

Abstract
The Kaposi's sarcoma-associated herpesvirus (KSHV) Mta protein, encoded by open reading frame 57, is a transactivator of gene expression that is essential for productive viral replication. Previous studies have suggested both transcriptional and posttranscriptional roles for Mta, but little is known regarding Mta's transcriptional function. In this study, we demonstrate that Mta cooperates with the KSHV lytic switch protein, Rta, to reactivate KSHV from latency, but Mta has little effect on reactivation when expressed alone. We demonstrate that the Mta and Rta proteins are expressed with similar but distinct kinetics during KSHV reactivation. In single-cell analyses, Mta expression coincides tightly with progression to full viral reactivation. We demonstrate with promoter reporter assays that while Rta activates transcription in all cell lines tested, Mta's ability to transactivate promoters, either alone or synergistically with Rta, is cell and promoter specific. In particular, Mta robustly transactivates the nut-1/PAN promoter independently of Rta in 293 and Akata-31 cells. Using nuclear run-on assays, we demonstrate that Mta stimulates transcriptional initiation in 293 cells. Rta and Mta physically interact in infected cell extracts, and this interaction requires the intact leucine repeat and central region of Rta in vitro. We demonstrate that Mta also binds to the nut-1/PAN promoter DNA in vitro and in infected cells. An Mta mutant with a lesion in a putative A/T hook domain is altered in DNA binding and debilitated in transactivation. We propose that one molecular mechanism of Mta-mediated transactivation is a direct effect on transcription by direct and indirect promoter association.
Reactivation of Kaposi's-sarcoma associated herpesvirus (KSHV) (also known as human herpesvirus 8) from latency is a crucial step in Kaposi's sarcoma development (7, 11, 27, 43, 65, 76, 81, 87, 104). It is likely that reactivation of KSHV contributes to Kaposi's sarcoma development by facilitating dissemination of the virus from its B-cell reservoir and permitting the expression of lytic cycle genes with direct roles in cancer progression. Most of the candidate pathogenic genes of KSHV (encoding proteins with growth-deregulatory and immunomodulatory functions) are expressed in the delayed early class of the lytic gene expression program (8, 14, 17, 25, 26, 28, 30, 41, 46, 49, 75, 86, 94). Therefore, a complete understanding of KSHV pathogenesis demands elucidation of the mechanisms that regulate viral reactivation and progression through the lytic cycle.
The members of the Herpesviridae all encode multiple lytic cycle transactivators that cooperate to promote viral replication. For herpes simplex virus type 1 (HSV-1) infection, the two essential transactivating proteins are called infected cell protein 4 (ICP4) and ICP27. ICP4 is a transcriptional transactivator necessary for activation of early and late genes (80, 103). ICP27 is a posttranscriptional and transcriptional activator that stimulates the switch from early- to late-gene expression (66, 67, 82, 84, 95). ICP27 interacts with the C-terminal domain (CTD) of RNA polymerase II, suggesting a role in coupling transcription to posttranscriptional regulation (21, 111). ICP27 is the only HSV immediate-early protein that is conserved across the Herpesviridae, and two of its homologs are Epstein-Barr virus (EBV) SM and KSHV Mta (9, 79). In EBV, the SM protein (also known as Mta) cooperates with the lytic switch protein ZEBRA (18) (also known as Zta or BZLF1). EBV SM functions at the posttranscriptional level (93) and, like ICP27, is essential for production of infectious virus (33). Previous studies have demonstrated that the Herpesviridae Mta homologs transactivate in a gene-specific fashion. Most notably, HSV-1 ICP27 and EBV SM are required for accumulation of delayed early transcripts that are essential for lytic DNA replication (31, 33).
We and others have demonstrated that the KSHV protein Rta (for “replication and transcriptional activator,” expressed from open reading frame 50 [ORF50]) is both necessary and sufficient for reactivation of KSHV in tissue culture models of latency (32, 56, 57, 92, 108). Rta directly transactivates viral and cellular promoters (17, 24, 25, 56, 57, 94), and a truncated mutant of Rta lacking the transactivation domain (called ORF50ΔSTAD) cannot reactivate the virus (56). Rta transactivates viral promoters by binding DNA, either independently or in combination with cellular transcription factors, (12, 13, 15, 16, 48, 49, 55, 83, 91, 99, 100), and is regulated by oligomerization and posttranslational modifications (10, 35, 50). Since less than 30% of primary effusion lymphoma (PEL) cells transfected with an Rta expression vector express viral lytic proteins (56, 57), it is likely that control of Rta's function following its expression is critical for regulating productive viral reactivation.
We have previously shown that the KSHV Mta protein dramatically synergized with ORF50/Rta in activating transcription of the nut-1/PAN and kaposin promoters in transiently transfected CV-1 cells (45). Mta did not simply enhance the expression levels of Rta mRNA or protein but synergized with Rta expressed at saturating concentrations (45). Synergy was abolished when Rta transactivation was eliminated by either (i) mutation of the Rta-responsive element (RRE) in the PAN promoter or (ii) cotransfection of the Rta-specific dominant-negative mutant ORF50ΔSTAD (45). Since Mta had little effect on the PAN and kaposin promoters when expressed alone, the data collectively suggested that synergism between Mta and Rta depended strictly upon Rta's ability to activate transcription.
Like its homologs in HSV-1 and EBV, KSHV Mta is required for production of infectious virions (37, 58). However, the mechanisms by which Mta synergizes with Rta remain unclear. Mta encodes protein domains with putative transcriptional and posttranscriptional functions, and Mta directly binds to proteins having established roles in multiple levels of gene expression control (59, 62, 63, 73). Mta and Rta bind directly to each other and can coprecipitate the Rta promoter in KSHV-infected cells (61). Mta enhances the accumulation of both viral (37, 45, 58-60, 72) and cellular (34) mRNAs in a gene-specific manner. This effect is independent of the promoter used to transcribe the messages and is more efficient for intronless RNAs (34, 45). Mta also shuttles between nuclei in heterokaryon assays (6) and promotes the nuclear export of unspliced RNAs (62). Mta may also function in translational control, since it stimulates translation of mRNAs containing internal ribosome entry sites (73).
