

Abstract
Apoptosis is generally accompanied by a late phase of ceramide (Cer) production, the significance of which is unknown. This study describes a previously unrecognized link between Cer accumulation and phosphatidylserine (PS) exposure at the cell surface, a characteristic of the execution phase of apoptosis resulting from a loss of plasma membrane phospholipid asymmetry. Using a fluorescent sphingomyelin (SM) analogue, N-(N-[6-[(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]caproyl]–sphingosylphosphorylcholine (C6-NBD-SM), we show that Cer is derived from SM, initially located in the outer leaflet of the plasma membrane, which gains access to a cytosolic SMase by flipping to the inner leaflet in a process of lipid scrambling paralleling PS externalization. Lipid scrambling is both necessary and sufficient for SM conversion: Ca2+ ionophore induces both PS exposure and SM hydrolysis, whereas scrambling-deficient Raji cells do not show PS exposure or Cer formation. Cer is not required for mitochondrial or nuclear apoptotic features since these are still observed in Raji cells. SM hydrolysis facilitates cholesterol efflux to methyl-β-cyclodextrin, which is indicative of a loss of tight SM–cholesterol interaction in the plasma membrane. We provide evidence that these biophysical alterations in the lipid bilayer are essential for apoptotic membrane blebbing/vesiculation at the cell surface: Raji cells show aberrant apoptotic morphology, whereas replenishment of hydrolyzed SM by C6- NBD-SM inhibits blebbing in Jurkat cells. Thus, SM hydrolysis, during the execution phase of apoptosis, results from a loss of phospholipid asymmetry and contributes to structural changes at the plasma membrane.
Keywords: CD95, cholesterol, PS exposure, DNA damage, blebbing
Introduction
Apoptosis can be induced by such diverse stimuli as triggering of death receptors (e.g., CD95 and tumor necrosis factor receptor 1), growth factor withdrawal, hypoxia, or DNA damage (e.g., etoposide or γ-radiation). These apoptotic stimuli initiate unique signal transduction events, but ultimately converge at the execution machinery of apoptosis, the caspase family of proteases. In addition to caspase activation, ceramide (Cer) formation is a universal phenomenon in cells undergoing apoptosis (Hannun 1996), but there is extensive debate about the significance and the possible role of Cer in apoptosis (Hofmann and Dixit 1998; Kolesnick and Hannun 1999). A rapid Cer response (in minutes), which is suggestive of a second messenger function, is observed in some cell systems using certain stimuli, whereas Cer formation at later time points (in hours) after apoptosis induction is more generally observed. The function of this late Cer response remains unknown. The mechanism of Cer generation in response to apoptotic stimuli has been the subject of extensive investigation. Sphingomyelin (SM) hydrolysis by either a neutral or acid sphingomyelinase (SMase) has been recognized as a universal pathway for Cer production (for review see Levade and Jaffrézou 1999). On the other hand, de novo Cer biosynthesis also has been implicated in apoptosis induction by anticancer drugs (Bose et al. 1995).
We have previously shown that CD95-, etoposide-, and γ-radiation–induced Cer formation in Jurkat T cells is a slow event, coinciding with nuclear fragmentation, and is secondary to caspase activation and cytochrome c release from the mitochondria (Tepper et al. 1997, Tepper et al. 1999). These findings argue against a triggering role for Cer in apoptosis, at least in this cell system, but leave open the possibility that Cer has some function in the apoptotic execution phase. One hallmark of the execution phase is the exposure of phosphatidylserine (PS) at the cell surface (Martin et al. 1995), which serves for recognition and subsequent phagocytosis by macrophages (Schroit et al. 1985; Fadok et al. 1992). PS exposure is the direct consequence of a loss of phospholipid (PL) asymmetry in the plasma membrane. The asymmetric PL distribution (i.e., SM and phosphatidylcholine [PC] in the outer leaflet and the aminophospholipids in the inner leaflet of the lipid bilayer) is maintained by an ATP-dependent aminophospholipid translocase that continuously transports PS and phosphatidylethanolamine (PE) to the inner leaflet (for reviews see Zachowski 1993; Zwaal and Schroit 1997). Cell-surface exposure of PS is believed to result from the coordinated inhibition of the aminophospholipid translocase and the activation of a Ca2+-induced, bidirectional PL scrambling (flip-flop) activity, causing the loss of lipid bilayer asymmetry (Verhoven et al. 1995; Bratton et al. 1997). Scrambling of PL is thought to be mediated by a distinct enzyme, PL scramblase (Zhou et al. 1997).
We were intrigued by the possibility that Cer formation and PL scrambling (PS exposure), two lipid events in the apoptotic execution phase, might be functionally linked. To address this possibility, we used scrambling-competent Jurkat T and SKW6.4 B cells and scrambling-deficient Raji B cells, as well as fluorescent 7-nitrobenz-2-oxa-1,3-diazol-4-yl-(NBD-) analogues of SM and PC. We show that, as a consequence of the loss of PL asymmetry, SM moves from the outer to the inner leaflet of the plasma membrane, where it serves as a substrate for an intracellular SMase. Thus, transbilayer movement of SM during apoptosis determines substrate availability and thereby controls Cer formation. Furthermore, we show that the breakdown of SM causes concomitant cholesterol efflux and, thus, significant alterations in the biophysical properties of the plasma membrane, which is a prerequisite for membrane blebbing and vesiculation at the surface of the apoptotic cell.
Materials and Methods
Materials
l-3-[14C]serine (54 mCi/mmol), methyl-[14C]-choline chloride (58 mCi/mmol) and 1α,2α(n)-[3H] cholesterol (49 Ci/mmol) were purchased from Amersham Pharmacia Biotech. Mouse anti-human CD95 mAb 7C11 was from Immunotech. Ionomycin, BSA (essentially fatty acid–free), acetyl-DEVD-aldehyde (DEVD-CHO) and N-benzoyloxycarbonyl-VAD-fluoroethylketone (zVAD-fmk) were from Calbiochem. Bacillus cereus sphingomyelinase, etoposide, propidium iodide (PI), fumonisin B1 (FB1), and methyl-β-cyclodextrin (MβCD) were obtained from Sigma Chemical Co. 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3)), N-(N-[6-[(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]caproyl]-sphingosine (C6-NBD-Cer), and N-(N-[6-[(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]caproyl]sphingosylphosphorylcholine (C6-NBD-SM) were from Molecular Probes, Inc. FITC-labeled annexin V (APOPTEST™-FITC) was from Nexins Research BV.
Cells
The J16 clone was derived by limiting dilution from the human T-acute lymphoblastic leukemia cell line Jurkat and selected for uniform T cell antigen receptor expression and CD95 sensitivity. J16 and SKW6.4 (Epstein-Barr virus-transformed) B cells and Raji Burkitt lymphoma B cells were cultured as previously described (Tepper et al. 1997). Before stimulation, cells were resuspended in serum-free Yssel's medium (Yssel et al. 1984).
Apoptosis Assays
To measure nuclear fragmentation (subdiploid DNA content), cells were lysed in 0.1% sodium citrate, 0.1% Triton X-100, and 50 μg/ml propidium iodide (PI; Nicoletti et al. 1991) as described earlier (Tepper et al. 1999). The loss of mitochondrial transmembrane potential (ΔΨm) was determined using DiOC6(3) (Zamzami et al. 1996). PS exposure was measured using FITC-labeled annexin V as previously described (Koopman et al. 1994). PI was added (5 μg/ml) before analysis to gate-out the cells that had lost membrane integrity.
