

Abstract
Synaptically released glutamate has been identified as a signal coupling excitatory neuronal activity to increased glucose utilization. The proposed mechanism of this coupling involves glutamate uptake into astrocytes resulting in increased intracellular Na+ (Nai+) and activation of the Na+/K+-ATPase. Increased metabolic demand linked to disruption of Nai+ homeostasis activates glucose uptake and glycolysis in astrocytes. Here, we have examined whether a similar neurometabolic coupling could operate for the inhibitory neurotransmitter γ-aminobutyric acid (GABA), also taken up by Na+-dependent transporters into astrocytes. Thus, we have compared the Nai+ response to GABA and glutamate in mouse astrocytes by microspectrofluorimetry. The Nai+ response to GABA consisted of a rapid rise of 4–6 mM followed by a plateau that did not, however, significantly activate the pump. Indeed, the GABA transporter-evoked Na+ influxes are transient in nature, almost totally shutting off within ≈30 sec of GABA application. The metabolic consequences of the GABA-induced Nai+ response were evaluated by monitoring cellular ATP changes indirectly in single cells and measuring 2-deoxyglucose uptake in astrocyte populations. Both approaches showed that, whereas glutamate induced a robust metabolic response in astrocytes (decreased ATP levels and glucose uptake stimulation), GABA did not cause any measurable metabolic response, consistent with the Nai+ measurements. Results indicate that GABA does not couple inhibitory neuronal activity with glucose utilization, as does glutamate for excitatory neurotransmission, and suggest that GABA-mediated synaptic transmission does not contribute directly to brain imaging signals based on deoxyglucose.
Astrocytes exert several important functions, including clearance of synaptically released glutamate and provision to neurons of metabolic substrates in register with synaptic activity (1). A tight link between transport of glutamate released at excitatory synapses and certain metabolic responses has been documented (2, 3) and is based on the fact that the Na+ load associated with glutamate transport causes an activation of the Na+/K+ ATPase that doubles its activity in the presence of extracellular glutamate concentrations in the low micromolar range, causing an increased energy demand in astrocytes. This mechanism links therefore excitatory neuronal transmission with an increase in astrocyte metabolism and glucose utilization (1, 2).
Astrocytes also contribute to clearing other neurotransmitters such as γ-aminobutyric acid (GABA) or glycine from the extracellular space by using Na+-coupled transport systems (4). The aim of the present study was to evaluate whether the mechanism of metabolic coupling proposed for the excitatory neurotransmitter glutamate could apply also to inhibitory neurotransmitters, in particular to GABA.
GABA is the major inhibitory neurotransmitter in the CNS. It has a widespread distribution in the adult brain, and high-affinity transport of GABA both in GABAergic neurons and surrounding glial cells is responsible for terminating GABA transmission (5). GABA transporters belong to a superfamily of Na+/Cl--dependent neurotransmitter transporters, with GAT1 and GAT3 being exclusively expressed in the brain, whereas BGT1, a lower-affinity subtype is found both in the brain and peripheral tissues. All three transporters appear to be expressed also by astrocytes, BGT1 being probably predominantly glial (5). Astrocytic GABA transporters neighboring GABAergic synapses are strategically positioned to take up GABA locally and contribute to terminating or modulating GABA-mediated neuronal transmission (4). In the present study, we compared the metabolic load induced by Na+ cotransported with GABA to that associated with glutamate uptake in primary cultures of mouse cortical astrocytes.
Materials and Methods
Cell Culture. Mouse cortical astrocytes in primary cultures were prepared as described (6) from 1- to 3-day-old OF1 mice. Astrocytes were plated on glass coverslips and cultured in DMEM (Sigma) plus 10% FCS. After 2–3 weeks of culture, culture media were supplemented with 250 μM dibutyryl cAMP for 7 days, a treatment that was described to maximize GABA transporter expression in mouse astrocytes (7), and that allowed us to obtain reproducible Na+ responses in all cells tested.
Intracellular Na+ (Nai+) and Intracellular Free Mg2+ Concentration Measurements. Intracellular ion imaging was performed on an inverted epifluorescence microscope (Zeiss) with a 40 × 1.3 numerical aperture oil-immersion objective lens. Fluorescence excitation wavelengths were selected by using a monochromator (TILL Photonics, Planegg, Germany), and fluorescence was detected by using a 12-bit cooled charge-coupled device camera (Princeton Instruments, Princeton). Image acquisition (at 0.1–1 Hz) and time series was computer-controlled by using the software METAF LUOR (Universal Imaging, Downingtown, PA) running on a Pentium computer. The acquisition rate of ratio images was varied. Up to ≈10 individual astrocytes were simultaneously analyzed in the selected field of view.
