

Abstract
We investigated the role of Fas ligand in murine silicosis. Wild-type mice instilled with silica developed severe pulmonary inflammation, with local production of tumor necrosis factor (TNF)-α, and interstitial neutrophil and macrophage infiltration in the lungs. Strikingly, Fas ligand–deficient generalized lymphoproliferative disease mutant (gld) mice did not develop silicosis. The gld mice had markedly reduced neutrophil extravasation into bronchoalveolar space, and did not show increased TNF-α production, nor pulmonary inflammation. Bone marrow chimeras and local adoptive transfer demonstrated that wild-type, but not Fas ligand–deficient lung macrophages recruit neutrophils and initiate silicosis. Silica induced Fas ligand expression in lung macrophages in vitro and in vivo, and promoted Fas ligand–dependent macrophage apoptosis. Administration of neutralizing anti-Fas ligand antibody in vivo blocked induction of silicosis. Thus, Fas ligand plays a central role in induction of pulmonary silicosis.
Keywords: Fas ligand, silicosis, macrophages, neutrophils, inflammation
Introduction
The deposition of silica particles in the lungs of man and experimental animals leads to silicosis, an industrial era disease that causes respiratory failure due to a fibrotic reaction 1 2 3. Occupations such as coal mining and quarrying increase the risk of developing silicosis 1 2. Silicosis starts with inflammation and tissue destruction, followed by tissue repair that leads to fibrosis 1 2 3. After reaching the alveoli, silica particles are ingested by pulmonary macrophages, which become activated and release inflammatory mediators, such as reactive oxygen intermediates, arachdonic acid metabolites, chemokines, and cytokines 1 2 3. Activated macro-phages attract neutrophilic granulocytes, which upregulate alveolitis and promote connective tissue destruction 3. The proinflammatory cytokine TNF-α plays a pivotal role in silicosis by mediating the widespread inflammatory reaction and inducing the late fibrogenic reaction 3 4.
Fas ligand (FasL) is a membrane bound and shed protein belonging to the TNF gene family, and the natural counterreceptor for the death-promoting Fas molecule expressed by a variety of lymphoid and nonlymphoid tissues 5. Lymphocyte apoptosis mediated by Fas/FasL interaction regulates immune responses 6 7, and FasL-mediated apoptosis of leukocytes prevents inflammatory reactions at immune-privileged sites 8. However, FasL also exerts a proinflammatory role. Tumors expressing FasL are destroyed by infiltrating granulocytes in vivo 9 10. Microenvironmental influence, such as TGF-β, appears to determine suppressive versus inflammatory effects of FasL 11. Here we show that silica-induced pulmonary inflammation requires FasL expression and that FasL-expressing pulmonary macrophages attract neutrophils and initiate silicosis. Blockade of FasL function by antibodies prevents development of silicosis in vivo.
Materials and Methods
Animals.
Wild-type (wt) BALB/c (BALB.wt) and BALB generalized lymphoproliferative disease mutant (gld)/gld (BALB. gld) mice of both sexes (aging 6–8 wk, weighing 25–30 g) were from the Oswaldo Cruz Institute Animal Care facility, Rio de Janeiro. BALB.gld mice were produced at the National Cancer Institute, National Institutes of Health, Bethesda, MD, by serially backcrossing the gld gene onto a BALB/c background for 15 generations. All animal work in this study was conducted in accordance with humane institutional guidelines.
Silica Instillation.
Mice were anesthetized by sevoflurane and tracheotomyzed. Mice received an intratracheal injection of 50 μl of either sterile saline or silica suspension (SiO2; 20 mg/50 μl in sterile saline, 1–5-μm sterile particles; Sigma-Aldrich).
Respiratory Mechanics.
After 15 d of silica instillation, mice were sedated (diazepam, 1.0 mg intraperitoneally), anesthetized (sodium pentobarbital, 2.0 mg/kg intraperitoneally), and a 20 G gauge cannula was introduced into the trachea. Air flow 12, volume, and total (ΔPtot), resistive (ΔP1), and viscoelastic/inhomogeneous (ΔP2) pressures 13 were measured. All data were analyzed using ANADAT software (RHT-Info Data Inc.). Data are presented as means ± SEM of 8–10 mice per group.
Histopathology, Immunohistochemistry, Immunofluorescence, and In Situ Detection of Apoptosis.
Animals were killed by cervical dislocation. The pulmonary circulation was flushed by intracardiac injection of saline, followed by formaline. Lungs were fixed in 10% formaline, embedded in paraffin, and sections were stained with hematoxylin and eosin for histopathological analysis. Alternatively, lung cryostat sections (4–10 μm) were embedded in OCT Tissue-Tek (Sakura Finetek Inc.), and mounted on poly-l-lysine–coated glass microscope slides. FasL and Fas were detected with anti–mouse FasL mAb MFL3, and anti–mouse Fas mAb Jo2 (both from BD PharMingen), incubated at 5 μg/ml, and developed with horseradish peroxidase (HRP)-conjugated goat anti–hamster Ig (Sigma-Aldrich). Negative controls were stained with 5 μg/ml control hamster IgG (anti-TNP mAb A19-3; BD PharMingen). For double staining immunofluorescence, cryostat sections were treated with 5 μg/ml Fc block (anti-Fcγ RII/III mAb 2.4G2; BD PharMingen), and stained with 5 μg/ml PE- or FITC-labeled control hamster IgG, PE-labeled anti–mouse FasL MFL3, and FITC-labeled anti–mouse F4/80 (a macrophage surface marker; Caltag Laboratories) mAbs. Slides were examined with an Axiophot-2 ZEISS fluorescence microscope equipped with differential interference contrast microscopy (DIC; reference 14). DNA fragmentation associated with apoptosis was detected by terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick-end labeling (TUNEL), using TdT-FragEL™ detection kit (Oncogene Research Products), according to the manufacturer's instructions. Slides were developed with streptavidin-HRP conjugate, counterstained with methyl green, and mounted for photomicroscopy. In addition, lungs were subjected to intrabronchial lavage (three washes of 500 μl saline each). Cells from bronchoalveolar lavage (BAL) fluid were cytocentrifuged, and stained with May-Grunwald stain (Sigma-Aldrich).
Myeloperoxidase Activity.