Measurement of SM Hydrolysis and Cer Quantification
Cells were metabolically labeled with 0.2 μCi/ml each of either [14C]serine or [14C]choline chloride for 40–48 h in Yssel's medium, washed, and further incubated for 3 h in Yssel's medium before stimulation. Lipids were extracted and radioactivity in SM and PC was analyzed after TLC (Silica G60 plates) using chloroform/methanol/acetic acid/water (60:30:8:5, vol/vol) as developing solvent. The water-soluble [14C]choline metabolites in cells were quantified after TLC separation (van Blitterswijk et al. 1991). In this case, radiolabeling of cells was followed by a 24-h chase period before stimulation. Cer levels were determined after [14C]serine labeling as described previously (Tepper et al. 1997), and were expressed relative to PS plus PE, which remained unaltered upon stimulation.
Analysis of NBD–Phospholipid Metabolism
Cells were washed once in Hanks' balanced salt solution with 10 mM Hepes (H/H), pH 7.4, and resuspended in the same medium supplemented with 1 mM CaCl2. NBD-lipid was dissolved in a minimum volume of ethanol. H/H was added to this solution under vigorous vortexing to yield a four times concentrated lipid mixture. Cells were preincubated for 10 min at 37°C with the NBD-lipid and subsequently stimulated. Lipids were extracted and separated by TLC in chloroform/methanol/NH4OH 25% (70:30:5 vol/vol). NBD-lipids were visualized by UV light (Eagle EyeII™; Stratagene) and quantitated by fluorescence spectrophotometry. In some experiments, cells were labeled with NBD-SM during 30 min at 37°C, followed by back extraction (i.e., two consecutive incubations with 5% BSA for 4°C at 5 min) to remove exoleaflet-associated NBD-SM. SKW6.4 cells were suspended in Yssel's medium, treated with etoposide (5 μg/ml) or irradiation (20 Gy) for 14 h (yielding PS exposure after approximately 10–12 h), washed, and resuspended in H/H. NBD-SM was added (4 μM final concentration) during the last 20 min of the incubation period, followed by lipid extraction and TLC analysis.
Efflux of Labeled Cholesterol
Cells were labeled for 2 d in complete medium containing 0.5 μCi/ml [3H]cholesterol, followed by a 24-h equilibration in Yssel's medium without radioactivity. Efflux of cholesterol from cells was determined in Hanks'/Hepes (H/H) containing 0.08% MβCD, added 3 min before addition of B. cereus SMase (150 mU/ml). Cholesterol efflux during apoptosis induction by anti-CD95 mAb (200 ng/ml) was measured at the indicated time points, after centrifugation (for 2 min at 14,000 g) and resuspension of cells in 1.25 ml H/H containing 0.08% MβCD to initiate cholesterol efflux at 37°C. Aliquots of the cell suspension were harvested at different time points and radioactivity in the supernatant was determined by scintillation counting. The extent of cholesterol efflux was calculated from the radioactivity initially present in the cell suspension.
Results
Correlation between PS Exposure and Cer Formation
Earlier work indicated that Cer accumulates relatively late after apoptosis induction of Jurkat T cells via CD95 stimulation or DNA-damaging regimens (Tepper et al. 1997, Tepper et al. 1999). A slow and sustained Cer response has been described for many different cell types using various apoptotic stimuli, suggesting that late Cer production is a general characteristic of the apoptotic execution phase. To elucidate the mechanism and relevance of this late phase of Cer formation, we set out to investigate a possible relationship between Cer production and PS exposure, another lipid event that is usually observed during the execution phase. Fig. 1 A shows a close temporal correlation between Cer formation, PS exposure, and nuclear fragmentation during CD95 stimulation of Jurkat T cells. Cer formation could be dissociated from nuclear fragmentation by inhibiting effector caspases using the tetrapeptide DEVD-CHO (Thornberry et al. 1997; Fig. 1 B). DEVD-CHO inhibited nuclear fragmentation, whereas it only marginally affected Cer formation and PS exposure in response to etoposide or CD95 stimulation. Abrogation of inducer and effector caspase activity using zVAD-fmk (Thornberry et al. 1997) abolished both PS exposure and Cer formation. The low level of inhibition of PS exposure and Cer formation by DEVD-CHO can be explained by the weak inhibitory effect of this compound on inducer caspases (Garcia-Calvo et al. 1998). The similar kinetics and caspase dependency of Cer formation and PS exposure suggest that there is a causal relationship between these events.
Figure 1.

Cer formation is associated with PS exposure. (A) Time course of CD95-induced Cer formation, PS exposure, and nuclear fragmentation in Jurkat cells. Sphingolipids were labeled to equilibrium with [14C]serine. Cells were stimulated with anti-CD95 mAb (200 ng/ml) for the indicated periods of time. Cer formation was assessed by TLC. PS exposure and nuclear fragmentation were analyzed by annexinV-FITC staining of intact cells or propidium iodide staining of nuclei, respectively. Data shown are of one experiment performed three times with similar results. Cer data points represent the mean ± SD of three individual measurements. The other data points represent individual measurements. (B) CD95- and etoposide-induced Cer formation, PS exposure, and nuclear fragmentation were determined in parallel in the presence and absence of caspase inhibitors. Jurkat cells were radiolabeled as described above, left untreated (black bars) or preincubated for 2 h with zVAD-fmk (50 μM; gray bars) or DEVD-CHO (100 μM; white bars), and then exposed to anti-CD95 mAb (200 ng/ml) during 4 h or etoposide (10 μg/ml) during 16 h. Data are individual measurements within one experiment representative of at least four separate experiments.
Cer Is Derived from Outer-leaflet SM
To define the source of Cer generated in CD95-stimulated cells, we first assessed whether it resulted from SM hydrolysis or de novo synthesis. Metabolic labeling of cells with [14C]serine allowed the detection and quantitation of sphingolipids by TLC. Fig. 2 A shows that CD95 stimulation induced a significant reduction in SM levels (∼20%) concomitant with Cer formation, which is indicative of SMase activity. Fig. 2 B shows that CD95-induced Cer formation was not affected by FB1, an inhibitor of de novo Cer biosynthesis (Schroeder et al. 1994). The same holds for other apoptotic stimuli, etoposide and γ-radiation (data not shown). We confirmed that FB1 efficiently blocked de novo sphingolipid biosynthesis (by >85%), as evidenced by the loss of [14C]serine incorporation into SM (Fig. 2 B, inset). Thus, CD95-induced Cer formation results from the breakdown of SM, without a role for Cer synthase activity.
Figure 2.

CD95-induced Cer is derived from SM, not from de novo biosynthesis. (A) Jurkat cells labeled with [14C]serine were stimulated with anti-CD95 mAb (200 ng/ml; closed symbols) for indicated time periods or left untreated (open symbols), after which lipids were extracted and analyzed by TLC. Data are expressed as a percentage of SM (circles) and multiple-fold increase in Cer (triangles) relative to an untreated control sample harvested just before the start of the experiment. (B) Effect of fumonisin B1 (FB1) on CD95-induced Cer formation. Jurkat cells labeled to equilibrium with [14C]serine were preincubated for 24 h without (medium) or with 25 μM FB1 (+FB1), washed, resuspended in medium with or without 25 μM FB1, and stimulated with anti-CD95 mAb (200 ng/ml; closed bars) or left untreated (open bars). After 3 h, lipids were extracted and Cer was quantified. Data (means ± SD) represent two independent experiments. The Cer response is indicated as a percentage of untreated control cells. Note that, as expected, FB1 reduces absolute [14C]Cer levels as defined in Materials and Methods. (Inset) Effect of FB1 on SM synthesis. Cells were preincubated with 25 μM FB1 for time periods indicated, after which [14C]serine was added for an additional 36 h. Lipids were extracted and SM was quantified. Results are expressed as a percentage of untreated control cells and are representative of two experiments. (C) The pool of SM hydrolyzed upon CD95 stimulation is fully accessible to an exogenous SMase. Jurkat cells metabolically labeled with [14C]choline were treated with B. cereus SMase (200 mU/ml) alone (closed circles) or with SMase plus anti-CD95 mAb (200 ng/ml; open squares). At various time points, samples were analyzed for radiolabeled SM and PC content. The ratio of SM relative to PC is expressed as a percentage of control. This experiment was performed twice with similar results.