In this paper, we establish that one mechanism by which KSHV Mta functions is by stimulating transcriptional initiation. The transactivation abilities of Mta, in the absence of Rta, are promoter and cell line specific. Among seven cell lines of different lineages, Mta transactivates the nut-1/PAN promoter in 293 and Akata-31 cells. We show that promoter-specific Mta transactivation mirrors promoter-specific synergy by Mta and Rta. However, Mta synergizes with Rta in cell lines in which Mta does not independently transactivate transcription. A DNA element that is conserved in multiple KSHV promoters is necessary but not sufficient for synergy. We demonstrate that Mta binds to both the Rta protein and promoter DNA in vitro and in infected cells and that Mta enhances complete productive KSHV lytic reactivation stimulated by Rta in PEL cultures. A putative A/T hook domain within Mta contributes to DNA binding and transactivation. Our data suggest that Mta synergizes with Rta using, at least in part, a transcriptional mechanism.
MATERIALS AND METHODS
Plasmids and bacmids.
All plasmids were propagated and purified as described previously (55). The plasmids described previously were the following: pcDNA3-FLg50, pcDNA3.1-ORF57-Hygro (genomic ORF57), pGL3-nut1 (−1467), pGL3-B3 (kaposin), pGL3-TK, pcDNA3.1-lacZ (57), pcDNA3-FLc50, pGem3-FLc50, pGem3-50ΔSTAD, pORF57-GL3, pK-bZIP-GL3 (56), pGL3-nut1 (−706; wild type [WT] and mutant) and pcDNA3-FLc57 (45), the ori-Lyt (L) reporter plasmid 13F (101), and pGem3-50ΔLR (10).
Reporter plasmids pNut-1 (−131)-GL3 and pNut-1 (−73)-GL3 contain nut-1/PAN promoter sequences spanning positions −131 to +24 and −73 to +24, respectively. Both were subcloned as PCR products synthesized with Phusion Polymerase (New England Biotechnology), a common reverse primer (5′-GCGAGATCTAGCCAAGGTGACT-3′), and either −131 forward primer (5′-GCGAGATCTGGATTATTAAGGGT-3′) or −73 forward primer (5′-GATCTAAAATGGGTGGCTAACCTGTCCAAAAA-3′). The WT or mutant Nut-1-GL3 (−706) was used as a template to generate the respective WT or mutant versions of the nut-1/PAN reporter plasmids. The PCR products were digested with BglII and cloned into pGL3-basic (Promega) that had been digested with BglII (for the −131 reporter) or SmaI/BglII (for the −73 reporter).
Reporter plasmid pNut-1 (−1467)-GL3-mutant was constructed by subcloning the insert from pNut-1 (−706)-GL3-mutant to pNut-1 (−1467)-GL3.
Reporter plasmid Pnut46-hspluc was constructed as described previously (55) by annealing two oligonucleotides (5′-GATCTTTCCAAAAATGGGTGGCTAACCTGTCCAAAATATGGGAACACTGGAA-3′ and 5′-GATCTTCCAGTGTTCCCATATTTTGGACAGGTTAGCCACCCATT TTTGGAAA-3′) and then cloning them into the plasmid hsp-luc that had been digested with BglII.
Reporter plasmid p50-1-GL3 was constructed by PCR amplification of nucleotide positions −910 to +50 of the ORF50 promoter, introducing an NcoI site at +50. The PCR product and pGL3-basic were digested with NcoI and joined by ligation.
Reporter plasmid pCMV-Luc was constructed by transferring the Nru I/XhoI fragment containing the cytomegalovirus (CMV) promoter from pcDNA3 (Invitrogen) to pGL3-basic (Promega) that had been digested with SmaI/XhoI.
pGem3-FL50ΔNA was constructed in multiple steps. pGem3-FLc50 was digested with PstI to eliminate extraneous restriction sites in the polylinker downstream of ORF50. The resulting plasmid was digested with NcoI and AvaI to remove the Rta cDNA sequence encoding amino acids (aa) 274 to 483. The deletion was repaired by ligating a double-stranded oligonucleotide linker containing an in-frame fusion between NcoI and AvaI sites. The resulting plasmid expresses the Rta cDNA containing aa 1 to 273 fused in frame to aa 484 to 691.
pGex5x-3-Mta was constructed by PCR amplification of the ORF57 cDNA template using primers that introduced 5′-EcoRI and 3′-XhoI restriction sites. The resulting product was cloned into pGex5x-3 (GE Biosciences) following digestion of the insert and vector with those enzymes.
pGex5x-3-MtaΔA/T was constructed by ligating two PCR amplicons. Amplicon 1 corresponded to Mta ORF nucleotides 463 to 1371 with a 3′ AgeI restriction site introduced by one primer. Amplicon 2 corresponded to Mta nucleotides 1 to 335, with a 5′ AgeI site introduced by one primer. Amplicon 1 was digested with SphI/Age I, amplicon 2 was digested with Age I/KpnI, and both were ligated with pGex5x-3-Mta that had been digested with SphI/KpnI.
pcDNA3-c57ΔA/T was constructed by generating a composite PCR amplicon of AgeI-digested amplicons 1 and 2 (see above), facilitated by the complementary AgeI overhangs in each amplicon. The resulting amplicon was digested with PshAI/XhoI (the XhoI site was introduced by one outside primer) and cloned into pcDNA3-FLc57 that had been digested with the same enzymes.
pET28a-Mta was constructed by PCR amplification of the full-length Mta cDNA using primers that introduced EcoRI/XhoI restriction sites, followed by cloning it into those sites in the pET28a vector (Novagen).
pET28a-MtaΔA/T was constructed by cloning the EcoRI/XhoI fragment of pGex5x-3-MtaΔA/T into pET28b that had been digested with the same enzymes.
pRSET A-RBP-Jk was constructed by PCR amplification of the complete human RBP-Jk cDNA (48), using primers that introduced a 5′ BamHI restriction site and a 3′ PstI restriction site. The PCR product and pRSET A (Invitrogen) were digested with those enzymes and joined by ligation.
pGem-7SK (71) encodes the 7SK cDNA (a gift of Hua Zhu).
pCR2.1-nut-1 (+) contains the TA-cloned nut-1/PAN cDNA product of PCR amplification.
BAC36 is the full-length clone of KSHV described previously (112).
Cell lines and transfections.
HH-B2 cells, a gift of George Miller, were propagated as described previously (32). HH-B2 and BCBL-1 cells were transfected by electroporation, as previously described (57), at 200 V and 150 V, respectively.
CV-1 cells were a gift of Harvey Ozer and were propagated in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 290 μg/ml glutamine, and antibiotics (penicillin/streptomycin [10 U/ml and 10 μg/ml, respectively] or 50 μg/ml gentamicin). CV-1 cells were transfected using TransIt LT-1 (Mirus) according to the manufacturer's suggestions, with a total of 2.5 μg of DNA, which included 0.25 μg each of luciferase reporter and pcDNA3.1-lacZ for quantitating transfection efficiency.