Neutrophil content in BAL fluid or pulmonary parenchyma was evaluated by measuring myeloperoxidase (MPO) activity, as described 15. After collecting BAL fluid, individual lungs were homogenized in 1.0 ml PBS-Triton X-100, 0.1%. Homogenized samples were centrifuged at 10,000 g, for 15 min at 4°C, and the supernatants (50 μl) were added to 50 μl substrate solution containing 5 mM o-phenylene diamine (Sigma-Aldrich) in 10 mM citrate buffer pH 5.0, and 8.8 mM H2O2. The reaction was stopped after 15 min with 50 μl H2SO4 4 N, and the absorbance was read at 492 nm. BAL cells from either individual (silica treated) or pooled (saline treated) mice were centrifuged, counted, and adjusted to a concentration of 107 cells/ml in PBS. Cells (50 μl) were added to 50 μl substrate solution, and the reaction was processed and read as above. Results shown are from individual mice, and each point represents the mean of triplicate readings, except for BAL from control mice (each point is the mean of pools from three mice).
Bone Marrow Radiation Chimeras.
Chimeras were generated as described 16. Male BALB.wt and BALB.gld mice, aging 8 wk, received acidified drinking water and ciprofloxacin (daily injection of 100 μg/mouse, intraperitoneally) for 15 d before irradiation. Immediately after irradiation (750 rad), mice received 5 × 106 bone marrow cells (in 0.1 ml DMEM) from male BALB.gld and BALB.wt donors, by intravenous route. After cell transfer, animals were maintained in sterile isolators, and received oral gentamicin (0.3 mg/ml in drinking water) throughout the experiment. The following chimeras were generated: BALB.wt→BALB.wt, BALB.gld→BALB.gld, BALB.wt→BALB.gld, and BALB.gld→ BALB.wt. After 30 d of bone marrow transfer, chimeras were instilled intratracheally with saline or silica. Development of silicosis was evaluated after 15 d, by lung MPO activity and histopathological analysis.
Local Adoptive Transfer of Macrophages.
Alveolar macrophages were obtained from BAL fluids of normal BALB.wt and BALB.gld donors (containing >95% macrophages). Male BALB. wt or BALB.gld mice were instilled intratracheally with donor macrophages (106 cells/animal in 50 μl PBS). After 5 d of macrophage transfer, animals were instilled again with either saline or silica. Development of silicosis was evaluated after 15 d of silica instillation, by interstitial lung MPO activity and histopathological analysis. Some BALB.wt and BALB.gld mice received only saline or silica instillation, and were used as negative and positive controls for lung MPO activity.
FasL Expression by Lung Macrophages In Vitro.
As internalized silica interferes with flow cytometry, we employed a cell ELISA assay for FasL expression. BAL cells from normal BALB.wt mice (2 × 105/ml in Hank's solution) were seeded (0.2 ml/well) on sterile flat bottom 96-well plates (Corning), and incubated for 2 h at 37°C and 5% CO2. Residual nonadherent cells were removed, and adherent lung macrophages were cultured overnight in DMEM supplemented with 5% heat-inactivated FCS, 2 mM l-glutamine, 5 × 10−5 M 2-ME, 10 μg/ml gentamicin sulfate, MEM nonessential aminoacids, 1 mM sodium pyruvate, and 10 mM HEPES. Macrophages were then treated with either medium alone, or 50 μg/ml silica particles (Sigma-Aldrich), in the presence or absence of 1 mM deferoxamine (Sigma-Aldrich), and cultured for additional 48 h. Plates were washed, fixed with 1% paraformaldehyde for 30 min, blocked with PBS-10% FCS for 60 min, washed with PBS-Tween 20 (0.05%), and incubated overnight with 5 μg/ml anti–mouse FasL mAb MFL3 (BD PharMingen) in PBS-FCS, at 4°C. Wells were washed, and the reaction was developed with secondary HRP-conjugated goat anti–hamster Ig (Sigma-Aldrich), and o-phenylene diamine as substrate. Results are mean and SE of absorbance readings of triplicate cultures.
Macrophage Apoptosis.
BAL cells from normal BALB.wt and BALB.gld mice (4 × 105) were seeded and cultured on 24-well Corning plates (0.25 ml/well) in triplicates, as above. Macrophages were treated or not with 100 μg/ml silica particles, in the presence of anti-FasL mAb MFL3 or a control hamster IgG (20 μg/ml), and cultured for 24 h. As apoptotic macrophages detach from the plates, the resulting non-adherent cells were harvested, counted, cytocentrifuged, and stained with May-Grunwald. Apoptotic cells were identified on oil immersion by dark, shrunk nuclei, or by nuclear segmentation 17. 200 cells were counted per replicate. To obtain the number of apoptotic macrophages, the percentage of apoptotic cells was applied to total nonadherent cell numbers.
Production of TNF-α.
The content of TNF-α in BAL fluid was measured by a sandwich ELISA technique according to a protocol provided by the manufacturer, using two cytokine-specific mAbs, one of which was biotinylated (BD PharMingen). The reaction was revealed with alkaline phosphatase–conjugated streptavidin (BD PharMingen) using paranitrophenol phosphate (Sigma-Aldrich) as substrate. Recombinant murine TNF-α (BD PharMingen) was used as positive control.
Administration of Anti-FasL In Vivo.
Affinity purified anti–mouse FasL mAb MFL4 18, or normal hamster IgG were injected intraperitoneally into BALB.wt mice (0.5 mg/mouse/day) on days 0, 3, 6, 9, and 12 after silica instillation. At day 15, animals were killed and analysed for silicosis induction by lung MPO activity, TNF-α content in BAL fluid, and lung histopathology.
Results
FasL-deficient gld Mice Do Not Develop Silicosis.
