

Abstract
The specific localization of L-type Ca2+ channels in skeletal muscle triads is critical for their normal function in excitation–contraction (EC) coupling. Reconstitution of dysgenic myotubes with the skeletal muscle Ca2+ channel α1S subunit restores Ca2+ currents, EC coupling, and the normal localization of α1S in the triads. In contrast, expression of the neuronal α1A subunit gives rise to robust Ca2+ currents but not to triad localization. To identify regions in the primary structure of α1S involved in the targeting of the Ca2+ channel into the triads, chimeras of α1S and α1A were constructed, expressed in dysgenic myotubes, and their subcellular distribution was analyzed with double immunofluorescence labeling of the α1S/α1A chimeras and the ryanodine receptor. Whereas chimeras containing the COOH terminus of α1A were not incorporated into triads, chimeras containing the COOH terminus of α1S were correctly targeted. Mapping of the COOH terminus revealed a triad-targeting signal contained in the 55 amino-acid sequence (1607–1661) proximal to the putative clipping site of α1S. Transferring this triad targeting signal to α1A was sufficient for targeting and clustering the neuronal isoform into skeletal muscle triads and caused a marked restoration of Ca2+-dependent EC coupling.
Keywords: calcium channel, dihydropyridine receptor, excitation–contraction coupling, immunofluorescence, skeletal muscle
Introduction
The precise localization of Ca2+ channels in specialized membrane domains is essential for their specific actions in multiple functions of excitable cells. In neurons, for example, voltage-gated Ca2+ channels located in the nerve terminal trigger neurotransmitter release and distinct populations of pre- and postsynaptic Ca2+ channels participate in different forms of synaptic plasticity (Berridge 1998). In muscle cells, voltage-gated Ca2+ channels are specifically located in the intracellular junctions between the Ca2+ stores of the sarcoplasmic reticulum (SR) and either the transverse tubules (t tubules) or the plasma membrane, called triads and peripheral couplings, respectively (Franzini-Armstrong and Jorgensen 1994), in which the Ca2+ channel initiates excitation–contraction (EC) coupling (Melzer et al. 1995). Even though voltage-gated Ca2+ channels play such important roles in vital cell functions, the signals and mechanisms that direct and immobilize the different voltage-gated Ca2+ channel isoforms into their specific membrane domains are not known.
The skeletal muscle L-type Ca2+ channel (also called dihydropyridine receptor, DHPR) is composed of four subunits, the pore-forming α1S and the accessory α2/δ, β1a, and γ1 subunits (Catterall 1996). The channel is concentrated in the junctional membranes of the triads (Jorgensen et al. 1989; Flucher et al. 1990), where it is in close contact with the Ca2+ release channels (ryanodine receptors, RyR) of the SR Ca2+ stores. Freeze-fracture analysis showed that in the triad junctions the DHPRs are arranged in square groups of four integral membrane particles called the tetrads and that their distribution pattern corresponds to that of the RyR arrays in the opposite SR membrane (Block et al. 1988). It is believed that the skeletal muscle DHPR functions as the voltage sensor for the gating of the SR Ca2+ release channel by a mechanism that is independent of Ca2+ influx through the L-type channel (Ríos et al. 1992). Thus, the highly orderly arrangement of DHPRs and RyRs in the triads is the structural basis for the depolarization-induced SR Ca2+ release in skeletal muscle EC coupling.
But what are the mechanisms by which the Ca2+ channels are specifically targeted into the triad junction and by which they achieve their characteristic organization? In skeletal muscle of the dysgenic mouse, which lacks the α1 subunit of skeletal muscle DHPR, triads form and RyRs are normally incorporated in these junctions in the absence of α1S (Powell et al. 1996). Conversely, in myotubes of a skeletal RyR knock-out mouse, triads are also formed and DHPRs aggregate in these junctions, despite the absence of the RyR (Takekura et al. 1995); however, their arrangement in tetrads fails (Protasi et al. 1998). This suggests that triad targeting and tetrad formation are two independent processes and that interactions with the RyR are not necessary for the targeting of the DHPR into the junctional membrane domain of the triad. Evidence from studies in heterologous expression systems showing that coexpression of the DHPR α1 subunit with β and α2/δ increases membrane insertion of functional Ca2+ channels (Chien et al. 1995; Brice et al. 1997; Neuhuber et al. 1998a; Walker et al. 1998) suggests a role of the accessory Ca2+ channel subunits in the targeting process. Moreover, Ca2+ currents and EC coupling are deficient in skeletal myotubes of β-null mice and both functions can be reconstituted by heterologous expression of β1a (Gregg et al. 1996; Beurg et al. 1997). Thus, expression of the β subunit is important for the efficient expression and functional insertion of the Ca2+ channel in the membrane, but not necessarily for its targeting into triads. Immunolocalization of α2/δ and of recombinant β1a expressed in dysgenic myotubes showed that, without the α1S subunit, both subunits failed to be localized in the junctions (Flucher et al. 1991; Neuhuber et al. 1998b). Instead, the β and α2/δ subunits required coexpression of α1S for their own incorporation into the triads, suggesting that their own triad targeting is secondary to that of the α1S subunit and that β and α2/δ do not posses an independent targeting signal. The role of the γ subunit of the skeletal muscle Ca2+ channel is still poorly understood. However, the fact that EC coupling is not perturbed in skeletal muscle of a γ knock-out mouse also suggests that this subunit is not required for a function as important as the targeting of the Ca2+ channel complex into the triad (Freise et al. 2000). Thus, considering that the RyR and the accessory DHPR subunits either play no role in triad targeting of the DHPR or depend on the α1S subunit for their own targeting into the junctions, the signal for triad targeting is likely to be contained within the α1S subunit itself.
Here, we used heterologous expression of different α1 subunit isoforms and isoform chimeras in dysgenic myotubes to identify the targeting signal in the skeletal muscle DHPR. Taking advantage of differential targeting properties of the skeletal muscle α1S and the neuronal α1A subunit, we generated a series of α1S/α1A chimeras and used them to localize the triad targeting signal in the COOH terminus of α1S. The 55 amino acid sequence of α1S, which is sufficient to confer triad targeting properties to α1A, is the first description of a targeting signal of a voltage-gated Ca2+ channel to its native membrane domain.
Materials and Methods
Cell Culture and Transfections
Myotubes of the homozygous dysgenic (mdg/mdg) cell line GLT were cultured as described in Powell et al. 1996. At the onset of myoblast fusion (2–4 d after addition of differentiation medium), GLT cultures were transfected using DOTAP or FuGene (Boehringer). Cultures were analyzed 3–4 d after transfection.
Construction of Chimeric α1 Subunits
The cDNA coding sequences of Ca2+ channel α1S/α1A subunit chimeras were inserted in-frame 3′ to the coding region of a green fluorescent protein (GFP), which was modified for thermal stability (Grabner et al. 1998) and contained in a proprietary mammalian expression vector (kindly provided by P. Seeburg, Zentrum für Molekulare Biologie, Heidelberg, Germany). Insertion of the cDNAs was accomplished as follows (nucleotide numbers, nt, are given in parentheses; asterisks indicate restriction sites introduced by PCR).
GFP-α1S, GFP-α1A.
Construction of the GFP-tagged α1S (Tanabe et al. 1987) and α1A (Mori et al. 1991) subunits that were used as maternal clones for chimerization are described elsewhere (Grabner et al. 1998).
GFP-α1SkNa.
The α1A (A) cDNA coding for the NH2 terminus was fused to the α1S (Sk) cDNA at position (nt A294/Sk154) using the “gene SOEing” technique (Horton et al. 1989). The SalI*-SacI fragment (nt 5′ polylinker-Sk651) of the cDNA product generated by the fusion PCR was coligated with the SacI–BglII fragment of Sk (nt 651–4488) into the corresponding SalI*/BglII restriction enzyme (RE) sites of plasmid GFP-α1S.
GFP-α1SkI-IIa.
The HindIII–EcoRI fragment of Sk (nt 5′ polylinker-1007) and the EcoRI*-SphI A/Sk SOE fusion product (nt A1085–Sk1735) with the A/Sk transition at position (nt A1461/Sk1297) were coligated into the HindIII/SphI-cleaved polylinker of plasmid pSV-Sport1 (Life Technologies.). The SbfI–SphI fragment (nt 5′ polylinker-Sk1735) of this intermediate clone was coligated with the SphI–XhoI fragment of Sk (nt 1735–2654) into the corresponding SbfI/XhoI RE sites of plasmid GFP-α1S.
