

Abstract
Hepatitis B virus (HBV) is a noncytopathic virus, and the recognition of infected hepatocytes by HBV-specific CD8 cells has been assumed to be the central mechanism causing both liver damage and virus control. To understand the role of cytotoxic T cells in the pathogenesis of HBV infection, we used functional assays that require T cell expansion in vitro and human histocompatibility leukocyte antigen (HLA)-peptide tetramers that allow direct ex vivo quantification of circulating and liver-infiltrating HBV-specific CD8 cells. Two groups of patients with persistent HBV infection were studied: one without liver inflammation and HBV replication, the other with liver inflammation and a high level of HBV replication. Contrary to expectation, a high frequency of intrahepatic HBV-specific CD8 cells was found in the absence of hepatic immunopathology. In contrast, virus-specific T cells were more diluted among liver infiltrates in viremic patients, but their absolute number was similar because of the massive cellular infiltration. Furthermore, inhibition of HBV replication was associated with the presence of a circulating reservoir of CD8+ cells able to expand after specific virus recognition that was not detectable in highly viremic patients with liver inflammation.
These results show that in the presence of an effective HBV-specific CD8 response, inhibition of virus replication can be independent of liver damage. When the HBV-specific CD8 response is unable to control virus replication, it may contribute to liver pathology not only directly but by causing the recruitment of nonvirus-specific T cells.
Keywords: hepatitis, tetramers, antiviral cytotoxic T lymphocytes, cell migration, immunopathology
Introduction
300 million people worldwide have a persistent infection with hepatitis B virus (HBV) and are at risk of developing chronic liver inflammation leading to cirrhosis and hepatocellular carcinoma 1. Although the pathogenesis of chronic liver disease is not well understood, there is a consensus that liver damage is immune mediated. Since the virus is preferentially hepatotropic and not cytopathic, it has been assumed that the recognition of HBV-infected hepatocytes by Ag-specific HLA class I–restricted CTLs is causing the liver damage 2. This hypothesis is supported by a transgenic mouse model of HBV infection 3, in which liver damage occurs after transfer of virus-specific CD8 cells. In human HBV infection, studies have been hampered by the inability of HBV to infect cells in vitro and by the difficulty of studying the intrahepatic compartment. For these reasons, knowledge of the HBV-specific CTL response has been mostly restricted to the circulating compartment, relying on in vitro cell culture after peptide stimulation and confined to HLA-A2+ patients (for a review, see reference 4). Overall, these data have shown a better correlation between the virus-specific CD8 response and protection, rather than liver damage. Indeed, patients with acute HBV infection, who clear circulating virus, present a strong and multispecific CTL response that persists in the circulation without further liver damage 5 6 7 8. In contrast, this response is barely detectable in the circulation of chronic patients despite clinical and histological signs of liver damage 9, except after resolution when viral replication is controlled 10.
The low level of virus-specific CD8 cells in the circulation of chronic patients has been partially reconciled with a model of liver damage mediated by HBV-specific cytotoxic CD8 cells by the assumption that HBV-specific CTLs are preferentially sequestered in the liver, where they cause persistent hepatic damage without complete virus eradication 2. HBV-specific CD4 and CD8 cells have been shown to infiltrate the liver 11 12, but it is not known whether they represent the majority of intrahepatic cells or whether their number is proportional to the extent of liver damage. In addition, reports that have correlated the number of intrahepatic T cells with hepatocyte damage have not investigated the specificity of these cells 13 14 15 16 17 18. In this study, we examined the relationships among the circulating and intrahepatic HBV-specific CD8 response, liver damage, and virus replication.
HBV-specific CD8 cells were visualized directly in different T cell populations using HLA–peptide tetrameric complexes. The tetramer consists of four biotinylated HLA class I molecules, each folded with a nominal peptide and multimerized by the addition of streptavidin. This multimeric HLA class I–peptide complex has a high avidity for T cells displaying the appropriate T cell receptor. Binding to specific cells is detectable by flow cytometry if a fluorochrome-labeled streptavidin reagent is used 19. Tetramer staining has facilitated the dissection of cell-mediated immunity in several viral infections 20 21 22 23 24, and we have recently reported the use of HBV-specific tetramers during acute HBV infection 25. This new approach allows direct quantification of virus-specific cells in lymphocytes of peripheral blood and, importantly, in intrahepatic lymphocyte infiltration.
Since the yield of T cells purified from diagnostic biopsies is very low (typically 0.5–5 × 105 cells), information about specific intrahepatic T cells has so far only been possible after extensive in vitro stimulation, which may alter the detection of the immune events taking place in vivo. To overcome this limitation, tetramers specific for known HLA-A2–restricted CTL epitopes were used to quantify the frequency of HBV-specific CD8 cells directly ex vivo from the circulation and liver of chronically infected HBV patients. This approach allowed us to compare the frequency, compartmentalization, and functional responsiveness of HBV-specific CD8 cells in patients differing in their extent of liver damage and viral control.
Materials and Methods
Patients and Controls.
A total of 49 (36 men and 13 women) patients with chronic HBV were included in this study. 33 of them were HLA-A2+. The remaining 16 were HLA-A2− and served as a control group. An additional control group comprised five HLA-A2+ healthy subjects without evidence of exposure to HBV (i.e., negative for HBV surface [HBs]Ag and anti-HBV core [HBc] Abs). All patients with chronic HBV infection were HBsAg and anti-HBc positive and were negative for Abs to hepatitis C virus (HCV), delta virus, and to HIV-1 and -2 (except patient 20, who was anti–HIV-1+, with a normal CD4 count). Patients on current or recent (last 6 mo) antiviral therapy were excluded from the study. HLA-A2+ patients were divided in two groups according to their serum HBV DNA level and alanine transaminase (ALT) level at the time of investigation (see Table ). Analysis of demographic characteristics (age, sex, source of infection) showed no significant differences between the two groups (data not shown). Patients with HBV DNA level <2 pg/ml and ALT <35 U/liter were all HBV e (HBe)Ag− and anti-HBe+, had no documented evidence of recent seroconversion (HBeAg+ to anti-HBe Ab) or flare, and had been tested for ALT normality on at least two occasions in the month before tetramer analysis. Patients with HBV DNA level >800 pg/ml and ALT >70 U/liter were all HBeAg+ and anti-HBe−. 10 patients underwent percutaneous needle liver biopsy as part of their diagnostic evaluation (see Table ). The biopsies were divided in two parts: one for histological examination, the other for research. The histological diagnosis of the patients with HBV DNA level <2 pg/ml and ALT <35 U/liter was “normal liver” (patients 9, 15, and 17) or liver with minimal evidence of liver inflammation (patients 16 and 18). The histological diagnosis of the patients with HBV DNA level >800 pg/ml and ALT >70 U/liter was chronic active hepatitis, with massive portal infiltration of mononuclear cells (patients 19, 20, 32, and 33). One patient (patient 31) also showed extensive parenchymal infiltration of mononuclear cells.
Table 1.
