

Abstract
The retinoblastoma (Rb) tumor suppressor controls cellular proliferation, survival, and differentiation and is functionally inactivated by mutations or hyperphosphorylation in most human cancers. Although activation of endogenous Rb is thought to provide an effective approach to suppress cell proliferation, long-term inhibition of apoptosis by active Rb may have detrimental consequences in vivo. To directly test these paradigms, we targeted phosphorylation-resistant constitutively active Rb alleles, RbΔKs, to the mouse mammary gland. Pubescent transgenic females displayed reduced ductal elongation and cell proliferation at the endbuds. Postpuberty transgenic mice exhibited precocious cellular differentiation and β-casein expression and extended survival of the mammary epithelium with a moderate but specific effect on the expression of E2F1, IGF1Rα, and phospho–protein kinase B/AKT. Remarkably, ∼30% RbΔK transgenic females developed focal hyperplastic nodules, and ∼7% exhibited full-blown mammary adenocarcinomas within 15 mo. Expression of the RbΔK transgene in these mammary tumors was reduced greatly. Our results suggest that transient activation of Rb induces cancer by extending cell survival and that the dual effects of Rb on cell proliferation and apoptosis impose an inherent caveat to the use of the Rb pathway for long-term cancer therapy.
Keywords: Rb; breast cancer; mammary gland; apoptosis; transgenic mice
Introduction
The retinoblastoma (Rb)* tumor suppressor exerts diverse effects on cell growth and differentiation by modulating the activity of transcription factors such as members of the E2F protein family (Dyson, 1998). Rb itself is regulated at the phosphorylation level by G1 cyclins, their associated cyclin-dependent kinases (Cdks), and specific inhibitors (e.g., p16Ink4a) (Sherr, 2000). During most of the G1 phase of the cell cycle, hypophosphorylated Rb interacts with E2F on cognate binding sites and actively represses transcription of genes required for cell cycle progression and DNA replication. Active repression involves, at least in part, the recruitment by Rb of chromatin-modifying enzymes such as the histone deacetylase HDAC1 (Lai et al., 1999; Harbour and Dean, 2000a). Mitotic signals propagated through G1 cyclins induce sequential phosphorylation and inactivation of Rb (Lundberg and Weinberg, 1998). Early in G1, Cdk4/6–cyclin D1 phosphorylates Rb at phosphoacceptor sites just downstream of the Rb pocket domain, which is involved in protein–protein interaction (Harbour et al., 1999). Phosphorylation of these sites induces intramolecular interaction between the negatively charged phosphate groups and basic residues in the pocket. This intramolecular interaction induces a conformation change that expels HDAC1 from Rb, thereby relieving active transcriptional repression. Later in G1, Cdk2–cyclin E phosphorylates Rb at Serine-567, resulting in dissociation of Rb from E2F, transcriptional depression of target genes, and cell cycle progression.
In addition to keeping the cell cycle in check, Rb also controls cell differentiation and survival. There is evidence that Rb can bind a plethora of differentiation factors and cooperatively induce terminal differentiation and transcriptional activation of differentiation genes in vitro (Macleod, 1999; DiCiommo et al., 2000). In addition, Rb mutant embryos exhibit incomplete differentiation in certain tissues and aberrant expression of specific markers, for example, NGF receptors during neurogenesis (Lee et al., 1994), muscle creatine kinase during myogenesis (Zacksenhaus et al., 1996b), and filensin/crystallin γB during lens development (Liu and Zacksenhaus, 2000).
A role for Rb as a survival factor is evident from the massive cell death observed in Rb-deficient mice in tissues where Rb is normally highly expressed (Lee et al., 1994; Zacksenhaus et al., 1996b; Jiang et al., 1997; Macleod, 1999). Furthermore, in vitro, loss of Rb increases cell susceptibility to cytotoxic drugs, whereas ectopic expression of Rb inhibits cell death (Almasan et al., 1995; Haas-Kogan et al., 1995; Kranenburg et al., 1996). The dual effects of Rb on cell proliferation and apoptosis are mediated in some cell types by E2F1, which controls the expression of genes required for cell cycle progression and cell death. For example, deregulated E2F1 can induce the transcription of apoptosis protease-activating factor (Apaf)-1 (Moroni et al., 2001) and p14/19ARF, which binds and inactivates MDM2, a p53 inhibitor, thereby linking loss of Rb to p53-induced apoptosis (Sherr, 2000). Consistent with these in vitro studies, apoptosis in Rb-deficient mice is mediated in certain tissues by an E2F1–p53-Apaf-1–dependent pathway (Morgenbesser et al., 1994; Macleod et al., 1996; Tsai et al., 1998; Guo et al., 2001).
Although germ-line mutations in Rb predispose individuals to retinoblastoma, somatic mutations in Rb are often associated with cancer progression (DiCiommo et al., 2000). However, in many types of cancer, such as breast cancer (Buckley et al., 1993), other components of the Rb pathway (i.e., Cdk4, cyclin D1, or p16Ink4a) are mutated preferentially or deregulated (Weinberg, 1995). In such tumors, Rb becomes hyperphosphorylated and inactive, but the protein is intact and amenable to therapeutical activation. Reactivation of endogenous Rb in such tumors (e.g., by Cdk inhibitors) may provide an effective approach to suppress cell proliferation. In support of this notion, introduction of phosphorylation-resistant constitutively active Rb alleles, RbΔKs, into many cell types in vitro (Mittnacht, 1998) and in vivo (Chang et al., 1995) efficiently suppresses cell proliferation. Moreover, several Cdk4/6 inhibitors that target the Rb pathway are currently in preclinical and clinical development (Bange et al., 2001).
However, the inherent ability of Rb to suppress both cell division and apoptosis points to a potential caveat. Activation of Rb may inadvertently protect cells, which have acquired oncogenic alterations or are programmed to die in response to normal developmental signals, from apoptosis and lead to detrimental consequences in vivo. Of relevance here is the observation that both overexpression and inactivation of E2F1 induce cancer in mouse models presumably by disrupting different aspects (proliferation versus apoptosis, respectively) of cell physiology (Field et al., 1996; Yamasaki et al., 1996; Pan et al., 1998; Pierce et al., 1998, 1999).
To directly address the consequences of activating Rb in vivo, we have targeted RbΔK alleles to the mammary gland of transgenic mice. We show that transgenic females initially display ductal growth suppression but later exhibit precocious differentiation, extended survival of the mammary epithelium, and ultimately develop hyperplastic lesions and mammary adenocarcinomas. The implications of these results to cancer therapy are discussed.
Results
Targeted expression of phosphorylation-resistant Rb alleles in the mammary gland
The interaction of Rb with transcription factors is dictated by cyclin D– and cyclin E–associated kinases that phosphorylate Rb in a cell cycle–dependent manner (Lundberg and Weinberg, 1998; Mittnacht, 1998). Rb contains 16 potential phosphoacceptor sites that are distributed along the protein (Lundberg and Weinberg, 1998; Mittnacht, 1998) (Fig. 1 A). Recent evidence indicates that a cluster of serine and threonine in exon 23 of Rb just downstream of the pocket plays a major role in regulating Rb activity (Harbour et al., 1999). In our study, we used two RbΔK alleles: RbΔp34 contains mutations in eight Cdk sites, four of which are within exon 23 (Hamel et al., 1992) (Fig. 1 A). Using RbΔp34 as a backbone, we generated three additional serine/threonine to alanine substitutions in exon 23, yielding RbΔK11, in which all phosphoacceptor sites in exon 23 are protected completely from Cdk-mediated phosphorylation (see Materials and methods) (Fig. 1 A). Due to their resistance to phosphorylation by Cdks, the RbΔKs outperform wild-type Rb in multiple assays and are viewed as constitutively active, underphosphorylated, native Rb (Hamel et al., 1992; Geng et al., 1996; Knudsen et al., 1998; Sellers et al., 1998; Brown et al., 1999; Harbour et al., 1999). To determine the effects of constitutive activation of Rb on mammary gland development and cancer progression, the two RbΔK alleles were targeted to the mammary gland under control of the mouse mammary tumor virus (MMTV) long terminal repeat (Matsui et al., 1990; Fig. 1 B). Four independent MMTV-RbΔp34 transgenic lines in mixed C57BL/6×SJL background and three MMTV-RbΔK11 lines in pure FVB background were established (Fig. 1 and Table I).
Figure 1.
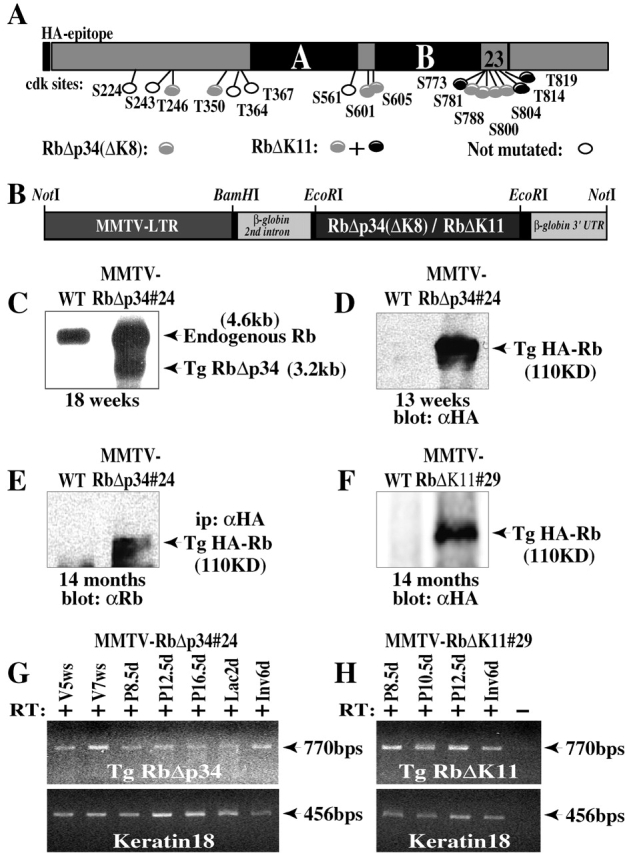
Targeted expression of constitutively active Rb to the mammary gland. (A) Schematic structure of Rb showing the pocket domain (A and B), relative location of 16 Cdk phosphoacceptor sites, and mutated residues in RbΔp34 and RbΔK11. (B) Schematic presentation of the MMTV-RbΔp34 and MMTV-RbΔK11 transgenes. (C) Detection of RbΔK transgene by Northern blot analysis. The transgene has a shorter 3′ UTR and migrates faster. (D and F) Western blots of protein lysates (50 μg) from mammary glands of (D) 13-wk-old or (F) 14-mo-old transgenic and wild-type females were developed with anti-HA monoclonal antibody. (E) Protein lysates (1.2 mg) from 14-mo-old transgenic and control mammary glands were immunoprecipitated with anti-HA antibodies and Western blotted with anti-Rb antibody. (G and H) RT-PCR analysis of the MMTV-RbΔp34#24 and MMTV-RbΔK11#29 transgenes relative to keratin 18 in virgin (V), pregnant (P), lactating (Lac), and involuting (Inv) glands. w, weeks. + and − signs indicate the presence or absence of reverse transcriptase.