Plasma membrane phospholipids are asymmetrically distributed and, depending on the cell type, up to 90% of the total cellular SM is localized in the outer leaflet (Koval and Pagano 1991). We quantified this pool of SM using bacterial sphingomyelinase (bSMase). Approximately 70% of total cellular SM, radiolabeled either in the polar headgroup or in the sphingoid backbone, was degraded by bSMase. The remaining intracellular pool was resistant to treatment, even upon prolonged incubation (Fig. 2 C). To define the pool of SM that is hydrolyzed during apoptosis, cells were treated simultaneously with bSMase and anti-CD95 mAb. If distinct cellular pools were involved, CD95 ligation should induce an additional breakdown of SM. However, the combined treatment of cells with bSMase and anti-CD95 mAb did not result in a greater loss of SM than that observed with bSMase alone (Fig. 2 C). CD95 signaling was effective under these conditions, as determined by nuclear fragmentation (results not shown). These findings suggest that the CD95-induced SMase activity and exogenous bSMase hydrolyze the same pool of SM (i.e., the one residing in the outer leaflet of the plasma membrane).
To further substantiate this notion, we monitored the fate of the fluorescent SM analogue N-(N-[6-[(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]caproyl]–sphingosylphosphorylcholine (NBD-SM), which inserts into the outer leaflet of the plasma membrane (Koval and Pagano 1990). We investigated whether cells undergoing apoptosis can metabolize NBD-SM. Indeed, CD95 stimulation resulted in a progressive hydrolysis of NBD-SM to NBD-Cer (Fig. 3A and Fig. B), with similar kinetics to those observed for endogenous SM conversion (Fig. 2 A). NBD-phosphatidylcholine (NBD-PC) was not susceptible to degradation to either lysoPC, phosphatidic acid, or diacylglycerol (data not shown), indicating that SM to Cer conversion is specific and does not reflect general lipid breakdown. The appearance of a small amount of NBD-Cer in untreated control cells (Fig. 3 A) most likely reflects hydrolysis of NBD-SM that has entered the cell via endocytosis (Koval and Pagano 1990, Koval and Pagano 1991). This pool of NBD-Cer was further metabolized by glycosylation to NBD-glucosylceramide, whereas the NBD-Cer formed during CD95 stimulation was not (Fig. 3 A). Apparently, NBD-Cer derived from endocytosed NBD-SM is further glycosylated in the Golgi apparatus, whereas CD95-induced NBD-Cer does not reach this site, but rather, Cer glycosylation seems to be inhibited in apoptotic cells (Fig. 3 C). To provide evidence that CD95-mediated Cer formation is derived from an SM pool in the outer leaflet of the plasma membrane and not from endocytosed NBD-SM, we labeled cells with NBD-SM for 30 min at 37°C to allow labeling of intracellular membranes, and then depleted exoleaflet NBD-SM via BSA back extraction (Van Meer et al. 1987; Koval and Pagano 1990) before CD95 stimulation. Fig. 3 C shows that, after back extraction, CD95 is no longer capable of inducing NBD-Cer formation, indicating that this SMase activity utilizes NBD-SM from the exoleaflet and not the pool of internalized NBD-SM. Of note, NBD-GlcCer was much less susceptible to BSA back extraction (Fig. 3 C), reflecting its predominant localization to intracellular membranes. To ensure that the phenomenon of outer leaflet NBD-SM hydrolysis was not restricted to Jurkat T cells and/or CD95-induced apoptosis, we also analyzed NBD-Cer production in SKW6.4 cells (Epstein-Barr virus–transformed B cells) during etoposide- or γ-radiation–induced apoptosis. Because apoptosis induction by these stimuli is much slower than via death receptor ligation, NBD-SM was presented to the cells for only 20 min to avoid high background levels of NBD-Cer. Similar to the Jurkat/CD95 system, SKW6.4 cells showed increased NBD-Cer production upon DNA damage–induced apoptosis (Fig. 3 D), which temporally associated with PS exposure (data not shown). Taken together, these results indicate that distinct apoptotic stimuli induce late Cer formation through hydrolysis of a SM pool that is derived from the outer leaflet of the plasma membrane.
Figure 3.


Cer is derived from the SM originally present in the outer leaflet of the plasma membrane. (A) CD95 induces hydrolysis of exogenous fluorescent NBD-SM. Jurkat cells were incubated with 4 μM NBD-SM 10 min before CD95 stimulation (200 ng mAb/ml) for the times indicated. NBD-lipids were extracted, separated by TLC, visualized under UV light, and photographed. The identities of NBD-Cer and NBD-GlcCer (NBD-glucosylceramide) were established using co-chromatography of commercially obtained lipid standards. NBD-Cer formation was most easily detected when the lipid probe was continuously present during stimulation, but significant NBD-SM hydrolysis in CD95-stimulated cells was also observed when NBD-SM was added during the last 15 min of incubation (results not shown). (B) Quantitation of the NBD-Cer spots shown in A by fluorescence spectrophotometry (expression in arbitrary units). (C) NBD-Cer is derived from outer leaflet NBD-SM. Jurkat cells were incubated with 4 μM NBD-SM during 30 min at 37°C to allow labeling of intracellular and plasma membranes, followed or not followed by BSA back extraction (see Materials and Methods). Cells resuspended in H/H were warmed to 37°C and exposed to anti-CD95 mAb (200 ng/ml; +) or left untreated (−). Lipids were extracted and analyzed by TLC after the indicated time periods. The BSA extraction protocol did not affect apoptosis induction, as verified by nuclear fragmentation (data not shown). (D) DNA damage–induced NBD-SM hydrolysis in SKW6.4 cells, 14 or 16 h after exposure to etoposide (5 μg/ml; Eto) or radiation (20 Gy; γ-IR), NBD-SM (4 μM) was presented during 20 min to SKW6.4. TLC analysis indicates increased NBD-SM hydrolysis to NBD-Cer in cells undergoing apoptosis relative to untreated control cells (con).
SM Is Hydrolyzed Intracellularly after Lipid Scrambling
If outer leaflet SM is the source of Cer, it has to redistribute within the lipid bilayer to gain access to the hydrolyzing enzyme, because SMases act intracellularly (Linardic and Hannun 1994; Andrieu et al. 1996; Liu et al. 1998). To confirm that SM is hydrolyzed intracellularly, we analyzed the production of water-soluble choline metabolites in the cytosol. Jurkat cells were labeled with [14C]choline, which incorporates into the polar headgroup of SM and PC, and then triggered to undergo apoptosis via the CD95 receptor. We detected no increase in the intracellular level of [14C]phosphocholine, a direct product of SMase activity (Table ). However, apoptotic cells showed increased levels of CDP-choline (Table ), which is likely to be derived from phosphocholine directly and intracellularly, by CTP:phosphocholine cytidylyltransferase, a regulatory enzyme in PC biosynthesis (Kent 1997). Since we found no PC hydrolysis (already noted above), the increased level of CDP-choline can only be derived from SMase-produced phosphocholine, which is directly channeled from inner-leaflet SM via the cytidylyltransferase into CDP-choline.
Table 1.