Na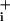 was measured by using the Na+-sensitive fluorescent dye sodium-binding benzofuran isophthalate (SBFI, Teflabs, Austin, TX) in single cells grown on glass coverslips. Cells were loaded at 37°C with 15 μM SBFI acetoxymethyl ester in a Hepesbuffered balanced solution (see below) and then placed in a thermostated chamber designed for rapid exchange of perfusion solutions (2) and superfused at 35°C. Fluorescence was sequentially excited at 340 and 380 nm and detected at 510 nm (80-nm bandwidth). Fluorescence excitation ratios (F340nm/F380nm) were computed for each image pixel and produced ratio images of cells that were proportional with Na
was measured by using the Na+-sensitive fluorescent dye sodium-binding benzofuran isophthalate (SBFI, Teflabs, Austin, TX) in single cells grown on glass coverslips. Cells were loaded at 37°C with 15 μM SBFI acetoxymethyl ester in a Hepesbuffered balanced solution (see below) and then placed in a thermostated chamber designed for rapid exchange of perfusion solutions (2) and superfused at 35°C. Fluorescence was sequentially excited at 340 and 380 nm and detected at 510 nm (80-nm bandwidth). Fluorescence excitation ratios (F340nm/F380nm) were computed for each image pixel and produced ratio images of cells that were proportional with Na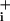 . At the end of each experiment an in situ calibration was performed as described (2) by cell permeabilization for monovalent cations by using gramicidin and monensin with simultaneous inhibition of the Na+/K+-ATPase with ouabain. Cells were then sequentially perfused with solutions containing 5, 10, 20, 50, and 100 mM Na+; a four-point calibration curve was computed for each selected cell in the field of view and used to convert fluorescence ratio values (F340nm/F380nm) into Na+ concentrations.
. At the end of each experiment an in situ calibration was performed as described (2) by cell permeabilization for monovalent cations by using gramicidin and monensin with simultaneous inhibition of the Na+/K+-ATPase with ouabain. Cells were then sequentially perfused with solutions containing 5, 10, 20, 50, and 100 mM Na+; a four-point calibration curve was computed for each selected cell in the field of view and used to convert fluorescence ratio values (F340nm/F380nm) into Na+ concentrations.
Intracellular free Mg2+ concentration was measured by using the fluorescent probe magnesium green (MgG, Molecular Probes). Cell loading was performed at 37°C by using 10 μM of MgG acetoxymethyl ester. Fluorescence was excited at 495 nm and detected at 535 nm (35-nm bandwidth).
Solutions contained 135 mM NaCl, 5.4 mM KCl, 25 mM NaHCO3, 1.3 mM CaCl2, 0.8 mM MgSO4, 0.78 mM NaH2PO4, 5 mM glucose, bubbled with 5% CO2/95% air. The solution for dye-loading contained 135 mM NaCl, 5.4 mM KCl, 20 mM Hepes, 1.3 mM CaCl2, 0.8 mM MgSO4, 0.78 mM NaH2PO4, and 20 mM glucose and was supplemented with 0.1% Pluronic F-127 (Molecular Probes) or 1% BSA (fraction V, Fluka).
[3H]2-Deoxy-Glucose (2DG) Uptake and Lactate Release. For determination of 2DG uptake (3) cell cultures were preincubated for 2 h at 37°C in serum-free DMEM supplemented with 5 mM glucose and 44 mM bicarbonate. 2DG (≈25 nm, 1 μCi) uptake was measured for 20 min in the presence or absence of GABA or glutamate, and the reaction was stopped with ice-cold PBS. Cells were lysed with 0.01 M NaOH + 0.1% Triton X-100. Lysates were counted in a scintillation β counter (Canberra Packard, Meriden, CT) and assayed for protein content by using the Bradford method (8). The portion of the counts that was not inhibited by the glucose transporter inhibitor cytochalasin B (25 μM), i.e., ≈20%, was subtracted from the total counts to yield the glucose transporter-mediated uptake. Lactate release from astrocytes was assayed enzymatically, as described (3), in the medium collected at the end of the 2DG uptake period.
Materials. SKF-89976A, CI-966, and nipecotic acid were from Tocris-Anawa Trading (Zurich). Ouabain was from Fluka. All other substances were from Sigma.
Expression of Data and Statistics. Data are presented as means ± SE. Student's t test was performed to assess the statistical significance of results with a P < 0.05 considered as significant. For estimation of EC50 values from concentration-response curves, the Levenberg-Marquardt algorithm implemented in the kaleidagraph package (Synergy Software, Reading, PA) was used for nonlinear curve fitting of the following equation:
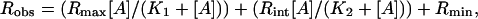 |
1 |
where Robs, is the observed response, Rmax, Rmin, and Rint are maximum, minimum, and intermediate (for two-site model) parameters, respectively, of the response. [A] is the concentration of agonist, and K1 and K2 are the agonist concentrations that yield half-maximum responses (i.e., EC50). Eq. 1 corresponds to a two-site noncooperative model.
Results
Glial GABA transport is coupled to Na+ (5) and should lead to an increased Na+ influx into astrocytes. Fig. 1 A and B shows fluorescence images of Na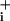 increases in response to GABA and typical traces of Na
increases in response to GABA and typical traces of Na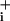 concentration changes after GABA application at different concentrations. The Na+ response to GABA appears to involve two components (Fig. 1C), of high and low affinity with apparent EC50 values of 12 μM and 3.9 mM, respectively. In subsequent experiments we focused on the high-affinity component as it is operative within the physiological range of GABA effects. Thus 500-μM GABA was considered as the maximally effective concentration of the high-affinity component. The maximum amplitude of observed Na
concentration changes after GABA application at different concentrations. The Na+ response to GABA appears to involve two components (Fig. 1C), of high and low affinity with apparent EC50 values of 12 μM and 3.9 mM, respectively. In subsequent experiments we focused on the high-affinity component as it is operative within the physiological range of GABA effects. Thus 500-μM GABA was considered as the maximally effective concentration of the high-affinity component. The maximum amplitude of observed Na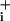 responses was ≈4–6 mM, compared with the 25to 30-mM increases evoked on average in response to glutamate at comparable concentrations (2).
responses was ≈4–6 mM, compared with the 25to 30-mM increases evoked on average in response to glutamate at comparable concentrations (2).
Fig. 1.