HLA-A2+ Patient Data
| Patients | HBV | ALT | No. of tetramer+ cells | Sequence core 18–27 | Expansion after stim. | Liver biopsy | ||
|---|---|---|---|---|---|---|---|---|
| Tc | Tp | Te | ||||||
| pg/ml | U/liter | 5 × 104 CD8 | ||||||
| HBV-ALT↓ | ||||||||
| 1 | <2 | 24 | 1 | 12 | 6 | Wt | Neg | No |
| 2 | <2 | 25 | 4 | 24 | 16 | Wt | Neg | No |
| 3 | <2 | 35 | 0 | 5 | 5 | ND | Neg | No |
| 4 | <2 | 18 | 5 | 40 | 4 | Wt | Pos | No |
| 5 | <2 | 17 | 14 | 8 | 14 | ND | Pos | No |
| 6 | <2 | 34 | 17 | 1 | 16 | Wt | Pos | No |
| 7 | <2 | 23 | 30 | 5 | 5 | Wt | Pos | No |
| 8 | <2 | 22 | 50 | 1 | 7 | 21T/24Y | Pos | No |
| 9 | <2 | 25 | 90 | 16 | 31 | N ampl | Pos | Yes |
| 10 | <2 | 20 | 91 | 15 | 5 | Wt | Pos | No |
| 11 | <2 | 29 | 8 | ND | ND | N ampl | Neg | No |
| 12 | <2 | 19 | 8 | ND | ND | ND | Pos | No |
| 13 | <2 | 21 | 16 | ND | ND | ND | Pos | No |
| 14 | <2 | 26 | 23 | ND | ND | N ampl | Pos | No |
| 15 | <2 | 26 | 23 | ND | ND | Wt | Pos | Yes |
| 16 | <2 | 34 | 10 | ND | ND | Wt | ND | Yes |
| 17 | <2 | 30 | 9 | ND | ND | Wt | ND | Yes |
| 18 | <2 | 28 | 10 | ND | ND | Wt | ND | Yes |
| HBV-ALT↑ | ||||||||
| 19 | 1,221 | 70 | 0 | 8 | 4 | Wt | Neg | Yes |
| 20 | >2,000 | 70 | 4 | 14 | 4 | Wt | Neg | Yes |
| 21 | >2,000 | 137 | 10 | 10 | 0 | Wt | ND | No |
| 22 | >2,000 | 108 | 2 | 10 | 10 | ND | ND | No |
| 23 | >2,000 | 234 | 2 | 10 | 10 | ND | ND | No |
| 24 | 835 | 282 | 4 | 8 | 0 | Wt | Neg | No |
| 25 | >2,000 | 200 | 5 | 1 | 1 | ND | Neg | No |
| 26 | >2,000 | 92 | 13 | 8 | 8 | I27 | Pos | No |
| 27 | >2,000 | 318 | 2 | 2 | 4 | Wt | ND | No |
| 28 | >2,000 | 85 | 5 | ND | ND | ND | Neg | No |
| 29 | >2,000 | 90 | 5 | ND | ND | ND | Neg | No |
| 30 | >2,000 | 211 | 11 | ND | ND | Wt | Neg | No |
| 31 | >2,000 | 465 | 15 | ND | ND | Wt | Neg | Yes |
| 32 | >2,000 | 75 | 4 | ND | ND | Wt | ND | Yes |
| 33 | >2,000 | 60 | 6 | ND | ND | I27 | Neg | Yes |
HBV-ALT↓ patients: HBsAg+, anti-HBc+, HBeAg−, anti-HBe+; HBV-ALT↑ patients: HBsAg+, anti-HBc+, HBeAg+, anti-HBe−. ND, not done; N ampl, HBV sequence not amplified.
Virological Assessment.
HBsAg, anti-HBs, total and IgM anti-HBc, HBeAg, anti-HBe, anti-delta, anti-HCV, anti–HIV-1, and anti–HIV-2 were determined by commercial enzyme immunoassay kits (Abbot Laboratories; Ortho Diagnostic System; and Sanofi Diagnostic Pasteur). HBV DNA level was quantified by commercial hybridization assay (Digene).
Tissue Typing.
Screening for the HLA-A2 haplotype was performed by staining PBMCs with an anti–HLA-A2 Ab (Incstar) followed by an FITC-conjugated goat anti–mouse IgG second layer and flow cytometric analysis. Patients were subsequently confirmed to have the HLA-A2.01 allele by PCR DNA typing.
Synthesis of HLA-A2–Peptide Tetrameric Complexes.
Soluble HLA-A2–peptide tetramers were produced as described previously 26. In brief, recombinant class I (HLA-A2) heavy chains and β2-microglobulin were produced in Escherichia coli cells transformed with the relevant expression vectors. Only the extracellular domain of class I heavy chain was expressed, after modification by replacement of the COOH-terminal domain with a substrate sequence for BirA biotinylation. Complexes were folded in vitro using 30 mg of HLA-A2 heavy chain protein, 25 mg of β2-microglobulin, and 10 mg of synthetic peptide. Sequences of HBV (genotype D) peptides used were FLPSDFFPSV (core 18–27), FLLSLGIHL (polymerase 575–583), and WLSLLVPFV (envelope 335–343). The HLA-A2–peptide complexes were biotinylated using purified BirA enzyme at a concentration of 5 μg/ml, 0.5 mM biotin, and 5 mM ATP. The reaction was incubated at room temperature for 16 h. Biotinylated HLA-A2–peptide complexes were recovered by fast protein liquid chromatography purification (using buffer containing 20 mM Tris, pH 8.0, and 50 mM NaCl) and ion exchange chromatography (0–0.5 M NaCl gradient). Tetramers were generated by mixing biotinylated protein complex with streptavidin-PE at a molar ratio of 4:1.
Synthetic Peptides.
Peptides corresponding to the sequence of core 18–27, envelope 335–343, and polymerase 575–583 region of HBV genotype D were purchased from Chiron Mimotopes.
HLA-A2 Tetramer and Ab Staining.
PBMCs were isolated from heparinized blood samples by density gradient centrifugation on Ficoll-Hypaque. Mononuclear cells were purified from biopsies according to previous methods 27. In brief, excess liver tissue not needed for diagnostic purposes was extensively washed in RPMI and then digested with collagenase (1 mg/ml; Sigma Chemical Co.) and DNase (25 μg/ml; Sigma Chemical Co.) for 1 h at 37°C. The cell suspension was washed twice, and mononuclear cells were recovered by centrifugation over a Ficoll-Hypaque density gradient.
0.5–1 × 106 PBMCs or variable numbers of liver-infiltrating mononuclear cells (always >0.05 × 106) were incubated for 30 min at 37°C with 1 μg of PE-labeled tetrameric complex in RPMI 1640, 10% FCS in round-bottomed polystyrene tubes (Becton Dickinson). Cells were washed in PBS and then incubated at 4°C for 30 min with saturating concentrations of directly conjugated anti-CD8–Cychrome (PE-Cy5) mAb (Sigma Chemical Co.) and one of a panel of FITC-conjugated Abs. For phenotyping experiments, these consisted of anti–HLA-DR, anti-CD45RA, anti-CD38, and anti-CD62L (PharMingen). Cells were washed twice and then analyzed immediately on a Becton Dickinson FACS® using CELLQuest™ software. For analysis of circulating tetramer+ cells, ∼0.4 × 106 cells were acquired within the live gate to ensure that at least 0.05 × 106 CD8 cells were available for analysis. For analysis of liver-infiltrating tetramer+ cells, all the cells available from a given biopsy were processed and acquired, to analyze at least 104 CD8 cells within the live gate.
Production of T Cell Lines.
T cell lines were produced as described previously 6. In brief, PBMCs were resuspended at a concentration of 3 × 106/ml in RPMI 1640, 10% FCS. Cells were stimulated with 1 μM of core 18–27 peptide in a 96-well plate. Recombinant IL-2 (50 IU/ml) was added on day 4 of culture, and cells were analyzed after a total of 10–12 d of culture.
Chromium-release Assays.
T cell lines were tested for cytotoxic activity using HLA-A2–matched EBV-B cells as targets, labeled with 100 μCi 51Cr (Na51CrO4; Amersham International plc) for 1 h at 37°C. After washing, targets were diluted in RPMI 1640, 10% FCS and pulsed or unpulsed for 1 h with 1 μM of the appropriate peptide before being added to effector T cell lines at the indicated E/T ratios in 96-well round-bottomed plates. Chromium release was measured in the supernatant after 5 h of incubation at 37°C, and percent specific lysis was calculated as described 9.
Intracellular IFN-γ Staining.
Core 18–27-specific T cell lines were incubated for 4 h at 37°C at 1 × 106 cells/ml in RPMI 1640, 10% FCS with PMA (5 ng/ml) and ionomycin (1 μM) in the presence of Brefeldin A (10 μg/ml; Sigma Chemical Co.). Cells were washed and surface stained with tetramer as above (tetramer staining was also performed before stimulation, giving equivalent results). After a further wash, cells were subjected to intracellular staining using Permeafix (Ortho Diagnostic Systems) to permeabilize and fix cells according to the manufacturer's instructions, followed by staining with FITC-conjugated anti–IFN-γ Ab and its isotope-matched control (PharMingen). Cells were washed twice and analyzed by flow cytometry. For the peptide stimulation experiment, fresh PBMCs were stimulated with core 18–27 peptide (1 μM) for 6 h, with addition of Brefeldin A (10 μg/ml) after the first 1 h of this incubation. Surface staining with tetramer, permeabilization, and intracellular staining were then carried out as above.