Table I. Incidence of focal hyperplastic nodules and adenocarcinomas in transgenic mice expressing constitutively active Rb in the mammary gland.
| Transgenic line | Number of mammary glands analyzedb |
Number of mammary glands with tumorsc |
Incidence per line |
Incidence combined |
|
|---|---|---|---|---|---|
| %
|
|||||
| MMTV-RbΔp34(K8) | |||||
| #24 | 38 | 8d | 21 | ||
| #16 | 7 | 4 | 57 | ||
| #8 | 16 | 1 | 6.25 | ||
| #27 | 2 | 1d | 50 | ||
| Total | 63 | 14 | 22 | ||
| Control littermatesa | 50 | 0 | 0 | 0 | |
| MMTV-RbΔK11 | |||||
| #29 | 18 | 43 d | 22 | 22 | |
| Control littermatesa | 7 | 0 | 0 | 0 | |
| WAP-RbΔp34(K8) | |||||
| #9 | 7 | 2 | 28.6 | ||
| #44 | 5 | 4d | 80 | ||
| #61 | 9 | 2d | 22.2 | ||
| Total | 21 | 8 | 38 | ||
| Control littermatesa | 21 | 0 | 0 | 0 | |
Control littermates were caged together with transgenic females and analyzed in parallel.
Focal hyperplastic nodules were detected by whole-mount staining of the right inguinal mammary glands of 10–15-mo-old females. Palpable adenocarcinomas were confirmed by histopathology.
Numbers include both hyperplastic nodules and adenocarcinomas.
Indicates one full-blown mammary adenocarcinoma in this group.
Transgenic transcripts were found in the mammary gland and the salivary gland and spleen but not in the brain, kidney, and liver (Fig. 1, C, G, and H; data not shown). The HA-tagged RbΔK transgenic protein was readily detected by immunoprecipitations and Western blots with antibodies specific to the HA epitope (Fig. 1, D–F). However, we have been unable so far to detect the transgenes by immunohistochemistry (IHC) with our anti-HA antibodies. Reverse transcribed (RT)-PCR analysis showed that the MMTV-RbΔp34 and MMTV-RbΔK11 transgenes (collectively referred to as MMTV-RbΔK) were expressed throughout mammary gland development, pregnancy, lactation, and involution (Fig. 1, G and H).
Regulated expression of endogenous Rb during mammogenesis in wild-type mice
To relate the expression of the RbΔK transgenes to endogenous Rb, we performed in situ hybridization and IHC analyses on wild-type mammary glands (Fig. 2). Both methods of detection revealed that endogenous Rb expression was below detection level or sporadic in nulliparous females, induced at mid-pregnancy, and peaked at lactation and involution (Fig. 2). Thus, compared with endogenous Rb the RbΔK transgenes are expressed at a relatively high level in nulliparous females (Fig. 1, G and H). Accordingly, we observed the strongest phenotypes in nulliparous MMTV-RbΔK transgenic mice as described below.
Figure 2.
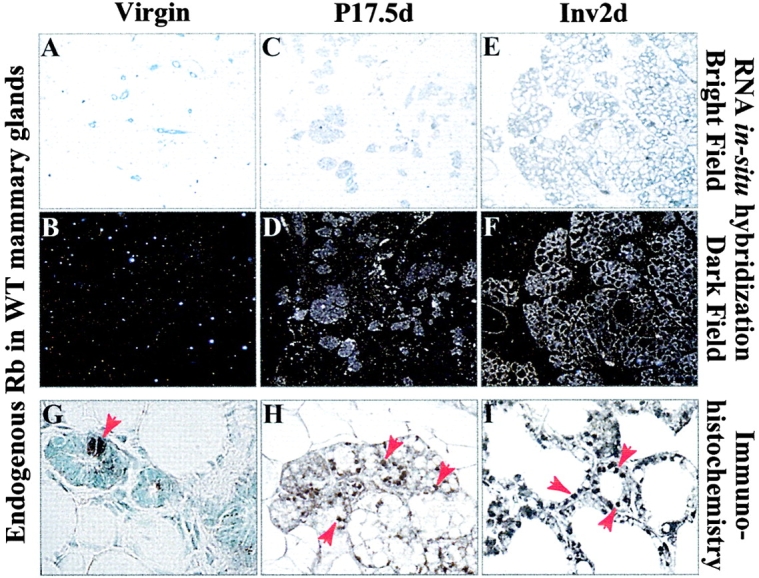
Temporal expression of endogenous Rb during mammogenesis in wild-type females. (A–F) RNA In situ hybridization analysis of endogenous Rb. Shown are bright field (A, C, and E) and dark field (B, D, and F) micrographs of representative sections. (G–I) IHC of endogenous Rb in wild-type mammary glands. Arrows indicate Rb-positive nuclei.
MMTV-RbΔK transgenes suppress ductal growth in pubescent females
During the ductal stage, the mammary epithelium infiltrates the fat pad by proliferation at the terminal endbuds (TEBs) and dichotomous side branching (Medina, 1996; Hennighausen and Robinson, 1998; Cardiff et al., 2000; Silberstein, 2001). Whole-mount analysis of pubescent MMTV-RbΔK transgenic females revealed a moderate delay in TEB progression (Fig. 3, A–D). The suppression of ductal growth coincided with reduced expression of proliferation cell nuclear antigen (PCNA), a marker for cell proliferation (Fig. 3, F–I). On average, MMTV-RbΔK TEBs exhibited three- to fourfold less PCNA-positive nuclei compared with control wild-type littermates (Fig. 3 J). The suppression of ductal growth was most evident at 5–7 wk of age and reduced thereafter (Fig. 3 E). Accordingly, there were no obvious histological differences between the mammary glands of transgenic and wild-type littermates during pregnancy and lactation, and transgenic females were able to nurse their young and raise normal size litters (unpublished data).
Figure 3.
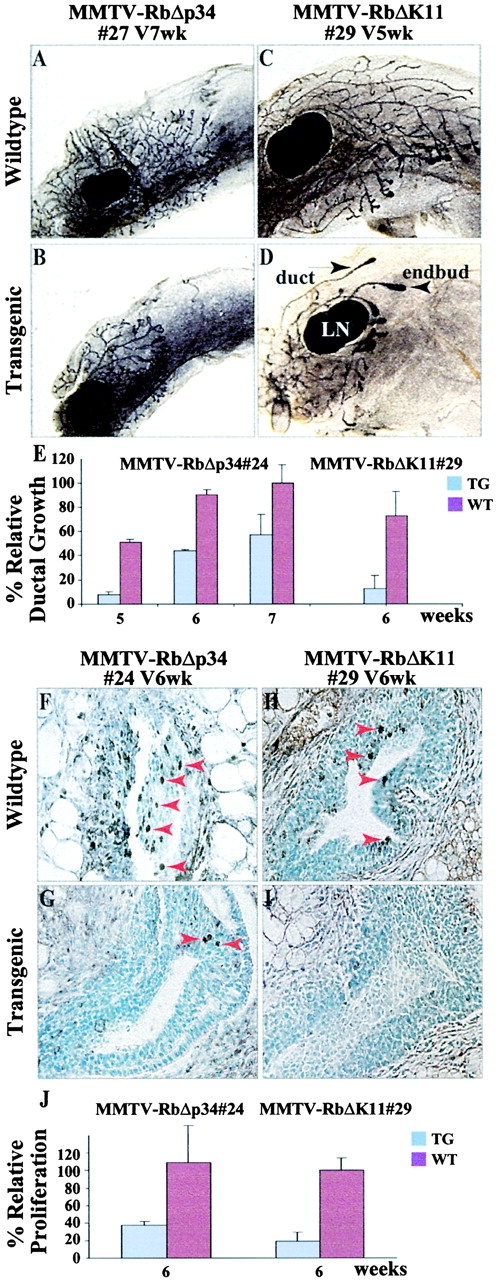
Suppression of ductal growth in pubescent MMTV-RbΔK transgenic mice. Whole-mount staining of 5- and 7-wk-old pubescent MMTV-RbΔp34#27 (A and B) and MMTV-RbΔK11#29 (C and D) transgenic and control females. LN, lymph node. (E) The distance from the endbuds to the lymph nodes in 7-wk-old wild-type females was set at 100%. The average growth rates of endbuds in three transgenic females and three wild-type littermates were plotted against this value for each time point. PCNA staining of sections through endbuds of 6-wk-old MMTV-RbΔp34#24 (F and G) and MMTV-RbΔK11#29 (H and I) transgenic females and control wild-type littermates. Arrows indicate PCNA-positive nuclei. (J) Quantitative analysis of the relative proliferation (PCNA-positive nuclei) in TEBs at the indicated stages. For each time point, two sections each from three transgenic and three wild-type mice were analyzed. The average number of PCNA-positive nuclei in the control wild-type females for MMTV-RbΔK11 was set at 100%. Bars in E and J indicate standard deviation.
MMTV-RbΔK transgenes induce alveolar outgrowth and precocious differentiation in nulliparous females
After puberty, the mammary epithelium undergoes cyclic proliferation, limited differentiation, and cell death during the estrous cycle (Robinson et al., 1995, 2000). Interestingly, we found that fully developed nulliparous MMTV-RbΔK transgenic females exhibited consistent alveolar outgrowth relative to control littermates (Fig. 4, A and B). PCNA-staining revealed very few cycling cells in both wild-type and transgenic mice (Fig. 4, C and D), indicating that enlargement of the alveolar compartment was not due to overt cellular proliferation. To test whether the alveolar outgrowth was indicative of precocious differentiation of the mammary epithelium, we determined the expression of β-casein, a milk protein. Remarkably, β-casein transcripts were detected by RT-PCR in ∼28% (10 of 35) of 9–20-wk-old nulliparous transgenic females but only rarely and at lower levels in wild-type littermates (Fig. 4 Q). Furthermore, IHC and Western blot analysis revealed that this milk protein was expressed in some nulliparous transgenic but in none of the wild-type females (Fig. 4, E, F, and R).