Mutant gld mice express a loss-of-function mutation in FasL, leading to lack of Fas-mediated apoptosis, autoimmunity, and lymphoproliferative disorder 19. To investigate the requirement of FasL on silica-induced pulmonary inflammation, BALB.wt and FasL-deficient BALB.gld mice were instilled with silica intratracheally. This model of silica exposure leads to acute pulmonary inflammation after 2 wk, followed by a chronic inflammatory response by 12 wk 20. Silica instillation in BALB.wt mice results in large granulomatous nodules, with increased collagen and elastic fiber content in the pulmonary parenchyma after 15 d 21. We studied the mice 15 d after silica instillation throughout the experiments. Silica-treated BALB.wt mice showed severe wasting, with loss of 43% of their body weight, compared with saline-instilled controls (Fig. 1 A). Silica-treated BALB.gld mice appeared healthy, without any weight loss (Fig. 1 A). A 3.3-fold increase in levels of TNF-α was detected in BAL fluid of silica-treated BALB.wt mice (Fig. 1 B). Treatment of BALB.gld mice with silica did not increase basal levels of TNF-α, which were elevated compared with wt mice (Fig. 1 B). Induction of acute silicosis was evaluated by respiratory mechanics (Fig. 1 C) and histopathology (Fig. 1D–F). Lungs from silica-treated BALB.wt mice showed increased elastance (not shown), and resistive (ΔP1) and viscoelastic/inhomogeneous (ΔP2) pressures, compared with saline-instilled controls (Fig. 1 C). Silica-treated BALB.gld mice showed no change in respiratory mechanics (Fig. 1 C). Histological evaluation of silica-treated BALB.wt mice demonstrated interstitial and alveolar edema, granulomatous nodules with large accumulations of inflammatory cells, mainly neutrophils and macrophages, as well as alveolar collapse and intrabronchial cellular infiltration obstructing the lumen (Fig. 1 E). Immunohistochemistry also revealed the presence of T cells in inflammatory infiltrates (not shown). No inflammatory change in pulmonary tissue was observed in silica-treated BALB.gld mice (Fig. 1 F, compared with Fig. 1 D). Examination of lungs after 30 d of silica instillation similarly failed to reveal any changes in gld mice (not shown). The cellular composition of BAL fluid was examined. BAL from control mice instilled with saline typically contained >95% macrophages (see Fig. 2 C). The differential count of BAL cells from control mice (n = 3, mean ± SE) was: 96.7 ± 0.3% macrophages, 1.5 ± 0.4% neutrophils, and 2.4 ± 0.3% lymphocytes. Silica-treated BALB.wt mice showed a large influx of macrophages (12-fold increase in number) and neutrophils (11,600-fold increase) into BAL fluid (Fig. 2A and Fig. C). There was also an influx of lymphocytes (84-fold increase), resulting in a differential cell count (n = 3, mean ± SE) of: 13.2 ± 1.6% macrophages, 84.3 ± 1.5% neutrophils, and 2.2 ± 0.4% lymphocytes. BAL fluid from control BALB.gld mice contained 86.0 ± 1.2% macrophages, 1.6 ± 0.3% neutrophils, and 14.0 ± 1.0% lymphocytes. In spite of the absence of silicosis, BAL fluid from silica-treated BALB.gld mice contained a large number of inflammatory cells, most of which were macrophages (25-fold increase in number), as shown in Fig. 2A and Fig. C. Strikingly, the number of neutrophils in BAL from silica-treated gld mice (506-fold increase) was reduced 6-fold, compared with wt mice (Fig. 2A and Fig. C). There was also an influx of lymphocytes (13-fold increase), resulting in a differential cell count of: 68.3 ± 2.2% macrophages, 28.3 ± 1.5% neutrophils, and 5.5 ± 1.6% lymphocytes. These results demonstrate that gld mice have both an absolute and relative deficiency in neutrophil extravasation in response to silica. Deficient neutrophil extravasation was also evident in assays of neutrophil MPO enzymic activity of gld BAL cells (Fig. 2 B, left). Different from wt mice, MPO activity failed to reveal any neutrophil accumulation in lung parenchyma from gld mice after BAL removal (Fig. 2 B, right). These results indicate that the reaction of gld mice to silica was not pathogenic. The results also indicate that functional FasL expression is required for massive neutrophil extravasation into the lungs, and for development of interstitial lung inflammation.
Figure 1.
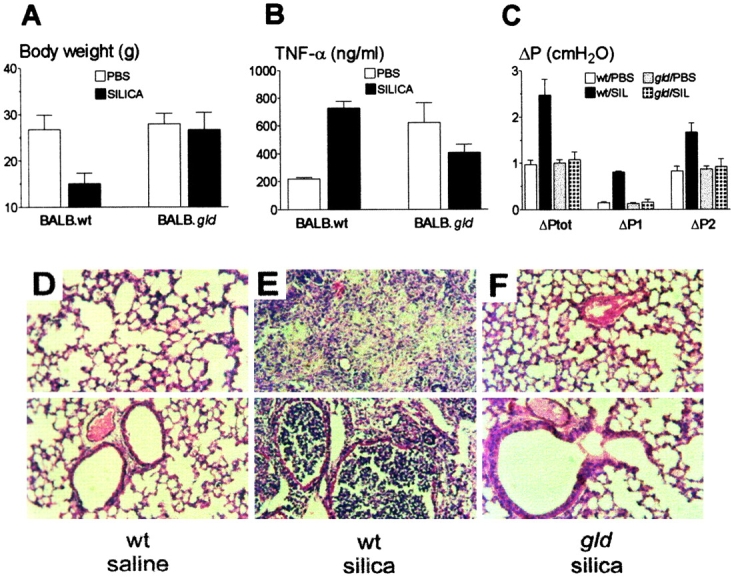
FasL-deficient mice do not develop silicosis. (A) Weight loss. Mice were instilled with saline (white bars) or silica (black bars) intratracheally. BALB.wt, but not FasL-deficient BALB.gld mice lost weight (P < 0.05, n = 6; NS, n = 6, respectively). (B) TNF-α production. BALB.wt, but not BALB.gld mice increased BAL levels of TNF-α after silica instillation (P < 0.05, n = 4; NS, n = 4, respectively). (C) Respiratory mechanics. BALB.wt, but not BALB.gld mice increased lung total (ΔPtot), resistive (ΔP1), and viscoelastic/inhomogeneous (ΔP2) pressures after silica instillation (P < 0.01, n = 6; NS, n = 6, respectively). SIL, silica. (D–F) Histopathology of lung sections from wt and gld mice. Top and bottom fields highlight areas of lung parenchyma and bronchial cross-sections, respectively. (D) Normal aspect of lung tissue from BALB.wt mice after saline instillation. (E) Intense alveolitis and heavy nodular infiltration of neutrophils and macrophages in lung from BALB.wt mice after silica instillation. (F) Normal aspect of lung tissue from BALB.gld mice after silica instillation. Hematoxylin and eosin staining. Original magnification: ×200.