PCR and HBV DNA Sequencing.
DNA was extracted from serum samples taken at the time of liver biopsy using QIAmp DNA Blood mini kit (QIAGEN). The HBV DNA was amplified with primers specific for the HBV core gene, as described previously 28. The amplicons were purified and the precore/core region was sequenced directly using an automated sequencer (model ABI 377; Applied Biosystems).
Immunohistochemistry of the Liver.
The distribution of CD8+ T lymphocytes in the liver was visualized by immunostaining in formalin-fixed, paraffin-embedded liver specimens. The liver sections were first microwaved in a citrate buffer (pH 6.0) for Ag retrieval, followed by incubation with an mAb to human CD8 molecule (clone C8/144B; Dako). The detection was with a sensitive immunoperoxidase kit (EnVision HRP system; Dako) with diaminobenzidine as a substrate, and the sections were counterstained with hematoxylin. The number of CD8+ T lymphocytes in the portal tracts and the intralobular areas was scored in equivalent fields (×400).
Hepatic expression of HBcAg was detected by immunostaining of formalin-fixed, paraffin-embedded liver specimens, as described previously 29. Polyclonal, rabbit anti-HBc (Dako) and an immunoalkaline phosphatase kit (En Vision System AP; Dako) were used in these experiments.
Results
Frequency of Circulating HBV-specific CD8 Cells.
HBV-infected patients were selected for study on the basis of the expression of the HLA-A2 allele, and were grouped according to serum levels of the liver enzyme ALT (as a measure of hepatic inflammation) and HBV DNA. The first group was composed of 10 HLA-A2+ (A2+) patients with chronic HBV infection who were negative for HBeAg, and whose serum contained normal levels of ALT and <2 pg/ml HBV DNA (hereafter indicated as HBV-ALT↓). The second group comprised nine HLA-A2+ patients with chronic HBV infection (HBeAg+) with >800 pg/ml HBV DNA and with raised serum ALT (hereafter HBV-ALT↑) (Table ). Six HLA-A2− (A2−) patients with chronic HBV infection and six A2+ non–HBV-infected subjects were also tested as negative controls.
PBMCs from patients and controls were double stained directly ex vivo with anti-CD8 mAbs, and with HLA-A2 tetrameric complexes able to visualize CD8 cells specific for core 18–27 (Tc 18–27), polymerase 575–583 (Tp 575–583), and envelope 335–343 (Te 335–343) 25. The number of tetramer-binding cells was different in the two groups of chronic HBV patients. In 9 out of 10 HBV-ALT↓ patients, the frequency of tetramer+ cells exceeded the level of 0.02% of circulating CD8+ representing the maximum staining observed in the controls (Table , top, and Fig. 1). The core 18–27-specific CD8 cells were the numerically dominant population of tetramer+ cells in six patients, reaching a frequency of 0.18% of circulating CD8+ cells in two patients. Polymerase tetramer+ cells were numerically dominant in three patients, while five out of nine patients had circulating CD8 cells specific for more than one epitope. In contrast, the ex vivo analysis of the frequency of tetramer+ cells in HBV-ALT↑ patients only showed numbers of tetramer+ cells above the control level in two patients (Table , bottom, and Fig. 1).
Figure 1.
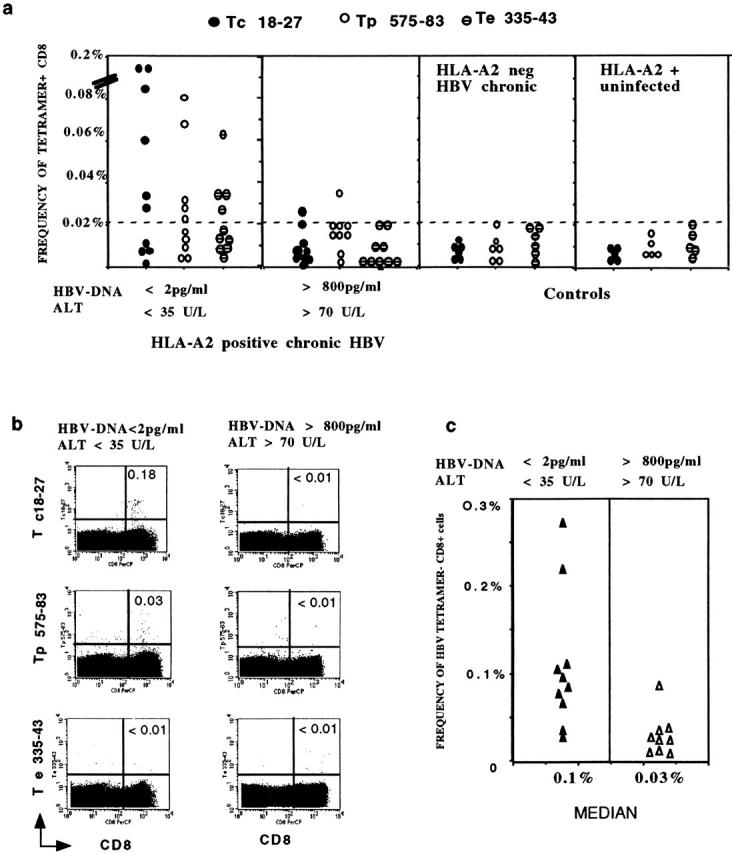
Quantification of tetramer+ CD8 cells in the circulation. (a) PBMCs from the two groups of A2+, HBV-infected patients were stained with HBV tetramers (Tc 18–27, Tp 575–583, Te 335–343) and anti-CD8 mAb immediately after isolation. The frequency of tetramer+ CD8 cells was calculated out of 50,000 CD8 cells analyzed. Results obtained in control patients (A2− patients chronically infected by HBV and A2+ HBV-uninfected subjects) are also shown. (b) FACScan™ dot plots of PBMCs stained with anti-CD8 mAb and the indicated HBV tetramers. The numbers in the upper right quadrants indicate the percent of tetramer+ cells out of total CD8 cells, calculated with CELLQuest™ software. (c) The total frequency of Tc 18–27, Tp 575–583, and Te 335–343 positive cells out of 50,000 CD8 cells was calculated. Results for the two groups of patients are presented. The sum of the frequency of tetramer+ cells for Tc 18–27, Tp 575–583, and Te 335–343 was significantly higher in the HBV-ALT↓ group (P < 0.01, Mann Whitney U test).
The difference in tetramer staining between the two groups of A2+ chronic HBV patients persisted when the overall level of tetramer staining with all three tetramers was calculated. The median level of tetramer+ cells was 54/50,000 CD8 cells in HBV-ALT↓ patients compared with 17/50,000 CD8 cells in HBV-ALT↑ patients (Fig. 1 c).
Frequency of Liver-infiltrating HBV-specific CD8 Cells.
Circulating HBV-specific CD8 cells may not accurately reflect the number of such cells sequestered in the liver. Thus, the HBV-ALT↑ patients may have lower circulating frequencies of HBV-specific CD8 cells as a result of their preferential compartmentalization within the liver. T cells purified from the livers of 20 HBV chronic patients (10 A2+, 10 A2−) were analyzed for the presence of Tc 18–27+ CD8 cells.
The frequency of Tc 18–27+ CD8 cells among intrahepatic T cells purified from biopsies performed in A2− control patients (4 HBV-ALT↓, 6 HBV-ALT↑) was always <0.01% (data not shown). Tc 18–27+ CD8 cells were detectable at a higher frequency in the liver than the circulation in 8 out of 10 A2+ HBV patients, consistent with preferential hepatic sequestration occurring in both groups of patients (Fig. 2). However, the proportion of Tc 18–27+ CD8 cells among the intrahepatic cellular infiltrate did differ between the two groups. Higher frequencies of Tc 18–27+ CD8 cells were observed in the HBV-ALT↓ group, in whom as many as 1 in 11 (patient 16) or 1 in 25 (patients 9 and 15) CD8 cells were specific for this single epitope (Fig. 2 b). In the HBV-ALT↑ patients, the highest frequency seen was 1 in 120 (patient 19), and in 2 patients there were no Tc 18–27+ CD8 cells detectable either in the periphery or in the liver (Fig. 2 b).