Figure 4.
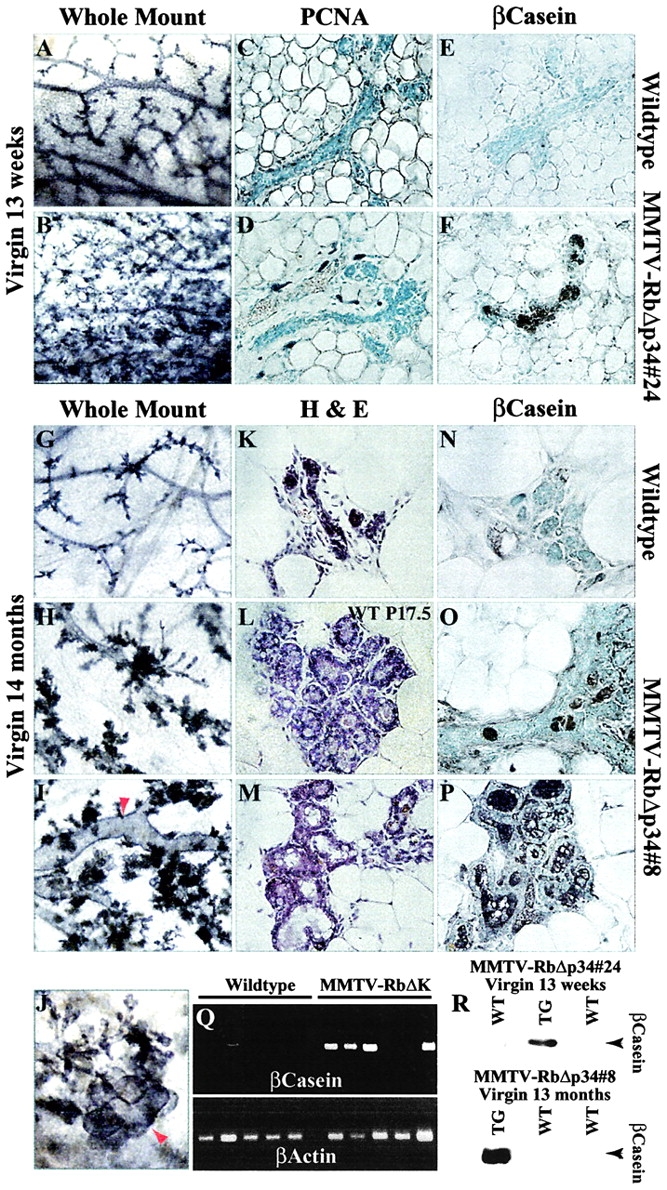
Precocious differentiation of the mammary epithelium in nulliparous MMTV-RbΔK transgenic females. Whole-mount analyses of 13-wk-old nulliparous transgenic and control females (A and B); PCNA staining (C and D); IHC for β-casein (E and F). Whole-mount hematoxylin staining of 14-mo-old control (G) and two nulliparous transgenic (H and I) females (40×). Note the dilated ducts (arrow). (J) Higher magnification (70×) of the mammary gland in I, showing a lobuloalveolar-like structure (arrow). (K and M) H-E staining of mammary glands of 14-mo-old transgenic (M) and wild-type (K) littermate female. (L) H-E staining of a mammary gland from a 17.5-d-old pregnant wild-type female (P17.5). Note the similarity between nulliparous transgenic (M) and pregnant wild-type glands (L). (N–P) IHC of β-casein in mammary glands of 14-mo-old nulliparous wild-type (N) and two transgenic (O and P) females. (Q) RT-PCR analysis of β-casein expression in 9–20-wk-old nulliparous transgenic and control females. (R) Western blot analysis of β-casein expression in transgenic (TG) and wild-type (WT) females.
The alveolar outgrowth and precocious expression of β-casein became significantly more pronounced in 12–14-mo-old nulliparous MMTV-RbΔK mice. Some transgenic females exhibited lactating-like glands that appeared distended with milk (unpublished data). Western blot analysis revealed a high level of β-casein in these glands (Fig. 4 R). Upon histological examination, some RbΔK transgenic females exhibited hyperplastic alveoli, dilated ducts, and enlarged lobules that resembled secretory lobuloalveoli in pregnant females with characteristic vacuoles and acinar morphology (Fig. 4, G–M). IHC analysis demonstrated high levels of β-casein expression in the mammary gland of four out of five transgenic females but not in control littermates (Fig. 4, N–P). RT-PCR analysis of another group of three 18-mo-old nulliparous transgenic and three control females revealed β-casein expression exclusively in the transgenic females; whey acidic protein (WAP), a later differentiation marker, was detected in two of the three transgenic mice (unpublished data). Thus, persistent expression of the RbΔK transgenes in nulliparous transgenic females induces precocious differentiation of the mammary epithelium both at the molecular and cellular levels.
MMTV-RbΔK transgenic females develop hyperplastic nodules
Intriguingly, whole-mount analysis of 10–15-mo-old MMTV-RbΔK transgenic females revealed the presence of focal hyperplastic nodules (Fig. 5). These nodules appeared in 22% of the transgenic females (18 out of 81) from independent MMTV-RbΔp34 and MMTV-RbΔK11 lines (Table I). Both nulliparous and multiparous transgenic females developed these lesions, indicating that pregnancy was not required for neoplastic transformation. In many cases there were multifocal lesions per gland (Fig. 5, B, E, and F), suggesting independent stochastic transformation of the mammary epithelium. The incidence of hyperplastic nodules may be underestimated because only one mammary gland per animal (of the ten glands in a female mouse) was subject to whole-mount analysis. No such lesions were observed in over 57 wild-type littermate females (Fig. 5, A and C; Table I).
Figure 5.
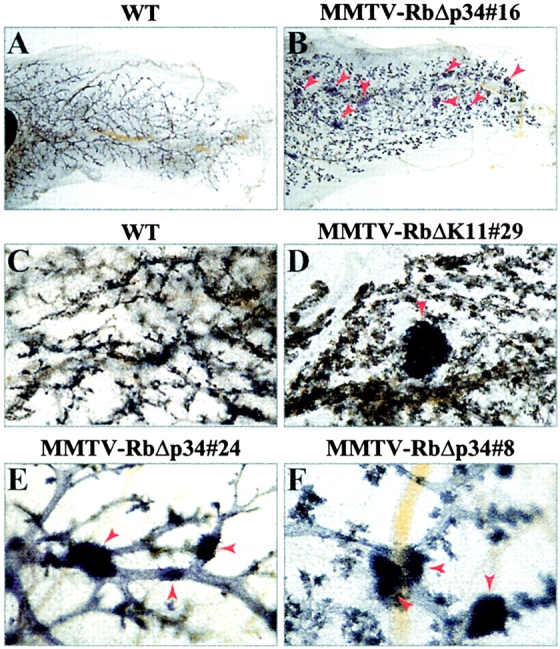
Hyperplastic nodules in the mammary gland of MMTV-RbΔK transgenic mice. Low power views (8×) of whole-mount staining of #4 right inguinal mammary glands from 13.5-mo-old nulliparous wild-type female (A) and MMTV-RbΔp34#16 transgenic littermate (B). Arrows point to hyperplastic nodules. (C and D) 13.5-mo-old multiparous wild-type and MMTV-RbΔK11#29 transgenic littermate gland with a hyperplastic nodule. (E and F) High power views of representative hyperplastic nodules (arrows) in independent MMTV-RbΔp34 transgenic lines.
WAP-RbΔp34 transgenic females also develop hyperplastic nodules
To further corroborate these observations, we targeted RbΔp34 to the mammary gland under control of the WAP promoter/3′ UTR (Fig. 6 A). The WAP regulatory unit directs linked transgenes exclusively to differentiated mammary epithelial cells during the estrous cycle and pregnancy, and WAP transgenes are expressed and exert phenotypic changes in both nulliparous and multiparous females (Jhappan et al., 1993; Sympson et al., 1994; Robinson et al., 1995). Three WAP-RbΔp34 transgenic lines were established in C57BL/6×SJL background. Expression of the WAP-RbΔp34 transgenes was low in all three lines, and transcripts could only be detected during pregnancy (Fig. 6 B). Further analysis revealed normal ductal elongation, pregnancy, and lactation in WAP-RbΔp34 transgenic females (unpublished data). However, remarkably 38% (8 of 21) of the transgenic females from the three independent WAP-RbΔp34 lines developed focal hyperplastic nodules within 10–15 mo (Fig. 6, D and E; Table I). As with the MMTV-RbΔK lines, both nulliparous and parous females developed these micro lesions. No similar lesions were found in >21 wild-type littermate females (Fig. 6 C; Table I). Thus, targeted activation of Rb by two independent promoters, MMTV and WAP, induces hyperplastic lesions in the mammary epithelium.
Figure 6.
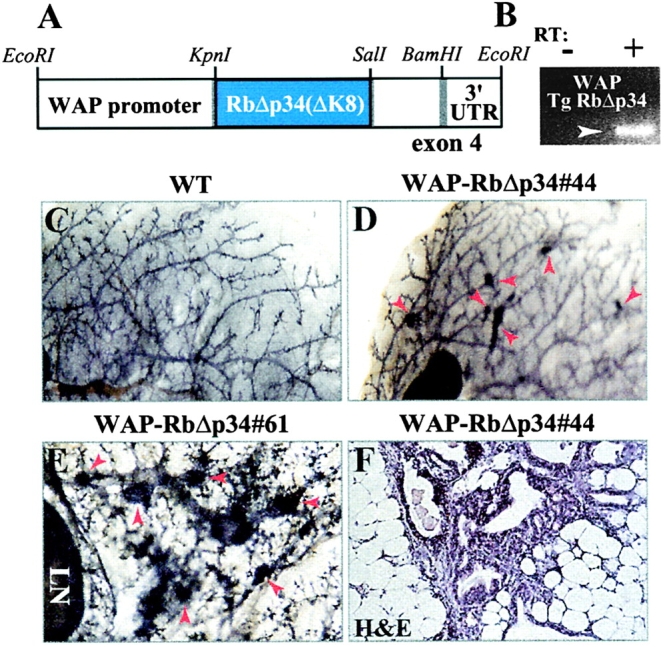
WAP-RbΔp34 transgenic mice also develop hyperplastic nodules. (A) Schematic structure of the WAP-RbΔp34 transgene. (B) RT-PCR analysis of WAP-RbΔp34 expression in an 18.5-d-old pregnant transgenic female. (C and D) Whole-mount analysis of 12-mo-old nulliparous wild-type (C) and WAP-RbΔp34#44 transgenic (D) glands (8×). (E) Representative high power view of hyperplastic nodules in a WAP-RbΔp34#61 transgenic female (40×). (F) H-E staining of cross-section through a hyperplastic nodule. Arrows in D and E point to hyperplastic nodules.