CD95-induced Increase in the Intracellular Level of CDP-choline
| Time | CDP-choline | Phosphocholine |
|---|---|---|
| min | ||
| 0 | 267 ± 65 (100%) | 8,007 ± 476 (100%) |
| 30 | 261 ± 25 (98%) | 7,442 ± 503 (93%) |
| 60 | 554 ± 110 (208%) | 8,484 ± 98 (106%) |
| 90 | 804 ± 155 (301%) | 5,813 ± 834 (73%) |
Jurkat cells were labeled to equilibrium with [14C]choline chloride, stimulated with anti-CD95 mAb for the indicated times, and cell-associated, water-soluble phosphocholine metabolites were extracted and separated by TLC, as described in Materials and Methods. Data, in [14C]arbitrary (PhosphorImager) units, show the mean ± range of two data points from a representative experiment (out of three). The apparent decrease in intracellular phosphocholine levels after 90 min of stimulation probably reflects the release of membrane vesicles, resulting in an overall loss of cell material that is pelleted at 14,000 g.
In principle, transbilayer movement of SM during lipid scrambling should be measurable with fluorescent NBD-SM, since internalized NBD-SM will be sequestered from BSA back extraction. However, CD95-stimulated Jurkat cells displayed no sequestration of NBD-SM, whereas they did show enhanced internalization of NBD-PC, which temporally paralleled PS exposure, indicative of enhanced nonspecific PL scrambling (Bratton et al. 1997; data not shown). Apparently, NBD-SM hydrolysis (Fig. 3A and Fig. B) occurs rapidly after its internalization, after which the neutral lipid product NBD-Cer can freely flip-flop to the outer leaflet (Pagano 1989) to become available again for back extraction by BSA. Indeed, we found that 87% of the NBD-Cer produced upon CD95 stimulation was susceptible to BSA back extraction (data not shown). Together, the data suggest that activation of nonspecific PL scrambling in the plasma membrane not only results in PS exposure and NBD-PC internalization, but also in internalization of NBD-SM, which is immediately hydrolyzed and back-extracted as NBD-Cer.
Phospholipid Scrambling Is Necessary and Sufficient for Cer Formation
To address the question of whether lipid scrambling activity is necessary for Cer formation, we used the Burkitt lymphoma cell line Raji, which is defective in PS exposure in response to a rise in intracellular Ca2+ levels (Zhao et al. 1998) or apoptotic stimuli (Frey 1997; Fadeel et al. 1999). CD95 stimulation of Raji cells did not result in Cer formation nor significant PS exposure up to 8 h after stimulation, whereas two other apoptotic features, loss of mitochondrial transmembrane potential (ΔΨm) and nuclear fragmentation, became evident after 2 h of incubation (Fig. 4 A). In addition, when NBD-SM was presented to Raji cells undergoing apoptosis in response to CD95 stimulation, no hydrolysis to NBD-Cer was observed (Fig. 4 B). Thus, consistent with the tight connection between scrambling of membrane phospholipids and Cer formation, analysis of the Raji cells revealed that phospholipid scrambling is required for Cer formation. Another important conclusion drawn from these results is that, at least in these cells, Cer formation is not required for apoptotic alterations in mitochondria and the nucleus to occur.
Figure 4.



Scrambling of plasma membrane phospholipids, monitored by annexin V staining (PS exposure), is necessary and sufficient for Cer formation. (A) Raji cells show no CD95-induced PS exposure, nor Cer formation, yet exhibit loss of mitochondrial transmembrane potential (ΔΨm) and nuclear fragmentation. Cells labeled with [14C]serine were exposed to anti-CD95 mAb (200 ng/ml) for the indicated time periods and [14C]Cer was quantified. PS exposure (annexin V staining), ΔΨm (measured with the fluorescent dye DiOC6(3)), and nuclear fragmentation were determined in parallel. (B) Absence of NBD-SM hydrolysis to NBD-Cer in CD95-stimulated Raji cells. Cells were incubated with 4 μM NBD-SM 10 min before the addition of anti-CD95 mAb (200 ng/ml). At the indicated time points, total lipids were extracted and separated by TLC. A commercial NBD-Cer standard was run in parallel. (C) Ionomycin induces PS exposure in Jurkat but not in Raji cells. Cells were exposed to 5 μM ionomycin for the indicated time periods or left untreated (control; con), and PS exposure was detected using FITC-labeled annexin V. Histograms are representative of at least three experiments. (D) Ionomycin induces NBD-SM hydrolysis to NBD-Cer in Jurkat but not in Raji cells. Cells preincubated for 10 min with 4 μM NBD-SM were exposed to 5 μM ionomycin or its solvent (control) for the times indicated, after which lipids were extracted and separated by TLC. NBD-Cer fluorescence was quantified using fluorescence spectrophotometry.
To determine whether SM hydrolysis to Cer is the immediate consequence of the loss of phospholipid asymmetry, lipid scrambling was induced directly by elevating intracellular Ca2+ levels (Williamson et al. 1992; Smeets et al. 1994; Zhou et al. 1998). Accordingly, the Ca2+ ionophore ionomycin, caused rapid surface exposure of PS in Jurkat but not in Raji cells (Fig. 4 C). Remarkably, ionomycin also induced the conversion of NBD-SM to NBD-Cer in Jurkat but not in Raji cells (Fig. 4 D). NBD-SM conversion was time-dependent, with kinetics similar to those of PS exposure. No metabolism of NBD-PC was observed under these conditions (data not shown). EGTA inhibited ionomycin-induced PS exposure and NBD-SM hydrolysis, indicating that both events require extracellular Ca2+ (results not shown). We conclude that the loss of phospholipid asymmetry is both necessary and sufficient for SM to be hydrolyzed intracellularly.
Plasma Membrane SM Content Affects Apoptotic Cell-surface Morphology
Given that SM conversion to Cer occurs during the execution phase of apoptosis, but seems to be dispensable for nuclear apoptosis or mitochondrial depolarization (see Fig. 4 A), the question arises whether it contributes to other apoptotic characteristics, such as altered cell-surface morphology. We noticed that Raji cells, undergoing apoptosis, barely displayed the typical small surface blebs and release of membrane vesicles that are characteristic of normal apoptotic lymphoid cells. Rather, Raji cells adopted elongated, sausagelike shapes (Fig. 5). To examine whether SMase activity but not Cer production is necessary for membrane blebbing during apoptosis, NBD-SM was replenished to CD95-stimulated Jurkat cells. Strikingly, the extensive cell-surface blebbing and vesicle shedding normally observed in Jurkat cells was completely inhibited when NBD-SM was present in the medium (Fig. 5). Moreover, the shape of the cells in the presence of NBD-SM resembled that of CD95-stimulated Raji cells. Both the polar headgroup and the sphingoid base moiety of NBD-SM were required for the inhibition of blebbing, since neither NBD-Cer nor NBD-PC affected the apoptotic morphology of Jurkat cells (data not shown). We conclude that the lack of plasma membrane SM hydrolysis in Raji cells, or replenishment of the SM hydrolyzed in Jurkat cells, prevents the formation of apoptotic blebs and shedding of membrane vesicles. Evidently, the physiological significance of SM hydrolysis relates to apoptotic cell-surface morphology, and lies in plasma membrane SM depletion, rather than the generation of Cer.
Figure 5.