Figure 2.

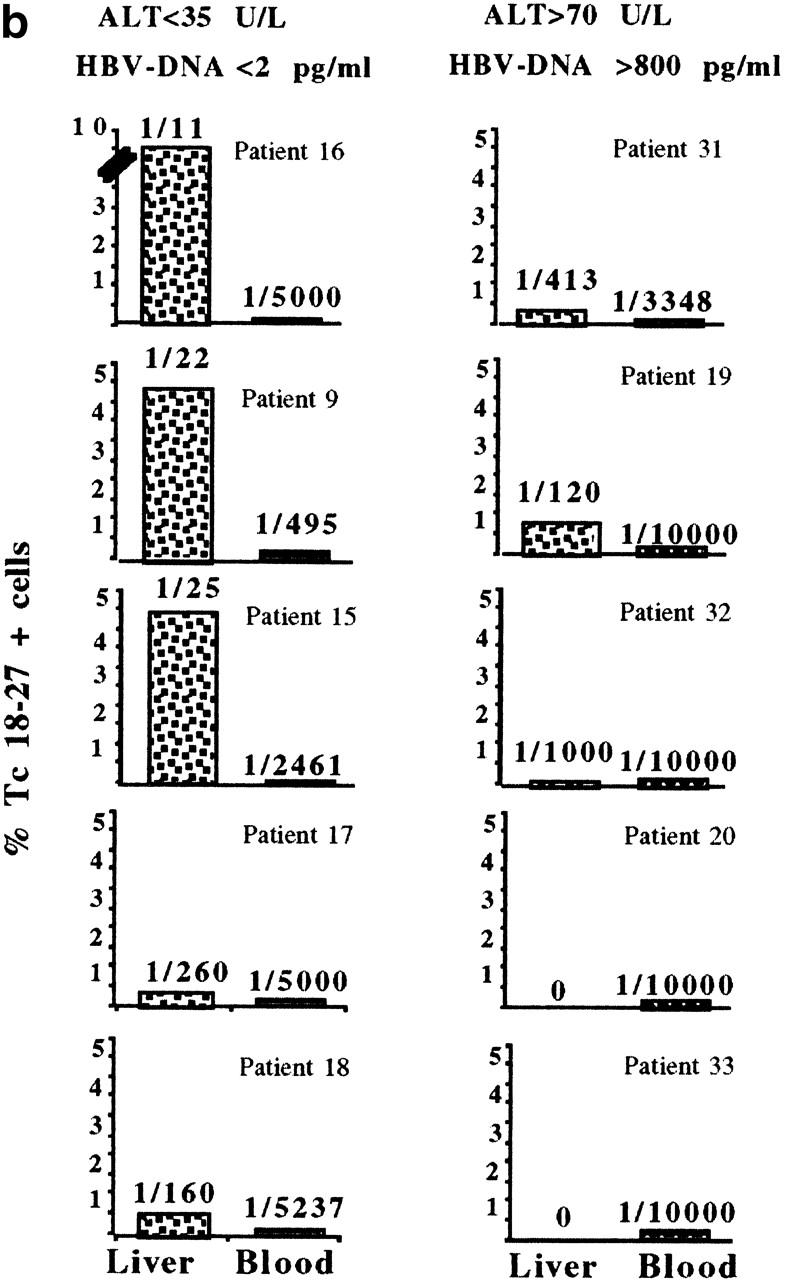
Preferential sequestration of Tc 18–27+ CD8 cells in the liver compartment. (a) Visualization of liver-infiltrating and circulating core 18–27-specific CD8+ cells by tetramer staining. Dot plots show live gated cells obtained from liver or blood of the indicated patients. a shows representative FACS® plots from two patients from each group, as indicated. Because of the differing yields of liver-infiltrating cells, total numbers of gated cells varied but were always >104 CD8 cells. Numbers in the upper right quadrants indicate the percentage of Tc18–27 CD8+ cells within the total CD8 population. (b) Frequencies of liver and circulating Tc 18–27+ CD8 cells assessed by flow cytometric analysis as illustrated in a are shown. Bars represent the percentage of Tc 18–27+ CD8 cells out of total CD8 cells. The numbers on top of the bars represent the ratio of Tc 18–27+ CD8 to total CD8+ cells.
The finding of high frequencies of Tc 18–27+ CD8 cells in the absence of biochemical evidence of hepatocyte lysis prompted us to examine whether recognition of the HBV core 18–27 sequence was still possible in these patients. Sequencing of the HBV genome across the core 18–27 epitope did not reveal any mutations that could abrogate T cell recognition (Table , top). Furthermore, ongoing active recognition of virus-infected cells was supported by phenotypic analysis of intrahepatic T cells in patient 16, where the high numbers of intrahepatic Tc 18–27 CD8+ cells allowed analysis of their activation state. The great majority (∼90%) of these lymphocytes had the phenotype of recently activated cells with high levels of HLA-DR expression (Fig. 3). Strikingly few of the remaining CD8 cells in the liver had this activated phenotype, making it unlikely that bystander cytokine production was the cause of their activation.
Figure 3.

Phenotypic analysis of liver-infiltrating Tc 18–27+ CD8 cells. Intrahepatic Tc 18–27+ CD8 cells of patient 16 were analyzed by three-color flow cytometry using the PE-conjugated Tc 18–27, cychrome-conjugated anti-CD8, and the FITC-conjugated anti–HLA-DR. The number in the upper right quadrant of the first dot plot indicates the percentage of Tc 18–27 CD8+ within the total CD8 population. The grid in the second dot plot shows the percentage of CD8+ cells in each quadrant.
Quantification of Liver-infiltrating Tc 18–27+ CD8 Cells.
We then estimated the total numbers of Ag-specific CD8 cells in the livers of patients with different clinical presentations. Different frequencies of Tc 18–27+ CD8 cells could merely reflect differing degrees of dilution in tetramer− CD8 cells, rather than a difference in the absolute number of HBV-specific CD8+ cells.
In line with this possibility, the yield of mononuclear cells obtained from liver biopsies was generally higher in patients with evidence of liver disease than in those without (data not shown). However, to better quantify the number of liver-infiltrating tetramer+ cells, an immunohistological comparison of the total CD8 infiltration in liver sections was performed. These numbers were then correlated with the frequency of Tc 18–27+ CD8 cells obtained in purified infiltrates. As expected, in patients with raised liver enzymes and histological evidence of “active hepatitis,” a large number of CD8 cells was present in the portal areas with some spreading into the intralobular area. By contrast, subjects with normal liver enzymes had relatively low numbers of infiltrating CD8 cells preferentially localized within the lobules among the hepatocytes (Fig. 4 a). Combining these findings with the frequencies of Tc 18–27+ CD8 cells found in the lymphomononuclear cells obtained from the liver of the same patients, we could estimate the absolute number of intrahepatic Tc 18–27+ CD8 cells. As illustrated in Fig. 4 a for patients 9 and 19, this calculation suggests that the frequency comparisons were misleading and that the total number of liver-infiltrating Tc 18–27+ CD8 cells may not differ between patients with or without liver damage. Patients 15 and 32 showed similar results (Table ).
Figure 4.

Analysis of liver-infiltrating CD8 cells. Liver biopsies from patient 9 (HBV DNA <2 pg/ml, ALT <35 U/liter) and patient 19 (HBV DNA 1221 pg/ml, ALT 70 U/liter) were stained with anti-CD8 mAbs. The distribution of CD8+ T lymphocytes in the liver was visualized by immunostaining in formalin-fixed, paraffin-embedded liver specimens. The number of CD8+ T lymphocytes in the portal tracts and intralobular areas was scored in equivalent histological fields (original magnification: ×400). To estimate the numbers of Tc 18–27+ cells present in each field, the number of CD8 cells was multiplied by the frequency of Tc 18–27+ cells obtained from CD8 cells purified from the same biopsy.