MMTV-RbΔK and WAP-RbΔp34 transgenic females develop full-blown mammary adenocarcinomas
A fraction (7 in 102 is ∼7%) of the MMTV-RbΔp34, MMTV-RbΔK11, and WAP-RbΔp34 transgenic females developed visible masses of 1–2 cm in diameter in their mammary glands after 10–15 mo (Table I). Pathology analysis revealed that these masses represented full-blown mammary adenocarcinomas (Fig. 7; see legend for pathology). None of >100 wild-type littermate females in our colony developed mammary tumors (Table I). The incidence of tumors was highest in the MMTV-RbΔK11#29 line (Table I and unpublished data). We have analyzed recently two retired MMTV-RbΔK11#29 transgenic and two wild-type breeders at 20 mo of age (not included in Table I). Both transgenic females but none of the control mice developed palpable tumors in several mammary glands. Histological analysis revealed that each of these mammary glands contained one to four tumors. Fig. 7 G shows one such gland with a small tumor (Fig. 7 G, letter H) and several additional lesions (Fig. 7 G, letter I–K). Taken together, the results indicate that some hyperplastic nodules in RbΔK transgenic mice progress into full-fledged mammary adenocarcinomas.
Figure 7.
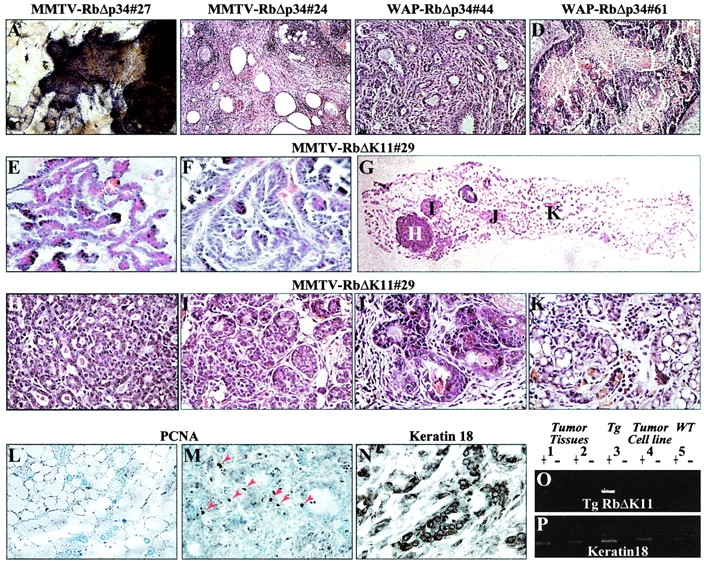
Mammary adenocarcinomas in MMTV-RbΔK and WAP-RbΔp34 transgenic mice and loss of transgene expression. Histology of mammary adenocarcinomas. (A) Whole-mount staining of a palpable mammary adenocarcinoma in a 13.5-mo-old nulliparous MMTV-RbΔp34#27 transgenic female, showing the periphery of a tumor with invasion into the ducts. (B) Mammary adenocarcinoma with multiple cystic areas in a 12-mo-old multiparous MMTV-RbΔp34#24 transgenic female. (C) Well-differentiated tubulo-acinar mammary adenocarcinoma in a 12-mo-old nulliparous WAP-RbΔp34#44 transgenic female. (D) Well-differentiated papillary mammary adenocarcinoma with peripheral growth of basaloid cells and radiating keratinization in a 14-mo-old multiparous WAP-RbΔp34#61 transgenic female. (E and F) Papillary mammary adenocarcinoma and mammary adenocarcinoma in the first and third glands, respectively, from a multiparous 18-mo-old MMTV-RbΔK11#29 transgenic female. (G) Multiple lesions in an MMTV-RbΔK11#29 retired breeder. (H and K) High power magnifications of the lesions shown in (G). H–J are mammary adenocarcinomas; K is an enlarged lobuloalveolar-like structure. Tumors in A–F were ∼1–2 cm in length and significantly larger than H. Analysis of mammary adenocarcinomas. (L–M) PCNA staining demonstrating high mitotic index (M, arrows) in a tumor relative to a tumor-free gland in the same transgenic female (L). (N) Positive staining of mammary adenocarcinomas with antikeratin antibody, an epithelial marker. (O and P) RT-PCR analysis of transgene (O) and keratin 18 (P) expression in tumor biopsies and a derived cell line. Adenocarcinoma biopsies from the tumors shown in E and F (1 and 2); mammary gland from a MMTV-RbΔK11 transgenic female (3); cell line derived from the mammary adenocarcinoma in F (4); mammary gland from a wild-type female (5). The + and – signs indicate the presence or absence of reverse transcriptase.
Transgene expression is lost in mammary adenocarcinomas from MMTV-RbΔK transgenic mice
IHC analysis for PCNA revealed a relatively high proliferation index in the mammary adenocarcinomas derived from MMTV-RbΔK and WAP-RbΔp34 mice (Fig. 7, L–M). This observation has prompted us to determine whether expression of RbΔK was lost during the progression of these tumors. To this end, we analyzed the expression of the RbΔK transgene in two tumor biopsies and tumor cells derived from the mammary adenocarcinoma shown in Fig. 7 F and successfully propagated in vitro. Remarkably, RbΔK transcripts were virtually lost in both the tumor biopsies and the cultured tumor cells (PFig. 7, O and P). These results suggest that downregulation of the transgene with its inhibitory effect on cell proliferation may precede tumor progression in MMTV-RbΔK transgenic mice.
MMTV-RbΔK transgenes extend the survival of differentiated mammary epithelial cells after single and multiple pregnancies
To directly test the effect of RbΔK on cell survival, we examined the first postlactational involution in MMTV-RbΔK transgenic mice. During this process, the extracellular matrix is degraded and the mammary epithelium undergoes massive apoptosis and remodeling back to a virgin-like state. After 6 d, most of the differentiated epithelial cells are eliminated, and the expression of milk genes is greatly reduced (Strange et al., 1992; Lund et al., 1996). At this stage, there were remarkably more β-casein– expressing lobuloalveoli in MMTV-RbΔK11 females compared with wild-type littermates (Fig. 8 D; unpublished data). RT-PCR and Western blot analyses demonstrated a modest yet consistent elevation in the expression of WAP and β-casein in the transgenic MMTV-RbΔK11 females relative to control littermates (Fig. 8, F and G). In situ apoptosis by the TdT-mediated dUtp-biotin nick end labeling (TUNEL) assay revealed that collapsed lobules that did not express β-casein exhibited some cell death (Fig. 8, A and B), whereas lobules that maintained β-casein expression contained very few apoptotic nuclei (Fig. 8, C and D). Overall, there was a 44% reduction in apoptosis in the involuting glands of transgenic females compared with wild-type littermates (Fig. 8 E). Combined, the results suggest that the persistent expression of milk genes at day 6 of involution was due to inhibition of lobuloalveolar cell death by activated Rb.
Figure 8.

Extended survival of the mammary epithelium in MMTV-RbΔK transgenic mice. (A–G) Effects of activated Rb on day 6 of the first postlactational involution. TUNEL analysis (A and C) and β-casein IHC (B and D) were performed on adjacent sections (A and B and C and D) from MMTV-RbΔK11#29 transgenic females at day 6 of first involution. Note the inverse correlation between the presence of apoptotic nuclei (A, black arrows) and the expression of β-casein (D, white arrows) in the same lobules. (E) Relative apoptosis was determined by counting TUNEL-positive nuclei in multiple random areas under 400× magnification. (F and G) RT-PCR and Western blot analyses of milk gene expression in transgenic and wild-type littermate females at day 6 of first involution. The level of milk genes was slightly higher in the transgenic females. (H and M) Enlarged lobuloalveolar-like structures in multiparous retired MMTV-RbΔK11#29 transgenic breeders but not in control littermates. (H–K) H-E staining (H and I, 10×; J and K, 40×). (L and M) Gomori trichrome staining for collagen (L, 126×; M, 63×). ECM is present in transgenic (black arrow) but not in wild-type females (white arrow). (N–P) Western blot analyses of the indicated proteins in multiparous retired breeders (N and P) and 4-mo-old nulliparous females (O).
Although RbΔK exerted only a moderate suppression of apoptosis during the first involution, we asked whether it might have a compounded effect after multiple pregnancies. To address this possibility, we performed serial cross-section analysis on multiparous, retired, transgenic MMTV-RbΔK breeders. Interestingly, nonregressing areas with enlarged and aberrant lobuloalveolar-like structures were detected in each of five transgenic females (Fig. 8, I and K) but not in five wild-type control breeders (Fig. 8, H and J). These lobuloalveolar-like structures expressed extracellular matrix as revealed by Gomori-trichrome, a specific stain for collagen (Fig. 8, L and M), and were often observed in multiparous transgenic females alongside hyperplastic nodules and mammary adenocarcinomas (Fig. 7 K).
Aberrant expression of survival factors in multiparous MMTV-RbΔK transgenic breeders
We next determined whether the expression pattern of known survival factors was altered in multiparous MMTV-RbΔK transgenic mice. Western blot analyses of whole mammary glands revealed that E2F1 was reduced moderately; IGFR-1Rα and phospho–protein kinase B (PKB)/AKT were elevated slightly; p21waf1/cip1 and BCl-Xl exhibited additional faster migrating forms of unknown functions (PFig. 8, N and P). Expression of cyclin D1 and BCl2 was not obviously affected (Fig. 8 N; unpublished data). The aberrant expression of E2F1, IGFR-1Rα, phospho-PKB/AKT, p21waf1/cip1, and BCl-XL was specific to the multiparous transgenic females and was not observed in 4-mo-old nulliparous transgenic mice (Fig. 8 O). Taken together, the results presented in Fig. 8 suggest that RbΔK suppress mammary epithelial cell death at least in part by modulating the level or activity of E2F1, IGFR-1Rα, and PKB/AKT.