GABA applied at different concentrations increased Na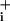 concentration in mouse astrocytes. (A) Images of sodium-binding benzofuran isophthalate-loaded astrocytes showing a raw fluorescence image excited at 380 nm (Upper Right) and false color excitation ratio images recorded under control conditions (Upper Left) and in the presence of 500 μM(Lower Left)and 1 mM (Lower Right) GABA, respectively. (Scale bar: 20 μm.) (B) Traces of two individual astrocytes are shown out of 82 from 11 experiments. (C) GABA concentration-response analysis by using the two site Michaelis–Menten equation (Eq. 1) yielded two apparent EC50 values of 12 μM and 3.9 mM, when amplitude of the response was considered, and 90 μM and 10.6 mM when initial rate of Na+ increase was considered (data not shown).
concentration in mouse astrocytes. (A) Images of sodium-binding benzofuran isophthalate-loaded astrocytes showing a raw fluorescence image excited at 380 nm (Upper Right) and false color excitation ratio images recorded under control conditions (Upper Left) and in the presence of 500 μM(Lower Left)and 1 mM (Lower Right) GABA, respectively. (Scale bar: 20 μm.) (B) Traces of two individual astrocytes are shown out of 82 from 11 experiments. (C) GABA concentration-response analysis by using the two site Michaelis–Menten equation (Eq. 1) yielded two apparent EC50 values of 12 μM and 3.9 mM, when amplitude of the response was considered, and 90 μM and 10.6 mM when initial rate of Na+ increase was considered (data not shown).
To assess the involvement of GABA transporters in these responses, transporter inhibitors were applied, namely β-alanine (500 μM), nipecotic acid (500 μM), SKF-89976A (10–100 μM), and CI-966 (10–100 μM). All compounds led to Na+ responses in the order of magnitude of those observed with GABA itself (data not shown). Fig. 2 shows that the responses to SKF-89976A and CI-966 were not additive to those of GABA on the same cell at maximally effective concentrations (high-affinity component), indicating that the Na+ influx evoked by these compounds and GABA shared the same pathway.
Fig. 2.

Effects of GABA transporter inhibitors on Na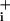 concentration. The responses of astrocytes to GABA transporter inhibitors SKF-89976A (A) and CI-966 (B) were examined in the presence and the absence of maximally effective GABA concentration at the high-affinity component. The absolute amplitudes of responses are presented. Statistical significance of differences compared with GABA alone by using a paired t test and the number of cells studied is indicated in the graphs. *, P < 0.05; **, P < 0.001; ns, not significant (P > 0.05).
concentration. The responses of astrocytes to GABA transporter inhibitors SKF-89976A (A) and CI-966 (B) were examined in the presence and the absence of maximally effective GABA concentration at the high-affinity component. The absolute amplitudes of responses are presented. Statistical significance of differences compared with GABA alone by using a paired t test and the number of cells studied is indicated in the graphs. *, P < 0.05; **, P < 0.001; ns, not significant (P > 0.05).
In the next phase, the activation of the Na+/K+ ATPase associated with this Na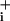 increase was evaluated and compared with that induced by glutamate. Two concentrations of GABA were tested for this purpose, namely 500 μM, corresponding to the upper physiological range, and 5 mM, a high concentration that yields robust responses. To assess how GABA influences Na+/K+ ATPase activity, GABA alone was first applied until stable elevated Na
increase was evaluated and compared with that induced by glutamate. Two concentrations of GABA were tested for this purpose, namely 500 μM, corresponding to the upper physiological range, and 5 mM, a high concentration that yields robust responses. To assess how GABA influences Na+/K+ ATPase activity, GABA alone was first applied until stable elevated Na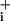 levels were obtained (Fig. 3 A and B). At this point, the Na+/K+ ATPase was inhibited with ouabain (1 mM). This maneuver led to a further Na
levels were obtained (Fig. 3 A and B). At this point, the Na+/K+ ATPase was inhibited with ouabain (1 mM). This maneuver led to a further Na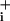 increase above the elevated steady state, which lasted as long as the pump was inhibited. The effect of ouabain was reversible, and upon GABA washout Na
increase above the elevated steady state, which lasted as long as the pump was inhibited. The effect of ouabain was reversible, and upon GABA washout Na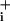 returned to baseline values. A final ouabain application was made at the end of the experiment, resulting in an increase in Na
returned to baseline values. A final ouabain application was made at the end of the experiment, resulting in an increase in Na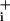 over baseline. With this protocol, the Na
over baseline. With this protocol, the Na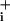 increase after pump blockade reveals the rate of Na+ influx, both the basal and the sum of basal plus GABA-evoked Na+ influx. Analysis of the slope (mM Na
increase after pump blockade reveals the rate of Na+ influx, both the basal and the sum of basal plus GABA-evoked Na+ influx. Analysis of the slope (mM Na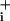 per min), performed by linear regression of the initial phase of Na
per min), performed by linear regression of the initial phase of Na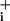 increase during ouabain application, indicated that GABA did not evoke an increase in Na+ influx in this steady-state condition. In fact, for a GABA concentration of 500 μM, the rate of Na
increase during ouabain application, indicated that GABA did not evoke an increase in Na+ influx in this steady-state condition. In fact, for a GABA concentration of 500 μM, the rate of Na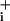 increase was even slightly lower than in the absence of GABA (Fig. 3D).
increase was even slightly lower than in the absence of GABA (Fig. 3D).
Fig. 3.