Table 2.
Estimation of Tc 18–27+ CD8 Cells Present in Equivalent Liver Sections
| Patients | HBV-ALT level | CD8 cells | Percent intrahepatic Tc 18–27+ CD8 cells | No. of Tc 18–27+ CD8 cells |
|---|---|---|---|---|
| 19 | HBV-ALT↑ | 138 | 0.8 | 1.1 |
| 32 | HBV-ALT↑ | 188 | 0.23 | 0.6 |
| 9 | HBV-ALT↓ | 40 | 4 | 1.6 |
| 15 | HBV-ALT↓ | 25 | 4 | 1 |
Immunostaining of liver sections for HBcAg showed, as expected, that HBcAg was undetectable in patients with successful control of HBV replication (patients 9 and 15), whereas patients with high levels of serum HBV DNA (patients 19 and 32) had HBcAg expression in >30% of hepatocytes (not shown).
Therefore, these data showed that similar numbers of intrahepatic core 18–27-specific CD8 cells can be associated with different clinical outcomes. Patients with good viral control have a relative paucity of CD8 infiltrate composed of a high proportion of Tc 18–27+ CD8 cells. By contrast, the absence of effective viral control is associated with a much greater infiltration of CD8 cells, in which Tc 18–27+ CD8 cells appear more diluted.
Core 18–27-specific CD8 Cell Responsiveness.
Mathematical models of the dynamics of the immune response during persistent infections have shown that there need not be any difference in the number of virus-specific CD8 cells between individuals despite varying abilities to control the virus 30. The principal variable determining viral control has been suggested to be the ability of virus-specific CD8 cells to clonally expand and exert their antiviral effector functions. Therefore, we investigated the capacity of circulating core 18–27-specific CD8 cells to expand after exposure to viral Ag and to exhibit antiviral activity.
PBMCs from 25 A2+ and 6 A2− HBV carriers were stimulated in vitro with core 18–27 synthetic peptide. After 10 d of culture, expansion of core 18–27 CD8 cells was quantified by staining short-term T cell lines with the appropriate tetramer. No expansion of core 18–27-specific CD8 cells was detectable in 6 A2− patients with chronic HBV infection (data not shown). 11 out of 15 of the HBV-ALT↓ but only 1 out of 10 HBV-ALT↑ patients demonstrated a vigorous expansion of core 18–27-specific CD8 cells (Fig. 5). The expanded cells from patients 7, 10, and 14 were capable of specific lysis and IFN-γ production (Fig. 6 a) when tested in vitro, suggesting that these cells should be able to exert antiviral function after virus challenge in vivo. In addition, the high circulating frequency of Tc 18–27+ CD8 cells present in patient 10 allowed us to demonstrate that core 18–27-specific CD8 cells produced IFN-γ when stimulated with the specific peptide directly ex vivo (Fig. 6 b).
Figure 5.


Ability of Tc 18–27+ CD8 cells to expand after specific peptide stimulation. PBMCs of the indicated patients were stained with Tc 18–27 and anti-CD8 mAbs before or after stimulation with core 18–27 peptide. Stimulated PBMCs were cultured for 10 d and then analyzed by flow cytometry for the presence of Tc 18–27+ cells (as shown in the dot plots). The numbers of Tc 18–27+ out of 5 × 104 cells were calculated directly (white bars) or after 10 d stimulation (hatched bars) by counting Tc 18–27+CD8+ cells out of CD8 cells within the live gate. The difference in expansion potential was significantly different between the two groups (P = 0.004, Fisher's exact test).
Figure 6.
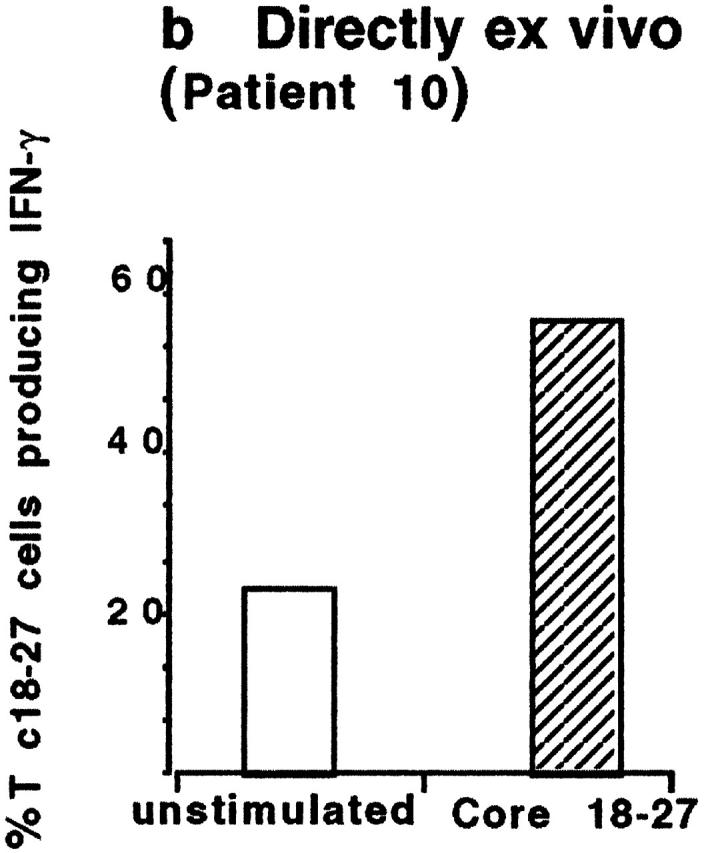

Functional characterization of circulating Tc 18–27+ CD8 cells. (a) Cytolytic activity and IFN-γ production of expanded Tc 18–27+ CD8 cells. Tc 18–27+ CD8 cells obtained after PBMC stimulation with core 18–27 peptide were tested for their ability to lyse HLA-A2+ target cells pulsed or unpulsed with 1 μM of core 18–27 peptide and to produce IFN-γ after stimulation with PMA and ionomycin for 4 h. Experiments with lines derived from patients 10, 7, and 14 are shown. For intracellular IFN-γ production, staining with an IgG isotype control allowed the threshold for IFN-γ–producing cells to be determined. (b) IFN-γ production of circulating Tc 18–27+ CD8 cells. Circulating core 18–27-specific CD8 cells (from patient 10) were tested for their ability to produce IFN-γ after specific peptide stimulation. PBMCs were incubated with or without core 18–27 peptide (1 μm) for 6 h with addition of Brefeldin A (able to block release of intracellular cytokines) after the first 1 h. Cells were stained with Tc 18–27, anti-CD8, and anti–IFN-γ or isotype-matched IgG. The number of Tc 18–27+ CD8 cells producing IFN-γ with or without specific stimulation was calculated gating on resting and activated lymphocytes by forward and side scatter, CD8+ and Tc 18–27+ cells.
Interestingly, the only HBV-ALT↑ patient with a population of HBV-specific CD8 cells capable of expansion (Fig. 5) was found to harbor a virus with Ile at position 27 (Table ). Position 27 is one of the two major anchor residues in the core 18–27 epitope, and Ile at position 27 is a naturally occurring variant known to reduce the binding affinity of the peptide to HLA-A2 9 31. Thus, the I27 sequence will not be efficiently presented on the surface of HBV-infected cells. Therefore, the core 18–27-specific CD8 cells that were expanded in vitro by stimulation with the prototype 18–27 sequence (presumably primed by previous exposure to the wild-type virus 32) would not control this patient's infecting virus efficiently.
The relatively high frequency of circulating Tc 18–27+ CD8 cells in patients 7 and 10 (HBV-ALT↓) allowed direct analysis of their phenotype by three-color flow cytometry. The Tc 18–27+ CD8 cells had low expression of T cell activation markers (HLA-DR, CD38), and were mainly negative for L-selectin (CD62L, the lymphocyte homing receptor) and for CD45RA (the high molecular weight isoform of CD45 conventionally associated with a naive phenotype). This combined phenotype is characteristic of nonactivated Ag-experienced T cells (20 34; Fig. 7). A population of Tc 18–27+ CD8 cells with similar phenotype and functional ability is present in patients resolving acute HBV infection 25.