Discussion
We report that targeted expression of phosphorylation-resistant constitutively active Rb proteins in the mammary gland leads to ductal growth suppression, precocious differentiation, extended survival of the mammary epithelium, and paradoxically to mammary tumors. The growth suppression is moderate, affects ductal but not alveolar development, and represents the only phenotype that is consistent with the role of Rb as a tumor suppressor. The precocious differentiation and mammary tumors were less expected and are discussed below.
Mechanisms of precocious differentiation in MMTV-RbΔK transgenic mice
Although the role of Rb in cell proliferation and apoptosis is well established, it is not clear whether Rb affects terminal differentiation directly through interaction with differentiation factors or indirectly by allowing proper cell cycle exit and cell survival (Dyson, 1998; Macleod, 1999; Harbour and Dean, 2000b). In this study, we show for the first time that active Rb can induce cellular and molecular differentiation in vivo. Precocious differentiation of the mammary gland has also been described in transgenic mice overexpressing activated β-catenine (Imbert et al., 2001), stromolysin (Sympson et al., 1994), dominant negative TGFβR (Gorska et al., 1998), E-cadherin (Delmas et al., 1999), and in mice lacking P-cadherin (Radice et al., 1997).
The coinduction of Rb and β-casein at mid-pregnancy (Fig. 2; unpublished data) suggests that Rb may be involved directly in transcriptional activation of milk genes (Fig. 9 B). This model is consistent with numerous examples, demonstrating a required role for Rb in terminal differentiation of certain cell types and transcriptional activation by MyoD (Gu et al., 1993; Sellers et al., 1998; Puri et al., 2001), C/EBP (Doppler et al., 1995), the glucocorticoid receptor (Stocklin et al., 1996), and other factors. From a cancer treatment point of view, the possibility that activation of Rb can induce differentiation is attractive, since it may allow the development of drugs that would propel breast tumor cells not only to exit the cell cycle but also to terminally differentiate. However, although the lack of Rb can be a limiting factor for differentiation, activation of the Rb pathway may not be sufficient to force terminal differentiation. Indeed, we have not been able so far to detect any effect of overexpressing RbΔKs on the β-casein promoter in transient reporter assays in Rb-positive mammary epithelial HC11 cells (unpublished data).
Figure 9.

Models for the effects of inactivation versus activation of the Rb pathway. (A) Loss of Rb leads to ectopic DNA synthesis, increased cell death, and incomplete differentiation. Unscheduled cell proliferation is often counteracted by apoptosis but in some cell types results in neoplastic transformation, that is, retinoblastoma in human and pituitary tumors in the mouse. Inhibition of proapoptotic pathways (e.g., inactivation of p53) cooperates with loss of Rb during cancer progression. Impaired differentiation in Rb-deficient mice may be an indirect effect due to the ectopic cell proliferation and apoptosis that perturb the differentiation program. Alternatively, Rb may be required for the onset of terminal differentiation via direct or indirect stimulation of differentiation factors (e.g., glucocorticoid receptor, C/EBP, MyoD, etc.). (B) As shown in this study, activation of Rb in the mammary gland results in suppression of cell proliferation, precocious differentiation, extended survival of the mammary epithelium, and mammary tumors. The precocious differentiation may reflect the ability of activate Rb to extend the survival of differentiated cells and/or to directly promote differentiation. We propose that inhibition of apoptosis by active Rb can lead to the accumulation of oncogenic alterations and neoplastic transformation in susceptible tissues such as the mammary epithelium.
In an alternative model, the RbΔK transgene may induce precocious development by extending the life span of differentiated mammary epithelial cells with each estrous cycle, hence leading to the accumulation of differentiated mammary epithelial cells and milk secreting lobuloalveoli. This “survival model” is consistent with other phenotypes observed in our transgenic mice, most notably the development of mammary tumors in the MMTV-RbΔK and WAP-RbΔp34 mice. We note that the two models are not mutually exclusive, and activated Rb may promote both the onset and survival of the differentiation state (Fig. 9 B).
Mechanisms of mammary adenocarcinoma in MMTV/WAP-RbΔK transgenic mice
Paradoxically, we found that transgene expression of constitutively active Rb in the mammary gland induces mammary adenocarcinoma. We propose that these tumors may be induced in three steps. First, growth suppression by RbΔK may exert a selection pressure in favor of transforming mutations that accelerate cell proliferation. Second, the suppression of cell death by the RbΔK transgenes may promote the survival and accumulation of transformed cells that would otherwise die by apoptosis. Third, notwithstanding the limited number of tumors analyzed so far (Fig. 7 O), our results suggest that downregulation of transgene expression may permit rapid clonal expansion of transformed cells and the development of full-blown mammary adenocarcinomas. Thus, in accordance with the “survival model” described above suppression of apoptosis by active Rb may extend the life span of mammary epithelial cells that have partly differentiated during the estrous cycle, leading to precocious differentiation, or have acquired transforming mutations, resulting in mammary adenocarcinoma (Fig. 9 B). There is a reciprocal relationship between the phenotypes of Rb knockout mice and E2F1 transgenic mice on the one hand (Fig. 9 A) and the RbΔK transgenic mice and E2F1-null mice on the other hand (Fig. 9 B) (Field et al., 1996; Yamasaki et al., 1996; Pierce et al., 1998, 1999). This symmetry is consistent with the antagonistic effects of Rb and E2F1 on cell cycle progression and apoptosis so that both activation and inactivation of these factors induce cancer in susceptible tissues (Fig. 9).
We note that transgenic expression of the survival factor Bcl2 in the mammary gland is not sufficient to induce mammary adenocarcinoma, although it accelerates tumorigenicity in double transgenic mice expressing Bcl2 and Myc (Jager et al., 1997). Similarly, a BCl2 transgene induces hyperplastic splenic follicles, which do not progress to B cell lymphoma in transgenic mice (McDonnell et al., 1989). In contrast, expression of BCl2 in the T cell lineage induces peripheral T cell lymphoma (Linette et al., 1995). Activation of PKB/AKT in the mammary gland also delays involution and promotes mammary adenocarcinomas in conjunction with polyomavirus middle T antigen but does not by itself induce tumors (Hutchinson et al., 2001). The moderate phenotypes of the MMTV-BCl2 and -PKB/AKT transgenic mice may reflect the expression levels of the transgenes relative to the endogenous proteins, or the differential sensitivity of different tissues to perturbations in distinct apoptotic and survival pathways.
Although unlikely, it is formally possible that by virtue of the mutations in the Cdk phosphoacceptor sites the RbΔK alleles have acquired activities that are not associated normally with wild-type Rb. We note that numerous other MMTV transgenic mice expressing dominant-active alleles have been created, for example, neu/erbB2 (Siegel et al., 1999), β-catenin (Imbert et al., 2001), TGFβ (Pierce et al., 1993), and PKB/AKT (Hutchinson et al., 2001). The RbΔK alleles have been used in diverse transcriptional and cellular contexts in vitro and in vivo and invariably demonstrated the same albeit superior functions as native unphosphorylated Rb (Hamel et al., 1992; Geng et al., 1996; Knudsen et al., 1998; Sellers et al., 1998; Brown et al., 1999; Harbour et al., 1999). Most importantly, using RbΔK11 as bait in exhaustive yeast two-hybrid interactive screens of cDNA libraries from mammary and prostate glands we have identified multiple known and novel Rb binding factors, all of which could also bind wild-type Rb (but not nonfunctional RbΔ22 mutant; unpublished data]). Thus, there is no evidence to our knowledge that supports the notion that the RbΔK alleles are neomorphs, though we cannot categorically rule out this possibility.
In addition to the inhibition of apoptosis, it is conceivable that constitutive expression of Rb may promote tumorigenicity by inducing DNA damage. Rb can modulate the progression through S and G2 (Karantza et al., 1993; Knudsen et al., 1998; Lukas et al., 1999; Knudsen et al., 2000), and unscheduled activation of Rb may interfere with DNA synthesis and/or DNA repair processes. Moreover, in response to DNA damage ATM phosphorylates and stabilizes E2F1, thereby inducing apoptosis through p53 and p73 (Irwin et al., 2000; Lin et al., 2001). Thus, constitutive activation of Rb may result in the accumulation of damaged DNA and the acquisition of transforming mutations. If this model is correct, the hyperplastic nodules in MMTV/WAP-RbΔK transgenic mice should exhibit uncharacteristically high chromosomal anomalies, a possibility that should be tested in future studies. Activation of endogenous Rb by Cdk inhibitors might induce similar chromosomal aberrations and elicit tumors as in our transgenic mice.
The results presented in Fig. 8 suggest that inhibition of apoptosis by active Rb involve an E2F1, IGFR1α, and PKB/AKT pathway. Recent analysis of anchorage-independent epithelial cell survival in vitro has indicated that the Rb–E2F1 complex regulates PKB/AKT activity via IGF-1 and suppresses rather than induces cell survival (Yu et al., 2001a). These observations establish a link between Rb, E2F1, and PKB/AKT, and conceivably this same pathway may be used by RbΔK to prolong cell survival via PKB in the mammary epithelium (Scheid and Woodgett, 2001). Future analysis of MMTV-RbΔK:E2F1−/− composite mice will directly establish the role of E2F1 in the development of neoplasia in our transgenic mice.
In contrast to the MMTV/WAP-RbΔK transgenic mice, cyclin D1–deficient mice do not succumb to mammary tumors but are rather protected from a subset of oncogenic pathways in the mammary gland (Sicinski et al., 1995; Yu et al., 2001b). However, cyclin D1 has other targets in addition to Rb that might inhibit tumor progression in cyclin D1−/− mice. For example, cyclin D1 binds and stimulates the estrogen receptor by acting as a bridging factor between the ER and steroid receptor coactivators (Neuman et al., 1997; Zwijsen et al., 1997, 1998). In addition, cyclin D1 induces the phosphorylation of all members of the Rb protein family, including p107 and p130, which are coexpressed during mammary gland development (our unpublished observations), and together may mediate a strong inhibition of proliferation in the cyclin D1−/− mammary epithelium. Thus, the activation of Rb cannot be equated with the loss of cyclin D1. Furthermore, the adverse effect of the Rb pathway may be unraveled only when activation is transient; constitutive activation of Rb in cyclin D1–null mice may not allow mammary epithelial cells that acquired transforming mutations to proliferate and progress into full-blown mammary adenocarcinomas.