293 cells (a gift of Abraham Pinter) were cultured as described previously (12) and transfected by the calcium phosphate method. The cells were plated at a density of 5 × 104 per well of a six-well plate. One hundred microliters of transfection buffer (consisting of 10 μl of 0.5 M HEPES, 81 μl distilled H2O [dH2O], 9 μl 2 M NaCl, 0.2 μl 1 M Na2HPO4) was mixed with 100 μl plasmid mixture (5 μg total DNA, 10 μl 10× NTE [150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, pH 8.0], 12.5 μl 2 M CaCl2, and dH2O to a final volume of 100 μl). The mixture was incubated at room temperature for 15 to 30 min and then added to the cells, and the plates were returned to the incubator. Twelve to 15 h later, the media were changed and the plates were returned to the incubator for 48 h.
All KSHV-negative Burkitt's and plasma cell lymphoma cell lines (BJAB, BL-41, RPMI8226, U266, and Akata-31) were electroporated at a density of 1 × 107 cells/electroporation, with 23 μg total DNA, which included 5 μg of luciferase reporter plasmid, 3 μg of pcDNA3.1-lacZ for quantitating electroporation efficiency, and titrations of Rta or Mta expression vectors. All were cultured identically to BJAB cells, as described previously (56). BJAB cells were electroporated at 150 V/0.975 μF. BL-41 cells (a gift of Y. Yuan [University of Pennsylvania]) were electroporated at 224 V/0.975 μF.
RPMI8226 and U266 (American Type Culture Collection) were electroporated at 200 V/0.975 μF and 175 V/0.975 μF, respectively. Akata-31 cells, an EBV-negative subclone of Akata cells, were a gift of Paul Farrell (40). They were electroporated at 200 V/0.975 μF.
Luciferase assays.
Luciferase assays were performed exactly as described previously (55). Luciferase activity was normalized for each transfection using β- galactosidase activity as an internal control for all cell lines except 293 (in which Mta transactivated the β-galactosidase plasmid). For all luciferase assays, corresponding transfections were performed in triplicate at least twice.
Luciferase RNA stability.
293 cells were cotransfected with 3.0 μg of the Mta expression vector or empty vector (as indicated) and the reporter plasmid pCMV-Luc or pGL3-nut1 (−706). On the following day, the cells were washed with 1× phosphate-buffered saline, half were treated with actinomycin D (ActD) (5 μg/ml; Sigma), and the cells were returned to the incubator for 18 additional hours. Total RNA was isolated using RNABee (Tel-Test), and 15 μg RNA from each transfected dish was analyzed by Northern blotting and quantitated with a phosphorimager.
Viral reactivation.
HH-B2 cells (107) were stimulated with tetradecanoyl phorbol acetate (TPA) and/or transfected with the plasmids indicated. After growth in 10 ml complete RPMI for 6 days, virions were harvested from the supernatant, and encapsidated viral DNA was quantitated by Southern blotting as described previously (44). The probe was derived from KSHV ORF6 (56). Alternatively, BCBL-1 cells were transfected with expression vectors for ORF50/Rta and/or ORF57/Mta and analyzed for reactivation by immunofluorescence, as described previously (10). At least 500 Rta-positive cells were screened.
Northern blotting.
Northern blotting was performed as described previously (56). Double-stranded probes were the EcoRI fragment of pCR2.1-nut-1 (+) (for nut-1/PAN), the AccI/KpnI fragment of pGem-7SK (7SK), and the NcoI/XbaI fragment of pGL3-basic (luciferase).
Nuclear run-on assays.
293 cells were seeded in six-well dishes and transfected with pNut-1 (−1467)-GL3 alone or together with pcDNA3.1-ORF57-Hygro. Forty-eight hours following transfection, the cells were washed twice with 1× PBS, scraped into 15-ml conical tubes, and pelleted by centrifugation for 5 min at 1,000 rpm. The cells were resuspended in three times the pellet volume of 10 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 10 mM NaCl, 0.1%NP-40, 0.5 mM dithiothreitol and vortexed gently for 5 to 10 seconds. Nuclei were pelleted at 1,000 × g and frozen in 27 mM Tris, pH 7.5, 13 mM MgCl2, 160 mM KCl, and 27% glycerol. ATP, CTP, GTP (0.5 mM final concentration [each]), and digoxigenin (DIG)-11-UTP (6 μl; Roche Applied Science) were added to the thawed nuclei, and transcription was continued by incubation at 37°C for 20 min. Ten microliters of RQ1 DNase (Promega) and 1 μl of 100 mM CaCl2 were added to the nuclei and incubated at 37°C for 30 min. One milliliter of RNABee (Tel-Test, Inc.) and 100 μl of chloroform were added to lyse the cells. The tubes were kept on ice for 5 min and then microcentrifuged at 10,000 rpm for 20 min. RNA was precipitated with an equal volume of isopropanol. Six micrograms of pGL3-basic or pGem-7SK plasmid DNA was denatured and suctioned onto a Hybond XL membrane (GE Biosciences) in a dot-blotting apparatus. Following UV cross-linking, the blot was prehybridized for 2 h in 20 mM TES [N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid], pH 7.4, 400 mM NaCl, 4 mM EDTA, 200 μg/ml yeast RNA, 2× Denhardt's solution, and 0.1% sodium dodecyl sulfate (SDS). The total RNA that had been purified from the nuclei of each transfected cell sample was added and hybridized overnight at 65°C. The following day, the blot was developed using the DIG Luminescent Detection Kit for Nucleic Acids (Roche Applied Science).
Protein expression and purification.
Glutathione-S-transferase (GST)-Mta protein was expressed by inoculating 50 ml of LB (containing 50 μg/ml ampicillin) with a glycerol stock of Rosetta Escherichia coli (Novagen) containing pGex5x-3-Mta. Following 12 to 15 h of growth at 37°C with agitation, the culture was diluted into 450 ml of fresh LB (supplemented as described above), and growth was continued at 37°C until the culture reached an optical density of 0.6. The flask was transferred to ice for 20 min, after which 0.9 mM isopropylthio-β-d-galactosidase (IPTG) was added. The culture was grown at 16°C for 18 additional hours with agitation. Bacteria were pelleted by centrifugation and resuspended in 10 ml 1× NETN+ (20 mM Tris buffer, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.1% Nonidet P-40, 10% glycerol, 5 mM β-mercaptoethanol, 0.1% Triton X-100, and protease inhibitors [Sigma]). The bacteria were lysed for 10 cycles by sonication using a large tip and 15% power for 8 seconds, and the supernatant was clarified by centrifugation for 30 min at 7,000 rpm. The supernatant was aliquoted and flash frozen in a dry-ice/ethanol bath and then transferred to −80°C storage for future use. At the time of use, aliquots were thawed slowly on ice, and protein was purified by the addition of 40 μl glutathione-Sepharose (Sigma; 50% [vol/vol] in NETN+)/ml, followed by nutation for 1 hour at 4°C and washing with 7 column volumes of fresh NETN+. The GST moiety alone was expressed and purified similarly using the plasmid pGex5x-3.