Figure 7.

Phenotypic analysis of Tc 18–27+ CD8 cells. Fresh PBMCs from patients 10 and 7 were analyzed by three-color flow cytometry using the PE-conjugated Tc 18–27, Cychrome–conjugated anti-CD8, and the following FITC-conjugated mAbs: anti–HLA-DR, anti-CD38, anti-CD62L, and anti-CD45RA. Dot plots show data after gating on CD8+ T cells.
These data show that there is a functional similarity between the core tetramer+ cells found in chronically infected patients with low viremia and those found in patients controlling acute HBV infection.
Discussion
Although several reports have demonstrated that HBV control does not require massive destruction of infected hepatocytes 18 35, inhibition of HBV replication in humans has always been linked to immune-mediated events leading to liver inflammation. Thus, subjects with persistent HBV infection but without signs of liver damage have generally been considered to lack an active cellular response to HBV 36 37. We demonstrate the presence of functionally active HBV-specific CD8 cells in the large majority of chronic HBV-infected patients lacking evidence of liver damage but controlling HBV replication. Furthermore, HBV-specific CD8 cells are not only present in the circulation but are within the liver in this group of patients, demonstrating that peripheral tolerance and T cell exhaustion of HBV-specific CD8 cells do not occur in this patient population.
HCV-specific CTLs are also present in the normal liver of chimpanzees 1 yr after resolution of acute HCV infection 38. Furthermore, our findings have parallels with a murine model of influenza virus infection in which efficient virus control is strictly dependent on the kinetics and distribution of the virus-specific CD8 response, but is not associated with pathological damage induced by the immune response 39 40. The pattern of the HBV-specific CD8 response is also similar to that seen in other human persistent virus infections, such as EBV 33 and HTLV-I 41 42, in which a powerful CTL response can play an important role in limiting virus replication without causing inflammatory disease.
Our data do not establish whether HBV-specific CD8 cells can control virus replication through the secretion of cytokines alone or whether direct lysis of infected cells is also involved. The sparsely scattered pattern of CD8+ cells within the liver parenchyma of patients with low level viral replication suggests that secreted cytokines may be playing a major role in antiviral control. The recent demonstration of the efficacy of IFN-γ in activating a pathway of intracellular virus inactivation in the hepatocytes reinforces this interpretation 35 43. However, some degree of direct hepatocyte lysis caused by HBV-specific CD8 cells may exist, which is not detectable by serum liver enzyme measurement.
Patients with evidence of liver inflammation and a high level of HBV replication show a different pattern of distribution of HBV-specific CD8 cells. The frequency of these cells is not above the background level in the circulation of the great majority of the subjects tested. However, HBV-specific CD8 cells are not completely deleted, as they are detectable in the liver compartment. Although HLA–peptide tetramers have the advantage of allowing direct and reproducible quantification of HBV-specific CD8 cells 25, they also have potential limitations. The detection of virus-specific CD8 cells is dependent on the expression of TCRs on their cell surface. Therefore, one interpretation of the absence of tetramer-binding cells could be TCR downregulation in the presence of high levels of Ag 21. However, the fact that we can detect HBV-specific CD8 cells in the liver, the site of maximal HBV replication, implies that TCR downregulation does not completely abrogate their detection, and that the absence of these cells in the periphery is due to a genuine compartmentalization.
Another limitation of the tetramer technology is that it can only be applied to the study of defined epitopes, and cannot quantify the entire spectrum of CD8+ cells specific for HBV. We therefore cannot rule out the possibility that HBV-specific CD8 responses in chronic patients could be directed against epitopes other than those covered by the HBV tetramers used in this study. However, these tetramers have been synthesized because they cover the most frequent CTL epitopes found in acutely and chronically infected patients after screening with multiple peptides 6 7 8 9. Furthermore, CD8 cells specific for core 18–27 appear to be numerically dominant in the immunoprotective response associated with the control of acute infection 25.
In the context of this dominant CD8 response, we found that frequencies of intrahepatic Tc 18–27+ CD8 cells are lower in the majority of patients with high virus load than in patients controlling the virus. However, the total number of intrahepatic Tc 18–27+ CD8 cells is likely to be of the same magnitude in the two groups, because the higher CD8 infiltration in the group of patients with liver inflammation could compensate for the lower frequency of tetramer-specific CD8 cells. If the quantity of core 18–27-specific cells is not the variable determining the liver pathology, hepatocyte damage may not be primarily due to lysis by HLA class I–restricted HBV-specific CD8 cells, but might be the consequence of the large infiltrate of T cells. It is tempting to speculate that this infiltration may be largely nonvirus specific 44. Such recruitment of nonantigen-specific CD8 cells mediated by IFN-γ has been demonstrated in a transgenic mouse model of fulminant hepatitis 45 and in the setting of poor viral control in a mouse model of influenza infection 39.
Support for the concept that the CD8 liver infiltrate may have a large nonvirus-specific component comes from several studies. In chronic hepatitis C, the frequency of liver-infiltrating HCV-specific CD8 cells is very low 46, suggesting that the bulk of intrahepatic CD8 cells are nonantigen specific. In a transgenic mouse model of fulminant hepatitis, liver damage is only seen when infiltration of nonantigen-specific CD8 cells follows the Ag-specific component 45. Furthermore, in recent results from chimpanzees infected with HBV, liver damage occurs concomitant with massive infiltration of CD8 cells 18. This sequestration occurs after clearance of most of the HBV, and is therefore unlikely to be composed primarily of HBV-specific CD8 cells.
The data also show that viral replication does not depend on the quantity of Tc 18–27+ CD8 cells. We found that completely different levels of HBV replication can coexist with slightly different numbers in the circulation and with comparable numbers of intrahepatic HBV-specific CD8 cells. Escape of CTL recognition due to mutations within the epitope is not the explanation of this finding. The presence of similar numbers of virus-specific CD8 cells despite large differences in viral load seems counterintuitive if CTLs have an important role in viral control. However, this conundrum has also been observed in HTLV-1 41 47 and fits a mathematical model where steady-state CTL numbers do not correlate with virus load 30. This model suggests instead that the primary factor controlling viral load is CTL responsiveness, which denotes the rate at which virus-specific CD8 cells expand and exert antiviral activity. Since efficient CTL responsiveness will result in a lowering of the viral load, virus-specific CD8 numbers would fall with the lower antigenic stimulus. Thus, at equilibrium, there may be little discernible difference in actual CTL abundance between patients with high and low levels of HBV replication.
Our data from the two groups of HBV patients are consistent with this model in that the most striking difference between them is their CTL responsiveness rather than actual numbers at equilibrium. Patients controlling the virus demonstrate circulating HBV-specific CD8 cells expressing the phenotype of Ag-experienced resting cells. These cells exhibit efficient proliferation after reencounter with the viral Ag, and can exert antiviral effector functions after such expansion, demonstrating that patients without liver inflammation and with low viral load are characterized by the potential to rapidly mount strong CD8 effector mechanisms. This reservoir of circulating HBV-specific CD8 cells able to expand after recognition of the specific virus sequence is not detectable in patients unable to control the virus. This prevents phenotypic and functional comparison of equivalent populations of HBV-specific CD8 between the two groups, which might reveal possible mechanisms contributing to chronicity. Whether these different outcomes of chronic infection result from differences in the efficiency, kinetics 48, and distribution 40 of the antiviral CD8 response, differences in CD4 T cell help 49, or differences in the size or fitness of the initial virus inoculum 21 39 remains to be determined. However, recent studies (Boni, C., manuscript in preparation) in HBeAg+ chronic patients undergoing lamivudine treatment have shown that a reduction in viral load allows repopulation with functionally active HBV-specific CD8 cells. This reconstitution of a circulating reservoir of HBV-specific CTLs, combined with the absence of hepatic inflammation mediated by these cells when viral load is low, supports an immunotherapeutic approach designed to boost HBV-specific CD8 responses.