Implications for cancer therapy
Since deregulated proliferation is the hallmark of tumor development, antiproliferative agents have become a major therapeutic approach to cancer treatment. However, as stated recently (Evan and Vousden, 2001) many growth deregulatory mutations have pleiotropic and tissue-specific effects on cell proliferation and apoptosis, and the therapeutic consequences of antiproliferative agents are thus difficult to predict.
Our results illustrate this potential problem. Although activation of Rb suppresses epithelial cell proliferation in pubescent females, the RbΔK transgenic mice eventually develop breast tumors. This scenario may also occur with antiproliferative drugs in the course of cancer therapy. Systemic activation of Rb by therapeutical drugs such as Cdk inhibitors may extend the survival of cells destined to die by apoptosis, leading to the accumulation of oncogenic alterations, clonal expansion, and ultimately cancer. Several Cdk inhibitors (e.g., flavopiridol) that target the Rb pathway are currently in preclinical and clinical development (Bange et al., 2001). Although such drugs have beneficial inhibitory effect on tumor growth, they have also been documented to suppress apoptosis through Rb (Osuga et al., 2000). Our results suggest that such drugs should be examined cautiously in long-term assays in vivo to determine their systemic effects on tissues with high turnover such as the mammary epithelium.
Combined, the coupled effects of Rb on cell proliferation and apoptosis appear to render this tumor suppressor oncogenic in certain tissues both when mutated and activated. This inherent caveat may limit the use of certain Cdk inhibitors, which restrict cancer cell proliferation by activating Rb, to short term treatments. Alternatively, inasmuch as that cancer progression often involves the inactivation of both Rb and p53 long-term cancer therapy may require combinatorial drugs that would induce the Rb pathway but also antagonize its survival effect.
Materials and methods
Plasmid construction and verification
pECE-HA-RbΔK11.
pECE-HA-RbΔp34 is a mouse Rb cDNA with an HA tag at the NH2 terminus and eight mutations in the indicated Cdk sites (a gift from Dr. P. Hamel, University of Toronto) (Hamel et al., 1992; Fig. 1 A). The plasmid was transformed into dcm− bacteria to allow digestion with StuI. The Cdk sites, S773, T814, and T819, in exon 23 of RbΔp34 were mutated to alanine by PCR-directed mutagenesis using Pfu polymerase. Complex PCR included a forward primer (EZ52, 5′-GCACCCACAAAAATGGCTCCGAGATCTAGAATCTTGG) and a reverse primer (EZ53, 5′-CGGAGCCATTTTTGTGGGTGCTGGCAGACCTTCTG). The flanking primers were as follows: a 5′ primer that overlapped the StuI site at the junction of exons 22 and 23 (EZ51, 5′-TAATAGGCCTCCTACCTTGGCACCAATACCTCACATTCC) and a 3′ reversed primer that overlapped the SauI site downstream of the Rb stop codon. The ∼450-bp PCR product was digested with StuI and SauI and cloned into pECE-HA-RbΔp34, partially digested with StuI, and further linearized with SauI. The PCR region in the resulting pECE-HA-RbΔK11 plasmid was sequenced on both strands to confirm the site directed. The ability of RbΔK11 to bind large T, localize to the nucleus, induce the flat cell phenotype in Saos-2 cells, and suppress E2F activation was confirmed by transfecting pECE-HA-RbΔK11 DNA into COS and Saos2 cells using standard methods (Zacksenhaus et al., 1993, 1996a).
pBS-MMTV-HA-RbΔp34 and pBS-MMTV-HA-RbΔK11.
Briefly, the linker in pBluescript was replaced by a double strand linker containing SacI, NotI, XhoI, NotI, and KpnI sites, yielding pBS-EZ15/16. A 2.684-kb XhoI fragment containing the MMTV-LTR and rabbit β-globin cassette was transferred from mmtv-KCR plasmid (Matsui et al., 1990) provided by Dr. Robert Coffey (Vanderbilt University, Nashville, TN) and subcloned into pBS-EZ15/16 (pBS-MMTV). The RbΔK alleles were subcloned into the unique EcoRI site in pBS-MMTV (Fig. 1B).
WAP-HA-RbΔp34.
pUC-mWAP, containing a 6.75 kb of the mouse WAP gene (Campbell et al., 1984) cloned into a unique EcoRI, was purchased from American Type Culture Collection. A linker KpnI, HindIII, SalI was used to replace a 1.5 kb between the first and third exons of the WAP gene, yielding pUC-mWAP(K, H, S). The pECE-HA-RbΔp34 plasmid was modified so that it contained a 5′ HindIII and 3′ SalI site, flanking the RbΔp34 coding cDNA. The HA-RbΔp34 was excised as a 2.7-kb HindIII-SalI fragment and subcloned into pUC-mWAP(K, H, S), partially digested with HindIII and further linearized with SalI. The junction fragments at the 5′ and 3′ ends in WAP-HA-RbΔp34 were confirmed by sequencing.
pBS-MMTV-HA-RbΔp34 and pBS-MMTV-HA-RbΔK11 were analyzed in vitro to confirm the expression of the Rb alleles, interaction with large T, and nuclear localization using antibodies specific to both Rb and HA. To induce the MMTV-LTR promoter, COS cells were transfected with pBS-MMTV-HA-RbΔp34 or pBS-MMTV-HA-RbΔK11 together with an expression plasmid for the glucocorticoid receptor (Giguere et al., 1986; Zacksenhaus et al., 1996a), and the cells were cultured in the presence of dexamethasone for 36 h before analysis.
Generation, genotyping, and analysis of transgenic mice
Microinjection of 2 ng/ml DNA into pronuclei of C57BL/6×SJL or FVB mice and generation of chimeric mice were done in the transgenic mouse facilities at the Hospital for Sick Children and Ontario Cancer Institute. Transgenic mice were initially identified by Southern blot hybridization and later genotyped by PCR. Primers used for PCR of MMTV-RbΔKs were as follows: forward primer EZ99 from exon 23 of Rb (5′-ATTTCAGAAGGTCTGCCAAC) and reverse primer EZ38 from the β-globin cassette (5′-GAATAAGGAATGGACAGCAGGG); WAP-RbΔp34 (EZ99 and EZ72, 5′-CATGTCATGACACAGTCGAC). Transgenic females and control wild-type littermates were caged together. For timed pregnancy, the morning of vaginal plug observation was considered as P0.5.
Southern, Northern, and Western blots, and RT-PCR analysis
Genotyping by Southern blot and PCR analyses was performed on tail DNA as described (Zacksenhaus et al., 1996b). To detect MMTV-RbΔKs, DNA was digested with SstI, Southern blotted, and hybridized with a 32P-dCTP SstI-EcoRI 1.0-kb Rb cDNA fragment from the plasmid. WAP-RbΔp34 transgene was detected after digestion of genomic DNA with PvuII and hybridization with a 32P-labeled KpnI-PvuII 1.9-kb probe from WAP-RbΔp34. Total RNA was isolated from mammary glands or other tissues with Trizol Reagent (GIBCO BRL). For Northern blots, PolyA+ RNA samples (2 μg) were resolved on 1% agarose formaldehyde gels, transferred to nylon membranes, and hybridized to a 32P-labeled mouse Rb KpnI-SstI 1.9 kb DNA probe. Protein lysates from mammary gland tissues were prepared as described (Yao et al., 1999). Anti-HA monoclonal antibody was used directly from hybridoma supernatant. Anti-Rb G3-245 antibody was from Pharmingen (14001A). E2F1 (C-20), IGF1Rα (sc-712), cyclin D1 (sc-450), BCl−Xl (sc-7195), and p21 (sc-397) were from Santa Cruz Biotechnology, Inc. Total AKT (#9272) and phospho-AKT (#9271) polyclonal antibodies were from NEB. Anti–keratin 18 was obtained from Sigma-Aldrich. The β-casein monoclonal antibodies were a gift from Dr. Charlotte Kaetzel (University of Kentucky, Lexington, KY) (Kaetzel and Ray, 1984).
MMTV-RbΔK transcripts were detected using reverse transcribed cDNA (0.3–0.4 μg) as a template with forward primer EZ99 and reverse primer EZ73 (5′-TAGCCAGAAGTCAGAT-GCTC). WAP-RbΔp34 was detected with EZ99 and EZ72. Primers for WAP were forward EZ178 (5′-TAGCAGCAGATTGAAAGCATTATG) and reverse EZ179 (5′-GACACCGGTACCAT-GCGTTG), for β-casein were forward EZ182 (5′-TCCTGGATCCAGCCTATTGCTCAAC) and reverse EZ183 (5′-AGTCCATGGGTCGAATTC), for β-actin were forward EZ70 (5′-CTTCTACAATGAGCTGCGTG) and reverse EZ71 (5′-TGGCATAGAGGTCTTTACGG), and for keratin 18 were forward EZ245 (5′-TGGTCTCAGCAGATTGAGG) and reverse EZ246 (5′-CTGGTGTCATTAGTCTCGG).
Whole-mount analysis
Right inguinal mammary glands (#4) were resected and flatten fixed in 4% paraformaldehyde, treated sequentially in 100% ethanol, 100% acetone, 100% ethanol, and 95% ethanol, and stained with iron hematoxylin. The glands were destained with 50% acidified ethanol followed by ascending ethanol, Xylenes, and finally stored in methyl salicylate.
Histology, in situ hybridization, and TUNEL
Left inguinal mammary glands (#4) were fixed in 4% paraformaldehyde, treated with 1×PBS, saline, 50% ethanol, and 70% ethanol (in saline), paraffin embedded, and sections were stained with hematoxylin and eosin (H-E) or Gomori-trichrome. In situ hybridization and TUNEL were performed exactly as described (Jiang et al., 1997, 2000).
Analysis of mammary adenocarcinomas
Mammary glands with visible palpable tumors were resected, fixed in 4% paraformaldehyde, and stained with H-E. Pathology analysis was performed by Dr. Colin McKerlie (Sunnybrook Hospital, Toronto, Canada). Some mammary tumors were cultured in α-MEM medium supplemented with 10% FCS, 10 ng/ml EGF, and 5 μg/ml insulin.