His6-Mta and His6-RBP-Jk proteins were purified as described above with the following modifications: inocula were glycerol stocks of E. coli BL21(DE3)-RIL (Stratagene) containing the respective plasmids. Ten milliliters of saturated culture was diluted into 500 ml of fresh LB (supplemented as described above), and protein was induced for 18 to 48 h. The bacterial pellet was resuspended in NTN+ (10 mM Tris buffer, pH 7.5, 100 mM NaCl, 0.5% Nonidet P-40, and protease inhibitors [Sigma]) and purified by liquid chromatography over a Ni-nitrilotriacetic acid-agarose column (QIAGEN; 1-ml bed volume, preequilibrated in NTN+). The beads were washed with 10 ml modified NTN+ and eluted stepwise in five fractions of 0.5 M imidizole in NTN+. Fractions containing either of the His6 proteins (identified by Western blots) were combined and dialyzed versus 1× DNA binding buffer (55).
Generation of anti-Mta serum.
Anti-Mta serum was generated exactly as described previously (57), using GST-Mta as an antigen.
GST pull-down assays.
Equal amounts of GST-Mta or GST protein, respectively (determined by Bradford analysis), were purified on GST-Sepharose (as described above). Beads were resuspended in 0.5 ml NETN+ and combined with equal volumes of rabbit reticulocyte lysate (RRL) (TNT Quick Coupled Translation kit [Promega]) that had been programmed in the presence of [l-35S]methionine with plasmids expressing Rta or mutants. Following incubation at 4°C with nutation, the beads were washed twice with 0.5 ml NETN+. The beads were mixed with 30 μl of 2× Laemmli buffer, boiled, and separated by SDS-polyacrylamide gel electrophoresis (PAGE). The gels were fixed in 10% acetic acid-50% methanol-40% dH2O, the signals were amplified by incubation in 0.1 M salicylic acid, the gels were dried, and the proteins were visualized by a PhosphorImager (Molecular Dynamics).
Immunoprecipitations and Western blotting.
BCBL-1 cells were induced with TPA (20 ng/ml) for 12 to 15 h or transfected with the indicated plasmids, and immunoprecipitations were performed as described previously (61). Anti-Mta and -Rta antisera were conjugated to horseradish peroxidase using EZ-Link Plus Activated Peroxidase (Pierce) prior to incubation with the indicated blots. Anti-alpha-actinin antibody was purchased from Sigma.
EMSAs.
Purified proteins were mixed with 1x DNA binding buffer (55) and incubated on ice for 15 min. All electrophoretic mobility shift assay (EMSA) reaction mixtures contained 10 ng of unlabeled salmon sperm DNA (50-fold molar excess over labeled DNA probe; Sigma) as a nonspecific competitor. Probe (1 × 105 cpm per reaction) labeled as described previously (12) was added and incubated for 15 min at 16°C. DNA-protein complexes were resolved by electrophoresis on 7.5% polyacrylamide-0.5× Tris-borate-EDTA gels at 4°C, as described previously (12). The gels were transferred to Whatman paper, dried, and exposed to Bio-Max MS autoradiography film (Kodak).
ChIP.
Chromatin immunoprecipitation (ChIP) was performed as described previously (12), with the exception that chromatin was cross-linked at 40 h after TPA addition. DNA was resuspended in 100 μl of double-distilled H2O, and 5 μl was used in real-time PCRs.
Real-time PCR.
Real-time PCR was performed using AmpliTaq Gold polymerase (Applied Biosystems) and a Corbett RotorGene 3000 instrument according to the manufacturer's suggestions. Cycling parameters were 5 min at 95°C and then 40 to 60 cycles of 20 s at 95°C/30 s at 55°C/40 s at 72°C.
Primer sequences (5′ to 3′) were as follows: Nut-1/PAN (forward, GTTTTCTTATGGATTATTAAGGGTC, and reverse, AGGTGAAGCGGCAGCCAAGGTGAC) and K6 (forward, CGCCTAATAGCTGCTGCTACGG, and reverse, TGCATCAGCTGCCTAACCCAG).
The ΔΔCt method was used (51) for quantitation, with the following modifications: ΔCt was calculated individually for each primer pair, for cells treated with TPA, and for cells left untreated, using the following formula: ΔCt = Ct (IP) − Ct (input chromatin), in which Ct is “threshold cycle” and IP is immunoprecipitated chromatin. Next, ΔΔCt was calculated using the following formula: ΔΔCt = ΔCt (for untreated cells) − ΔCt (for TPA-treated cells). Enrichment (n-fold) was thus calculated as 2−ΔΔCt.
Design and synthesis of molecular beacons.
Design and synthesis of molecular beacons were performed as described previously (12). Molecular-beacon sequences were as follows (lowercase letters denote stems, and uppercase letters denote regions complementary to the genomic sequences of the indicated genes): nut-1/PAN (cgctcgGTTAATGACATAAAGGGGCGTGGcgagcg) and K6 (cccctccCACCCACCGCCCGTCCAAATTCggagggg).
Immunofluorescence.
Immunofluorescence was determined exactly as described previously (57). Secondary antibodies raised against rabbit immunoglobulin (Ig) were tetramethyl rhodamine isothiocyanate (TRITC) conjugated, and those against mouse Ig were fluorescein isothiocyanate (FITC) conjugated (ICN).
RESULTS
ORF57/Mta and ORF50/Rta synergize to reactivate KSHV from latency.
Using firefly luciferase as a reporter gene in transiently transfected CV-1 cells, we previously reported that ORF57/Mta and ORF50/Rta synergized to transactivate the KSHV Nut-1/PAN and kaposin promoters (45). To determine whether Rta and Mta also synergized to reactivate KSHV from latency, we measured reactivation by ectopically expressing both proteins in latently infected PEL cells. First, we harvested virions from the media of transfected HH-B2 cells and measured encapsidated viral DNA using Southern dot blots. As shown in Fig. 1A, virus released from cells transfected with the empty expression vector was barely detectable, similar to untransfected HH-B2 cells. Mta expressed alone resulted in a slight increase in virus detected in the supernatant. Rta expressed alone, as expected, resulted in about a fivefold increase in virus induction relative to the empty vector. Cotransfection of the Mta and Rta expression vectors, however, yielded a synergistic induction (approximately 18-fold) of productive reactivation of KSHV from latency.