GFP-α1SkII-IIIm.
The EcoRI–BalI fragment of Sk (nt 1007–1973) was coligated with the BalI–NdeI fragment (nt 1982–2296) from the muscle α1 subunit (M) of Musca domestica (Grabner et al. 1994) into plasmid pSP72 (Promega) using the internal NdeI site (plasmid nt 2379) and the EcoRI site of the polylinker. The NdeI/EcoRI RE sites of pSP72 were also used to coligate two cDNA fragments, the NdeI*/XhoI fragment that was PCR generated from clone SkLC, a GFP-α1S with a cardiac (C) II-III loop (nt C2716–Sk2654) (Grabner et al. 1999) plus the XhoI/BglII fragment of Sk (nt 2654–4488). The NdeI* primer was designed to introduce downstream of the NdeI* site additional Musca residues, A907G and S908T. In a subsequent step fragments EcoRI–NdeI (nt Sk1007–M2297) and NdeI*–BglII (C2716–Sk4488) were isolated from the pSP72 subclones and coligated into the EcoRI/BglII-cleaved pSP72 vector. Finally, the SalI–EcoRI fragment of Sk (nt 5′ polylinker-1007) was coligated with the EcoRI–BglII fragment (nt Sk1007–Sk4488) from the last pSP72 subclone into the SalI/BglII sites of plasmid GFP-α1S.
GFP-α1SkIII-IVa.
The III-IV loop of the A cDNA was inserted into the corresponding Sk cDNA by a three-fragment SOE fusion PCR, thereby generating the transitions Sk/A (nt Sk3195/A4561) and A/Sk (nt A4725/Sk3355). The final PCR product was cleaved at its peripheral Sk XhoI/BglII RE sites and the resulting fragment (nt 2654–4488) was ligated into the corresponding XhoI/BglII sites of plasmid GFP-α1S.
GFP-α1Sa.
The XhoI–SmaI fragment of Sk (nt 2654–4038) and the SmaI–BglII Sk/A cDNA fusion fragment (nt Sk4038–A5891) with the Sk/A transition (nt Sk4143/A5461) created by SOE PCR were coligated into the XhoI/BglII RE sites of plasmid GFP-α1A (nt 1395/5891). Note that the XhoI sites are not corresponding RE sites and were used for subcloning only. Finally, the HindIII–XhoI fragment of Sk (nt 5′ polylinker-2654) was inserted into this HindIII/XhoI (nt 5′ polylinker/A1395, Sk2654) opened subclone to yield plasmid GFP-α1Sa.
GFP-α1As.
The Xho–AccI fragment of A (nt 1395–4504) was coligated with the A/Sk SOE fusion fragment AccI–BglII (nt A4504–Sk4488) carrying its A/Sk transition at nt A5460/Sk4144, into the XhoI/BglII (nt 2654/4488) cleaved plasmid GFP-α1S. Again, the A and Sk XhoI sites are not corresponding RE sites and were only used for subcloning. To yield GFP-α1As, the SalI–EcoRI fragment from A (nt 5′ polylinker-1567) was coligated with the EcoRI–BglII fragment (nt A1567–Sk4488) after isolation from the subclone into the SalI/BglII (nt 5′ polylinker/4488) cleaved plasmid GFP-α1S.
GFP-α1Aas.
The PCR generated BglII*–XbaI* fragment of Sk (nt 4566–4991) was inserted into the corresponding BglII/XbaI RE sites of plasmid GFP-α1A (nt 5891/3′ polylinker). Upstream from the artificial XbaI* site of the Sk fragment, two stop codons (nt 4984–4989) were introduced to terminate the reading frame at residue T1661, which is close to the physiological clipping site of the α1S carboxyl terminus (De Jongh et al. 1991).
GFP-α1Aas(1524-1591).
The BglII*–XbaI* Sk/A cDNA fusion fragment (nt Sk4566–A6347) with the Sk/A transition (nt Sk4773/A6118) generated by SOE PCR was ligated into the corresponding BglII/XbaI RE sites of plasmid GFP-α1A (nt 5891/3′ polylinker). Again, two stop codons were introduced upstream of the artificial XbaI* site of the A portion of the fusion product (nt 6340–6345) to terminate the reading frame at residue G2113.
GFP-α1Aas(1592-clip).
The BglII–XbaI* A/Sk SOE fusion fragment (nt A5891–Sk4991) with the A/Sk transition at nt A6117/Sk4774 was ligated into the corresponding BglII/XbaI RE sites of plasmid GFP-α1A (nt 5891/3′ polylinker).
GFP-α1A-clip.
The BglII–XbaI* fragment of A (nt 5891–6347) was ligated into the corresponding BglII/XbaI RE sites of plasmid GFP-α1A (nt 5891/3′ polylinker). Stop codons were introduced as in plasmid GFP-α1Aas(1524–1591).
GFP-α1Aas(1607-clip).
The BglII–XbaI* A/Sk SOE fusion fragment (nt A5891–Sk4991) with the A/Sk transition at nt A6165/Sk4819 was ligated into the corresponding BglII/XbaI RE sites of plasmid GFP-α1A (nt 5891/3′ polylinker).
All cDNA portions modified by PCR were checked for sequence integrity by sequence analysis (sequencing facility of MWG Biotech).
GFP and Immunofluorescence Labeling
Differentiated GLT cultures were fixed and immunostained as previously described (Flucher et al. 1994), using the monoclonal antibody 1A against the DHPR α1S subunit at a final concentration of 1:1,000 (Morton and Froehner 1987), the affinity purified antibody #162 against the type 1 RyR at a dilution of 1:5,000 (Giannini et al. 1995), and a monoclonal or an affinity purified anti–GFP antibody at a dilution of 1:2,000 and 1:4,000, respectively (Molecular Probes, Inc.). Alexa- and fluorescein-conjugated secondary antibodies were used with the anti–GFP antibodies so that the antibody label and the intrinsic GFP signal were both recorded in the green channel. Texas red–conjugated antibodies were used in double-labeling experiments to achieve a wide separation of the excitation and emission bands. Controls (for example, the omission of primary antibodies and incubation with inappropriate antibodies) were routinely performed. Images were recorded on an Axiophot microscope (Carl Zeiss, Inc.) using a cooled CCD camera and Meta View image processing software (Universal Imaging, Corp.).
Quantitative analysis of the labeling patterns was performed by systematically screening the coverslips for transfected myotubes using a 63× objective. The labeling pattern of positive myotubes with more than two nuclei were classified as either “clustered,” “ER/SR,” or “other” in the case that the labeled compartment was not clearly identifiable. The counts were obtained from several samples of at least three different experiments for each condition.
Patch-Clamp and Fluorescent Ca2+ Recording
Whole cell patch-clamp recordings were performed with an Axopatch 200A amplifier controlled by pClamp 8.0 software (Axon Instruments, Inc.). The bath solution contained (mM): 10 CaCl2 or 3 CaCl2 plus 7 MgCl2, 145 tetraethylammonium chloride, and 10 HEPES (pH 7.4 with TEA-OH). Patch pipettes had resistances of 2–4 MΩ when filled with 145 Cs-aspartate, 2 MgCl2, 10 HEPES, 0.1 Cs-EGTA, 2 Mg-ATP (pH 7.4 with Cs-OH). Leak currents were digitally subtracted by a P/4 prepulse protocol. Recordings were low-pass Bessel filtered at 2 kHz and sampled at 5 kHz. Currents were determined with 200-ms depolarizing steps from a holding potential of −80 mV to test potentials between −40 and +80 mV in 10-mV increments. Test pulses were preceded by a 1-s prepulse to −30 mV to inactivate endogenous T-type Ca2+ currents (Adams et al. 1990).
Action potential–induced Ca2+ transients were recorded in cultures incubated with 5 μM Fluo-4-AM plus 0.1% Pluronic F-127 (Molecular Probes, Inc.) in HEPES and bicarbonate-buffered DME for 45 min at room temperature, as previously described (Flucher et al. 1994; Powell et al. 1996). Action potentials were elicited by passing 1-ms pulses of 30 V across the 19-mm incubation chamber. 0.5 mM Cd2+ and 0.1 mM La3+ were added to block Ca2+ influx and therefore allow discrimination between Ca2+-induced Ca2+ release and skeletal-type EC coupling. Application of 6 mM caffeine proved the functionality of SR Ca2+ release.