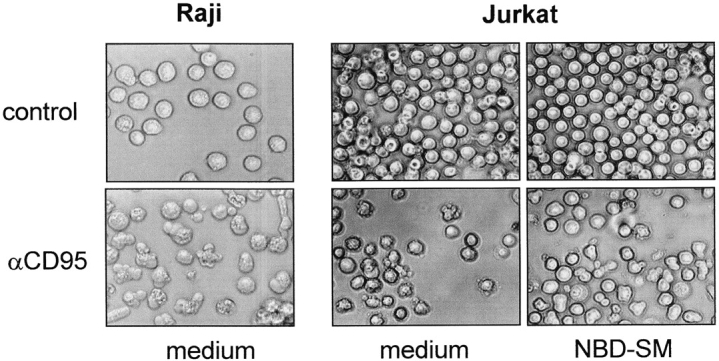
Plasma membrane SM content determines surface morphology of apoptotic cells. Raji and Jurkat cells suspended at 2.5 × 106 cells per ml in H/H medium without (medium) or with 4 μM NBD-SM (NBD-SM) were left untreated (control) or exposed to anti-CD95 mAb (200 ng/ml) for 6 h (Raji) or 1.5 h (Jurkat). Cells were viewed and photographed using a light transmission microscope with a 40× objective.
Why Is Plasma Membrane SM Content Important?
To understand how plasma membrane SM content can affect cell-surface morphology, it is important to recognize that SM has a high affinity for cholesterol and retains cholesterol in the plasma membrane (Wattenberg and Silbert 1983; van Blitterswijk et al. 1987). Both lipids are major determinants for the biophysical properties of the lipid bilayer, such as membrane fluidity and structural integrity, also in functional membrane domains (Simons and Ikonen 1997; Anderson 1998). SM hydrolysis by exogenous SMase disturbs the physical association with cholesterol, resulting in the redistribution of cholesterol from the plasma membrane to intracellular compartments (Neufeld et al. 1996; Ohvo et al. 1997) or to extracellular acceptors (Rothblat et al. 1999). We used MβCD, a specific and well established cholesterol acceptor (Yancey et al. 1996), to examine whether CD95-induced SM hydrolysis altered cholesterol packing with plasma membrane PL, resulting in increased cholesterol efflux from that membrane towards MβCD. Treatment of [3H]cholesterol-labeled cells with bacterial SMase resulted in enhanced MβCD-mediated efflux of [3H]cholesterol (Fig. 6 A). Similarly, cholesterol was more readily removed from cells undergoing CD95-induced apoptosis when compared with time-matched control cells (Fig. 6 B). The effect became discernible between 1 and 2 h of stimulation, which is consistent with the kinetics of SM hydrolysis. From these data, we conclude that degradation of plasma membrane SM, either upon exposure to bacterial SMase or by endogenous SMase during CD95 stimulation, decreases plasma membrane cholesterol content. The concomitant loss of SM and cholesterol during apoptosis will fluidize and destabilize (domains in) the plasma membrane, enabling membrane blebbing (zeiosis) and the shedding of membrane vesicles.
Figure 6.

Enhanced efflux of [3H]cholesterol from the plasma membrane during apoptosis induction. (A) Depletion of SM from the plasma membrane by exogenous SMase enhances efflux of membrane cholesterol to methyl-β-cyclodextrin (MβCD), which is a specific cholesterol acceptor. Jurkat cells were labeled with [3H]cholesterol and resuspended in 0.08% MβCD. After a 3-min incubation at 37°C, cells were treated with 150 mU/ml B. cereus SMase (bSMase; closed squares) or left untreated (open squares). At different time intervals, the radioactivity that released into the extracellular medium was determined by liquid scintillation counting. Date shown are from one experiment with single point determinations, representative of at least five independent experiments. (B) Cells induced to undergo apoptosis (200 ng/ml; anti-CD95 mAb, closed symbols) or left untreated (open symbols) were pelleted after 1-, 2-, 3-, or 4-h incubation (indicated) and resuspended in medium containing 0.08% MβCD. After different incubation periods at 37°C, radioactivity which was released into the medium, was determined by liquid scintillation counting. Data are expressed as a percentage of radioactive cholesterol initially present in the cells and represent one out of three independent experiments with similar results.
Discussion
This study provides novel insights into both the mechanism and the physiological relevance of Cer formation in the execution phase of apoptosis. We found a previously unrecognized connection between two apoptotic features: Cer formation and cell-surface PS exposure. Both events are the immediate consequence of loss of plasma membrane PL asymmetry. This holds for several apoptotic systems, T and B lymphoid cells challenged by CD95 ligation, etoposide, or γ-radiation. Furthermore, we show that this late Cer formation is not required for major characteristics of apoptosis, such as a loss of mitochondrial transmembrane potential and nuclear fragmentation. Rather, the physiological relevance of Cer formation in the effector phase of apoptosis relates to the changes of cell-surface morphology. Not the production of Cer as such, but the depletion of its precursor SM in the plasma membrane is responsible for cholesterol efflux and, thus, major changes in membrane structure and fluidity, which allow membrane blebbing and vesicle formation at the cell surface.
This study does not resolve the controversial issue about a possible existence and the function of rapid Cer formation (in minutes) in apoptosis induction. Despite extensive lipid analyses, using three different assays as well as inhibitors of Cer clearance, we have not been able to detect rapid Cer formation in Jurkat T cells or JY (Epstein-Barr virus–transformed) B cells in response to either CD95 ligation, etoposide, or γ-radiation (Tepper et al. 1997, Tepper et al. 1999; Boesen-de Cock et al. 1998). Yet, it can not be excluded that (in other systems) such a rapid Cer response exists.
Cer Formation Results from a Loss of Plasma Membrane Phospholipid Asymmetry
Our data strongly support the causal relationship between plasma membrane PL scrambling and the hydrolysis of SM to Cer. PL scrambling is both sufficient and necessary for SM hydrolysis: Ca2+-induced scrambling (PS exposure) associates with Cer formation, whereas scrambling-deficient Raji cells fail to hydrolyze SM. The defect in Raji cells that renders them scrambling deficient is unknown. The mere reconstitution of PL scramblase expression by retroviral gene transduction is not sufficient to restore PS exposure in these cells (Fadeel et al. 1999; Tepper, A.D., unpublished data).
Fig. 7 illustrates the sequence of events in the plasma membrane as suggested by our data: apoptotic stimuli or elevated Ca2+ induce the counterbalancing flip-flop (scrambling) of PS and PE, originally residing in the inner leaflet, and SM and PC, originally located in the outer leaflet. Internalized SM becomes accessible to cytosolic SMase, leading to the formation of Cer. This scenario is supported by three pieces of evidence. First, CD95 stimulation induces Cer formation from an SM pool that is accessible to an exogenous SMase. Second, exogenous NBD-SM that, by definition, first inserts into the outer leaflet, is hydrolyzed to NBD-Cer upon an apoptotic stimulus or ionomycin. Thus, NBD-SM has to flip to the inner leaflet to be degraded to NBD-Cer by an SMase inside the cell. NBD-SM internalized otherwise (e.g., by endocytosis) represents a different pool, which is not inducibly hydrolyzed to NBD-Cer. The third piece of evidence is the elevation of intracellular CDP-choline levels in apoptotic cells, reflecting liberation of the SMase product phosphocholine in the cytosol (Table ). Since levels of other choline metabolites remained unchanged, we interpret these data as a compartmentalized (channeled) reaction in which SM-derived phosphocholine is directly converted into CDP-choline by a cytidylyltransferase (Kent 1997). Accumulation of CDP-choline does not occur in normal viable cells, but has been encountered in apoptotic cells, also by others (Anthony et al. 1999), and indicates a block in PC synthesis at the level of CDP-choline:1,2-diacylglycerol cholinephosphotransferase.
Figure 7.