FIG. 1.
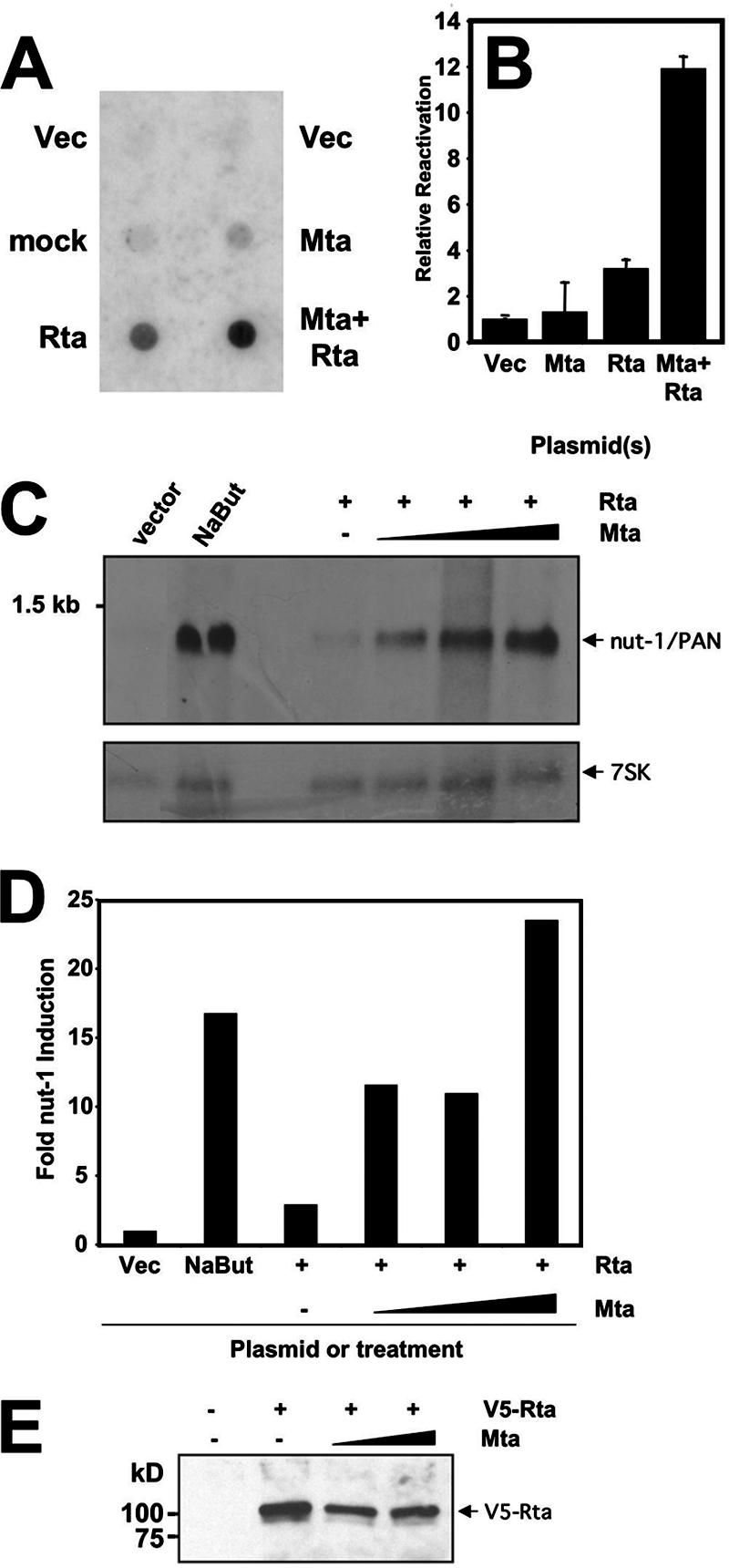
Mta cooperates with Rta to reactivate KSHV from latency. (A) Virion production. HH-B2 (PEL) cells were electroporated with the indicated plasmids or without DNA (mock). Virion-associated DNA was purified 6 days postelectroporation, dot-blotted, and probed with 32P-labeled ORF6 DNA. Signal was detected by autoradiography. Vec, pcDNA3 plasmid. (B) Late protein production in single cells. BCBL-1 (PEL) cells were electroporated in duplicate with the indicated plasmids; 48 h postelectroporation, the cells were analyzed by indirect immunofluorescence to detect expression of the KSHV proteins ORF50/Rta and K8.1. At least 500 cells were quantitated manually by fluorescence microscopy. The percentage of Rta-positive cells that were also K8.1 positive was determined for each transfection; the value calculated for vector-transfected cells (Vec; pcDNA3, i.e., spontaneously reactivating cells) was subtracted from each of the others, and the resulting difference is graphed. The error bars indicate standard deviations. (C and D) Delayed early transcript production. HH-B2 cells were electroporated with the indicated plasmids or treated with sodium butyrate (NaBut), and total RNA was purified 24 h postelectroporation. RNA was analyzed by Northern blotting, sequentially probing for nut-1/PAN or 7SK transcripts. Signals were quantitated by phosphorimager; nut-1/PAN signals were normalized to 7SK signals and are graphed in panel D. (E) Mta does not stimulate Rta expression from a plasmid in infected cells. BCBL-1 cells were electroporated with expression plasmids for V5-Rta (10 μg) and Mta (2.5 or 10 μg), alone or together. Proteins immunoprecipitated by the V5-specific antiserum were analyzed by Western blotting using the V5 antiserum as the primary probe.