Figure 2.

Deficient neutrophil extravasation in FasL-deficient mice. (A) Macrophage and neutrophil numbers in BAL fluid from mice instilled with PBS or silica intratracheally. Silica exposed BALB.gld mice (black bars) showed a 5.8-fold reduction in neutrophil number (P < 0.01, n = 4), and a 4.0-fold increase in macrophage number (P < 0.01, n = 4), compared with BALB.wt mice (white bars). SIL, silica. (B) MPO activity. Mean MPO activity of both BAL cells and lung parenchyma of wt and gld mice instilled with PBS alone (n = 4 pools of 3 mice each) was taken as 100% of control value. Left: MPO activity of BAL cells from mice instilled with silica. Cells from BALB.gld (▪) mice had much less MPO activity than cells from BALB.wt (•) mice (P < 0.05). Right: after BAL removal, lung parenchyma from BALB.wt (•), but not from BALB.gld (▪) mice, had significant MPO activity, compared with PBS-instilled controls (P < 0.01, and NS, respectively). (C) Cytospin aspect of BAL cells. BAL cells were cytocentrifuged and stained with May-Grunwald. Top: BAL cells from BALB.wt mice after saline instillation were unstimulated alveolar macrophages. Middle: BAL cells from BALB.wt mice after silica instillation were mainly neutrophils, a minority were large macrophages. Bottom: BAL cells from BALB.gld mice after silica instillation were mainly large macrophages, a minority were neutrophils. Original magnification: ×630.
Pulmonary Macrophages Expressing FasL Trigger Silicosis.
The cell type initiating silicosis could be either of bone marrow origin, or derived from lung parenchyma. To investigate this issue, we made bone marrow radiation chimeras. 1 mo after reconstitution, chimeras were instilled with silica. As shown in Fig. 3 A, silicosis developed in wt→gld chimeras expressing nonfunctional tissue FasL, but failed to develop in gld→wt chimeras expressing nonfunctional FasL on bone marrow cells. The results with interstitial MPO activity were confirmed by histological analysis (not shown). These data indicate that cells of bone marrow origin expressing FasL, most likely pulmonary macrophages, initiate silicosis. To confirm the role of macrophages, intratracheal adoptive transfer of wt alveolar macrophages into gld recipients was performed 5 d before silica instillation (Fig. 3 B). Transfer of 106 BAL macrophages from normal BALB.wt donors, but not from normal BALB.gld donors, allowed silicosis to develop in gld recipients (Fig. 3 B). FasL and Fas expression were immunolocalized in tissue sections from silica-exposed lungs (Fig. 4), and compared with sections stained with control hamster IgG (Fig. 4 A). Both FasL (Fig. 4 B) and, to a lesser extent, Fas (Fig. 4 C) staining were concentrated in granulomatous nodules containing macrophages that had ingested silica particles. Moreover, FasL expression was nearly absent in control saline-exposed lungs (Fig. 4 D), but was markedly upregulated in silica-exposed lungs (Fig. 4 E). Fas expression was detected in control lungs, but was also upregulated in silica-exposed lungs (Fig. 4F and Fig. G). We investigated FasL expression in macrophages by double staining immunofluorescence (Fig. 5 A). In control lung sections, F4/80+ macrophages did not express FasL (Fig. 5 A, left), while in silica-exposed lungs, F4/80+ macrophages were positive for FasL (Fig. 5 A, right). In silica-exposed sections, a small percentage (near 5%) of FasL+ cells were negative for F4/80 staining, and morphologically resembled activated lymphocytes. In addition, silica-exposed macrophages were also positive for Fas staining. As a control, after treatment with Fc block, macrophages from saline- and silica-exposed lung sections did not stain for labeled control hamster IgG (data not shown). FasL staining was absent in parenchymal cells and in infiltrating neutrophils from silica-exposed lungs (not shown). We also investigated induction of FasL expression in lung macrophages in vitro. Exposure to silica particles induced FasL expression in lung macrophages from control wt mice (Fig. 5 B). FasL expression could be blocked by simultaneous addition of deferoxamine, an inhibitor of iron-dependent free radical reactions (22; Fig. 5 B). Exposure to silica in vitro induced apoptosis in lung macrophages from wt, but not from gld donors (Fig. 5 C). Blockade of FasL with neutralizing anti-FasL mAb prevented macrophage apoptosis almost completely (Fig. 5 C). Taken together, the results indicate that interaction with silica particles induces FasL expression and FasL-mediated apoptosis of pulmonary macrophages.
Figure 3.