Results and Discussion
Reconstitution of Triad Targeting in Dysgenic Myotubes Transfected with the Skeletal Muscle Ca2+ Channel α1S Subunit
Normal skeletal muscle in culture forms junctions between the SR and t tubules (triads and diads) and between the SR and the plasma membrane (peripheral couplings). These types of junctions are equivalent in function in that they support skeletal muscle type EC coupling, in molecular composition in that they contain RyRs in the SR and DHPRs in t tubules and plasma membrane, and in structure in that the two types of Ca2+ channels are organized in characteristic arrays of feet and tetrads, respectively. Therefore, we will henceforth use the terms “triad” and “triad targeting” in a generic sense to include all these types of junctions.
Dysgenic muscle lacks the α1S subunit of the L-type Ca2+ channel, but still forms regular junctions containing RyRs (Powell et al. 1996). Transient transfection of myotubes of the dysgenic cell line GLT with an expression plasmid encoding a fusion protein of the GFP and the α1S subunit (GFP-α1S) restores expression of the α1S subunit in the triads, L-type Ca2+ currents, and skeletal muscle EC coupling (present study, and Grabner et al. 1998). Reconstitution of dysgenic myotubes with α1S is well established and the properties observed here with the NH2-terminal GFP-fusion protein are similar to those previously reported with a COOH-terminal GFP-fusion protein (Flucher et al. 2000) or to wild-type α1S expressed in dysgenic myotubes (Tanabe et al. 1988).
The triad localization of GFP-α1S can be detected in double immunofluorescence labeling experiments with antibodies against the α1S subunit and against the RyR (Fig. 1, a and b). Immunolabeling results in a punctate distribution pattern of anti–α1S that is colocalized with similar clusters of anti–RyR. This clustered distribution pattern is characteristic of triad proteins in developing myotubes, and the coexistence of the t-tubule protein α1S with the SR protein, the RyR, is indicative of their localization in junctions between the two membrane systems (Flucher et al. 1994). Myotubes not expressing GFP-α1S form RyR clusters (Fig. 1 b), which have previously been shown to correspond to t tubule/SR junctions by electron microscopy (Powell et al. 1996).
Figure 1.

Normal incorporation of heterologously expressed GFP-α1S in t tubule/SR junctions of dysgenic myotubes. (a and b) Double immunofluorescence labeling of α1S subunits (a) and RyRs (b) shows that GFP-α1S is colocalized with RyRs in clusters (examples indicated by arrows), presumably representing triad junctions and peripheral couplings. A nontransfected myotube in the same field (#) shows RyR clusters but no α1S labeling. (c and d) Double staining of GFP-α1S with antibodies against α1S and against GFP shows that the fusion protein can be localized using either one of the antibodies. (e and f) As with anti–α1S (a), clusters labeled with anti–GFP (e) are colocalized with RyR clusters (examples indicated by arrows). In poorly differentiated myotubes that lack RyR clusters (*), GFP-α1S accumulates in a reticular membrane system with densities in the perinuclear region, presumably the ER/SR network. N, nuclei. Bar, 10 μm.
Double immunofluorescence labeling of GFP-α1S with anti–GFP and anti–α1S (Fig. 1c and Fig. d) or with anti–GFP and anti–RyR (e and f) results in the same clustered distribution pattern in equally large fractions of GFP-α1S–expressing myotubes (57 and 58%, respectively). Therefore, we continued using anti–GFP for the immunolocalization of GFP-α1 constructs, because it recognizes both GFP-α1S and GFP-α1A, allowing the direct comparison of the labeling patterns of all chimeras used in this study. Fig. 1 e also shows a myotube in which the GFP-α1S is expressed but its labeling pattern is not clustered. Instead it is distributed throughout a tubular membrane system that is very dense in the perinuclear region and has previously been identified as the ER/SR (Powell et al. 1996; Flucher et al. 2000). ER/SR distribution of heterologously expressed α1 subunits can be observed with all constructs and occurs preferentially in poorly differentiated cells; i.e., myotubes in which triads are not formed. The myotube shown in Fig. 1e and Fig. f, for example, lacks RyR clusters, indicating that triad junctions, and thus the target for GFP-α1S, was missing and therefore GFP-α1S was retained in the biosynthetic apparatus. However, in addition to myotubes lacking the target for the α1 subunit, retention in the ER/SR system can also be observed in normally differentiated myotubes if they are transfected with α1 constructs lacking the triad targeting signal (see below).
Differential Targeting of the Skeletal and the Neuronal Ca2+ Channel α1 Subunits Expressed in Dysgenic Myotubes
Fig. 2 shows a direct comparison of the labeling patterns of the skeletal muscle GFP-α1S and the neuronal GFP-α1A expressed in dysgenic myotubes. Whereas double immunolabeling with anti–GFP and anti–RyR reveals the colocalization of GFP-α1S and RyR in the junctions (Fig. 2, a–c), GFP-α1A is localized exclusively in the ER/SR network (d), even though the presence of RyR clusters (e) indicates the normal differentiation of junctions. The merged color images of GFP-α1 in green and RyR in red further emphasizes the differential targeting of the skeletal and neuronal channels. Here, colocalization of GFP-α1S with RyR shows up as yellow clusters (c), whereas the distinct localization of GFP-α1A in the ER/SR and RyR in clusters is seen as separate green and red label, respectively (f). Quantification of the labeling patterns in at least six independent experiments showed that, while the overall expression of both constructs was the same, clusters were observed in 58% (n = 967) of the myotubes transfected with GFP-α1S, but never in myotubes expressing GFP-α1A (n = 418). Thus, GFP-α1A fails to be incorporated into triad junctions.
Figure 2.

Differential targeting of GFP-α1S and GFP-α1A in transiently transfected dysgenic myotubes. Double immunofluorescence labeling of the skeletal GFP-α1S and the neuronal GFP-α1A (using anti–GFP for both) and of the RyR shows that only GFP-α1S is colocalized with RyR clusters (a and b; examples indicated by arrows). GFP-α1A is not colocalized with RyR clusters (d and e), but is retained in a reticular cytoplasmic membrane system, the ER/SR (d). (c and f) The differential subcellular distribution of GFP-α1S and GFP-α1A is highlighted in a color overlay of the images of a and b, and d and e, respectively (insets at twofold magnification). Colocalization of GFP-α1S (green) with RyRs (red) results in yellow clusters. In contrast, separate green GFP-α1A and red RyR labeling indicates the lack of colocalization of GFP-α1A and RyRs. Bar, 10 μm.
To exclude the possibility that the absence of GFP-α1A resulted from improper folding or lack of plasma membrane incorporation of the GFP-α1A construct rather than lack of a triad targeting signal, we performed patch-clamp recordings of myotubes expressing this construct. Even though a plasma membrane stain was not detected with immunocytochemistry in GFP-α1A–transfected myotubes, the whole-cell recordings showed large Ca2+ currents with the macroscopic properties of class-A Ca2+ channels expressed in heterologous mammalian expression systems (example shown in Fig. 6, below) (Adams et al. 1994). Thus, GFP-α1A expressed in dysgenic myotubes formed functional channels in the cell membrane. But instead of becoming locally concentrated in the triads, GFP-α1A was distributed diffusely in the plasma membrane at densities below detectability with immunocytochemistry.
Figure 6.

Targeting properties and current properties of wild-type α1 subunit isoforms and COOH-terminal chimeras. (a) Isoform sequence composition of COOH termini in the studied chimeras, with sequences of α1S in gray and α1A in black. Bar graph indicates the percentages of transfected myotubes showing triad targeting in immunofluorescence analysis. Alignment of the 55 α1S amino acids containing the targeting signal with the corresponding sequences of α1C and α1A. (b) Representative current traces recorded from dysgenic myotubes transfected with wild-type GFP-α1S (in 10 mM Ca2+), with GFP-α1A or with the targeted chimera GFP-α1As (both in 3 mM Ca2+). Currents from GFP-α1Sa (in 10 mM Ca2+) were too small for systematic analysis; see frequency distribution of current densities. (c) Representative current trace of the targeted chimera GFP-α1Aas(1592-clip) and comparison of peak current densities recorded from GFP-α1A, GFP-α1Aas(1592-clip), and GFP-α1Aas(1524-1591). Substituting COOH-terminal α1A sequence with the skeletal sequence 1592–1661 results in a twofold increase of current density compared with GFP-α1A, whereas substituting for skeletal sequence 1524–1591 reduces the current densities to near the detection level and could be analyzed only after increasing the Ca2+ concentration to 10 mM (n = 7–17).