We previously used a single-cell reactivation assay to demonstrate that Rta was necessary and sufficient to reactivate KSHV from latency (56, 57). In those assays, our marker for induction of reactivation was the K8.1 protein, which is expressed in PEL cells with true late kinetics (i.e., K8.1 expression requires viral DNA replication) (54, 57, 107). To test Mta and Rta for synergistic induction of the KSHV lytic cycle, we scored reactivation similarly 48 h after electroporating BCBL-1 cells. Cells were scored positive if Rta expression was detected by visual inspection by fluorescence microscopy; for quantitation, we counted the percentage of Rta-positive cells that were also K8.1 positive (Rta+/K8.1+ divided by Rta+). At least 500 doubly positive cells were counted for each. Cells transfected with empty expression vector were scored similarly. Results from the empty-vector transfections (spontaneous reactivation) were subtracted from those in which the Mta or Rta vectors were transfected, and the results were plotted as relative reactivation. As shown in Fig. 1B, ectopic expression of Rta alone resulted in a slight increase in the number of doubly positive cells relative to vector-transfected cells. However, cotransfection of the Mta and Rta vectors resulted in a dramatic induction of viral reactivation. Ectopic expression of Mta alone did not produce a significant increase in Rta/K8.1-positive cells, suggesting that Mta alone is unable to induce Rta expression or complete viral reactivation in PEL cells. When these cells were scored by immunofluorescence for Mta/K8.1 doubly positive cells, the data also showed no difference in progression to K8.1 expression, comparing transfected vector to Mta-expressing cells (not shown). This single-cell assay agreed with Fig. 1A in showing that ectopic expression of Mta alone has little effect on viral reactivation but potently synergizes with ectopically expressed Rta.
Induction of nut-1/Pan expression by Rta has been used previously to measure viral reactivation (92). We repeated the approach shown in Fig. 1A, but we isolated total RNA from transfected HH-B2 cells 24 h after electroporation. As shown in Fig. 1C, vector-transfected cells showed little nut-1/PAN expression, while treatment with sodium butyrate robustly induced nut-1 expression (approximately 17-fold [Fig. 1D]). Ectopic expression of Rta alone resulted in about a threefold induction of nut-1 expression. Cotransfection of the same amount of Rta vector with increasing amounts of Mta vector demonstrated a dramatic, dose-responsive synergy of the two proteins, from 12- to 24-fold relative to vector-transfected cells. To confirm that ectopic Mta was not simply transactivating ectopic Rta expression, we coelectroporated BCBL-1 cells with the Mta vector and an expression vector for Rta fused to the V5 epitope so that we could distinguish ectopic Rta from Rta expressed from the endogenous virus. As shown in Fig. 1E, Mta did not increase ectopic Rta expression under the conditions used for Fig. 1A to D. Taken together, Fig. 1 demonstrates, using three independent assays, that Mta and Rta synergized posttranslationally to reactivate KSHV from latency.
Mta and Rta proteins are expressed with similar kinetics during KSHV reactivation in PEL cells.
Many studies have examined the kinetics of ORF57/Mta transcript expression during KSHV reactivation (22, 54, 56, 75), but relatively little is known regarding the kinetics of ORF57/Mta protein expression. To address this question, cellular protein extracts were prepared from KSHV-infected BCBL-1 cells at various times during viral reactivation following addition of TPA to the cultures. As seen in the upper part of Fig. 2A, Mta protein was detectable as a 49-kDa major species within 4 h following TPA treatment, slightly smaller than the predicted molecular mass for ORF57/Mta of 51.1 kDa (ExPASY). In other experiments, we have detected Mta protein as early as 1 h following TPA treatment (data not shown). Two minor species migrating with smaller apparent masses than the major Mta protein were also detected. Since Mta protein was not expressed in untreated, latently infected cells, these data agreed with previous classifications of Mta as a lytic viral protein. Rta protein was detectable prior to Mta protein, but both proteins increased in abundance until 24 h after TPA addition (Fig. 2A). By 48 h post-TPA, both Mta and Rta proteins were barely detectable by Western blotting (data not shown). Mta and Rta proteins were thus expressed with kinetically similar patterns during reactivation, but Rta protein expression preceded that of Mta, similar to expression of their transcripts (56).
FIG. 2.

ORF57/Mta expression is tightly linked to K8.1 expression at the single-cell level. (A) Kinetics of ORF50/Rta and ORF57/Mta protein expression are similar but distinct. BCBL-1 (PEL) cells were treated with TPA or left untreated (0), and total protein was harvested at the indicated times after TPA addition (hpi, hours postinduction). Equivalent amounts of total protein for each time point (as determined by Bradford assay) were displayed by SDS-PAGE and then transferred to nitrocellulose. The membrane was analyzed by immunoblotting it sequentially with the indicated primary antibodies. Alpha-actinin served as a loading control. (B) ORF57/Mta is expressed in the nucleus in reactivating PEL cells. BCBL-1 (PEL) cells were treated with TPA for 24 h and then analyzed by indirect immunofluorescence using primary antibodies specific for Mta or K8.1 and secondary antibodies conjugated to FITC or TRITC, respectively. DNA was stained with DAPI for visualization of nuclei. Fluorescent signals were digitally converted to grayscale. (C) The percentage of reactivating cells detected by Rta or Mta expression increases with kinetically distinct patterns during reactivation. BCBL-1 (PEL) cells were analyzed as in panel B using primary antibodies specific for Rta or Mta in parallel cultures and secondary antibody conjugated to FITC. The percentage of total cells expressing either Rta or Mta was quantitated at the indicated times post-TPA addition. The error bars indicate standard deviations. (D) ORF57/Mta expression is tightly linked to K8.1 expression at the single-cell level. The cells shown in panel C were simultaneously analyzed by immunofluorescence using primary antibody specific for K8.1 and secondary antibody conjugated to TRITC. The number of Mta/K8.1 double-positive cells was divided by the number of Mta single-positive cells and plotted. The calculation and plot were computed for Rta and K8.1 identically.
Figure 2B, left, shows that the Mta-specific antiserum detected Mta exclusively in the nucleus in reactivating BCBL-1 cells (compare to Fig. 2B, right, DNA labeled with DAPI [4′,6′-diamidino-2-phenylindole]). Figure 2B, center, serves as a reference for cytoplasmic localization of the K8.1 glycoprotein.
To evaluate single-cell expression of Mta and Rta, we performed indirect immunofluorescence assays of tandem populations of BCBL-1 cells untreated or induced to reactivate with TPA. Approximately 1,000 cells were scored, in duplicate or triplicate, and the percentage of the total cells expressing each protein was determined. As shown in Fig. 2C, both Mta and Rta were detected in less than 1% of latently infected cells, representing the small population of cells that spontaneously reactivated the virus. At 24 h post-TPA addition, the percentages of Mta- and Rta-expressing cells remained identical. From 24 to 72 h post-TPA addition, the percentages of cells expressing Rta and Mta continued to increase, although at different rates. The percentage of Rta-positive cells increased linearly up to 72 h post-TPA in nearly 20% of reactivating cells. However, over the same time course, the rate of increase of Mta-expressing cells steadily declined and reached a plateau of about 12% of the population by 72 h post-TPA. Importantly, the total number of cells per ml of culture did not change significantly over the time course of this experiment. Since Western blotting showed that expression of both proteins was barely detectable at 48 h (data not shown), total Mta or Rta protein was not proportional to the total number of cells expressing each protein.