Degree of Na+/K+ ATPase activity stimulation induced by GABA and glutamate. GABA (500 μM) (A), 5 mM GABA (B), or 2.5 μM glutamate (C) were applied to cells until a plateau of elevated Na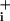 was obtained. At this point the Na+/K+ ATPase was transiently inhibited by using 1 mM ouabain for 1–2 min. After release of pump inhibition, Na
was obtained. At this point the Na+/K+ ATPase was transiently inhibited by using 1 mM ouabain for 1–2 min. After release of pump inhibition, Na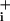 recovered to the levels observed in the presence of GABA or glutamate alone. Ouabain alone was then perfused to reveal the basal Na+ influx. The traces are representative of 46 cells from six experiments (A), 48 cells from seven experiments (B), and 32 cells from four experiments (C). Dotted rectangles highlight Na
recovered to the levels observed in the presence of GABA or glutamate alone. Ouabain alone was then perfused to reveal the basal Na+ influx. The traces are representative of 46 cells from six experiments (A), 48 cells from seven experiments (B), and 32 cells from four experiments (C). Dotted rectangles highlight Na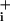 increases during Na+/K+ ATPase blockade. (D) The respective rates of Na
increases during Na+/K+ ATPase blockade. (D) The respective rates of Na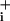 rise (mM·min-1) during pump blockade under basal conditions or in the presence of GABA or glutamate were measured from experiments presented in A–C, and from experiments performed with 200 μM glutamate (32 cells from four experiments, data not shown). Ratios of the two rates (stimulated over basal) are presented. The statistical significance of the rate change between stimulated and basal conditions is indicated in the graph. ns, Not significant (P > 0.05). (E and F) Rapid kinetics of Na+ entry during GABA and glutamate applications. The rate of Na
rise (mM·min-1) during pump blockade under basal conditions or in the presence of GABA or glutamate were measured from experiments presented in A–C, and from experiments performed with 200 μM glutamate (32 cells from four experiments, data not shown). Ratios of the two rates (stimulated over basal) are presented. The statistical significance of the rate change between stimulated and basal conditions is indicated in the graph. ns, Not significant (P > 0.05). (E and F) Rapid kinetics of Na+ entry during GABA and glutamate applications. The rate of Na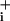 change (mM·sec-1) is plotted against time and shows a first acceleration of Na
change (mM·sec-1) is plotted against time and shows a first acceleration of Na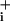 rise when 1 mM ouabain is added at t = 0, followed by a stable Na+ influx rate. When GABA (500 μM) was applied, a sudden acceleration of Na
rise when 1 mM ouabain is added at t = 0, followed by a stable Na+ influx rate. When GABA (500 μM) was applied, a sudden acceleration of Na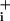 increase is evidenced by slope change that returns to the initial value within <30 sec, whereas without GABA application (dotted line), this transient was absent. (F) Glutamate application using the same protocol also led to transient acceleration of Na+ influx rate that, however, reached higher values and lasted longer than after GABA application. This analysis rate was obtained by averaging the responses of 8–10 cells in the field of view, applying a locally weighted least-squared error fit for high-frequency noise reduction, and computing the first-order derivative. The recordings of Na
increase is evidenced by slope change that returns to the initial value within <30 sec, whereas without GABA application (dotted line), this transient was absent. (F) Glutamate application using the same protocol also led to transient acceleration of Na+ influx rate that, however, reached higher values and lasted longer than after GABA application. This analysis rate was obtained by averaging the responses of 8–10 cells in the field of view, applying a locally weighted least-squared error fit for high-frequency noise reduction, and computing the first-order derivative. The recordings of Na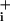 responses in the presence and the absence of applied GABA or glutamate were obtained consecutively during the same experiment and graphically presented with t = 0 corresponding to the start of ouabain application. This experimental protocol has been repeated on three (GABA) and four (glutamate) separate experiments with a total of 23 cells (GABA) and 29 cells (glutamate).
responses in the presence and the absence of applied GABA or glutamate were obtained consecutively during the same experiment and graphically presented with t = 0 corresponding to the start of ouabain application. This experimental protocol has been repeated on three (GABA) and four (glutamate) separate experiments with a total of 23 cells (GABA) and 29 cells (glutamate).