Recordings of cytoplasmic Ca2+ transients in response to electrical field stimulation showed that GFP-α1S regularly restored skeletal muscle EC coupling in dysgenic myotubes (see Fig. 7, and Powell et al. 1996; Flucher et al. 2000). In contrast, restoration of EC coupling by GFP-α1A was only rarely observed (see below). Thus, both the skeletal muscle GFP-α1S isoform and the neuronal GFP-α1A isoform were functionally expressed in dysgenic myotubes, but only GFP-α1S was targeted into the triad junctions and efficiently restored EC coupling. The differential distribution of GFP-α1S and GFP-α1A as well as their functional differences are in general agreement with observations from a previous study comparing the expression of GFP-α1S, GFP-α1A, and a cardiac GFP-α1C construct in primary cultured dysgenic myotubes (Grabner et al. 1998). In that study, GFP-α1A differed from the muscle isoforms in that its distribution patterns were restricted to near the injection site and that only GFP-α1A failed to respond in a contraction assay. Together, these data support our hypothesis that the skeletal muscle α1S contains a signal for its targeting and selective incorporation into triads, but that such a triad targeting signal is missing from the neuronal α1A subunit isoform.
Figure 7.
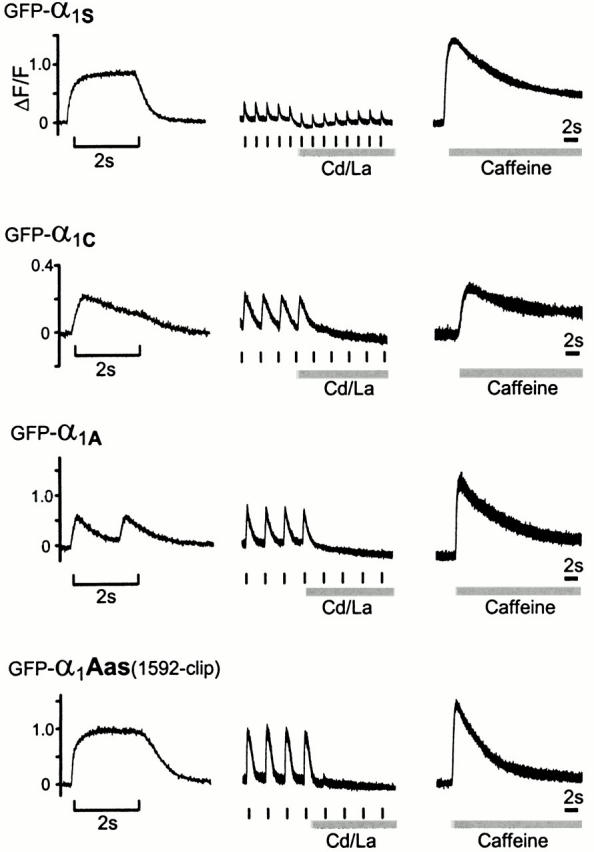
Restoration of EC coupling by targeted and nontargeted Ca2+ channel isoforms. Action potential–induced Ca2+ transients were recorded in transfected dysgenic myotubes loaded with the fluorescent Ca2+ indicator Fluo4-AM, using tetanic electrical stimulation (left, 20 Hz, 2 s, bracket) or low frequency stimulation (center, 0.3–0.5 Hz as marked). 0.5 mM Cd2+/0.1 mM La3+ (gray bar) was applied to block the Ca2+ influx during the low-frequency stimulation protocol. Ca2+ release from the SR could be triggered by the application of 6 mM caffeine (gray bar) to the bath after current block. Myotubes transfected with the skeletal GFP-α1S responded to electrical stimulation with Ca2+ transients independently of Ca2+ influx. The cardiac GFP-α1C also reconstituted EC coupling in dysgenic myotubes; however, Ca2+ transients stopped when the Ca2+ influx was blocked. Cardiac-type Ca2+ transients in response to electrical stimulation were rarely observed in dysgenic myotubes transfected with GFP-α1A (see Table ) and about nine times more often with the targeted GFP-α1Aas(1592-clip). Example traces for each construct were recorded from the same myotubes in sequential order.
Localization of the Triad Targeting Site to the COOH Terminus of the α1S Subunit
With a properly targeted GFP-α1S and a nontargeted GFP-α1A in our hands, we decided to start screening for the location of the targeting signal by replacing the prominent cytoplasmic portions of GFP-α1S with the corresponding sequences of α1A. GFP-α1S/α1A chimeras were generated with the following portions of α1A (Fig. 3): the NH2 terminus (GFP-α1SkNa), the cytoplasmic loop connecting repeats I and II (GFP-α1SkI-IIa) or that connecting repeats III and IV (GFP-α1SkIII-IVa), and the COOH terminus (GFP-α1Sa). Because the II-III loop of α1A is more than three times the size of that of α1S, we were concerned that it might impede appropriate incorporation of a chimera, not because of lacking a triad targeting signal, but because of sterical hindrance. Instead, the II-III loop of the house fly (M. domestica) α1 subunit (Grabner et al. 1994) was used for constructing a II-III chimera (GFP-α1SkII-IIIm). Its II-III loop has similar size as that of the rabbit skeletal muscle α1S but shows very little sequence homology to α1S and, like α1A, the Musca α1 subunit failed to be targeted into the junctions. Double immunolabeling with anti–GFP and anti–RyR and subsequent analysis of the targeting properties revealed that all of these chimeras with the exception of GFP-α1Sa were clustered together with the RyR. The clustering efficiencies were between 50 and 65% (n = 200) for each construct, which was similar to that of GFP-α1S (Fig. 3). Thus, neither the NH2 terminus nor any of the major cytoplasmic loops of α1S are essential for triad targeting.
Figure 3.
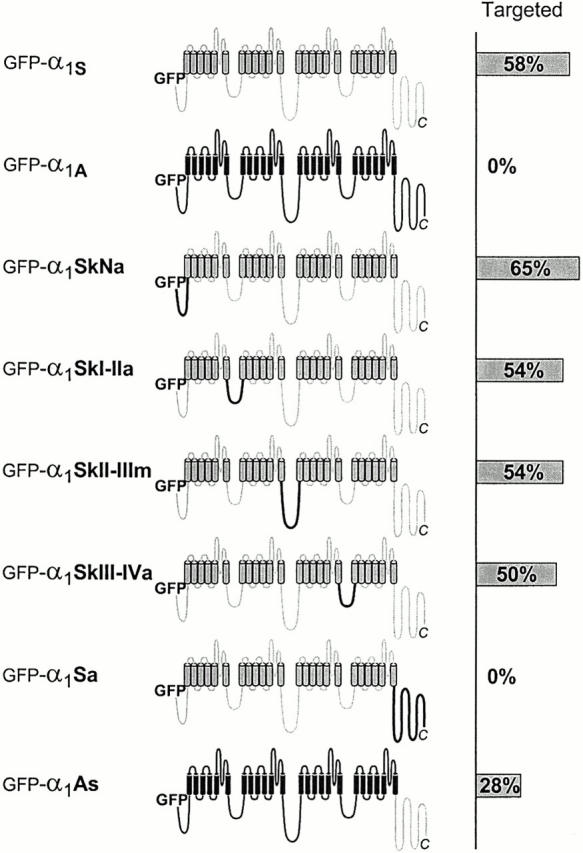
Screening for the molecular location of the triad targeting signal in α1S/α1A chimeras. Schematic representation of the membrane topology of the α1 subunit isoforms and chimeras with α1S sequences in gray and α1A sequences in black. Large cytoplasmic portions of α1S were systematically replaced by the corresponding sequences of α1A (or in the case of GFP-α1SkII-IIIm of the Musca α1 sequence). The bar graph at the right shows the percentage of transfected myotubes in which the expressed α1 subunit isoform/chimera achieved a clustered distribution indicative of correct triad targeting.