Immunohistochemistry
IHC analysis was performed with a DAKO streptABComplex/HRP kit. PCNA monoclonal antibody (P8825; Sigma-Aldrich) was used at 1:300 dilution. β-casein monoclonal antibody was diluted 1:800. Rabbit antikeratin wide spectrum screening polyclonal antibody (DAKOZ0622) was diluted 1:200. Rb (C-15) polyclonal antibodies from Santa Cruz Biotechnology, Inc. was diluted 1:50. HA monoclonal antibodies from tissue culture supernatant (at 1:20–1:200) or from Boehringer (1583816) (at 4 μg/ml) were used as in Chang et al. (1995).
Acknowledgments
We thank Dr. Paul Hamel for provision of pECE-HA-RbΔp34, Drs. Robert Coffey and Sean Egan for mmtv-KCR, Dr. Charlotte Kaetzel for the RβC1 anti–β-casein antibody, Andrew Ho for advice on Gomori-trichrome staining, Dr. Colin McKerlie for pathology analysis, and Drs. Jim Fata and Brenda Gallie for critical comments on the article.
E. Zacksenhaus is a recipient of a scholarship award from the Cancer Research Society Inc./Canadian Institute of Health Research. This study was supported by the Canadian Breast Cancer Research Initiative (08469) and the U.S. Army Breast Cancer Research Program (DAMD17-97-1-7321).
Footnotes
Abbreviations used in this paper: Cdk, cyclin-dependent kinase; H-E, hematoxylin and eosin; IHC, immunohistochemistry; MMTV, mouse mammary tumor virus; PCNA, proliferation cell nuclear antigen; PKB, protein kinase B; Rb, retinoblastoma; RT, reverse transcribed; TEB, terminal endbud; TUNEL, TdT-mediated dUtp-biotin nick end labeling; WAP, whey acidic protein.
References
- Almasan, A., Y. Yin, R.E. Kelly, E.Y. Lee, A. Bradley, W. Li, J.R. Bertino, and G.M. Wahl. 1995. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc. Natl. Acad. Sci. USA. 92:5436–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bange, J., E. Zwick, and A. Ullrich. 2001. Molecular targets for breast cancer therapy and prevention. Nat. Med. 7:548–552. [DOI] [PubMed] [Google Scholar]
- Brown, V.D., R.A. Phillips, and B.L. Gallie. 1999. Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol. Cell. Biol. 19:3246–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley, M.F., K.J. Sweeney, J.A. Hamilton, R.L. Sini, D.L. Manning, R.I. Nicholson, A. deFazio, C.K. Watts, E.A. Musgrove, and R.L. Sutherland. 1993. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 8:2127–2133. [PubMed] [Google Scholar]
- Campbell, S.M., J.M. Rosen, L.G. Hennighausen, U. Strech-Jurk, and A.E. Sippel. 1984. Comparison of the whey acidic protein genes of the rat and mouse. Nucleic Acids Res. 12:8685–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardiff, R.D., M.R. Anver, B.A. Gusterson, L. Hennighausen, R.A. Jensen, M.J. Merino, S. Rehm, J. Russo, F.A. Tavassoli, L.M. Wakefield, et al. 2000. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene. 19:968–988. [DOI] [PubMed] [Google Scholar]
- Chang, M.W., E. Barr, J. Seltzer, Y.Q. Jiang, G.J. Nabel, E.G. Nabel, M.S. Parmacek, and J.M. Leiden. 1995. Cytostatic gene therapy for vascular proliferative disorders with a constitutively active form of the retinoblastoma gene product. Science. 267:518–522. [DOI] [PubMed] [Google Scholar]
- Delmas, V., P. Pla, H. Feracci, J.P. Thiery, R. Kemler, and L. Larue. 1999. Expression of the cytoplasmic domain of E-cadherin induces precocious mammary epithelial alveolar formation and affects cell polarity and cell-matrix integrity. Dev. Biol. 216:491–506. [DOI] [PubMed] [Google Scholar]
- DiCiommo, D., B.L. Gallie, and R. Bremner. 2000. Retinoblastoma: the disease, gene and protein provide critical leads to understand cancer. Semin. Cancer Biol. 10:255–269. [DOI] [PubMed] [Google Scholar]
- Doppler, W., T. Welte, and S. Philipp. 1995. CCAAT/enhancer-binding protein isoforms beta and delta are expressed in mammary epithelial cells and bind to multiple sites in the beta-casein gene promoter. J. Biol. Chem. 270:17962–17969. [DOI] [PubMed] [Google Scholar]
- Dyson, N. 1998. The regulation of E2F by pRB-family proteins. Genes Dev. 12:2245–2262. [DOI] [PubMed] [Google Scholar]
- Evan, G.I., and K.H. Vousden. 2001. Proliferation, cell cycle and apoptosis in cancer. Nature. 411:342–348. [DOI] [PubMed] [Google Scholar]
- Field, S.J., F.Y. Tsai, F. Kuo, A.M. Zubiage, W.G. Kaelin, D.M. Livingston, S.H. Orkin, and M.E. Greenberg. 1996. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 85:549–561. [DOI] [PubMed] [Google Scholar]
- Geng, Y., E.N. Eaton, M. Picon, J.M. Roberts, A.S. Lundberg, A. Gifford, C. Sardet, and R.A. Weinberg. 1996. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene. 12:1173–1180. [PubMed] [Google Scholar]
- Giguere, V., S.M. Hollenberg, M.G. Rosenfeld, and R.M. Evans. 1986. Functional domains of the human glucocorticoid receptor. Cell. 46:645–652. [DOI] [PubMed] [Google Scholar]
- Gorska, A.E., H. Joseph, R. Derynck, H.L. Moses, and R. Serra. 1998. Dominant-negative interference of the transforming growth factor beta type II receptor in mammary gland epithelium results in alveolar hyperplasia and differentiation in virgin mice. Cell Growth Differ. 9:229–238. [PubMed] [Google Scholar]
- Gu, W., J.W. Schneider, G. Condorelli, S. Kaushal, V. Mahdavi, and B. Nadal-Ginard. 1993. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 72:309–324. [DOI] [PubMed] [Google Scholar]
- Guo, Z., Y. Shi, Y. Hiroki, T. Mak, and E. Zacksenhaus. 2001. Inactivation of the retinoblastoma tumor suppressor induces Apaf-1 dependent and independent apoptotic pathways during embryogenesis. Cancer Res. 61:8395–8400. [PubMed] [Google Scholar]
- Haas-Kogan, D.A., S.C. Kogan, D. Levi, P. Dazin, A. T'Ang, Y.K. Fung, and M.A. Israel. 1995. Inhibition of apoptosis by the retinoblastoma gene product. EMBO J. 14:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel, P.A., R.M. Gill, R.A. Phillips, and B.L. Gallie. 1992. Transcriptional repression of the E2-containing promoters EIIaE, c-myc and RB1 by the product of the RB1 gene. Mol. Cell. Biol. 12:3431–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour, J.W., and D.C. Dean. 2000. a. Chromatin remodeling and Rb activity. Curr. Opin. Cell Biol. 12:685–689. [DOI] [PubMed] [Google Scholar]
- Harbour, J.W., and D.C. Dean. 2000. b. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 14:2393–2409. [DOI] [PubMed] [Google Scholar]
- Harbour, J.W., R.X. Luo, A. Dei Santi, A.A. Postigo, and D.C. Dean. 1999. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 98:859–869. [DOI] [PubMed] [Google Scholar]
- Hennighausen, L., and G.W. Robinson. 1998. Think globally, act locally: the making of a mouse mammary gland. Genes Dev. 12:449–455. [DOI] [PubMed] [Google Scholar]
- Hutchinson, J., J. Jin, R.D. Cardiff, J.R. Woodgett, and W.J. Muller. 2001. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol. Cell. Biol. 21:2203–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert, A., R. Eelkema, S. Jordan, H. Feiner, and P. Cowin. 2001. Delta N89 beta-catenin induces precocious development, differentiation, and neoplasia in mammary gland. J. Cell Biol. 153:555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin, M., M.C. Marin, A.C. Phillips, R.S. Seelan, D.I. Smith, W. Liu, E.R. Flores, K.Y. Tsai, T. Jacks, K.H. Vousden, and W.G. Kaelin. 2000. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 407:645–648. [DOI] [PubMed] [Google Scholar]
- Jager, R., U. Herzer, J. Schenkel, and H. Weiher. 1997. Overexpression of Bcl-2 inhibits alveolar cell apoptosis during involution and accelerates c-myc-induced tumorigenesis of the mammary gland in transgenic mice. Oncogene. 15:1787–1795. [DOI] [PubMed] [Google Scholar]
- Jhappan, C., A.G. Geiser, E.C. Kordon, D. Bagheri, L. Hennighausen, A.B. Roberts, G.H. Smith, and G. Merlino. 1993. Targeting expression of a transforming growth factor b1 transgene to the pregnant mammary gland inhibits alveolar development and lactation. EMBO J. 12:1835–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Z., E. Zacksenhaus, B.L. Gallie, and R.A. Phillips. 1997. The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene. 14:1789–1797. [DOI] [PubMed] [Google Scholar]
- Jiang, Z., P. Liang, R. Leng, Z. Guo, Y. Liu, X. Liu, S. Bubnic, A. Keating, D. Murray, P.E. Goss, and E. Zacksenhaus. 2000. E2F1 and p53 are dispensable whereas p21Waf1/Cip1 cooperates with Rb to restrict endoreduplication and apoptosis during skeletal myogenesis. Dev. Biol. 227:28–41. [DOI] [PubMed] [Google Scholar]
- Kaetzel, C.S., and D.B. Ray. 1984. Immunochemical characterization of three major caseins and alpha-lactalbumin from rat milk using monoclonal antibodies. J. Dairy Sci. 67:64–75. [DOI] [PubMed] [Google Scholar]
- Karantza, V., A. Maroo, D. Fay, and J.M. Sedivy. 1993. Overproduction of Rb protein after the G1/S boundary causes G2 arrest. Mol. Cell. Biol. 13:6640–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, E.S., C. Buckmaster, T.T. Chen, J.R. Feramisco, and J.Y. Wang. 1998. Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev. 12:2278–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, K.E., D. Booth, S. Naderi, Z. Sever-Chroneos, A.F. Fribourg, I.C. Hunton, J.R. Feramisco, J.Y. Wang, and E.S. Knudsen. 2000. RB-dependent S-phase response to DNA damage. Mol. Cell. Biol. 20:7751–7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg, O., A.J. van der Eb, and A. Zantema. 1996. Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J. 15:46–54. [PMC free article] [PubMed] [Google Scholar]