The same strategy was applied to assess the effect of glutamate (2). We first performed a series of preliminary experiments (data not shown) that allowed us to select a glutamate concentration (2.5 μM) that yielded Na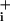 amplitudes comparable to those observed with GABA (Fig. 3C). In contrast to the results obtained with GABA, the slope of Na
amplitudes comparable to those observed with GABA (Fig. 3C). In contrast to the results obtained with GABA, the slope of Na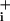 increase evoked by ouabain in the presence of 2.5 μM glutamate was dramatically enhanced, by ≈2.3- ± 0.1-fold. In the presence of 200 μM glutamate, a concentration that yields a larger Na
increase evoked by ouabain in the presence of 2.5 μM glutamate was dramatically enhanced, by ≈2.3- ± 0.1-fold. In the presence of 200 μM glutamate, a concentration that yields a larger Na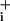 response (2), the slope ratio was larger (≈3.6- ± 0.3-fold). In this series of experiments, the amplitudes of Na
response (2), the slope ratio was larger (≈3.6- ± 0.3-fold). In this series of experiments, the amplitudes of Na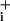 rise before application of ouabain were 2.4 ± 0.2 mM and 3.05 ± 0.25 mM for GABA 500 μM and 5 mM, respectively, whereas they were 5.1 ± 0.2 mM and 23.2 ± 1.1 mM for 2.5 and 200 μM glutamate, respectively. The modest responses to GABA could indicate a low transporter density at the membrane. If this were the case, the expected response would be a slow and steady Na
rise before application of ouabain were 2.4 ± 0.2 mM and 3.05 ± 0.25 mM for GABA 500 μM and 5 mM, respectively, whereas they were 5.1 ± 0.2 mM and 23.2 ± 1.1 mM for 2.5 and 200 μM glutamate, respectively. The modest responses to GABA could indicate a low transporter density at the membrane. If this were the case, the expected response would be a slow and steady Na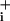 increase up to an elevated steady state, rather than the observed rapid but transient rise followed by a plateau. Mechanistically, such response pattern is more compatible with a fast regulation, e.g., desensitization, of transporter activity similar to what is observed with ionotropic receptors (9) or other transporters (10). To test this hypothesis, we analyzed the initial phase of the response to GABA having first inhibited Na+ extrusion by the Na+/K+ ATPase. Fig. 3E shows the result of these experiments, in which the Na+/K+ ATPase was inhibited continuously by 1 mM ouabain starting 25–30 sec before the application of GABA (500 μM). The slope of Na
increase up to an elevated steady state, rather than the observed rapid but transient rise followed by a plateau. Mechanistically, such response pattern is more compatible with a fast regulation, e.g., desensitization, of transporter activity similar to what is observed with ionotropic receptors (9) or other transporters (10). To test this hypothesis, we analyzed the initial phase of the response to GABA having first inhibited Na+ extrusion by the Na+/K+ ATPase. Fig. 3E shows the result of these experiments, in which the Na+/K+ ATPase was inhibited continuously by 1 mM ouabain starting 25–30 sec before the application of GABA (500 μM). The slope of Na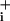 changes plotted against time shows a first increase to a plateau corresponding to the linear basal Na+ entry, then as GABA reached the cells, the rate suddenly increased. However, within 20–30 sec the rate of Na
changes plotted against time shows a first increase to a plateau corresponding to the linear basal Na+ entry, then as GABA reached the cells, the rate suddenly increased. However, within 20–30 sec the rate of Na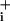 increase returned close to its initial level even though GABA was still present. In comparison, on the same cells, the rate of Na
increase returned close to its initial level even though GABA was still present. In comparison, on the same cells, the rate of Na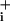 increase during pump blockade remained stable when no GABA was added. The initial Na+ increase was not accelerated by pump blockade, indicating that the pump is not significantly activated during the initial phase of Na+ rise, as observed before with glutamate (2). The same protocol was carried out with 200 μM glutamate. Fig. 3F shows that the maximal rate of Na
increase during pump blockade remained stable when no GABA was added. The initial Na+ increase was not accelerated by pump blockade, indicating that the pump is not significantly activated during the initial phase of Na+ rise, as observed before with glutamate (2). The same protocol was carried out with 200 μM glutamate. Fig. 3F shows that the maximal rate of Na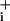 change was 2- to 4-fold higher than with GABA. Moreover, the slope of Na+ change in the presence of glutamate decreased more slowly and stabilized at a rate higher than before glutamate application. These experiments indicate that the main reason for the modest Na
change was 2- to 4-fold higher than with GABA. Moreover, the slope of Na+ change in the presence of glutamate decreased more slowly and stabilized at a rate higher than before glutamate application. These experiments indicate that the main reason for the modest Na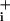 level changes after GABA application is a short-lived GABA-induced Na+ entry, likely caused by transporter deactivation.
level changes after GABA application is a short-lived GABA-induced Na+ entry, likely caused by transporter deactivation.
Overall, these experiments tend to support the idea that, although Na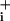 responses to GABA are clearly measurable, they should not cause, unlike glutamate, any significant activation of the Na+/K+ ATPase. To test this prediction at the metabolic level, two approaches were carried out. First, intracellular levels of ATP were assessed in single cells by monitoring changes in intracellular free Mg2+ concentration changes as described (11–13). Free Mg2+ provides an indirect assessment of intracellular ATP concentration, as ATP displays an ≈10-fold higher affinity for Mg2+ than ADP and binds a large proportion of cellular Mg2+ (12). Fig. 4 depicts a typical trace of MgG fluorescence changes measured in single astrocytes, where 200 μM glutamate induced an increase in MgG signal, corresponding to a decrease of ATP levels, that reached its maximum after ≈2 min and returned to baseline after glutamate washout. In comparison, GABA at either 500 μM or 5 mM did not evoke any detectable MgG fluorescence increase. At the end of the experiment, to verify the reactivity of the probe, the mitochondrial uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (1–2 μM) was applied to abolish ATP production through oxidative phosphorylation and induce maximal ATP hydrolysis by reversal of mitochondrial F0F1-ATPase activity (14). This maneuver massively increased the MgG signal, demonstrating the responsiveness of the probe. These experiments indicated that ATP levels, while being significantly diminished during glutamate uptake, are not altered by GABA transport activity.
responses to GABA are clearly measurable, they should not cause, unlike glutamate, any significant activation of the Na+/K+ ATPase. To test this prediction at the metabolic level, two approaches were carried out. First, intracellular levels of ATP were assessed in single cells by monitoring changes in intracellular free Mg2+ concentration changes as described (11–13). Free Mg2+ provides an indirect assessment of intracellular ATP concentration, as ATP displays an ≈10-fold higher affinity for Mg2+ than ADP and binds a large proportion of cellular Mg2+ (12). Fig. 4 depicts a typical trace of MgG fluorescence changes measured in single astrocytes, where 200 μM glutamate induced an increase in MgG signal, corresponding to a decrease of ATP levels, that reached its maximum after ≈2 min and returned to baseline after glutamate washout. In comparison, GABA at either 500 μM or 5 mM did not evoke any detectable MgG fluorescence increase. At the end of the experiment, to verify the reactivity of the probe, the mitochondrial uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (1–2 μM) was applied to abolish ATP production through oxidative phosphorylation and induce maximal ATP hydrolysis by reversal of mitochondrial F0F1-ATPase activity (14). This maneuver massively increased the MgG signal, demonstrating the responsiveness of the probe. These experiments indicated that ATP levels, while being significantly diminished during glutamate uptake, are not altered by GABA transport activity.