The I-II loop contains the interaction domain for the β subunit (Pragnell et al. 1994). Association of the β subunit with this loop has been implicated in an important early step in membrane insertion of Ca2+ channels (Bichet et al. 2000). The I-II loop at least of α1A seems to contain an ER retention signal that is blocked upon association with β to release the complex from the ER. Since the β interaction domain in the I-II loop is shared by all known α1 subunits and a β subunit is endogenously expressed in dysgenic myotubes, a negative effect on triad targeting due to this mechanism was not to be expected with the GFP-α1SkI-IIa chimera. The II-III loop of α1S contains the sequence important for the interaction with the RyR. It specifies the tissue-specific mode of EC coupling (Tanabe et al. 1990a) and the amplification of Ca2+ currents by association with the skeletal type 1 RyR (Nakai et al. 1996; Grabner et al. 1999). Therefore, it is quite remarkable that replacement of the α1S II-III loop by a loop as different as that of Musca's α1 subunit had no adverse effect on triad targeting of chimera GFP-α1SkII-IIIm. On the other hand, the finding that the molecular domain responsible for DHPR-RyR interactions is not essential for triad targeting is consistent with the observations showing that, in the RyR1 knock-out mouse, α1S clusters in the junctions despite the lack of RyR1 (Takekura et al. 1995).
Replacing the COOH terminus of GFP-α1S with that of α1A (GFP-α1Sa) disrupted triad targeting (Fig. 4, a–c). Similar to the distribution pattern described above for GFP-α1A, GFP-α1Sa was found in the ER/SR system, but never in clusters together with the RyR. Ca2+ currents in GFP-α1Sa-transfected cells were very small and frequently below detectability. This is not surprising for skeletal Ca2+ channels not localized in the triad junctions. Nakai et al. 1996 and Grabner et al. 1999 have shown that skeletal muscle α1S requires the specific interaction with type 1 RyR in the junctions for the expression of normal current densities. Both the absence of RyR1 in dyspedic myotubes and the interruption of the interaction between α1S and the RyR resulted in a considerable attenuation of skeletal Ca2+ currents. Similarly, it is to be expected that in addition to decreased membrane insertion, the failure of triad targeting of GFP-α1Sa and the associated lack of interactions with the RyR would result in the attenuation of Ca2+ currents.
Figure 4.
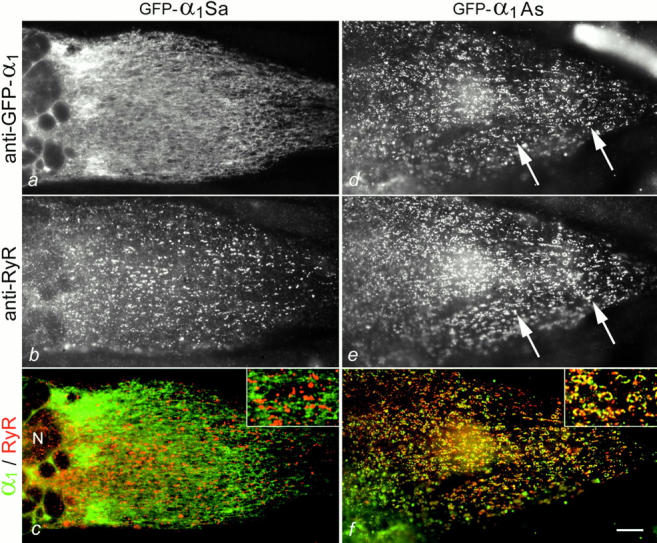
Exchange of the targeting properties between GFP-α1S and GFP-α1A by swapping their COOH-terminal tails. (a and b) Expression of an α1S chimera with the COOH terminus of α1A (GFP-α1Sa) in dysgenic myotubes results in a loss of triad targeting; instead, GFP-α1Sa was consistently localized in the ER/SR system. (d and e) The converse chimera, GFP-α1As, has gained the ability to become coclustered with RyRs in the junctions (examples indicated by arrows). (c and f) The color overlays show the lack of colocalization of GFP-α1Sa and RyR clusters, but a high degree of colocalization of clustered GFP-α1As and RyRs (insets at twofold magnification). Red and green clusters in f are mostly due to differences in labeling intensities and not due to differential distribution of GFP-α1As and RyR (compare d and e). N, nuclei. Bar, 10 μm.
The failure of triad targeting in the GFP-α1Sa chimera suggests that the triad targeting signal may be contained within the COOH terminus of α1S. To verify this interpretation, the corresponding reverse chimera, GFP-α1A with the COOH terminus of α1S (GFP-α1As) was constructed. When expressed in dysgenic myotubes, GFP-α1As was found coclustered with the RyR (Fig. 4, d–f). The efficiency of clustering was somewhat reduced compared with wild-type GFP-α1S (28% of 1,479 transfected myotubes from eight separate experiments; Fig. 3); however, it was clear that by replacing its COOH terminus with that of α1S, this otherwise class-A channel gained the ability to be targeted into the triad junctions. Ca2+ currents with properties similar to those of GFP-α1A were expressed (see Fig. 6 b), indicating that this channel chimera was functional. Since swapping the COOH termini of α1S and α1A conferred triad targeting properties to the neuronal isoform (GFP-α1As) and disrupted triad targeting in the skeletal muscle Ca2+ channel isoform (GFP-α1Sa), it was evident that the signal responsible for this specific localization must be contained in the COOH terminus of α1S.
Localization of the Triad Targeting Signal within the COOH Terminus of α1S
The skeletal muscle α1S subunit isolated from muscle preparations exists in two size forms, the minor fraction corresponding to the full-length α1S sequence and the major fraction corresponding to a COOH-terminally truncated α1S, lacking the sequences distal to approximately residue 1661 (De Jongh et al. 1991). Although truncation occurs after incorporation of α1S into the junctions (Flucher et al. 2000), the truncated form by itself is sufficient to restore skeletal muscle EC coupling in dysgenic myotubes (Beam et al. 1992), suggesting that it is correctly inserted into the junctions. Therefore, it seemed unlikely that the triad targeting signal resides in the part of the COOH terminus distal to the putative clipping site. Sequence comparison of different α1 subunit isoforms showed that the first 140 residues of the COOH terminus are highly homologous, followed by a stretch of similar length with much lower sequence homology. Thus, we concentrated our search for the triad targeting signal on the stretch in between the highly homologous region and the putative clipping site of α1S. We created one chimera (GFP-α1Aas) with the skeletal sequence 1524–1661 substituted for the corresponding region of α1A, and two daughter chimeras, each containing one half of that region from α1S (see Fig. 6 a). GFP-α1Aas(1524-1591), which contains the proximal half of this region from α1S (1524–1591) and the distal half from α1A, ends at an arbitrary site of α1A corresponding to the location of the clipping site in α1S, because α1A does not contain this putative clipping site itself. GFP-α1Aas(1592-clip) is neuronal up to residue 2039 of α1A, with the distal part of α1S, from residue 1592 to the putative clipping site (1661).
Fig. 5, a–c, shows that GFP-α1Aas was correctly targeted into the triad junctions. 38% (n = 1,602) of the transfected myotubes showed a clustered distribution of GFP-α1Aas colocalized with RyR immunolabel. This confirmed that the region beyond residue 1661, which can be subject to truncation, is not necessary for triad targeting. Rather, the triad targeting signal is contained in the sequence between residues 1524 and 1661 of α1S. Within this region, the proximal half did not confer triad targeting properties to α1A. Not a single myotube out of 887 GFP-α1Aas(1524-1591)–transfected myotubes showed a clustered distribution pattern of this α1 chimera; instead, GFP-α1Aas(1524-1591) was regularly found in the ER/SR (Fig. 5, d–f). In contrast, its sister chimera GFP-α1Aas(1592-clip) was efficiently targeted to the junctions (Fig. 5, g–i). 41% (n = 1,076) of the transfected myotubes showed a clustered distribution of GFP-α1Aas(1592-clip) colocalized with the RyR, indicating that this 70 amino acid sequence contains the triad targeting signal. To further restrict the region containing the triad targeting signal, one more chimera was created with only the last 55 residues proximal to the putative clipping site from α1S (GFP-α1Aas(1607-clip)). This construct was also found in clusters in 47% (n = 603) of the transfected myotubes (Fig. 6 a; immunofluorescence image not shown), demonstrating that the 55 amino acid segment between residues 1607 and 1661 of the skeletal muscle α1S is sufficient to confer triad targeting properties to the neuronal α1A subunit.