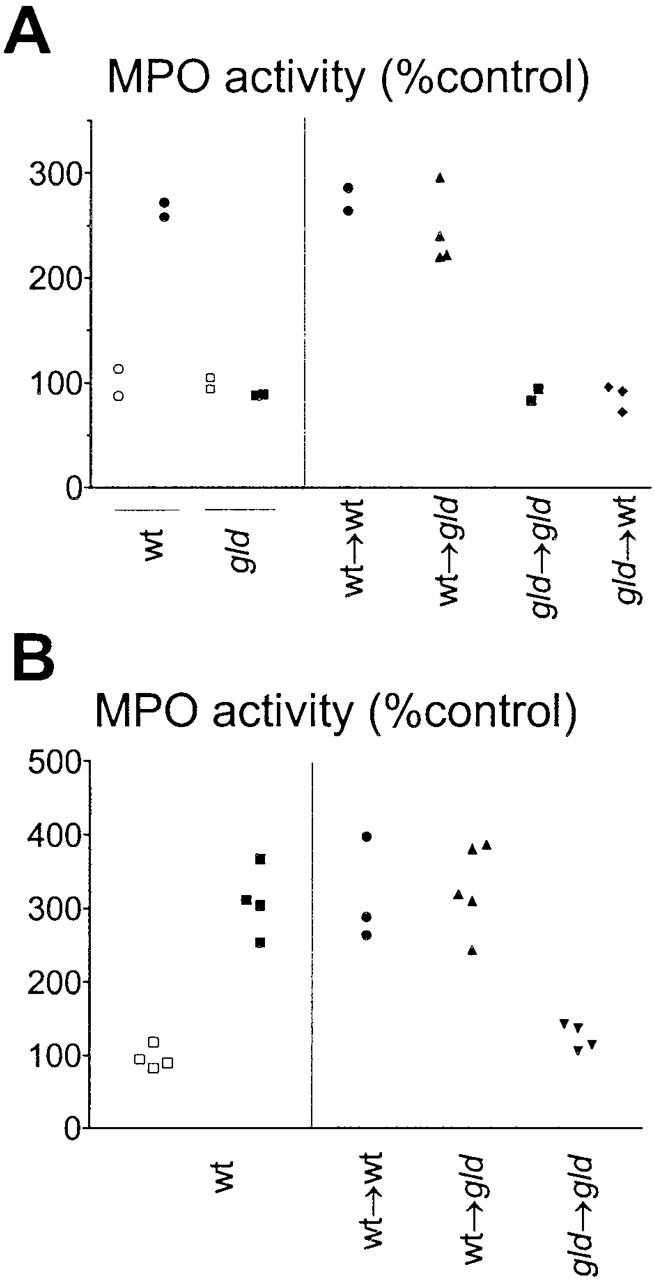
FasL-expressing macrophages initiate silicosis. (A) The cell type initiating silicosis is of bone marrow origin. MPO activity in the lungs of BALB.wt, BALB.gld mice, and the indicated chimeras, instilled with either saline (○,□) or silica (•,▪). Only mice with functional (wt) FasL on bone marrow cells accumulated neutrophils in the lung (P < 0.01). (B) FasL-expressing lung macrophages initiate silicosis. Left: MPO activity of BALB.wt mice treated with saline (□) or silica (▪) were taken as controls. Right: MPO activity in the lung parenchyma of wt (•) or FasL deficient gld mice (▴,▾), adoptively transferred intratracheally with wt (•,▴) or gld (▾) lung macrophages before silica instillation. Local transfer of wt (P < 0.01), but not of gld macrophages (NS), restored the ability of gld mice to develop silicosis.
Figure 4.

FasL and Fas expression in vivo. (A–C) Immunohistochemistry of lung tissue from BALB.wt mice instilled with silica. Lung sections were stained with control hamster IgG (A), anti-FasL (B), or anti-Fas (C) mAbs, and revealed by immunoperoxidase. Silica deposition induces FasL (B) and increases Fas (C) expression, compared with control hamster IgG (A). FasL staining is restricted to granulomatous nodules containing silica; both unaltered areas of parenchyma and epithelia were negative. Original magnification: ×200. (D–G) Higher magnification aspects of FasL (D and E) and Fas (F and G) expression in lung tissue from BALB.wt mice instilled with either saline (D and F) or silica (E and G). FasL expression is absent, and Fas expression is low in saline-instilled lung sections. Original magnification: ×630.
Figure 5.

Silica induces FasL expression in macrophages. (A) Macro-phages express FasL in vivo. Double staining immunofluorescence of lung macrophages from wt mice instilled with saline (left) or silica (right). Top: differential image contrast (DIC) microscopy of selected fields. Middle: staining of FITC-labeled F4/80+ macrophages in the same fields. Bottom: simultaneous staining of PE-labeled FasL+ macrophages in the same fields. Only silica-exposed F4/80+ macrophages stained for FasL expression. After Fc block, normal and silica-exposed macrophages did not stain for PE-labeled control hamster IgG (not shown). Original magnification: ×630. (B) Macrophages express FasL in vitro. FasL expression in normal wt BAL macrophages cultured with medium or silica in the absence (white bars) or in the presence (black bars) of deferoxamine (DFO). FasL expression was measured by cell ELISA. Silica induced FasL expression (P < 0.05), which was prevented by DFO (P < 0.05). (C) Silica induces FasL-dependent macrophage apoptosis. Apoptosis in BAL macrophages from normal gld and wt mice treated with silica in the presence of control hamster IgG or anti-FasL. Silica induced apoptosis in wt (P < 0.01), but not in gld (NS) macrophages; apoptosis was prevented by anti-FasL (P < 0.01). SIL, silica.
Administration of Anti-FasL mAb In Vivo Blocks Development of Silicosis.
We sought evidence that in vivo blockade of FasL would prevent silicosis (Fig. 6). BALB.wt mice were instilled with silica and treated with 2.5 mg of neutralizing anti-FasL mAb MFL4 18 divided into five intraperitoneal injections at 3 d intervals. Control BALB.wt mice were treated with a similar protocol using normal hamster IgG. Treatment with anti-FasL Ab prevented almost completely neutrophil accumulation into lung parenchyma, compared with control mice, as measured by MPO activity (Fig. 6 A). Furthermore, the increase in TNF-α levels was completely prevented by in vivo treatment with anti-FasL Ab (Fig. 6 B). These results were confirmed by histopathological analysis. Except for a discrete macrophage infiltrate, mice treated with anti-FasL had lungs with morphology comparable to negative controls (Fig. 6C and Fig. E), and different from the heavily inflamed lungs of mice treated with control IgG (Fig. 6 D). Taken together, these results demonstrate a pivotal role of FasL in induction of silicosis in vivo, and suggest that apoptotic cell death is required for neutrophil extravasation and pulmonary inflammation.
Figure 6.