Schematic representation of the proposed mechanism and relevance of Cer formation during the execution phase of apoptosis. In viable cells, SM (red) and PC (blue) localize to the exoplasmic leaflet of the plasma membrane, while PS and PE (white) are sequestered in the inner leaflet. Cholesterol (Chol; green) partitions between both leaflets, but only the preferential clustering with outer leaflet SM is indicated, for reasons of clarity. An apoptotic stimulus or elevated calcium induces loss of the asymmetric phospholipid distribution, and SM appears in the inner leaflet, where it is immediately hydrolyzed to Cer by an intracellular neutral sphingomyelinase (nSMase). Hydrolysis of SM disturbs its tight interaction with Chol, resulting in redistribution of Chol from the plasma membrane towards the cell interior or an extracellular acceptor. Reduced SM and Chol content alters biophysical properties of the lipid bilayer, allowing morphological changes such as membrane blebbing and vesicle shedding.
From our present and previous work (Boesen-de Cock et al. 1998; Tepper et al. 1999), it follows that a neutral SMase activity must be responsible for late Cer formation in Jurkat cells. As argued above and by other authors (Linardic and Hannun 1994; Andrieu et al. 1996; Liu et al. 1998), SMase acts at the cytosolic side of the plasma membrane. However, contrary to results of Andrieu et al. 1996 on tumor necrosis factor α–stimulated fibroblasts, we find that SM hydrolysis inside the cell is tightly linked to preceding transbilayer movement of SM, since depletion of exoleaflet NBD-SM by BSA prevented CD95-induced NBD-Cer formation. Although we can not exclude that an activation step is needed to hydrolyze internalized SM, we did not detect increased in vitro SMase activity in lysates of Jurkat cells after CD95 stimulation or treatment with ionomycin (data not shown). Thus, our data are consistent with the possibility that SM to Cer conversion is solely controlled by substrate availability.
The signal transduction underlying PL scrambling in apoptotic cells remains to be elucidated. PS exposure induced by many different apoptotic stimuli has been shown to require inducer caspase activation (Martin et al. 1996; Vanags et al. 1996; Zhuang et al. 1998; this study). While both inactivation of aminophospholipid translocase and activation of PL scramblase may, in concert, determine PL scrambling, there is no evidence that these enzymes are direct targets for caspases (Verhoven et al. 1999; Tepper, A.D., unpublished data). A role for intracellular Ca2+ as a regulator of PS exposure on apoptotic cells was proposed in a number of studies (Verhoven et al. 1995, Verhoven et al. 1999; Hampton et al. 1996; Bratton et al. 1997; this study). Mitochondrial changes were suggested to act upstream of PS exposure during apoptosis induction (Martin et al. 1995; Zhuang et al. 1998), and recent work has implicated the active participation of mitochondria in cellular Ca2+ signaling (Szalai et al. 1999; Zhu et al. 1999). Therefore, it is tempting to speculate that Ca2+ release from mitochondria plays a role in activation of PL scrambling.
Structural and Morphological Changes at the Plasma Membrane
While our results do not support (but neither exclude) a role for Cer in the apoptotic process, they implicate SM depletion to facilitate membrane blebbing and vesiculation since these features are absent in Raji cells, where PL scrambling and SM hydrolysis do not take place. Consistent with this notion, in Jurkat cells, these morphological changes are prevented when hydrolyzed SM is replenished by NBD-SM. In activated platelets, PS exposure serves to provide a procoagulant surface (for review see Zwaal and Schroit 1997). In these cells, a causal relationship between PL scrambling and the shedding of vesicles (microparticles) from the plasma was earlier suggested by Sims et al. 1989. The onset of NBD-PS exposure at the surface of activated platelets coincided with the onset of plasma membrane vesiculation (Chang et al. 1993), and shed microparticles from erythrocytes were reported to have a more random PL distribution compared with the remnant cells (Comfurius et al. 1990).
We propose the following explanation for the observed relationship between SM breakdown and vesiculation at the plasma membrane. SM preferentially associates with cholesterol, which is a major determinant of the structural order (i.e., reciprocal of fluidity) and integrity of the plasma membrane (van Blitterswijk et al. 1982, van Blitterswijk et al. 1987; Wattenberg and Silbert 1983). Depletion of SM causes redistribution of cholesterol to intracellular sites, and/or it enhances cholesterol efflux to natural or artificial external acceptors, such as serum lipoproteins and MβCD, respectively (Slotte and Bierman 1988, Ohvo et al. 1997; Rothblat et al. 1999; Fig. 7). We found that SM breakdown during apoptosis resulted in an increased efflux of cholesterol, which, conceivably, leads to increased fluidity of the plasma membrane (van Blitterswijk et al. 1987). Such membrane destabilization may facilitate processes like endocytosis from coated pits (Montesano et al. 1979), membrane blebbing (this study), and shedding of vesicles (van Blitterswijk et al. 1982; Chang et al. 1993). Membrane blebbing during apoptosis was suggested to involve contraction of cortical actin, and it was proposed that the bleb protrusions result from focal regions where the plasma membrane is no longer anchored to the cytoskeleton (Mills et al. 1999). Recent findings implicated sphingolipid/cholesterol microdomains (rafts) in the regulation of plasma membrane–cytoskeleton interactions (Oliferenko et al. 1999). It can be envisioned that plasma membrane SM depletion destabilizes raft integrity, and that the disturbance of the interaction between raft proteins and the underlying cytoskeleton contributes to membrane blebbing.
In conclusion, we propose a new mechanism that controls SM hydrolysis to Cer during the execution phase of apoptosis. This mechanism relies on the loss of PL asymmetry in the plasma membrane. While attention has long been focused solely on Cer as a putative apoptotic second messenger, our data introduce a new twist by suggesting that the breakdown of the SM is essential for at least one morphological characteristic of apoptosis, membrane blebbing, and vesicle formation at the cell surface.
Acknowledgments
The authors thank René Raggers (Academic Medical Center, Amsterdam, The Netherlands) for quantitative analysis of fluorescent lipids after TLC separation, and Dr. W. Moolenaar for critical reading of the manuscript.
This work was supported by the Dutch Cancer Foundation (grant No. NKI 96-1266) and the National Institutes of Health (grant No. HL63819).
Footnotes
Abbreviations used in this paper: Cer, ceramide; DEVD-CHO, Ac-Asp-Glu-Val-Asp aldehyde; DiOC6(3), 3,3'-dihexyloxacarbocyanine iodide; FB1, Fumonisin B1; γ-IR, γ-radiation; MβCD, methyl-β-cyclodextrin; NBD, N-[6-[(7-nitro-benz-2-oxa-1,3-diazo-4-yl)amino] caproyl]; NBD-Cer, N-(C6-NBD)-sphingosine; NBD-SM, C6-NBD-sphingosylphosphorylcholine; PC, phosphatidylcholine; PL, phospholipids; PS, phosphatidylserine; SM, sphingomyelin; SMase, sphingomyelinase; zVAD-fmk, N-benzoyloxycarbonyl-Val-Ala-Asp-fluoromethylketone.