In conclusion, measurement of circulating and intrahepatic HBV-specific CD8 cells in patients with differing viral load and liver pathology has provided new insights into the pathogenesis of HBV infection. Our data show an active HBV-specific CD8 response in patients controlling HBV replication. The presence of liver-infiltrating HBV-specific CD8 cells in the absence of liver inflammation suggests that control of HBV replication and liver damage may be independent events in these patients. Since comparable quantities of core 18–27-specific CD8 cells are demonstrable in the liver with a variable extent of damage, it is plausible that hepatocyte lysis might be the consequence of the dense infiltrate of nonantigen-specific T cells.
Acknowledgments
A. Bertoletti, C. Ferrari, and M.K. Maini conceived the experiments that were carried out by C. Boni, C.K. Lee, J.R. Larrubia, and S. Reignat. G.S. Ogg and A.S. King synthetized the tetramers, and J. Herberg sequenced HBV; N.V. Naoumov carried out the immunohistology and together with R. Gilson, R. Williams, A. Alisa, and M.K. Maini recruited the patients. M.K. Maini, D. Vergani, C. Ferrari, and A. Bertoletti co-wrote the paper. We thank Charles Bangham for critical reading of the manuscript.
M.K. Maini is funded by a Collaborative Research Grant from The Edward Jenner Institute for Vaccine Research. J.R. Larrubia is supported by the Fondo de Investigaciones Sanitarias (BEFI 98/9155) from the Ministerio de Sanidad y Consumo of Spain. J. Herberg is funded by a student fellowship from The Wellcome Trust. This work is supported in part by a University College London Clinical Research and Development Committee grant and by a Project Grant of The Wellcome Trust.
Footnotes
Abbreviations used in this paper: ALT, alanine transaminase; HBc, HBV core; HBe, HBV e; HBs, HBV surface; HBV, hepatitis B virus; HCV, hepatitis C virus; Tc 18–27, core 18–27 HLA-A2 tetrameric complex; Te 335–343, envelope 335–343 HLA-A2 tetrameric complex; Tp 575–583, polymerase 575–583 HLA-A2 tetrameric complex.
References
- Di Marco V., Lo Iacono O., Camma C., Vaccaro A., Giunta M., Martorana G., Fuschi P., Almasio P.L., Craxi A. The long-term course of chronic hepatitis B. Hepatology. 1999;30:257–264. doi: 10.1002/hep.510300109. [DOI] [PubMed] [Google Scholar]
- Chisari F., Ferrari C. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- Moriyama T., Guilhot S., Klopchin K., Moss B., Pinkert C.A., Palmiter R.D., Brinster R.L., Kanagawa O., Chisari F.V. Immunobiology and pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice. Science. 1990;248:361–364. doi: 10.1126/science.1691527. [DOI] [PubMed] [Google Scholar]
- Chisari F.V. Cytotoxic T cells and viral hepatitis. J. Clin. Invest. 1997;99:1472–1477. doi: 10.1172/JCI119308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayersina R., Fowler P., Guilhot S., Missale G., Cerny A., Schlicht H.J., Vitiello A., Chesnut R., Person J.L., Redeker A.J., Chisari F.V. HLA-A2 restricted cytotoxic T lymphocyte responses to multiple hepatitis B surface antigen epitopes during hepatitis B virus infection. J. Immunol. 1993;150:4659–4671. [PubMed] [Google Scholar]
- Penna A., Chisari F., Bertoletti A., Missale G., Fowler P., Giuberti T., Fiaccadori F., Ferrari C. Cytotoxic T lymphocytes recognize an HLA-A2–restricted epitope within the hepatitis B virus nucleocapsid antigen. J. Exp. Med. 1991;174:1565–1570. doi: 10.1084/jem.174.6.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehermann B., Person J., Redeker A., Fowler P., Brown M., Moss B., Sette A., Chisari F.V. The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J. Exp. Med. 1995;181:1047–1058. doi: 10.1084/jem.181.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehermann B., Ferrari C., Pasquinelli C., Chisari F.V. The hepatitis B virus persists for decades after patients' recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 1996;2:1104–1108. doi: 10.1038/nm1096-1104. [DOI] [PubMed] [Google Scholar]
- Bertoletti A., Costanzo A., Chisari F.V., Levrero M., Artini M., Sette A., Penna A., Giuberti T., Fiaccadori F., Ferrari C. Cytotoxic T lymphocyte response to a wild-type hepatitis B virus epitope in patients chronically infected by variant viruses carrying substitutions within the epitope. J. Exp. Med. 1994;180:933–943. doi: 10.1084/jem.180.3.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehermann B., Lau D., Hoofnagle J.H., Chisari F.V. Cytotoxic T lymphocyte responsiveness after resolution of chronic hepatitis B virus infection. J. Clin. Invest. 1996;97:1655–1665. doi: 10.1172/JCI118592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari C., Mondelli M., Penna A., Fiaccadori F., Chisari F. Functional characterization of cloned intrahepatic, hepatitis B virus nucleoprotein-specific helper T cell lines. J. Immunol. 1987;139:539–544. [PubMed] [Google Scholar]
- Barnaba V., Franco A., Alberti A., Balsano C., Benvenuto R., Balsano F. Recognition of hepatitis B envelope proteins by liver-infiltrating T lymphocytes in chronic HBV infection. J. Immunol. 1989;143:2650–2655. [PubMed] [Google Scholar]
- Colucci G., Colombo M., Ninno E.D., Paronetto F. In situ characterization by monoclonal antibodies of the mononuclear cell infiltrate in chronic active hepatitis. Gastroenterology. 1983;85:1138–1145. [PubMed] [Google Scholar]
- Dienes H.P., Hutteroth T., Hess G., Meuer S.C. Immunoelectron microscopic observation on the inflammatory infiltrates and HLA antigens in hepatitis B and non-A, non-B. Hepatology. 1987;7:1317–1325. doi: 10.1002/hep.1840070623. [DOI] [PubMed] [Google Scholar]
- Pape G.R., Rieber E.P., Eisenburg J., Hoffmann R., Balch C.M., Paumgartner G., Riethmuller G. Involvement of the cytotoxic/suppressor T-cell subset in liver tissue injury of patients with acute and chronic liver diseases. Gastroenterology. 1983;85:657–662. [PubMed] [Google Scholar]
- Yang P.-M., Su I.-J., Lai M.-Y., Huang G.-T., Hsu H.-C., Chen D.-S., Sung J.-L. Immunohistochemical studies on intrahepatic lymphocyte infiltrates in chronic type B hepatitis, with special emphasis on the activation status of the lymphocytes. Am. J. Gastroenterol. 1988;83:948–953. [PubMed] [Google Scholar]
- Montano L., Aranguibel F., Boffill M., Goodal A.H., Janossy G., Thomas H. An analysis of the composition of the inflammatory infiltrate in autoimmune and hepatitis B virus-induced chronic liver disease. Hepatology. 1983;3:292–296. doi: 10.1002/hep.1840030303. [DOI] [PubMed] [Google Scholar]
- Guidotti L.G., Rochford R., Chung J., Shapiro M., Purcell R., Chisari F.V. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- Ogg G.S., McMichael A.J. HLA-peptide tetrameric complexes. Curr. Opin. Immunol. 1998;10:393–396. doi: 10.1016/s0952-7915(98)80110-6. [DOI] [PubMed] [Google Scholar]