Fig. 4.

Intracellular free Mg2+ changes during glutamate and GABA uptake. A typical trace of intracellular free Mg2 concentration changes measured by using the MgG fluorescent probe (presented in fluorescence arbitrary units). The paradigm of glutamate and GABA addition is indicated in the graph. Toward the end of the experiment, ATP levels were maximally diminished by application of the mitochondrial uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), which induced a rapid and massive MgG signal increase. The trace is representative of 50 cells from 10 experiments using the same protocol.
Finally, the metabolic load of neurotransmitter transport was evaluated by measuring glucose uptake in astrocytes by using the 2DG technique (3). Fig. 5 shows that 2DG uptake was augmented by ≈42% by 200 μM glutamate whereas GABA at both 500 μM and 5 mM had no effect. At the end of experiments, the amount of lactate released into the medium was measured as an indication of the degree of glycolysis stimulation and showed that lactate release was increased in the presence of glutamate, whereas it was unaffected by GABA.
Fig. 5.

2DG uptake and lactate release after glutamate and GABA application. Astrocytes were exposed to glutamate (200 μM) or GABA (500 μM and 5 mM) for a 20-min incubation period, at the end of which the amounts of 2DG uptake (hatched bars) and lactate release in the medium (open bars) were determined. Results are means ± SEM of four separate determinations. Statistical analysis was performed by using ANOVA followed by Dunnett's test. **, P < 0.001 vs. control (CTRL). ns, Not significant (P > 0.05).
Discussion
Astrocytes avidly take up glutamate from the extracellular space at excitatory synapses. This glutamate uptake plays a pivotal role as a signal coupling neuronal activity with glucose metabolism in astrocytes (1). Glutamate uptake is accompanied by a substantial astrocytic Na+ load, leading to increased Na+/K+ ATPase activity, ATP consumption, and an enhancement of glycolysis with a concomitant astrocytic lactate production (2, 3). It has been postulated that lactate produced by such a mechanism could then serve as metabolic fuel for activated neurons (1).
The present study aimed at clarifying whether a similar mechanism could exist for the inhibitory neurotransmitter GABA, because it is also transported by astrocytes in a Na+-dependent manner. Data reported in this article indicate that even though astrocytes do respond to GABA by increased Na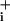 this does not lead to an enhanced metabolic response in astrocytes.
this does not lead to an enhanced metabolic response in astrocytes.
This conclusion was reached by two distinct sets of data: first by monitoring in situ changes in ATP levels, as determined by intracellular free Mg2+ with the fluorescent probe MgG (12). These experiments indicated that intracellular free Mg2+ was not altered by GABA uptake, whereas in comparison glutamate induced significant increases, an indication that ATP levels were decreased. It is noteworthy that the kinetics of MgG response to glutamate are different from the response obtained for intracellular Na+ after glutamate application, which reaches its maximum faster (2).
The second metabolic parameter tested was glucose utilization determined by 2DG uptake and the associated lactate release. Previous studies have demonstrated that glutamate stimulates aerobic glycolysis in astrocytes (3). Data reported here indicate that under the same conditions GABA does not mimic the effects of glutamate, consistent with the results obtained with MgG experiments documenting the lack of ATP changes.
The stoichiometry of GABA transport is thought to be two Na+/one Cl-/one GABA (5) whereas for glutamate it is three Na+ per glutamate (15). This difference in stoichiometry could partially explain the less robust Na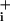 response of astrocytes to GABA. However, in a previous study on glutamate transporter-mediated Na+ response, we have shown that one of the most critical parameters to determine elevated Na
response of astrocytes to GABA. However, in a previous study on glutamate transporter-mediated Na+ response, we have shown that one of the most critical parameters to determine elevated Na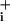 steady state and therefore Na+/K+-ATPase activation was glutamate transporter deactivation (2). The Na
steady state and therefore Na+/K+-ATPase activation was glutamate transporter deactivation (2). The Na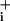 response to GABA is characterized by a steep rise, which abruptly ceases when Na
response to GABA is characterized by a steep rise, which abruptly ceases when Na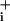 increases by 4–6 mM. This type of response can hardly be accounted for solely by low transporter density at the membrane; it is rather more compatible with a rapid regulation of transport activity. Indeed, we have been able to show that the GABA-stimulated Na+ influx is transient, lasting <30 sec. Although GABA transporter inactivation was not observed in Xenopus oocyte-excised membrane patches containing GAT-1, intracellular Cl- was found to potently inhibit transporter function (16). Because GABA transporters are also coupled to Cl- (5), a rapid local Cl- rise inhibiting the transporter could explain the transient nature of the response to GABA. Similarly, because of rapid desensitization, astrocytic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptors do not cause Na+ load and the associated metabolic response, which is solely caused by glutamate transporter activity both in vitro and in vivo (2, 17).
increases by 4–6 mM. This type of response can hardly be accounted for solely by low transporter density at the membrane; it is rather more compatible with a rapid regulation of transport activity. Indeed, we have been able to show that the GABA-stimulated Na+ influx is transient, lasting <30 sec. Although GABA transporter inactivation was not observed in Xenopus oocyte-excised membrane patches containing GAT-1, intracellular Cl- was found to potently inhibit transporter function (16). Because GABA transporters are also coupled to Cl- (5), a rapid local Cl- rise inhibiting the transporter could explain the transient nature of the response to GABA. Similarly, because of rapid desensitization, astrocytic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptors do not cause Na+ load and the associated metabolic response, which is solely caused by glutamate transporter activity both in vitro and in vivo (2, 17).