Expression of Mta is tightly associated with complete viral reactivation.
In Fig. 1, we demonstrated that ectopic coexpression of Mta and Rta resulted in synergistic induction of KSHV reactivation; we quantitated expression of the true late protein K8.1 as one measurement of synergistic reactivation. To compare coexpression of K8.1 with Mta or Rta when expressed in single cells from the endogenous viral genome, we performed indirect immunofluorescence assays in BCBL-1 cells uninduced or induced by TPA treatment. As shown in Fig. 2D, about 50% of untreated cells that expressed either Rta or Mta, representing the spontaneously reactivating population, also expressed K8.1. At 72 h post-TPA treatment, nearly 80% of Mta-expressing cells also expressed K8.1, while never more than about 20% of Rta-expressing cells expressed K8.1. Although Rta is necessary and sufficient to reactivate KSHV, these data suggest that expression of Mta predicted successful, full progression through the lytic gene cascade. Therefore, the expression of Mta seems to be required to commit a reactivating cell to the entire lytic cascade.
The RRE is necessary but not sufficient for Mta to synergize with Rta in transactivation of the Nut-1/PAN promoter.
To determine the function of Mta, we initially cotransfected CV-1 cells with increasing amounts of Mta or Rta, alone or together, and a plasmid containing the KSHV nut-1/PAN (−1463) promoter driving luciferase expression as a reporter (Fig. 3A shows a promoter schematic). The magnitude of transactivation for each was determined by comparison to transfection of the reporter with empty expression vector. Rta transactivated the nut-1 promoter to about 22-fold, but Mta had very little effect on the promoter alone (Fig. 3B). These effects of Rta and Mta alone were very similar to their effects on viral reactivation, as shown in Fig. 1. As we showed previously (45), when Rta and Mta were coexpressed, the two proteins dramatically synergized to transactivate the nut-1 promoter (Fig. 3B). In this experiment, the synergistic activation reached nearly 3,700-fold over basal promoter activity (and 143-fold greater than Rta transactivation alone). The optimal amounts of each expression vector (0.5 μg Rta expression vector and 3 μg of Mta expression vector) were identical to that which we previously published (45).
FIG. 3.

Mta and Rta synergize to transactivate the nut-1/PAN promoter in a sequence-specific fashion. (A) Schematic of the nut-1/PAN promoter. The double line at the top shows the nut-1/PAN promoter, extending to position −1467 from the transcriptional start site, which was cloned into the firefly luciferase reporter vector. The lines below the promoter represent the deletion mutants of the promoter cloned similarly. Nut-1-46 is the 46-bp region cloned into the heterologous reporter plasmid hsp-luc. EMSA DNA represents the double-stranded DNA used in the EMSA shown in Fig. 10B. The boxed letters along the promoter schematic represent relative locations of consensus cellular protein binding sites as determined by searching TransFac (105) at high stringency or published previously (48, 100). M, c-myc; A, AP-1; C, CAAT box; R, RBP-Jk; O, Oct-1; S, Sp1. (B) ORF50/Rta and ORF57/Mta synergize to transactivate the nut-1/PAN promoter. CV-1 cells were cotransfected with pGL3-nut1 (−1467) and empty pcDNA3 vector (Vector) or increasing amounts of pcDNA3.1-FLg50 plasmid (0 to 4 μg) to determine the amount of ORF50 plasmid that yielded the greatest magnitude of transactivation relative to empty vector. The experiment was then repeated using increasing amounts of pcDNA3.1-ORF57 Hygro alone (0 to 3 μg) or together with the optimal amount of pcDNA3.1-FLg50 (0.5 μg). The maximal amounts of transactivation for each condition are plotted; the error bars indicate standard deviations. pcDNA3.1-His-lacZ was cotransfected in all experiments to normalize transfection efficiency by determination of β-galactosidase activity. (C) Alignment of RREs from KSHV promoters. RREs from the indicated promoters were aligned with the mutant RRE analyzed in panel D and Fig. 6. (D) The RRE is necessary but not sufficient for Mta-Rta synergy. The experiments shown in panel B were repeated with each of the nut-1/PAN reporter vectors shown. The maximal amount of synergy of cotransfected Mta and Rta expression plasmids was divided by the amount of transactivation by Rta alone and graphed.
We and others have previously described an RRE in the nut-1/PAN promoter (15, 45, 91). Mutation of that element by replacing 9 of its 13 bp severely impaired activation of the nut-1/PAN promoter by Rta, and coexpression of Mta had no additional effect (45). The 13-bp element is conserved in the kaposin/ori-Lyt (R) and ori-Lyt (L) promoters (5, 15, 89, 101); Fig. 3C shows alignment of the three elements with the mutant nut-1/PAN element.
To characterize the cis requirements for Mta/Rta synergy on the nut-1/PAN promoter, we tested a series of promoter deletion mutants in transfected CV-1 cells. Figure 3D shows that deletion of the nut-1 promoter from −1467 to −706 resulted in an enhancement of Mta/Rta synergy, from 143-fold to 243-fold. Additional deletions of the promoter to −131 and −73 resulted in sequential reduction of synergy (71- and 22-fold, respectively). The deletion to −73 truncates the promoter just upstream of the RRE (Fig. 3A). These reductions in synergy were independent of the magnitude of Rta transactivation (data not shown). Thus, synergy was generally proportional to the size of the promoter, except for deletion of the sequences between −1467 and −706, which enhanced synergy.
As previously demonstrated, mutation of the 13-bp RRE (Fig. 3C) completely eliminated Rta activation and Mta-Rta synergy (Fig. 3D, Nut-1-1467 mut), affirming that the RRE is necessary for Mta-Rta synergy. To determine whether the RRE was sufficient for Mta-Rta synergy, we cloned the 46-bp DNA that included the RRE, the putative TATA box, and the intervening sequences (Fig. 3A) upstream of the heterologous hsp70 promoter. Although the resulting promoter was well activated by Rta alone (27-fold) (not shown), Mta did not synergize with Rta in this context. Therefore, the nut-1/PAN RRE was necessary but not sufficient for Mta-Rta synergy. Instead, sequences upstream and downstream of the RRE seemed to be required for optimal synergy of Mta with Rta.
Mta protein transcriptionally cooperates with Rta in a promoter-specific manner.