References
- Anderson R.G.W. The caveolae membrane system. Annu. Rev. Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- Andrieu N., Salvayre R., Levade T. Comparative study of the metabolic pools of sphingomyelin and phosphatidylcholine sensitive to tumor necrosis factor. Eur. J. Biochem. 1996;236:738–745. doi: 10.1111/j.1432-1033.1996.00738.x. [DOI] [PubMed] [Google Scholar]
- Anthony M.L., Zhao M., Brindle K.M. Inhibition of phosphatidylcholine biosynthesis following induction of apoptosis in HL-60 cells. J. Biol. Chem. 1999;274:19686–19692. doi: 10.1074/jbc.274.28.19686. [DOI] [PubMed] [Google Scholar]
- Boesen-de Cock J.G.R., Tepper A.D., de Vries E., van Blitterswijk W.J., Borst J. CD95 (Fas/APO-1) induces ceramide formation and apoptosis in the absence of a functional acid sphingomyelinase. J. Biol. Chem. 1998;273:7560–7565. doi: 10.1074/jbc.273.13.7560. [DOI] [PubMed] [Google Scholar]
- Bose R., Verheij M., Haimovitz-Friedman A., Scotto K., Fuks Z., Kolesnick R.N. Ceramide synthase mediates daunorubicin-induced apoptosisan alternative mechanism for generating death signals. Cell. 1995;82:405–414. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- Bratton D.L., Fadok V.A., Richter D.A., Kailey J.M., Guthrie L.A., Henson P.M. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J. Biol. Chem. 1997;272:26159–26165. doi: 10.1074/jbc.272.42.26159. [DOI] [PubMed] [Google Scholar]
- Chang C.P., Zhao J., Wiedmer T., Sims P. Contribution of platelet microparticle formation and granule secretion to the transmembrane migration of phosphatidylserine. J. Biol. Chem. 1993;268:7171–7178. [PubMed] [Google Scholar]
- Comfurius P., Senden J.M.G., Tilly R.H.J., Schroit A.J., Bevers E.M., Zwaal R.F.A. Loss of membrane phospholipid asymmetry in platelets and red cells may be associated with calcium-induced shedding of plasma membrane and inhibition of aminophospholipid translocase. Biochim. Biophys. Acta. 1990;1026:153–160. doi: 10.1016/0005-2736(90)90058-v. [DOI] [PubMed] [Google Scholar]
- Fadeel B., Gleiss B., Hogstrand K., Chandra J., Wiedmer T., Sims P.J., Henter J.I., Orrenius S., Samali A. Phosphatidylserine exposure during apoptosis is a cell-type-specific event and does not correlate with plasma membrane phospholipid scramblase expression. Biochim. Biophys. Res. Commun. 1999;266:504–511. doi: 10.1006/bbrc.1999.1820. [DOI] [PubMed] [Google Scholar]
- Fadok V.A., Voelker D.R., Campbell P.A., Cohen J.J., Bratton D.L., Henson P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992;48:2207–2216. [PubMed] [Google Scholar]
- Frey T. Correlated flow cytometric analysis of terminal events in apoptosis reveals the absence of some changes in some model systems. Cytometry. 1997;28:253–263. doi: 10.1002/(sici)1097-0320(19970701)28:3<253::aid-cyto10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M., Peterson E.P., Leiting B., Ruel R., Nicholson D.W., Thornberry N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Hampton M.B., Vanags D.M., Porn-Ares M.I., Orrenius S. Involvement of extracellular calcium in phosphatidylserine exposure during apoptosis. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1996;399:277–282. doi: 10.1016/s0014-5793(96)01341-5. [DOI] [PubMed] [Google Scholar]
- Hannun Y.A. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- Hofmann K., Dixit V.M. Ceramide in apoptosis-does it really matter? Trends Biochem. Sci. 1998;23:374–377. doi: 10.1016/s0968-0004(98)01289-4. [DOI] [PubMed] [Google Scholar]
- Kent C. CTPphosphocholine cytidylyltransferase. Biochim. Biophys. Acta. 1997;1348:79–90. doi: 10.1016/s0005-2760(97)00112-4. [DOI] [PubMed] [Google Scholar]
- Kolesnick R., Hannun Y.A. Ceramide and apoptosis. Trends Biochem. Sci. 1999;24:224–225. doi: 10.1016/s0968-0004(99)01408-5. [DOI] [PubMed] [Google Scholar]
- Koopman G., Reutelingsperger C.P.M., Kuijten G.A.M., Keehnen R.M.J., Pals S.T., Van Oers M.H.J. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- Koval M., Pagano R.E. Sorting of an internalized plasma membrane lipid between recycling and degradative pathways in normal and Niemann-Pick, type A fibroblasts. J. Cell Biol. 1990;111:429–442. doi: 10.1083/jcb.111.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval M., Pagano R.E. Intracellular transport and metabolism of sphingomyelin. Biochim. Biophys. Acta. 1991;1082:113–125. doi: 10.1016/0005-2760(91)90184-j. [DOI] [PubMed] [Google Scholar]
- Levade T., Jaffrézou J.-P. Signalling sphingomyelinaseswhich, where, how and why? Biochim. Biophys. Acta. 1999;1438:1–17. doi: 10.1016/s1388-1981(99)00038-4. [DOI] [PubMed] [Google Scholar]
- Linardic C.M., Hannun Y.A. Identification of a distinct pool of sphingomyelin involved in the sphingomyelin cycle. J. Biol. Chem. 1994;269:23530–23537. [PubMed] [Google Scholar]
- Liu B., Hassler D.F., Smith G.K., Weaver K., Hannun Y.A. Purification and characterization of a membrane bound neutral pH optimum magnesium-dependent and phosphatidylserine-stimulated sphingomyelinase from rat brain. J. Biol. Chem. 1998;273:34472–34479. doi: 10.1074/jbc.273.51.34472. [DOI] [PubMed] [Google Scholar]
- Martin S.J., Reutelingsperger C.P.M., McGahon A.J., Rader J.A., van Scie R.C., LaFace D.M., Green D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the stimulusinhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.J., Finucane D.M., Amarante-Mendes G.P., O'Brien G.A., Green D.R. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J. Biol. Chem. 1996;271:28753–28756. doi: 10.1074/jbc.271.46.28753. [DOI] [PubMed] [Google Scholar]
- Mills J., Stone N.L., Pittman R.N. Extranuclear apoptosisthe role of the cytoplasm in the execution phase. J. Cell Biol. 1999;146:703–707. doi: 10.1083/jcb.146.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesano R., Perrelet A., Vassalli P., Orci L. Absence of filipin-sterol complexes from large coated pits on the surface of culture cells. Proc. Natl. Acad. Sci. USA. 1979;76:6391–6395. doi: 10.1073/pnas.76.12.6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld E.B., Cooney A.M., Pitha J., Dawidowicz E.A., Dwyer N.K., Pentchev P.G., Blanchette-Mackie E.J. Intracellular trafficking of cholesterol monitored with a cyclodextrin. J. Biol. Chem. 1996;271:21604–21613. doi: 10.1074/jbc.271.35.21604. [DOI] [PubMed] [Google Scholar]
- Nicoletti I., Migliorati G., Pagliacci M.C., Grignani F., Riccardi C.A. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. 1991;Methods. 139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- Ohvo H., Olsio C., Slotte J.P. Effects of sphingomyelin and phosphatidylcholine degradation on cyclodextrin-mediated cholesterol efflux in cultured fibroblasts. Biochim. Biophys. Acta. 1997;1349:131–141. doi: 10.1016/s0005-2760(97)00126-4. [DOI] [PubMed] [Google Scholar]
- Oliferenko S., Paiha K., Harder T., Gerke V., Schwarzler C., Schwarz H., Beug H., Gunthert U., Huber L.A. Analysis of CD44-containing lipid raftsrecruitment of annexin II and stabilization by the actin cytoskeleton. J. Cell Biol. 1999;146:843–854. doi: 10.1083/jcb.146.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano R.E. A fluorescent derivative of ceramidephysical properties and use in studying the Golgi apparatus of animal cells. Methods Cell Biol. 1989;29:75–85. doi: 10.1016/s0091-679x(08)60188-0. [DOI] [PubMed] [Google Scholar]