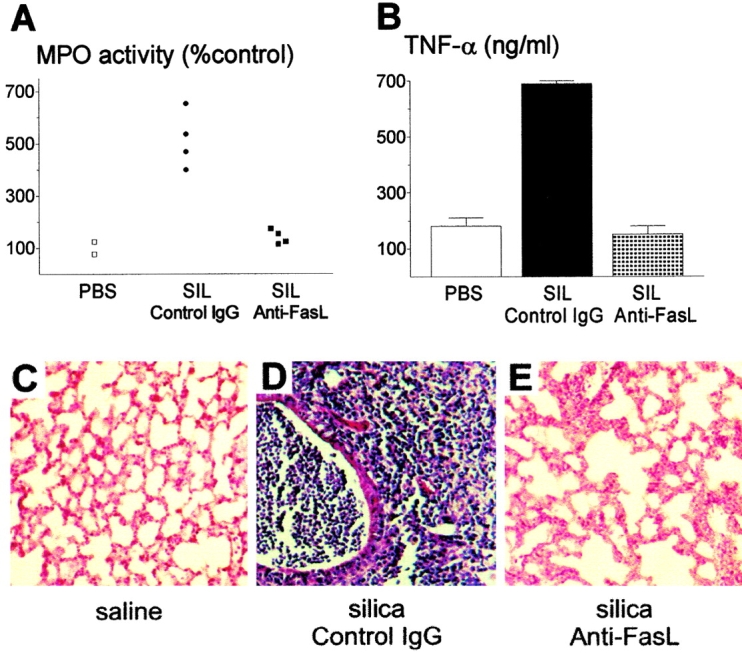
FasL blockade prevents silica-induced inflammation. (A) MPO activity in mice instilled with silica. Treatment with anti-FasL mAb MFL4 (NS compared with saline; n = 4), but not with control IgG (P < 0.01, n = 4), prevented increased MPO activity induced by silica in lung parenchyma. SIL, silica. (B) TNF-α production. Treatment with anti-FasL (NS compared with saline, n = 4), but not with control IgG (P < 0.01, n = 4), prevented increased TNF-α production in BAL fluid from silica exposed mice. (C–E) Histopathology of lung sections. (C) Normal aspect of lung tissue after saline instillation. (D) Intense pulmonary inflammation in mice instilled with silica and treated with control hamster IgG. (E) Lung tissue from mice instilled with silica and treated with anti-FasL mAb. Morphology comparable to C, except for a discrete macrophage infiltrate. Hematoxylin and eosin staining. Original magnification: ×200.
Discussion
Our data with pulmonary silicosis provide the first evidence of a FasL-mediated inflammatory disease mounted by the innate immune response. Previous studies have demonstrated that T lymphocytes modulate, but are not absolutely required in murine silicosis 23. We have used a model of silica instillation 20 21, where the dosage of silica can be controlled. The role of FasL should also be confirmed using silica inhalation 24, which more closely resembles occupational silica exposure. Silica instillation into mice leads to acute and chronic pulmonary inflammation with some characteristics resembling human silicosis, including acute neutrophilic extravasation and increased protein and phospholipid content of BAL fluid 23, as well as progressive fibrosis 20 21 23. In this study, we investigated the role of FasL in lung inflammation after 15 and 30 d of silica exposure. The role of FasL in chronic silicosis remains to be investigated. However, it should be noted that long term (1 yr) alterations in lung have been described even after a single administration of silica 25.
TNF-α plays a central role in pulmonary inflammation 3 and fibrosis 4 induced by silica deposition. Here we demonstrate a pivotal role for FasL in a mouse model of pulmonary silicosis, and locate its action upstream of TNF-α. FasL is also proinflammatory in other conditions. Forced expression of FasL in tumors and grafts induces a host neutrophilic infiltration that accelerates rejection 9 10 26. Moreover, FasL is chemotactic for neutrophils in vitro 27 and in vivo 28 29, and transduces neutrophil p38 mitogen-activated protein kinase (MAPK) activation 11. Lack of silicosis in gld mice indicates that functional FasL triggers the molecular and cellular pathways of pulmonary inflammation. Further support for this concept is that in vivo administration of a neutralizing anti-FasL mAb prevents development of silicosis.
FasL expression can be induced in nonlymphoid tissue 30. However, the cell type initiating murine silicosis is of bone marrow origin. These results exclude a major role for epithelial cell damage or other contribution from lung parenchyma in early steps of silicosis. Adoptive transfer of silicosis to gld mice by wt alveolar macrophages confirmed their essential role, and implied that activated macrophages must express FasL. We showed that silica exposure induces FasL expression in lung macrophages in vivo and in vitro. It is known that reactive oxygen intermediates induce FasL expression 31, and that treatment with a free radical scavenger compound alleviates silicosis 32. Deferoxamine, an inhibitor of reactive oxygen intermediate production 22, prevented FasL expression induced by silica. In agreement with previous studies 17, silica induced macrophage apoptosis in vitro. We showed that macrophage apoptosis is mediated by FasL. First, gld macrophages did not apoptose in response to silica. Second, wt macrophage apoptosis could be blocked with anti-FasL mAb. These results indicate that FasL expression is induced after oxidant injury provoked by silica, and that FasL expression results in macrophage apoptosis. Exposure of macrophages to other particulate stimuli also upregulates Fas and FasL expression, and triggers release of soluble FasL, which affects the viability of macrophages and bystander neutrophils 33 34. Recently, alveolar and inflammatory cell apoptosis mediated by FasL were described in acute lung injury induced in mice by bacterial LPS 35. A pathogenic role for FasL was suggested, as administration of anti-Fas antibody reduced albumin leakage into BAL fluid 35.
The gld mice had markedly impaired neutrophil extravasation, and lacked pulmonary inflammation. These data support a major chemotactic role for FasL 9 10 26 29, and indicate that neutrophils are the effector cells of lung injury. However, FasL does not account for all cellular changes in lungs exposed to silica. Macrophage recruitment into BAL fluid is increased in gld mice, suggesting that it is FasL independent. As the pulmonary parenchyma of gld mice was normal, these results indicate that in the absence of a FasL signaling cascade, leukocytes migrating from the blood locate in the alveolar space and are eliminated.
The factors that drive the FasL reaction to either silent apoptosis or acute neutrophilic infiltration are unclear. Local levels of either TGF-β 11 or soluble FasL 29 could suppress the neutrophilic response, and lead to immune privilege. On the other hand, Fas signaling is proinflammatory, as it results in activation of caspase-1 36. Caspase-1 cleaves and activates the proinflammatory cytokines IL-1β and IL-18 36. In fact, apoptosis mediated by FasL releases IL-1β from dying cells 28. Furthermore, FasL induces IL-18 secretion from macrophages, leading to acute liver injury 37. In the absence of immune privilege inducing factors, IL-1, and possibly other cytokines and chemokines released by apoptosis, could be responsible for neutrophil extravasation 29. Thus, one interpretation for our results is that continual interaction with silica particles induces FasL-mediated macrophage apoptosis, releasing chemotactic factors for neutrophils. Neutrophils in turn, would phagocytose dying cells and damage the lung parenchyma by releasing toxic oxygen metabolites, hydrolytic enzymes, and TNF-α. In support of this model, we found apoptotic cells in silica induced inflammatory infiltrates, and in vivo administration of caspase inhibitors alleviated neutrophil infiltration (data not shown). There was a small percentage of FasL+ cells resembling lymphocytes in silica-induced inflammatory nodules. However, the critical role of lung macrophages is supported by the adoptive transfer of silicosis they mediate, and by previous studies showing that induction of experimental silicosis does not require T cells 23.