Figure 5.
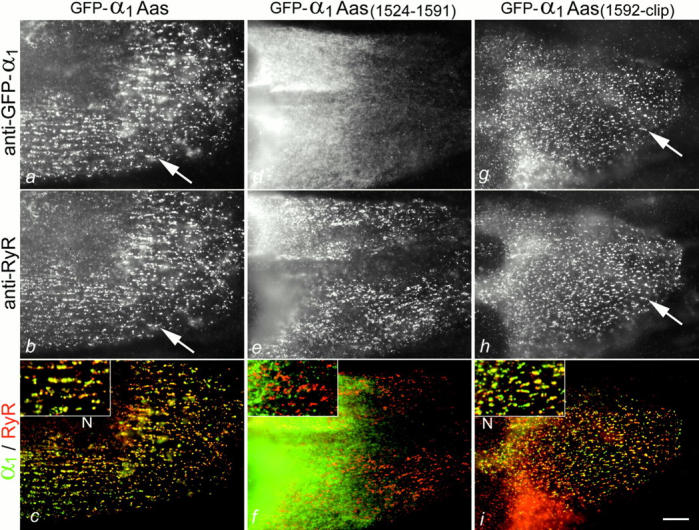
Localization of the triad targeting signal within the COOH-terminal tail of α1S. (a–c) Chimera GFP-α1Aas consisting of the body plus the first 145 COOH-terminal residues of α1A and the remaining COOH terminus of α1S ending at the putative clipping site at position 1661 is readily targeted into the junctions when expressed in dysgenic myotubes (example indicated by arrows). (d–f) Chimera GFP-α1Aas(1524-1591) containing only the proximal half of this COOH-terminal α1S sequence fails to be targeted into the junctions. (g–i) However, chimera GFP-α1Aas(1592-clip) containing the distal half of this COOH-terminal α1S sequence is efficiently targeted into junctions of t tubules and plasma membrane with the SR (example indicated by arrows). (c, f, and i) Color overlays of images shown above; insets at twofold magnification. N, nuclei. Bar, 10 μm.
Comparing this sequence of α1S with the corresponding sequences of the cardiac α1C, which is also targeted to triads (Grabner et al. 1998), and with that of the nontargeted α1A reveals surprisingly few residues that are conserved between α1S and α1C but distinct from α1A (Fig. 6 a). However, replacing individual of these residues with alanin did so far not result in a loss of targeting properties (data not shown). Either a targeting motif within this 55 amino acid stretch of α1S is made up of more than those residues conserved between α1S and α1C, or the signal that is contained in this sequence stretch of α1S is located outside the corresponding region of α1C (see below). To distinguish between these possibilities, the corresponding targeting signal in α1C and other α1 isoforms needs to be localized and extensive single and combinatorial amino acid mapping needs to be performed.
The importance of COOH-terminal sequences in targeting and immobilization in specialized neuronal membrane domains has been demonstrated for ligand- and voltage-gated ion channels (Sheng and Pak 1999; Lim et al. 2000). To our knowledge, the sequence between 1607 and 1661 of α1S contains only one known consensus protein binding site. The sequence SPV in position 1640–1642 corresponds to the PDZ-binding motif S/TXV; however, it is not positioned at the very COOH terminus as is the case in the majority of reported PDZ-binding proteins (Sheng 1996). Using a yeast–two-hybrid assay, Proenza et al. 2000 recently observed that this motif is part of a highly reactive region in the COOH terminus of α1S and that substitution of the valine within this motif by aspartate abolished the high reactivity. This makes the PDZ-binding motif within the region demonstrated to contain the triad targeting signal in the present study a good candidate for protein–protein interactions that may contribute to triad targeting. However, the corresponding sequence in α1C lacks the critical valine of this motif. Thus, if binding to PDZ proteins were the mechanism of triad targeting, other PDZ-binding motifs located elsewhere in the channel had to be responsible for the same function in the cardiac isoform. Two such motifs exist in the COOH terminus of α1C, however, immediately distally to the putative clipping site. Other evidence for the importance of the COOH terminus in membrane targeting comes from heterologous expression of the cardiac α1C in tsA201 cells (Gao et al. 2000). However, the region identified in that study to be important for functional membrane expression of α1C is in the proximal, highly homologous part of the COOH terminus, ending 69 amino acids upstream of the region corresponding to the triad targeting signal identified in our study using a gain-of-function approach. Assuming that the loss of function in response to COOH-terminal truncations and deletions arose from specific effects on the targeting process rather than from nonspecific damage to the expressed channel, this domain is most likely not involved in the highly specific insertion of the channel into triad junctions, but may be important for a more general aspect like the export from the ER or membrane incorporation. Our present results do not exclude the possible contribution of other signals, shared by α1S and α1A, to the complex process that culminates in triad targeting.
Effects of Triad Targeting of GFP-α1Aas(1592-clip) on Ca2+ Currents and EC Coupling
Comparison of the current properties of GFP-α1A and GFP-α1Aas(1524-1591), both of which are not targeted into the triad, with that of the targeted GFP-α1Aas(1592-clip) provides additional evidence for the existence of distinct mechanisms for membrane and triad targeting (Fig. 6b and Fig. c). Compared with GFP-α1A, GFP-α1Aas(1524-1591) exhibited strongly reduced current densities, even though the immunolabeling experiments gave no indication that the transfection efficiency or the amount of protein expressed had been decreased. It was necessary to increase the concentration of the charge carrier from 3 to 10 mM Ca2+ (Fig. 6 c) to show that this chimera did in fact express functional channels in the membrane; however, at strongly reduced levels. This suggested that in this chimera a distinct signal important for membrane expression of α1A had been abolished, but that this loss had not been compensated by the addition of the skeletal muscle triad targeting signal. To find out whether this putative membrane insertion signal of α1A resides in the region replaced by the skeletal sequence (1524–1591) or whether it is contained in the distal portion of the COOH terminus that had been truncated in this chimera, we generated a truncated GFP-α1A and expressed it in the dysgenic myotubes (data not shown). Current expression of this GFP-α1A-clip was also low, suggesting that the distal COOH terminus of α1A contains a separate signal that is important for its efficient membrane expression, but is not sufficient for triad targeting.
Considering the fact that our final triad-targeting chimeras lack this α1A membrane-targeting signal, it is even more astonishing that the skeletal residues 1592–1661 not only fully compensated the loss of current expression caused by the truncation of α1A, but that current densities of the targeted GFP-α1Aas(1592-clip) were increased by approximately twofold over those of the wild-type α1A (Fig. 6 c). Among several possible causes for this effect, an increase in current density accompanying triad targeting is consistent with a model by which the specific incorporation into the triadic complex stabilizes the channel in the membrane, thus leading to an increased total number of functional channels.
Finally, to the question of how the localization of a Ca2+ channel in skeletal muscle affects EC coupling. The two muscle isoforms, α1S and α1C, which are both targeted into triads, have repeatedly been shown to rescue EC coupling in dysgenic myotubes (Tanabe et al. 1988 and Tanabe et al. 1990b; Grabner et al. 1998; Neuhuber et al. 1998b; Flucher et al. 2000). However, the mechanism by which the skeletal muscle α1S and the cardiac α1C activate SR Ca2+ release in dysgenic myotubes differs. In α1S, it functions independently of Ca2+ influx, whereas α1C does require Ca2+ influx for the activation of EC coupling. In Fig. 7, we show that dysgenic myotubes transfected with GFP-α1S or GFP-α1C respond to electrical stimulation with strong Ca2+ transients, which have previously been described as action potential–induced Ca2+ transients based on their all-or-none characteristics (Flucher et al. 1994; Powell et al. 1996). With GFP-α1S, these Ca2+ transients continued after blocking the Ca2+ currents by the addition of Cd2+/La3+ to the bath solution, whereas with GFP-α1C the Ca2+ transients ceased, confirming that this SR Ca2+ release was Ca2+-induced. Caffeine induced strong Ca2+ transients even after the Cd2+/La3+ block, indicating that the cessation of Ca2+ transients was not due to depletion of the SR Ca2+ stores or damage to the release mechanism.
