes by T cells via immune interferon and its effect on antigen presentation. J. Immunol. 1985;134:3785–3793. [PubMed] [Google Scholar]
- Ghosh N., Piskurich J.F., Wright G., Hassani K., Ting J.P., Wright K.L. A novel element and a TEF-2-like element activate the major histocompatibility complex class II transactivator in B-lymphocytes. J. Biol. Chem. 1999;274:32342–32350. doi: 10.1074/jbc.274.45.32342. [DOI] [PubMed] [Google Scholar]
- Steiniger B., Falk P., van der Meide P.H. Interferon-gamma in vivo. Induction and loss of class II MHC antigens and immature myelomonocytic cells in rat organs. Eur. J. Immunol. 1988;18:661–669. doi: 10.1002/eji.1830180502. [DOI] [PubMed] [Google Scholar]
- Steiniger B., van der Meide P.H., Falk P., Lempnauer J.K. Induction of class II MHC antigen expression in rat organs after systemic application of recombinant gamma interferon. Adv. Exp. Med. Biol. 1988;237:795–799. doi: 10.1007/978-1-4684-5535-9_119. [DOI] [PubMed] [Google Scholar]
- Momburg F., Koch N., Moller P., Moldenhauer G., Butcher G.W., Hammerling G.J. Differential expression of Ia and Ia-associated invariant chain in mouse tissues after in vivo treatment with IFN-gamma. J. Immunol. 1986;136:940–948. [PubMed] [Google Scholar]
- Skoskiewicz M.J., Colvin R.B., Schneeberger E.E., Russell P.S. Widespread and selective induction of major histocompatibility complex–determined antigens in vivo by gamma interferon. J. Exp. Med. 1985;162:1645–1664. doi: 10.1084/jem.162.5.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz J.S., Auchincloss H., Jr., Grusby M.J., Glimcher L.H. Class II-positive hematopoietic cells cannot mediate positive selection of CD4+ T lymphocytes in class II-deficient mice. Proc. Natl. Acad. Sci. USA. 1993;90:2779–2783. doi: 10.1073/pnas.90.7.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocker T., Riedinger M., Karjalainen K. Targeted expression of major histocompatibility complex (MHC) class II molecules demonstrates that dendritic cells can induce negative but not positive selection of thymocytes in vivo. J. Exp. Med. 1997;185:541–550. doi: 10.1084/jem.185.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G.S., Malin M., Vremec D., Chang C.H., Boyd R., Benoist C., Mathis D. Mice lacking the transcription factor CIITA—a second look. Int. Immunol. 1998;10:1957–1967. doi: 10.1093/intimm/10.12.1957. [DOI] [PubMed] [Google Scholar]
- Itoh-Lindstrom Y., Piskurich J.F., Felix N.J., Wang Y., Brickey W.J., Platt J.L., Koller B.H., Ting J.P. Reduced IL-4-, lipopolysaccharide-, and IFN-gamma-induced MHC class II expression in mice lacking class II transactivator due to targeted deletion of the GTP-binding domain. J. Immunol. 1999;163:2425–2431. [PubMed] [Google Scholar]
- Piskurich J.F., Wang Y., Linhoff M.W., White L.C., Ting J.P. Identification of distinct regions of 5′ flanking DNA that mediate constitutive, IFN-gamma, STAT1, and TGF-beta-regulated expression of the class II transactivator gene. J. Immunol. 1998;160:233–240. [PubMed] [Google Scholar]
- Morris A.C., Spangler W.E., Boss J.M. Methylation of class II trans-activator promoter IVa novel mechanism of MHC class II gene control. J. Immunol. 2000;164:4143–4149. doi: 10.4049/jimmunol.164.8.4143. [DOI] [PubMed] [Google Scholar]
- Rigaud G., De Lerma B.A., Nicolis M., Cestari T., Ramarli D., Riviera A.P., Accolla R.S. Induction of CIITA and modification of in vivo HLA-DR promoter occupancy in normal thymic epithelial cells treated with IFN-gammasimilarities and distinctions with respect to HLA-DR-constitutive B cells. J. Immunol. 1996;156:4254–4258. [PubMed] [Google Scholar]
- Anderson K.L., Moore N.C., McLoughlin D.E., Jenkinson E.J., Owen J.J. Studies on thymic epithelial cells in vitro. Dev. Comp. Immunol. 1998;22:367–377. doi: 10.1016/s0145-305x(98)00011-1. [DOI] [PubMed] [Google Scholar]
- Huang S., Hendriks W., Althage A., Hemmi S., Bluethmann H., Kamijo R., Vilcek J., Zinkernagel R.M., Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Dalton D.K., Pitts-Meek S., Keshav S., Figari I.S., Bradley A., Stewart T.A. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- Shull M.M., Ormsby I., Kier A.B., Pawlowski S., Diebold R.J., Yin M., Allen R., Sidman C., Proetzel G., Calvin D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser A.G., Letterio J.J., Kulkarni A.B., Karlsson S., Roberts A.B., Sporn M.B. Transforming growth factor beta 1 (TGF-beta 1) controls expression of major histocompatibility genes in the postnatal mouseaberrant histocompatibility antigen expression in the pathogenesis of the TGF- beta 1 null mouse phenotype. Proc. Natl. Acad. Sci. USA. 1993;90:9944–9948. doi: 10.1073/pnas.90.21.9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa P.T., ter Meulen V., Fontana A. Hyperinducibility of Ia antigen on astrocytes correlates with strain-specific susceptibility to experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 1987;84:4219–4223. doi: 10.1073/pnas.84.12.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collawn J.F., Benveniste E.N. Regulation of MHC class II expression in the central nervous system. Microbes Infect. 1999;1:893–902. doi: 10.1016/s1286-4579(99)00228-2. [DOI] [PubMed] [Google Scholar]
- Li S., Kurts C., Kontgen F., Holdsworth S.R., Tipping P.G. Major histocompatibility complex class II expression by intrinsic renal cells is required for crescentic glomerulonephritis. J. Exp. Med. 1998;188:597–602. doi: 10.1084/jem.188.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger K., Howes E., Gallina M., Huang X.J., Travis G.H., Sarvetnick N. Transgenic mice expressing IFN-gamma in the retina develop inflammation of the eye and photoreceptor loss. Invest. Ophthalmol. Vis. Sci. 1994;35:2667–2681. [PubMed] [Google Scholar]
- Sarvetnick N., Liggitt D., Pitts S.L., Hansen S.E., Stewart T.A. Insulin-dependent diabetes mellitus induced in transgenic mice by ectopic expression of class II MHC and interferon-gamma. Cell. 1988;52:773–782. doi: 10.1016/0092-8674(88)90414-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton G.D., Calcutt N.A., Garrett R.S., Gu D., Sarvetnick N., Campana W.M., Powell H.C. Necrotizing myopathy induced by overexpression of interferon-gamma in transgenic mice. Muscle Nerve. 1999;22:156–165. doi: 10.1002/(sici)1097-4598(199902)22:2<156::aid-mus3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Horwitz M.S., Evans C.F., McGavern D.B., Rodriguez M., Oldstone M.B. Primary demyelination in transgenic mice expressing interferon-gamma. Nat. Med. 1997;3:1037–1041. doi: 10.1038/nm0997-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin J.G., Kelly D., Rath E.M., Baerwald K.D., Suzuki K., Popko B. Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol. Cell Neurosci. 1996;7:354–370. doi: 10.1006/mcne.1996.0026. [DOI] [PubMed] [Google Scholar]