- Callan M.F., Tan L., Annels N., Ogg G.S., Wilson J.D., O'Callaghan C.A., Steven N., McMichael A.J., Rickinson A.B. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus in vivo. J. Exp. Med. 1998;187:1395–1402. doi: 10.1084/jem.187.9.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallimore A., Glithero A., Godkin A., Tissot A.C., Pluckthun A., Elliott T., Hengartner H., Zinkernagel R. Induction and exhaustion of lymphocytic choriomeningitis virus–specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I–peptide complexes. J. Exp. Med. 1998;187:1383–1393. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali-Krishna K., Altman J.D., Suresh M., Sourdive D.J.D., Zajac A.J., Miller J.D., Slansky J., Ahmed R. Counting antigen-specific CD8 T cellsa reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- Kuroda J., Schmitz J.E., Barouch D.H., Craiu A., Allen T.M., Sette A., Watkins D.I., Forman M.A., Letvin N.L. Analysis of Gag-specific cytotoxic T lymphocytes in simian immunodeficiency virus–infected rhesus monkeys by cell staining with a tetrameric major histocompatibility complex class I–peptide complex. J. Exp. Med. 1998;187:1373–1381. doi: 10.1084/jem.187.9.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg G.S., Jin X., Bonhoeffer S., Dunbar P.R., Nowak M.A., Monard S., Segal J.P., Cao Y., Rowland-Jones S.L., Cerundolo V. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science. 1998;279:2103–2106. doi: 10.1126/science.279.5359.2103. [DOI] [PubMed] [Google Scholar]
- Maini M., Boni C., Ogg G., King A., Reignat S., Lee C., Larrubia J., Webster G., McMichael A., Ferrari C. Direct ex vivo analysis of hepatitis B virus-specific CD8+ T cells associated with the control of infection. Gastroenterology. 1999;117:1–13. doi: 10.1016/s0016-5085(99)70289-1. [DOI] [PubMed] [Google Scholar]
- Altman J.D., Moss P.A.H., Goulder P.J.R., Barouch D.H., McHeyzer-Williams M.G., Bell J.I., McMichael A.J., Davis M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- Bertoletti A., D'Elios M.M., Boni C., De Carli M., Zignego A.L., Durazzo M., Missale G., Penna A., Fiaccadori F., Del Prete G., Ferrari C. Different cytokine profiles of intrahepatic T cells in chronic hepatitis B and hepatitis C virus infections. Gastroenterology. 1997;112:193–199. doi: 10.1016/s0016-5085(97)70235-x. [DOI] [PubMed] [Google Scholar]
- Naoumov N., Thomas M., Mason A., Chokshi S., Bodicky C., Farzaneh F., Williams R., Perrillo R. Genomic variations in the hepatitis B virus core genea possible factor influencing response to interferon α treatment. Gastroenterology. 1995;108:505–514. doi: 10.1016/0016-5085(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Naoumov N., Portmann B., Tedder R., Ferns B., Eddleston A., Alexander G., Williams R. Detection of hepatitis B virus antigens in liver tissue. Gastroenterology. 1990;99:1248–1253. doi: 10.1016/0016-5085(90)90811-e. [DOI] [PubMed] [Google Scholar]
- Nowak M.A., Bangham C.R. Population dynamics of immune responses to persistent viruses. Science. 1996;272:74–79. doi: 10.1126/science.272.5258.74. [DOI] [PubMed] [Google Scholar]
- Bertoletti A., Sette A., Chisari F.V., Penna A., Levrero M., De Carli M., Fiaccadori F., Ferrari C. Natural variants of cytotoxic epitopes are T-cell receptor antagonists for antiviral cytotoxic T cells. Nature. 1994;369:407–410. doi: 10.1038/369407a0. [DOI] [PubMed] [Google Scholar]
- Klenerman P., Zinkernagel R.M. Original antigenic sin impairs cytotoxic T lymphocyte responses to viruses bearing variant epitopes. Nature. 1998;394:482–485. doi: 10.1038/28860. [DOI] [PubMed] [Google Scholar]
- Tan L., Gudgeon N., Annels N., Hansasuta P., O'Callaghan C., Rowland-Jones S., McMichael A., Rickinson A., Callan M. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J. Immunol. 1999;162:1827–1835. [PubMed] [Google Scholar]
- Walker P.R., Ohteki T., Lopez J.A., MacDonald H.R., Maryanski J.L. Distinct phenotypes of antigen-selected CD8 T cells emerge at different stages of an in vivo immune response. J. Immunol. 1995;155:3443–3452. [PubMed] [Google Scholar]
- Guidotti L.G., Ishikawa T., Hobbs M.V., Matzke B., Schreiber R., Chisari F.V. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- Hoofnagle J.H., Shafritz D.A., Popper H. Chronic type B hepatitis and the “healthy” HBsAg carrier state. Hepatology. 1987;7:758–763. doi: 10.1002/hep.1840070424. [DOI] [PubMed] [Google Scholar]
- Dudley F.J., Fox R.A., Sherlock S. Cellular immunity and hepatitis-associated, australia antigen liver disease. Lancet. 1972;1:723–726. doi: 10.1016/s0140-6736(72)90234-6. [DOI] [PubMed] [Google Scholar]
- Cooper S., Erickson A., Adams E., Kansopon J., Weiner A., Chien D., Houghton M., Parham P., Walker C. Analysis of a successful immune response against hepatitis C virus. Immunity. 1999;10:439–449. doi: 10.1016/s1074-7613(00)80044-8. [DOI] [PubMed] [Google Scholar]
- Moskophidis D., Kioussis D. Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J. Exp. Med. 1998;188:223–232. doi: 10.1084/jem.188.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerwenka A., Morgan T.M., Harmsen A.G., Dutton R.W. Migration kinetics and final destination of type 1 and type 2 CD8 effector cells predict protection against pulmonary virus infection. J. Exp. Med. 1999;189:423–434. doi: 10.1084/jem.189.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiesk S., Daenke S., Parker C.E., Taylor G., Weber J., Nightingale S., Bangham C.R. The transactivator gene of human T-cell leukemia virus type I is more variable within and between healthy carriers than patients with tropical spastic paraparesis. J. Virol. 1994;68:6778–6781. doi: 10.1128/jvi.68.10.6778-6781.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery K., Usuku K., Hall S., Matsumoto W., Taylor G., Procter J., Bunce M., Ogg G., Welsh K., Weber J. HLA alleles determine human T-lymphotropic virus-I (HTLV-I) proviral load and the risk of HTLV-I-associated myelopathy. Proc. Natl. Acad. Sci. USA. 1999;96:3348–3353. doi: 10.1073/pnas.96.7.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti L.G., Borrow P., Brown A., McClary H., Koch R., Chisari F.V. Noncytopathic clearance of lymphocytic choriomeningitis virus from the hepatocyte. J. Exp. Med. 1999;189:1555–1564. doi: 10.1084/jem.189.10.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrignani S. Bystander activation by cytokines of intrahepatic T cells in chronic viral hepatitis. Semin. Liver Dis. 1997;17:319–322. doi: 10.1055/s-2007-1007208. [DOI] [PubMed] [Google Scholar]
- Ando K., Moriyama T., Guidotti L.G., Wirth S., Schreiber R., Schlicht H., Huang S., Chisari F.V. Mechanisms of class I–restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J. Exp. Med. 1993;178:1541–1554. doi: 10.1084/jem.178.5.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.-S., Rehermann B., Lopez-Labrador F.X., Boisvert J., Cheung R., Mumm J., Wedemeyer H., Berenguer M., Wright T.L., Davis M.M., Greenberg H.B. Quantitative analysis of hepatitis C virus-specific CD8+ T cells in peripheral blood and liver using peptide-MHC tetramers. Proc. Natl. Acad. Sci. USA. 1999;96:5692–5697. doi: 10.1073/pnas.96.10.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C., Nightingale S., Taylor G., Weber J., Bangham C. Circulating anti-Tax cytotoxic T lymphocytes from human T-cell leukemia virus type I-infected people, with and without tropical spastic paraparesis, recognize multiple epitopes simultaneously. J. Virol. 1994;68:2860–2868. doi: 10.1128/jvi.68.5.2860-2868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehl S., Klenerman P., Aichele P., Hengartner H., Zinkernagel R.M. A functional and kinetic comparison of antiviral effector and memory cytotoxic T lymphocyte populations in vivo and in vitro . Eur. J. Immunol. 1997;27:3404–3413. doi: 10.1002/eji.1830271240. [DOI] [PubMed] [Google Scholar]
- Zajac A.J., Blattman J.N., Murali-Krishna K., Sourdive D.J.D., Suresh M., Altman J.D., Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]