Previous attempts to rescue EC coupling in dysgenic myotubes by expressing the neuronal α1A failed to display (Grabner et al. 1998), or only very rarely displayed, evoked contractions (Adams et al. 1994), despite the fact that α1A expressed sizable Ca2+ currents. Using a more direct method to monitor SR Ca2+ release with the fluorescent Ca2+ indicator fluo-4, we did observe rare action potential–induced Ca2+ transients in GFP-α1A–transfected myotubes (Table and Fig. 7). While >25 responsive myotubes were found in almost all of GFP-α1S– and GFP-α1C–transfected cultures, on average only every fifth culture transfected with GFP-α1A contained one or two responsive myotubes. The degree of restoration of EC coupling increased significantly (P = 0.002) when the class-A channel was targeted into the triad. In myotubes transfected with GFP-α1Aas(1592-clip), action potential–induced Ca2+ transients were observed in 60% of the cultures (Table ). As expected, these Ca2+ transients could be blocked with Cd2+/La3+, indicating that the mechanism by which GFP-α1Aas(1592-clip) restored EC coupling was Ca2+-induced Ca2+ release (Fig. 7).
Table 1.
Restoration of Action Potential-induced Ca2+ Transients in Dysgenic Myotubes Expressing Different α1 Subunit Isoforms and the Targeted Chimera GFP-α1Aas(1592-clip)
| Construct | Cultures showing action potential-induced Ca2+ transients* | Responsive myotubes per dish‡ | ||
|---|---|---|---|---|
| % | n | n | ||
| GFP-α1S | 95 | 22 | 26.6 ± 22.2 | 14 |
| GFP-α1C | 100 | 22 | 27.9 ± 23.5 | 22 |
| GFP-α1A | 22 | 23 | 0.2 ± 0.4 | 22 |
| GFP-α1Aas(1592-clip) | 60 | 43 | 1.8 ± 2.4 | 40 |
*Percentages of culture dishes with one or more responsive myotubes of number of dishes (n) tested. ‡Standard deviation of the mean of n dishes tested.
The enhanced restoration of EC coupling by the targeting of a class-A channel into the triads shows the importance of the correct localization of the Ca2+ channel in close proximity to the SR Ca2+ release channel. Apparently, the large Ca2+ influx through α1A distributed throughout the plasma membrane was not sufficient to induce Ca2+-induced Ca2+ release except in a few myotubes. However, concentrating the Ca2+ current to the restricted spaces of the triadic compartments strongly improved the chance of reaching the threshold for Ca2+-induced Ca2+ release. This interpretation is consistent with other cellular processes where the close proximity of a Ca2+ source and a Ca2+ target is required for normal function (e.g., the association of the RyR and the Ca2+-activated potassium channel in smooth muscle cells; Jaggar et al. 2000) and highlights the importance of specific targeting mechanisms for Ca2+ channels.
The more we learn about Ca2+ channels in their native environments, the more it appears to be the rule rather than the exception that they are specifically localized in functional domains. The signal contained in the 55 amino acid sequence of the COOH terminus of α1S is the first description of a targeting signal of a voltage-gated Ca2+ channel for a specific membrane domain. It may share its anchoring mechanism with other ion channels that have been shown to interact with proteins containing PDZ domains, but for which the importance of this protein–protein interaction in the targeting process has yet to be shown. The complex passage of the skeletal muscle Ca2+ channel from the biosynthetic apparatus into the triad junction involves multiple steps. At the beginning of the journey, the interaction of the β subunit with the I-II loop and with the COOH terminus seems to play an important role in the export of the channel from the ER and for its functional expression in the plasma membrane. At the end of the journey, the specific interaction with the RyR determines the tetradic organization of the α1S subunit that structurally sets it apart from the cardiac α1C. But between these two events occurs the essential targeting of the Ca2+ channel into the junctional domain of the triad, and the signal contained within residues 1607–1661 of the skeletal muscle α1S subunit is necessary and sufficient to confer this targeting property to a neuronal α1 subunit.
Acknowledgments
We thank Dr. J. Hoflacher and E. Emberger for their excellent experimental help, and Dr. H. Glossmann for generously providing support for the pursuit of this project.
This work was supported in part by the Fonds zur Förderung der wissenschaftlichen Forschung, Austria, grants P12653-MED (B.E. Flucher), and P13831-GEN (M. Grabner), and by the European Commission's Training and Mobility of Researchers Network grant ERBFMRXCT960032 (B.E. Flucher).
Footnotes
Abbreviations used in this paper: DHPR, dihydropyridine receptor; EC, excitation–contraction; GFP, green fluorescent protein; nt, nucleotides; RE, restriction enzyme; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; t tubule, transverse tubule.
References
- Adams B.A., Tanabe T., Mikami A., Numa S., Beam K.G. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 1990;346:569–572. doi: 10.1038/346569a0. [DOI] [PubMed] [Google Scholar]
- Adams B.A., Mori Y., Kim M.S., Tanabe T., Beam K.G. Heterologous expression of BI Ca2+ channels in dysgenic skeletal muscle. J. Gen. Physiol. 1994;104:985–996. doi: 10.1085/jgp.104.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beam K.G., Adams B.A., Niidome T., Numa S., Tanabe T. Function of a truncated dihydropyridine receptor as both voltage sensor and calcium channel. Nature. 1992;360:169–171. doi: 10.1038/360169a0. [DOI] [PubMed] [Google Scholar]
- Berridge M.J. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Beurg M., Sukhareva M., Strube C., Powers P.A., Gregg R.G., Coronado R. Recovery of Ca2+ current, charge movements, and Ca2+ transients in myotubes deficient in dihydropyridine receptor β1 subunit transfected with β1 cDNA. Biophys. J. 1997;73:807–818. doi: 10.1016/S0006-3495(97)78113-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichet D., Cornet V., Geib S., Carlier E., Volsen S., Hoshi T., Mori Y., De Waard M. The I-II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the β subunit. Neuron. 2000;25:177–190. doi: 10.1016/s0896-6273(00)80881-8. [DOI] [PubMed] [Google Scholar]
- Block B.A., Imagawa T., Campbell K.P., Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brice N.L., Berrow N. S., Campbell V., Page K.M., Brickley K., Tedder I., Dolphin A.C. Importance of the different β subunits in the membrane expression of the α1A and α2 calcium channel subunitsstudies using a depolarization-sensitive α1A antibody. Eur. J. Neurosci. 1997;9:749–759. doi: 10.1111/j.1460-9568.1997.tb01423.x. [DOI] [PubMed] [Google Scholar]
- Catterall W.A. Molecular properties of sodium and calcium channels. J. Bioenerg. Biomembr. 1996;28:219–230. doi: 10.1007/BF02110697. [DOI] [PubMed] [Google Scholar]
- Chien A.J., Zhao X., Shirokov R.E., Puri T.S., Chang C.F., Sun D., Ríos E., Hosey M.M. Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J. Biol. Chem. 1995;270:30036–30044. doi: 10.1074/jbc.270.50.30036. [DOI] [PubMed] [Google Scholar]
- De Jongh K.S., Warner C., Colvin A.A., Catterall W.A. Characterization of the two size forms of the α1 subunit of skeletal muscle L-type calcium channels. Proc. Natl. Acad. Sci. USA. 1991;88:10778–10782. doi: 10.1073/pnas.88.23.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher B.E., Morton M.E., Froehner S.C., Daniels M.P. Localization of the α1 and α2 subunits of the dihydropyridine receptor and ankyrin in skeletal muscle triads. Neuron. 1990;5:339–351. doi: 10.1016/0896-6273(90)90170-k. [DOI] [PubMed] [Google Scholar]