The difference between the metabolic effects of glutamate and GABA does not simply reside in the magnitude of the Na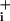 response, because a glutamate concentration that evokes similar Na
response, because a glutamate concentration that evokes similar Na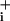 increases as GABA was able to more than double Na+/K+-ATPase activity (Fig. 3). The fact that the rate of Na
increases as GABA was able to more than double Na+/K+-ATPase activity (Fig. 3). The fact that the rate of Na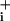 rise from the elevated plateau value was not enhanced in the presence of GABA is consistent with the transient nature of the Na+ influx evoked by GABA. However, it is not clear why under these conditions the Na+/K+-ATPase was not able to bring Na
rise from the elevated plateau value was not enhanced in the presence of GABA is consistent with the transient nature of the Na+ influx evoked by GABA. However, it is not clear why under these conditions the Na+/K+-ATPase was not able to bring Na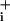 down to baseline values. One appealing possibility would be that GABA is somehow able to interfere with Na+/K+-ATPase activation.
down to baseline values. One appealing possibility would be that GABA is somehow able to interfere with Na+/K+-ATPase activation.
The absence of significant metabolic activation after GABA uptake into astrocytes could be of significance for our understanding of the cellular and molecular basis of brain imaging. Recently, evidence has emerged for a direct link between excitation (represented by local field potentials and involving glutamatergic neurotransmission) and metabolic/hemodynamic changes used as signals by positron emission tomography and functional MRI (18, 19). The putative cellular and molecular mechanism to explain such a link with glucose utilization has been proposed to involve intracellular Na+ changes in astrocytes via glutamate transporter activation (3) and has been substantiated in vivo (17). In contrast, no specific consensus exists concerning the effect of inhibition on brain imaging signals, as discussed (20, 21). Although certain studies have documented an increase in local glucose utilization induced by electrical stimulation of known inhibitory pathways in the rat (22) or by GABA agonist administration in humans (23), a decrease was found after administration of GABA agonists either in rat autoradiographic studies (24–26) or human positron emission tomography studies (27). Based on the presence or not of the blood oxygenation level-dependent signal (BOLD) observed with functional MRI, one group has proposed that inhibition is less metabolically demanding than excitation (28) whereas an other group found no difference (29).
In addition to these experimental data, anatomical and physiological arguments have been put forward to support the concept that inhibition might involve lower metabolic demands. Based on their reduced number, strategic position, and increased efficiency, inhibition provided by synapses from GABAergic interneurons in the cortex might simply involve less energy expenditure upon activation than the excitatory drive (30). Indeed, it is estimated that ≈15–30% of neurons in cortex are nonspiny nonpyramidal cells, which subdivide into different classes of inhibitory interneurons (31, 32), and that the number of cortical inhibitory synapses is ≈5- to 6-fold lower than excitatory synapses (33). A strategic targeting of inhibitory synapses at the proximity of the cell body of postsynaptic neurons ensures a more economical and yet efficient function. The element provided by the present study is the fact that, in addition to these considerations, the recycling mechanism for GABA is intrinsically not coupled to glucose utilization in astrocytes, in contrast to glutamate recycling. In other words, GABA uptake does not represent a metabolic signal and would not contribute directly to local glucose utilization, the parameter observed by brain imaging positron emission tomography approaches. This does not mean that activation of GABAergic neurons per se does not require energy. As proposed by Tagamets and Horwitz (21), it is more likely that neuronal inhibition would contribute rather indirectly to overall metabolic signals (increase or decrease) by modulating local recurrent excitatory circuits. Consequently, the major (and direct) trigger for local glucose utilization would remain glutamate. Consistent with this view is the fact that most GABAergic neurons are locally acting interneurons activated by glutamatergic inputs (34). Thus glutamate, by activating GABA neurons, could lead to an overall inhibitory activity in a given cortical area; yet glutamate release and uptake by astrocytes would stimulate glycolysis in astrocytes and constitute a sufficient signal coding for increased activity of GABAergic neurons as well. It would therefore appear that the most relevant, and possibly sufficient signal, linking neuronal activity (both excitatory and inhibitory) with metabolism is glutamate, which would mediate the appropriate metabolic coupling both for excitatory and inhibitory neurons.
Acknowledgments
We thank Corinne Moratal and Mauricette Maillard for their excellent technical assistance and Pierre Marquet for fruitful discussions. This work was supported by Swiss National Science Foundation Grant 31-67116.01 (to J.-Y.C.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GABA, γ-aminobutyric acid; Na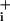 , intracellular Na+; MgG, magnesium green; 2DG, [3H]2-deoxy-glucose.
, intracellular Na+; MgG, magnesium green; 2DG, [3H]2-deoxy-glucose.