- Lai, A., J.M. Lee, W.M. Yang, J.A. DeCaprio, W.G. Kaelin, Jr., E. Seto, and P.E. Branton. 1999. RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins. Mol. Cell. Biol. 19:6632–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, E.Y.-H.P., N. Hu, S.-S.F. Yuan, L.A. Cox, A. Bradley, W.-H. Lee, and K. Herrup. 1994. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev. 8:2008–2021. [DOI] [PubMed] [Google Scholar]
- Lin, W.C., F.T. Lin, and J.R. Nevins. 2001. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- Linette, G.P., J.L. Hess, C.L. Sentman, and S.J. Korsmeyer. 1995. Peripheral T-cell lymphoma in lckpr-bcl-2 transgenic mice. Blood. 86:1255–1260. [PubMed] [Google Scholar]
- Liu, Y., and E. Zacksenhaus. 2000. E2F1 mediates ectopic proliferation and stage-specific p53-dependent apoptosis but not aberrant differentiation in the ocular lens of Rb deficient fetuses. Oncogene. 19:6065–6073. [DOI] [PubMed] [Google Scholar]
- Lukas, C., C.S. Sorensen, E. Kramer, E. Santoni-Rugiu, C. Lindeneg, J.M. Peters, J. Bartek, and J. Lukas. 1999. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature. 401:815–818. [DOI] [PubMed] [Google Scholar]
- Lund, L.R., J. Romer, N. Thomasset, H. Solberg, C. Pyke, M.J. Bissell, K. Dano, and Z. Werb. 1996. Two distinct phases of apoptosis in mammary gland involution: proteinase-independent and -dependent pathways. Development. 122:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg, A.S., and R.A. Weinberg. 1998. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 18:753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod, K. 1999. pRb and E2f-1 in mouse development and tumorigenesis. Curr. Opin. Genet. Dev. 9:31–39. [DOI] [PubMed] [Google Scholar]
- Macleod, K.F., Y. Hu, and T. Jacks. 1996. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- Matsui, Y., S.A. Halter, J.T. Holt, B.L.M. Hogan, and R.J. Coffey. 1990. Development of mammary hyperplasia and neoplasia in MMTV-TGFa transgenic mice. Cell. 61:1147–1155. [DOI] [PubMed] [Google Scholar]
- McDonnell, T.J., N. Deane, F.M. Platt, G. Nunez, U. Jaeger, J.P. McKearn, and S.J. Korsmeyer. 1989. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 57:79–88. [DOI] [PubMed] [Google Scholar]
- Medina, D. 1996. The mammary gland: a unique organ for the study of development and tumorigenicity. J. Mammary Gland Biol. Neoplasia. 1:5–19. [DOI] [PubMed] [Google Scholar]
- Mittnacht, S. 1998. Control of pRB phosphorylation. Curr. Opin. Genet. Dev. 8:21–27. [DOI] [PubMed] [Google Scholar]
- Morgenbesser, S.D., B.O. Williams, T. Jacks, and R.A. DePinho. 1994. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature. 371:72–74. [DOI] [PubMed] [Google Scholar]
- Moroni, M.C., E.S. Hickman, E.L. Denchi, G. Caprara, E. Colli, F. Cecconi, H. Muller, and K. Helin. 2001. Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 3:552–558. [DOI] [PubMed] [Google Scholar]
- Neuman, E., M.H. Ladha, N. Lin, T.M. Upton, S.J. Miller, J. DiRenzo, R.G. Pestell, P.W. Hinds, S.F. Dowdy, M. Brown, and M.E. Ewen. 1997. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol. Cell. Biol. 17:5338–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuga, H., S. Osuga, F. Wang, R. Fetni, M.J. Hogan, R.S. Slack, A.M. Hakim, J.E. Ikeda, and D.S. Park. 2000. Cyclin-dependent kinases as a therapeutic target for stroke. Proc. Natl. Acad. Sci. USA. 97:10254–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, H., C. Yin, N.J. Dyson, E. Harlow, L. Yamasaki, and T. Van Dyke. 1998. Key roles for E2F1 in signaling p53-dependent apoptosis and in cell division within developing tumors. Mol. Cell. 2:283–292. [DOI] [PubMed] [Google Scholar]
- Pierce, A.M., S.M. Fisher, C.J. Conti, and D.G. Johnson. 1998. Deregulated expression of E2F1 induces hyperplasia and cooperates with ras in skin tumor development. Oncogene. 16:1267–1276. [DOI] [PubMed] [Google Scholar]
- Pierce, A.M., R. Schneider-Broussard, I.B. Gimenez-Conti, J.L. Russell, C.J. Conti, and D.G. Johnson. 1999. E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol. Cell. Biol. 19:6408–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce, D.F.J., M.D. Johnson, Y. Matsui, S.D. Robinson, L.I. Gold, A.F. Purchio, C.W. Daniel, B.L.M. Hogan, and H.L. Moses. 1993. Inhibition of mammary duct development but not alveolar outgrowth during pregnancy in transgenic mice expressing active TGF-b1. Genes Dev. 7:2308–2317. [DOI] [PubMed] [Google Scholar]
- Puri, P.L., S. Iezzi, P. Stiegler, T.T. Chen, R.L. Schiltz, G.E. Muscat, A. Giordano, L. Kedes, J.Y. Wang, and V. Sartorelli. 2001. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell. 8:885–897. [DOI] [PubMed] [Google Scholar]
- Radice, G.L., M.C. Ferreira-Cornwell, S.D. Robinson, H. Rayburn, L.A. Chodosh, M. Takeichi, and R.O. Hynes. 1997. Precocious mammary gland development in P-cadherin–deficient mice. J. Cell Biol. 139:1025–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, G.W., R.A. McKnight, G.H. Smith, and L. Hennighausen. 1995. Mammary epithelial cells undergo secretory differentiation in cycling virgins but require pregnancy for the establishment of terminal differentiation. Development. 121:2079–2090. [DOI] [PubMed] [Google Scholar]
- Robinson, G.W., L. Hennighausen, and P.F. Johnson. 2000. Side-branching in the mammary gland: the progesterone-Wnt connection. Genes Dev. 14:889–894. [PubMed] [Google Scholar]
- Scheid, M.P., and J.R. Woodgett. 2001. Phosphatidylinositol 3′ kinase signaling in mammary tumorigenesis. J. Mammary Gland Biol. Neoplasia. 6:83–99. [DOI] [PubMed] [Google Scholar]
- Sellers, W.R., B.G. Novitch, S. Miyake, A. Heith, G.A. Otterson, F.J. Kaye, A.B. Lassar, and W.G.J. Kaelin. 1998. Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 12:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr, C.J. 2000. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 60:3689–3695. [PubMed] [Google Scholar]
- Sicinski, P., J.L. Donaher, S.B. Parker, T. Li, A. Fazeli, H. Gardner, S.Z. Haslam, R.T. Bronson, S.J. Elledge, and R.A. Weinberg. 1995. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 82:621–630. [DOI] [PubMed] [Google Scholar]
- Siegel, P.M., E.D. Ryan, R.D. Cardiff, and W.J. Muller. 1999. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 18:2149–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein, G.B. 2001. Postnatal mammary gland morphogenesis. Microsc. Res. Tech. 52:155–162. [DOI] [PubMed] [Google Scholar]
- Stocklin, E., M. Wissler, F. Gouilleux, and B. Groner. 1996. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 383:726–728. [DOI] [PubMed] [Google Scholar]
- Strange, R., F. Li, S. Saurer, A. Burkhardt, and R.R. Friis. 1992. Apoptotic cell death and tissue remodelling during mouse mammary gland involution. Development. 115:49–58. [DOI] [PubMed] [Google Scholar]
- Sympson, C.J., R.S. Talhouk, C.M. Alexander, J.R. Chin, S.M. Clift, M.J. Bissell, and Z. Werb. 1994. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J. Cell Biol. 125:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, K.Y., Y. Hu, K.F. Macleod, D. Crowley, L. Yamasaki, and T. Jacks. 1998. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol. Cell. 2:293–304. [DOI] [PubMed] [Google Scholar]
- Weinberg, R.A. 1995. The retinoblastoma protein and the cell cycle. Cell. 81:323–330. [DOI] [PubMed] [Google Scholar]
- Yamasaki, L., T. Jacks, R. Bronson, E. Goillot, E. Harlow, and N.J. Dyson. 1996. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 85:537–548. [DOI] [PubMed] [Google Scholar]
- Yao, Y., E.D. Slosberg, L. Wang, H. Hibshoosh, Y.J. Zhang, W.Q. Xing, R.M. Santella, and I.B. Weinstein. 1999. Increased susceptibility to carcinogen-induced mammary tumors in MMTV-Cdc25B transgenic mice. Oncogene. 18:5159–5166. [DOI] [PubMed] [Google Scholar]
- Yu, J.T., R.G. Foster, and D.C. Dean. 2001. a. Transcriptional repression by RB-E2F and regulation of anchorage-independent survival. Mol. Cell. Biol. 21:3325–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Q., Y. Geng, and P. Sicinski. 2001. b. Specific protection against breast cancers by cyclin D1 ablation. Nature. 411:1017–1021. [DOI] [PubMed] [Google Scholar]
- Zacksenhaus, E., R. Bremner, R.A. Phillips, and B.L. Gallie. 1993. A bipartite nuclear localization signal in the retinoblastoma gene product and its importance for biological activity. Mol. Cell. Biol. 13:4588–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacksenhaus, E., Z. Jiang, R.A. Phillips, and B.L. Gallie. 1996. a. Dual mechanisms of transcriptional repression of E2F1 activity by the retinoblastoma gene product. EMBO J. 15:5917–5927. [PMC free article] [PubMed] [Google Scholar]
- Zacksenhaus, E., Z. Jiang, D. Chung, J. Marth, R.A. Phillips, and B.L. Gallie. 1996. b. pRb controls cell proliferation, differentiation and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 10:3051–3064. [DOI] [PubMed] [Google Scholar]
- Zwijsen, R.M., E. Wientjens, R. Klompmaker, J. van der Sman, R. Bernards, and R.J. Michalides. 1997. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 88:405–415. [DOI] [PubMed] [Google Scholar]
- Zwijsen, R.M., R.S. Buckle, E.M. Hijmans, C.J. Loomans, and R. Bernards. 1998. Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes Dev. 12:3488–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]