Our data, and previous studies 9 10 26 28 29, demonstrate a proinflammatory role for apoptosis. In contrast, there is evidence that phagocytosis of apoptotic cells plays an antiinflammatory role 38 39, and downregulates nitric oxide production by macrophages 39. The reason for these differences is not clear. However, lung macrophages are deficient in the phagocytosis of apoptotic cells 40, suggesting that further changes in apoptotic cell membrane and release of intracellular mediators are allowed.
An important issue is whether the role of FasL can be extended to human silicosis. Increased levels of soluble FasL were reported in patients with silicosis 41. As locally produced soluble FasL can induce bystander neutrophil apoptosis 34, and blocks further neutrophil extravasation in mice 29, soluble FasL could play a role in modulation of silica-induced inflammation in humans. The finding of a central role of FasL in experimental silicosis could provide clues for the pathogenesis and treatment of this common and life-threatening occupational disease.
Acknowledgments
We thank Dr. Marlene Benchimol (Universidade Santa Ursula, Rio de Janeiro) for the use of fluorescence microscopy, and Dr. Nitza A. Gomes for enthusiastic help. We also thank Jose Carlos Gomes Domingos and Jose Nilson dos Santos for help with histopathology.
This work was financed by the Brazilian agencies Conselho Nacional de Pesquisas, Fundacão de Amparo à Pesquisa do Estado do Rio de Janeiro, Financiadora de Estudos e Projectos, and Programa de Núcleos de Excelencia-Ministério de Ciéncia e Tecnologia.
Footnotes
Abbreviations used in this paper: BAL, bronchoalveolar lavage; HRP, horseradish peroxidase; MPO, myeloperoxidase; wt, wild-type.
References
- Weill H., Jones R.N., Parkes W.R. Silicosis and related diseases. In: Parkes W., editor. Occupational Lung Disorders. Butterworths; London: 1994. pp. 285–332. [Google Scholar]
- Mossman B.T., Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am. J. Respir. Crit. Care Med. 1998;157:1666–1680. doi: 10.1164/ajrccm.157.5.9707141. [DOI] [PubMed] [Google Scholar]
- Fujimura N. Pathology and pathophysiology of pneumoconiosis. Curr. Opin. Pulmon. Med. 2000;6:140–144. doi: 10.1097/00063198-200003000-00010. [DOI] [PubMed] [Google Scholar]
- Piguet P.F., Collart M.A., Grau G.E., Sappino A.P., Vassalli P. Requirement of tumour necrosis factor for development of silica-induced pulmonary fibrosis. Nature. 1990;344:245–247. doi: 10.1038/344245a0. [DOI] [PubMed] [Google Scholar]
- Nagata S. Fas ligand-induced apoptosis. Annu. Rev. Genet. 1999;33:29–55. doi: 10.1146/annurev.genet.33.1.29. [DOI] [PubMed] [Google Scholar]
- Lenardo M., Chan K.M., Hornung F., McFarland H., Siegel R., Wang J., Zheng L. Mature lymphocyte apoptosisimmune regulation in a dynamic and unpredictable antigenic environment. Annu. Rev. Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- Krammer P.H. CD95's deadly mission in the immune system. Nature. 2000;407:789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- Griffith T.S., Brunner T., Fletcher S.M., Green D.R., Ferguson T.A. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- Seino K., Kayagaki N., Okumura K., Yagita H. Antitumor effect of locally produced CD95 ligand. Nat. Med. 1997;3:165–170. doi: 10.1038/nm0297-165. [DOI] [PubMed] [Google Scholar]
- Arai H., Gordon D., Nabel E.G., Nabel G.J. Gene transfer of Fas ligand induces tumor regression in vivo . Proc. Natl. Acad. Sci. USA. 1997;94:13862–13867. doi: 10.1073/pnas.94.25.13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.J., Sun Y., Nabel G.J. Regulation of the proinflammatory effects of Fas ligand (FasL) Science. 1998;282:1714–1717. doi: 10.1126/science.282.5394.1714. [DOI] [PubMed] [Google Scholar]
- Mortola J.P., Noworaj A. Two-sidearm tracheal cannula for respiratory airflow measurement in small animals. J. Appl. Physiol. 1983;55:250–253. doi: 10.1152/jappl.1983.55.1.250. [DOI] [PubMed] [Google Scholar]
- Bates J.H.T., Baconnier P., Milic-Emili J. A theoretical analysis of the interrupter technique for measuring respiratory mechanics. J. Appl. Physiol. 1988;64:2204–2214. doi: 10.1152/jappl.1988.64.5.2204. [DOI] [PubMed] [Google Scholar]
- Abramowitz M. Contrast Methods in Microscopy; Transmitted Light 1987. Olympus Corp., Precision Instrument Div; Lake Success, New York: pp. 31 pp [Google Scholar]
- Schneider T., Issekutz A.C. Quantification of eosinophil and neutrophil infiltration into rat lung by specific assays for eosinophil peroxidase and myeloperoxidase. J. Immunol. Methods. 1996;198:1–14. doi: 10.1016/0022-1759(96)00143-3. [DOI] [PubMed] [Google Scholar]
- Spangrude G.J. Assessment of lymphocyte development in radiation bone marrow chimeras Coligan J., Kruisbeek A., Margulies D., Shevach E., Strober W. Current Protocols in Immunology, Vol. 1 1994. Wiley and Sons; New York, NY: 4.6.1–4.6.8 [Google Scholar]
- Iyer R., Holian A. Involvement of the ICE family of proteases in silica-induced apoptosis in human alveolar macrophages. Am. J. Physiol. 1997;273:L760–L767. doi: 10.1152/ajplung.1997.273.4.L760. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Yamaguchi N., Nagao F., Matsuo S., Maeda H., Okumura K., Yagita H. Polymorphism of murine Fas ligand that affects the biological activity. Proc. Natl. Acad. Sci. USA. 1997;94:3914–3919. doi: 10.1073/pnas.94.8.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Tanaka M., Brannan C.I., Jenkins N.A., Copeland N.G., Suda T., Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- Callis A.H., Sohnle P.G., Mandel G.S., Wiessner J., Mandel N.S. Kinetics of inflammatory and fibrotic pulmonary changes in a murine model of silicosis. J. Lab. Clin. Med. 1985;105:547–553. [PubMed] [Google Scholar]