References
- 1.Magistretti, P. J., Pellerin, L., Rothman, D. L. & Shulman, R. G. (1999) Science 283, 496-497. [DOI] [PubMed] [Google Scholar]
- 2.Chatton, J.-Y., Marquet, P. & Magistretti, P. J. (2000) Eur. J. Neurosci. 12, 3843-3853. [DOI] [PubMed] [Google Scholar]
- 3.Pellerin, L. & Magistretti, P. J. (1994) Proc. Natl. Acad. Sci. USA 91, 10625-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masson, J., Sagn, C., Hamon, M. & Mestikawy, S. E. (1999) Pharmacol. Rev. 51, 439-464. [PubMed] [Google Scholar]
- 5.Gadea, A. & Lopez-Colome, A. M. (2001) J. Neurosci. Res. 63, 461-468. [DOI] [PubMed] [Google Scholar]
- 6.Sorg, O. & Magistretti, P. J. (1992) J. Neurosci. 12, 4923-4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsson, O. M., Hertz, L. & Schousboe, A. (1986) J. Neurosci. Res. 16, 699-708. [DOI] [PubMed] [Google Scholar]
- 8.Bradford, M. M. (1976) Anal. Biochem. 72, 248-254. [DOI] [PubMed] [Google Scholar]
- 9.Madden, D. R. (2002) Nat. Rev. Neurosci. 3, 91-101. [DOI] [PubMed] [Google Scholar]
- 10.Barbour, B., Brew, H. & Attwell, D. (1991) J. Physiol. (London) 436, 169-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Budinger, G. R., Duranteau, J., Chandel, N. S. & Schumacker, P. T. (1998) J. Biol. Chem. 273, 3320-3326. [DOI] [PubMed] [Google Scholar]
- 12.Leyssens, A., Nowicky, A. V., Patterson, L., Crompton, M. & Duchen, M. R. (1996) J. Physiol. (London) 496, 111-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoue, M., Fujishiro, N., Imanaga, I. & Sakamoto, Y. (2002) J. Physiol. (London) 539, 145-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Budd, S. L. & Nicholls, D. G. (1996) J. Neurochem. 66, 403-411. [DOI] [PubMed] [Google Scholar]
- 15.Danbolt, N. C. (2001) Prog. Neurobiol. 65, 1-105. [DOI] [PubMed] [Google Scholar]
- 16.Lu, C. C. & Hilgemann, D. W. (1999) J. Gen. Physiol. 114, 429-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voutsinos-Porche, B., Bonvento, G., Tanaka, K., Steiner, P., Welker, E., Chatton, J. Y., Magistretti, P. J. & Pellerin, L. (2003) Neuron 37, 275-286. [DOI] [PubMed] [Google Scholar]
- 18.Bonvento, G., Sibson, N. & Pellerin, L. (2002) Trends Neurosci. 25, 359-364. [DOI] [PubMed] [Google Scholar]
- 19.Logothetis, N. K., Pauls, J., Augath, M., Trinath, T. & Oeltermann, A. (2001) Nature 412, 150-157. [DOI] [PubMed] [Google Scholar]
- 20.Arthurs, O. J. & Boniface, S. (2002) Trends Neurosci. 25, 27-31. [DOI] [PubMed] [Google Scholar]
- 21.Tagamets, M. A. & Horwitz, B. (2001) Brain Res. Bull. 54, 267-273. [DOI] [PubMed] [Google Scholar]
- 22.Ackermann, R. F., Finch, D. M., Babb, T. L. & Engel, J., Jr. (1984) J. Neurosci. 4, 251-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peyron, R., Le Bars, D., Cinotti, L., Garcia-Larrea, L., Galy, G., Landais, P., Millet, P., Lavenne, F., Froment, J. C., Krogsgaard-Larsen, P., et al. (1994) Epilepsy Res. 19, 45-54. [DOI] [PubMed] [Google Scholar]
- 24.Kelly, P. A., Ford, I. & McCulloch, J. (1986) Neuroscience 19, 257-265. [DOI] [PubMed] [Google Scholar]
- 25.Kelly, P. A. & McCulloch, J. (1982) J. Neurochem. 39, 613-624. [DOI] [PubMed] [Google Scholar]
- 26.Palacios, J. M., Kuhar, M. J., Rapoport, S. I. & London, E. D. (1982) J. Neurosci. 2, 853-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roland, P. E. & Friberg, L. (1988) J. Cereb. Blood Flow Metab. 8, 314-323. [DOI] [PubMed] [Google Scholar]
- 28.Waldvogel, D., van Gelderen, P., Muellbacher, W., Ziemann, U., Immisch, I. & Hallett, M. (2000) Nature 406, 995-998. [DOI] [PubMed] [Google Scholar]
- 29.Heeger, D. J., Boynton, G. M., Demb, J. B., Seidemann, E. & Newsome, W. T. (1999) J. Neurosci. 19, 7162-7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koos, T. & Tepper, J. M. (1999) Nat. Neurosci. 2, 467-472. [DOI] [PubMed] [Google Scholar]
- 31.DeFelipe, J. & Farinas, I. (1992) Prog. Neurobiol. 39, 563-607. [DOI] [PubMed] [Google Scholar]
- 32.Fairen, A., DeFelipe, J. & Regidor, J. (1984) in Cerebral Cortex, eds. Peters, A. & Jones, E. G. (Plenum, New York), Vol. 1, pp. 201-253. [Google Scholar]
- 33.Beaulieu, C. & Colonnier, M. (1985) J. Comp. Neurol. 231, 130-1389. [DOI] [PubMed] [Google Scholar]
- 34.Houser, C. R., Vaughn, J. E., Hendry, S. H. C., Jones, E. G. & Peters, A. (1984) in Cerebral Cortex, eds. Jones, E. G. & Peters, A. (Plenum, New York), Vol. 2, pp. 63-89. [Google Scholar]