- Flucher B.E., Phillips J.L., Powell J.A. Dihydropyridine receptor α subunits in normal and dysgenic muscle in vitroexpression of α1 is required for proper targeting and distribution of α2 . J. Cell Biol. 1991;115:1345–1356. doi: 10.1083/jcb.115.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher B.E., Andrews S.B., Daniels M.P. Molecular organization of transverse tubule/sarcoplasmic reticulum junctions during development of excitation–contraction coupling in skeletal muscle. Mol. Biol. Cell. 1994;5:1105–1118. doi: 10.1091/mbc.5.10.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher B.E., Kasielke N., Gerster U., Neuhuber B., Grabner M. Insertion of the full-length calcium channel α1S subunit into triads of skeletal muscle in vitro. FEBS Lett. 2000;474:93–98. doi: 10.1016/s0014-5793(00)01583-0. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C., Jorgensen A.O. Structure and development of E-C coupling units in skeletal muscle. Annu. Rev. Physiol. 1994;56:509–534. doi: 10.1146/annurev.ph.56.030194.002453. [DOI] [PubMed] [Google Scholar]
- Freise D., Held B., Wissenbach U., Pfeifer A., Trost C., Himmerkus N., Schweig U., Freichel M., Biel M., Hofmann F. Absence of the γ subunit of the skeletal muscle dihydropyridine receptor increases L-type Ca2+ currents and alters channel inactivation properties. J. Biol. Chem. 2000;275:14476–14481. doi: 10.1074/jbc.275.19.14476. [DOI] [PubMed] [Google Scholar]
- Gao T., Buenemann M., Gerhardstein B.L., Ma H., Hosey M.M. Role of the C-terminus of the α1C (CaV1.2) subunit in membrane targeting of cardiac L-type calcium channels. J. Biol Chem. 2000;275:25436–25444. doi: 10.1074/jbc.M003465200. [DOI] [PubMed] [Google Scholar]
- Giannini G., Conti A., Mammarella S., Scrobogna M., Sorrentino V. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J. Cell Biol. 1995;128:893–904. doi: 10.1083/jcb.128.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner M., Bachmann A., Rosenthal F., Striessnig J., Schulz C., Tautz D., Glossmann H. Insect calcium channelsmolecular cloning of an α1 subunit from housefly (Musca domestica) muscle. FEBS Lett. 1994;339:189–194. doi: 10.1016/0014-5793(94)80413-3. [DOI] [PubMed] [Google Scholar]
- Grabner M., Dirksen R.T., Beam K.G. Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non–L-type Ca2+ channels expressed in dysgenic myotubes. Proc. Natl. Acad. Sci. USA. 1998;95:1903–1908. doi: 10.1073/pnas.95.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner M., Dirksen R.T., Suda N., Beam K.G. The II-III loop of the skeletal muscle dihydropyridine receptor is responsible for the bi-directional coupling with the ryanodine receptor. J. Biol. Chem. 1999;274:21913–21919. doi: 10.1074/jbc.274.31.21913. [DOI] [PubMed] [Google Scholar]
- Gregg R.G., Messing A., Strube C., Beurg M., Moss R., Behan M., Sukhareva M., Haynes S., Powell J.A., Coronado R., Powers P.A. Absence of the β subunit (cchβ1) of the skeletal muscle dihydropyridine receptor alters expression of the α1 subunit and eliminates excitation–contraction coupling. Proc. Natl. Acad. Sci. USA. 1996;93:13961–13966. doi: 10.1073/pnas.93.24.13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton R.M., Hunt H.D., Ho S.N., Pullen J.K., Pease L.R. Engineering hybrid genes without the use of restriction enzymesgene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Jaggar J.H., VA.Porter W.J., Lederer, Nelson M.T. Calcium sparks in smooth muscle. Am. J. Physiol. Cell Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- Jorgensen A.O., Shen A.C.-Y., Arnold W., Leung A.T., Campbell K.P. Subcellular distribution of the 1,4-dihydropyridine receptor in rabbit skeletal muscle in situan immunofluorescence and immunocolloidal gold-labeling study. J. Cell Biol. 1989;109:135–147. doi: 10.1083/jcb.109.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S.T., Antonucci D.E., Scannevin R.H., Trimmer J.S. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron. 2000;25:385–397. doi: 10.1016/s0896-6273(00)80902-2. [DOI] [PubMed] [Google Scholar]
- Melzer W., Herrmann-Frank A., Luttgau H.C. The role of Ca2+ ions in excitation–contraction coupling of skeletal muscle fibres. Biochim. Biophys. Acta. 1995;1241:59–116. doi: 10.1016/0304-4157(94)00014-5. [DOI] [PubMed] [Google Scholar]
- Mori Y., Friedrich T., Kim M.-S., Mikami A., Nakai J., Ruth P., Bosse E., Hofmann F., Flockerzi V., Furuichi T. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- Morton M.E., Froehner S.C. Monoclonal antibody identifies a 200-kDa subunit of the dihydropyridine-sensitive calcium channel. J. Biol. Chem. 1987;262:11904–11907. [PubMed] [Google Scholar]
- Nakai J., Dirksen R.T., Nguyen H.T., Pessah I.N., Beam K.G, Allen P.D. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- Neuhuber B., Gerster U., Mitterdorfer J., Glossmann H., Flucher B.E. Differential effects of Ca2+ channel β1a and β2a subunits on complex formation with α1S and on current expression in tsA201 cells J. Biol. Chem. 273 1998. 9110 9118a [DOI] [PubMed] [Google Scholar]
- Neuhuber B., Gerster U., Doring F., Glossmann H., Tanabe T., Flucher B.E. Association of calcium channel α1S and β1a subunits is required for the targeting of β1a but not of α1S into skeletal muscle triads Proc. Natl. Acad. Sci. USA. 95 1998. 5015 5020b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell J.A., Petherbridge L., Flucher B.E. Formation of triads without the dihydropyridine receptor α subunits in cell lines from dysgenic skeletal muscle. J. Cell Biol. 1996;134:375–387. doi: 10.1083/jcb.134.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pragnell M., De Waard M., Mori Y., Tanabe T., Snutch T.P., Campbell K.P. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- Proenza C., Wilkens C., Lorenzon N.M., Beam K.G. A carboxyl-terminal region important for the expression and targeting of the skeletal muscle dihydropyridine receptor. J. Biol. Chem. 2000;275:23169–23174. doi: 10.1074/jbc.M003389200. [DOI] [PubMed] [Google Scholar]
- Protasi F., Franzini-Armstrong C., Allen P.D. Role of ryanodine receptors in the assembly of calcium release units in skeletal muscle. J. Cell Biol. 1998;140:831–842. doi: 10.1083/jcb.140.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos E., Pizarro G., Stefani E. Charge movement and the nature of signal transduction in skeletal muscle excitation–contraction coupling. Annu. Rev. Physiol. 1992;54:109–133. doi: 10.1146/annurev.ph.54.030192.000545. [DOI] [PubMed] [Google Scholar]
- Sheng M. PDZs and receptor/channel clusteringrounding up the latest suspects. Cell. 1996;17:575–578. doi: 10.1016/s0896-6273(00)80190-7. [DOI] [PubMed] [Google Scholar]
- Sheng M., Pak D.T. Glutamate receptor anchoring proteins and the molecular organization of excitatory synapses. Ann. NY Acad. Sci. 1999;868:483–493. doi: 10.1111/j.1749-6632.1999.tb11317.x. [DOI] [PubMed] [Google Scholar]
- Takekura H., Nishi M., Noda T., Takeshima H., Franzini-Armstrong C. Abnormal junctions between surface membrane and sarcoplasmic reticulum in skeletal muscle with a mutation targeted to the ryanodine receptor. Proc. Natl. Acad. Sci. USA. 1995;92:3381–3385. doi: 10.1073/pnas.92.8.3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe T., Takeshima H., Mikami A., Flockerzi V., Takahashi H., Kangawa K., Kojima M., Matsuo H., Hirose T., Numa S. Primary structure of the receptor for calcium channel blocker from skeletal muscle. Nature. 1987;328:313–318. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- Tanabe T., Beam K.G., Powell J.A., Numa S. Restoration of excitation–contraction coupling and slow Ca2+ current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 1988;336:134–139. doi: 10.1038/336134a0. [DOI] [PubMed] [Google Scholar]
- Tanabe T., Beam K.G., Adams B.A., Niidome T., Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation–contraction coupling Nature 346 1990. 567 569a [DOI] [PubMed] [Google Scholar]
- Tanabe T., Mikami A., Numa S., Beam K.G. Cardiac-type excitation–contraction coupling in dysgenic skeletal muscle injected with cardiac dihydropyridine receptor cDNA Nature. 344 1990. 451 453b [DOI] [PubMed] [Google Scholar]
- Walker D., Bichet D., Campbell K.P., De Waard M. A β4 isoform-specific interaction site in the carboxyl-terminal region of the voltage-dependent Ca2+ channel α1A subunit. J. Biol. Chem. 1998;173:1361–1367. doi: 10.1074/jbc.273.4.2361. [DOI] [PubMed] [Google Scholar]