- Faffe D.S., Silva G.H., Kurtz P.M., Negri E.M., Capelozzi V.L., Rocco P.M., Zin W.A. Lung tissue mechanics and extracellular matrix composition in a murine model of silicosis. J. Appl. Physiol. 2001;90:1400–1406. doi: 10.1152/jappl.2001.90.4.1400. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Protection against tissue damage in vivo by desferrioxaminewhat is the mechanism of action? Free Radic. Biol. Med. 1989;7:645–651. doi: 10.1016/0891-5849(89)90145-7. [DOI] [PubMed] [Google Scholar]
- Suzuki N., Ohta K., Horiuchi T., Takizawa H., Ueda T., Kuwabara M., Shiga J., Ito K. T lymphocytes and silica-induced pulmonary inflammation and fibrosis in mice. Thorax. 1996;51:1036–1042. doi: 10.1136/thx.51.10.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis G.S., Leslie K.O., Hemenway D.R. Silicosis in miceeffects of dose, time and genetic strain. J. Environ. Pathol. Toxicol. Oncol. 1998;17:81–97. [PubMed] [Google Scholar]
- Reiser K.M., Haschek W.M., Hesterberg T.W., Last J.A. Experimental silicosis. II. Long-term effects of intratracheally instilled quartz on collagen metabolism and morphologic characteristics of rat lungs. Am. J. Pathol. 1983;110:30–41. [PMC free article] [PubMed] [Google Scholar]
- Kang S.M., Schneider D.B., Lin Z., Hanahan D., Dichek D.A., Stock P.G., Baekkeskov S. Fas ligand expression in islets of Langerhans does not confer immune privilege and instead targets them for rapid destruction. Nat. Med. 1997;3:738–743. doi: 10.1038/nm0797-738. [DOI] [PubMed] [Google Scholar]
- Seino K., Iwabuchi K., Kayagaki N., Miyata R., Nagaoka I., Matsuzawa A., Fukao K., Yagita H., Okumura K. Chemotactic activity of soluble Fas ligand against phagocytes. J. Immunol. 1998;161:4484–4488. [PubMed] [Google Scholar]
- Miwa K., Asano M., Horai R., Iwakura Y., Nagata S., Suda T. Caspase 1-independent IL-1 beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat. Med. 1998;4:1287–1292. doi: 10.1038/3276. [DOI] [PubMed] [Google Scholar]
- Hohlbaum A.M., Moe S., Marshak-Rothstein A. Opposing effects of transmembrane and soluble Fas ligand expression on inflammation and tumor cell survival. J. Exp. Med. 2000;191:1209–1219. doi: 10.1084/jem.191.7.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E., Stuart P.M., Brunner T., Lin T., Griffith T.S., Gao Y., Nakajima H., Henkart P.A., Ferguson T.A., Green D.R. Inducible nonlymphoid expression of Fas ligand is responsible for superantigen-induced peripheral deletion of T cells. Immunity. 1998;9:711–720. doi: 10.1016/s1074-7613(00)80668-8. [DOI] [PubMed] [Google Scholar]
- Bauer M.K.A., Vogt M., Los M., Siegel J., Wesselborg S., Schulze-Osthoff K. Role of reactive oxygen intermediates in activation-induced CD95 (APO-1/Fas) ligand expression. J. Biol. Chem. 1998;273:8048–8055. doi: 10.1074/jbc.273.14.8048. [DOI] [PubMed] [Google Scholar]
- Gossart S., Cambon C., Orfila C., Séguélas M.H., Lepert J.C., Rami J., Carré P., Pipy B. Reactive oxygen intermediates as regulators of TNF-α production in rat lung inflammation induced by silica. J. Immunol. 1996;156:1540–1548. [PubMed] [Google Scholar]
- Oddo M., Renno T., Attinger A., Bakker T., MacDonald H.R., Meylan P.R. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis . J. Immunol. 1998;160:5448–5454. [PubMed] [Google Scholar]
- Brown S.B., Savill J. Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander leukocytes. J. Immunol. 1999;162:480–485. [PubMed] [Google Scholar]
- Kitamura Y., Hashimoto S., Mizuta N., Kobayashi A., Kooguchi K., Fujiwara I., Nakajima H. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am. J. Respir. Crit. Care Med. 2001;163:762–769. doi: 10.1164/ajrccm.163.3.2003065. [DOI] [PubMed] [Google Scholar]
- Restifo N.P. Not so Fasre-evaluating the mechanisms of immune privilege and tumor escape. Nat. Med. 2000;6:493–495. doi: 10.1038/74955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H., Kayagaki N., Kuida K., Nakano H., Hayashi N., Takeda K., Matsui K., Kashiwamura S., Hada T., Akira S. Caspase 1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity. 1999;11:359–367. doi: 10.1016/s1074-7613(00)80111-9. [DOI] [PubMed] [Google Scholar]
- Savill J., Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- Freire-de-Lima C.G., Nascimento D.O., Soares M.B.P., Bozza P.T., Castro-Faria-Neto H.C., de Mello F.G., DosReis G.A., Lopes M.F. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature. 2000;403:199–203. doi: 10.1038/35003208. [DOI] [PubMed] [Google Scholar]
- Hu B., Sonstein J., Christensen P.J., Punturieri A., Curtis J.L. Deficient in vitro and in vivo phagocytosis of apoptotic T cells by resident murine alveolar macrophages. J. Immunol. 2000;165:2124–2133. doi: 10.4049/jimmunol.165.4.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomokuni A., Otsuki T., Isozaki Y., Kita S., Ueki H., Kusaka M., Kishimoto T., Ueki A. Serum levels of soluble Fas ligand in patients with silicosis. Clin. Exp. Immunol. 1999;118:441–444. doi: 10.1046/j.1365-2249.1999.01083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]