- Rothblat G.H., de la Llera-Moya M., Atger V., Kellner-Weibel G., Williams D.L., Phillipis M.C. Cell cholesterol effluxintegration of old and new observations provides new insights. J. Lipid Res. 1999;40:781–796. [PubMed] [Google Scholar]
- Schroit A.J., Madsen J.W., Tanaka Y. In vivo recognition and clearance of red blood cells containing phosphatidylserine in their plasma membranes. J. Biol. Chem. 1985;260:5131–5138. [PubMed] [Google Scholar]
- Schroeder J.J., Crane H.N., Xia J., Liotta D.C., Merrill A.H., Jr. Disruption of sphingolipid metabolism and stimulation of DNA synthesis by fumonisin B1. A molecular mechanism for carcinogenesis associated with Fusarium moniliforme . J. Biol. Chem. 1994;269:3475–3481. [PubMed] [Google Scholar]
- Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Sims P.J., Wiedmer T., Esmon C.T., Weiss H.J., Shattil S.J. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott syndromean isolated defect in platelet procoagulant activity. J. Biol. Chem. 1989;264:17049–17057. [PubMed] [Google Scholar]
- Slotte J.P., Bierman E.L. Depletion of plasma-membrane sphingomyelin rapidly alters the distribution of cholesterol between plasma membrane and intracellular cholesterol pools. Biochem. J. 1988;250:653–658. doi: 10.1042/bj2500653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets E.F., Comfurius P., Bevers E.M., Zwaal R.F.A. Calcium-induced transbilayer scrambling of fluorescent phospholipid analogs in platelets and erythrocytes. Biochim. Biophys. Acta. 1994;1195:281–286. doi: 10.1016/0005-2736(94)90268-2. [DOI] [PubMed] [Google Scholar]
- Szalai G., Krishnamurthy R., Hajnóczky G. Apoptosis driven by IP3-linked mitochondrial calcium signals EMBO (Eur . Mol. Biol. Organ.) J. 1999;18:6349–6361. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper A.D., Boesen-de Cock J.G.R., de Vries E., Borst J., van Blitterswijk W.J. CD95/Fas-induced ceramide formation proceeds with slow kinetics and is not blocked by caspase-3/CPP32 inhibition. J. Biol. Chem. 1997;272:24308–24312. doi: 10.1074/jbc.272.39.24308. [DOI] [PubMed] [Google Scholar]
- Tepper A.D., de Vries E., van Blitterswijk W.J., Borst J. Ordering of ceramide formation, caspase activation and mitochondrial changes during CD95- and DNA-damage-induced apoptosis. J. Clin. Investig. 1999;103:971–978. doi: 10.1172/JCI5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry N.A., Rano T.A., Peterson E.P., Rasper D.M., Timkey T., Garcia-Calvo M., Houtzager V., Nordstrom P.A., Roy S., Vallaincourt J.P., Chapman K.T., Nicholson D.W. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- Vanags D.M., Porn-Ares M.I., Coppola S., Burgess D.H., Orrenius S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J. Biol. Chem. 1996;271:31075–31085. doi: 10.1074/jbc.271.49.31075. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk W.J., de Veer G., Krol J.H., Emmelot P. Comparative lipid analysis of purified plasma membranes and shed extracellular membrane vesicles from normal murine thymocytes and leukemic GRSL cells. Biochim. Biophys. Acta. 1982;688:495–504. doi: 10.1016/0005-2736(82)90361-3. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk W.J., van der Meer B.W., Hilkmann H. Quantitative contributions of cholesterol and the individual classes of phospholipids and their degree of fatty acyl (un)saturation to membrane fluidity measured by fluorescence polarization. Biochemistry. 1987;26:1746–1756. doi: 10.1021/bi00380a038. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk W.J., Hilkmann H., de Widt J., van der Bend R.L. Phospholipid metabolism in bradykinin-stimulated human fibroblasts. J. Biol. Chem. 1991;266:10344–10350. [PubMed] [Google Scholar]
- Van Meer G., Stelzer E.H.K., Wijnaendts-van-Resandt R.W., Simons K. Sorting of sphingolipids in epithelial (Madin-Darby canine kidney) cells. J. Cell Biol. 1987;91:872–877. doi: 10.1083/jcb.105.4.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoven B., Schlegel R.A., Williamson P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal on apoptotic T lymphocytes. J. Exp. Med. 1995;182:1597–1601. doi: 10.1084/jem.182.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoven B., Krahling S., Schlegel R.A., Williamson P. Regulation of phosphatidylserine exposure and phagocytosis of apoptotic T lymphocytes. Cell Death Differ. 1999;6:262–270. doi: 10.1038/sj.cdd.4400491. [DOI] [PubMed] [Google Scholar]
- Wattenberg B.W., Silbert D.F. Sterol partitioning among intracellular membranes. J. Biol. Chem. 1983;258:2284–2289. [PubMed] [Google Scholar]
- Williamson P., Kulick A., Zachowski A., Schlegel R.A., Devaux P.F. Ca2+ induces transbilayer redistribution of all major phospholipids in human erythrocytes. Biochemistry. 1992;31:6355–6360. doi: 10.1021/bi00142a027. [DOI] [PubMed] [Google Scholar]
- Yancey P.G., Rodrigueza W.V., Kilsdonk E.P.C., Stoudt G.W., Johnson W.J., Phillips M.C., Rothblat G.H. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration of kinetic pools and mechanism of efflux. J. Biol. Chem. 1996;271:16026–16034. doi: 10.1074/jbc.271.27.16026. [DOI] [PubMed] [Google Scholar]
- Yssel H., de Vries J.E., Koken M., van Blitterswijk W.J., Spits H. Serum-free medium for the generation and the propagation of functional human cytotoxic and helper T cell clones. J. Immunol. Methods. 1984;72:219–227. doi: 10.1016/0022-1759(84)90450-2. [DOI] [PubMed] [Google Scholar]
- Zachowski A. Phospholipids in animal eukaryotic membranestransverse asymmetry and movement. Biochem. J. 1993;294:1–14. doi: 10.1042/bj2940001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N., Susin S.A., Marchetti P., Hirsch T., Gomez-Monterrey I., Castedo M., Kroemer G. Mitochondrial control of apoptosis. J. Exp. Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Zhou Q., Wiedmer T., Sims P.J. Level of expression of phospholipid scramblase regulates induced movement of phosphatidylserine to the cell surface. J. Biol. Chem. 1998;273:6603–6606. doi: 10.1074/jbc.273.12.6603. [DOI] [PubMed] [Google Scholar]
- Zhou Q., Zhao J., Stout J.G., Luhm R.A., Wiedmer T., Sims P.J. Molecular cloning of human plasma membrane phospholipid scramblase. J. Biol. Chem. 1997;272:18240–18244. doi: 10.1074/jbc.272.29.18240. [DOI] [PubMed] [Google Scholar]
- Zhou Q., Sims P.J., Wiedmer T. Identity of a conserved motif in phospholipid scramblase that is required for Ca2+ accelerated transbilayer movement of membrane phospholipids. Biochemistry. 1998;37:2356–2360. doi: 10.1021/bi972625o. [DOI] [PubMed] [Google Scholar]
- Zhu L., Ling S., Yu X.-D., Venkatesch L.K., Subramanian T., Chinnadural G., Kuo T.H. Modulation of mitochondrial Ca2+ homeostasis by Bcl-2. J. Biol. Chem. 1999;274:33267–33273. doi: 10.1074/jbc.274.47.33267. [DOI] [PubMed] [Google Scholar]
- Zhuang J., Ren Y., Snowden R.T., Zhu H., Gogvadze V., Savill J.S., Cohen G.M. Dissociation of phagocyte recognition of cells undergoing apoptosis from other features of the apoptotic program. J. Biol. Chem. 1998;273:15628–15632. doi: 10.1074/jbc.273.25.15628. [DOI] [PubMed] [Google Scholar]
- Zwaal R.F.A., Schroit A.J. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]