We extended these studies in CV-1 cells by testing six other viral promoters in a fashion similar to that for Fig. 3 (shown in Fig. 4). Rta, but not Mta, activated each of these promoters alone in CV-1 cells (data not shown). Figure 4 shows the level of additional activation over Rta alone when Mta was coexpressed. Among these promoters, those from ori-Lyt (L) and kaposin/ori-Lyt (R) were synergized to the highest magnitudes, 38- and 24-fold, respectively. Synergistic activations of the other promoters, in declining amounts, were 13.8 for ORF57, 13.3 for TK, 6.8 for K-bZIP, and 2.8 for ORF50. Mta-Rta synergy thus differs by promoter. Synergistic activation of the promoters shown in Fig. 4 never reached the magnitude of synergy of the nut-1/PAN −706 promoter (243-fold) (Fig. 3B). However, the two promoters that were preferred for Mta/Rta synergy were those that share the 13-bp homology with nut-1/PAN [i.e., ori-Lyt (L) and kaposin] (Fig. 3C).
FIG. 4.
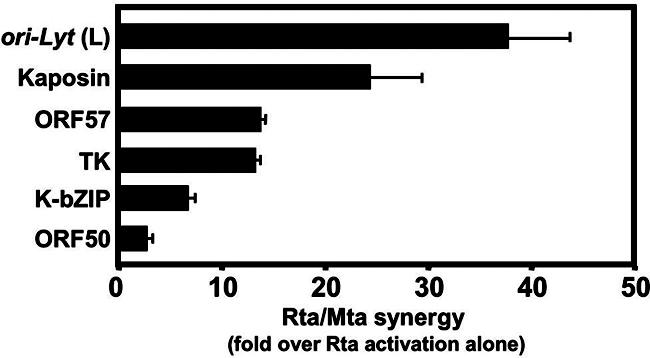
Mta and Rta synergize in a promoter-specific fashion. Each of the indicated reporter plasmids was analyzed in transfections of CV-1 cells as in Fig. 3D. The maximal amount of synergy of cotransfected Mta and Rta expression plasmids was divided by the amount of transactivation by Rta alone and graphed. The error bars indicate standard deviations.
Independent transactivation by Mta, and synergy with Rta, is cell line specific.
Vero cells support productive reactivation of KSHV (37, 96), so it is likely that CV-1 cells would also be permissive. However, to determine Mta's function in an uninfected cell line that is more similar to human B cells, the natural KSHV reservoir, we repeated the CV-1 experimental strategy (as in Fig. 3 and 4) in a series of human cell lines. Each was transfected with a range of plasmid amounts, and the maximal value for Rta alone was quantitated. Maximal synergy was then quantitated by cotransfecting the optimum amount of Rta expression vector with a range of Mta expression vector.
The top row of Table 1 shows the values for independent and synergistic transactivation of the nut-1/PAN promoter by Mta and Rta in CV-1 cells that were graphed in Fig. 3B. The next two rows show results from two Burkitt's lymphoma cell lines, BL-41 and BJAB. Similar to CV-1 cells, Rta transactivated the nut-1 promoter in both cell lines, while Mta did not. However, as shown in the right column, Mta synergized with Rta only in BL-41, but not BJAB, cells. The synergistic effect in BL-41 cells was quantitatively similar to that in CV-1 cells. Clearly, there was a cell-specific difference in synergy in B cells.
TABLE 1.
Transcriptional Synergy between Rta and Mta is cell line specific
| Cell line | Transactivationa
|
||
|---|---|---|---|
| Rta | Mta | Rta + Mta/Rta aloneb | |
| CV-1 | 22.0 (3.5) | 2.9 (0.3) | 142.7 (8.1) |
| BL-41 | 30.8 (6.6) | 2.4 (0.7) | 129.5 (29.8) |
| BJAB | 25.9 (1.6) | 1.4 (0.1) | 1.4 (0.1) |
| RPMI 8226 | 9.1 (6.3) | 1.4 (1.0) | 85.5 (20.0) |
| U266 | 188.5 (9.5) | 4.2 (1.0) | 30.5 (4.0) |
| Akata-31 | 47.9 (18.0) | 87.7 (0.5) | 33.6 (4.9) |
| 293 | 64.6 (9.9) | 20.0 (0.8) | 15.1 (0.1) |
The numbers represent activation (n-fold) of the nut-1/PAN (−1467) promoter by pcDNA3-FLg50 (Rta) or pcDNA3.1-ORF57-Hygro (Mta), alone or together, for each of the cell lines. Standard deviations are shown in parentheses.
Rta-Mta cooperation was calculated as described in the legend to Fig. 3D.
A number of publications have compared the cellular transcriptome of PEL cells to that of a series of lymphoid cell lines representing different stages of B-cell differentiation. These studies found that the PEL transcriptome clusters most closely with that of plasma cell lymphomas or multiple myelomas (42, 47). Therefore, we tested Mta-Rta synergy in the transfected plasma cell lymphoma lines RPMI 8266 and U266. As shown in Table 1, both cell lines reflected our CV-1 data: Rta activated the nut-1 promoter alone, Mta did not, and both proteins synergized. However, the levels of synergy of Rta and Mta were lower than in CV-1 or BL-41 cells. Independent transactivation by Rta was also clearly more robust in U266 cells.
Finally, in Akata-31 and 293 cells, Mta robustly transactivated the nut-1/PAN promoter independently of Rta (Table 1). Rta also independently transactivated the nut-1/PAN promoter in both cell lines. Coexpression of Mta with Rta also enhanced Rta transactivation alone, but the magnitude of cooperation was not synergistic (i.e., it did not exceed the level of transactivation calculated by multiplying the values of independent transactivation by Mta and Rta). Akata-31 cells are a Burkitt's lymphoma line that is a subclone of Akata cells that has been cured of latent EBV (40). 293 cells are human embryonic kidney epithelial cells that have been used extensively for genetic studies of KSHV replication. Together, these data demonstrate that transactivation of the nut-1/PAN promoter by Mta and synergy between Rta and Mta are cell line specific in human cells.
To confirm that Mta was expressed regardless of its ability to synergize with Rta, we compared its expression in transfected BJAB and BL-41 cells by immunofluorescence. As shown in Fig. 5A, Mta was well expressed in the nuclei of both cell lines. Furthermore, Mta did not increase expression of Rta in BL-41 cells transfected under the conditions used for Table 1 (Fig. 5B), demonstrating that Mta-Rta synergy was not the result of Mta increasing the intracellular Rta concentration. Finally, Mta expressed in transfected 293 cells (Fig. 5C) migrated with an apparent mass similar to that in infected PEL cells (Fig. 2A).
FIG. 5.
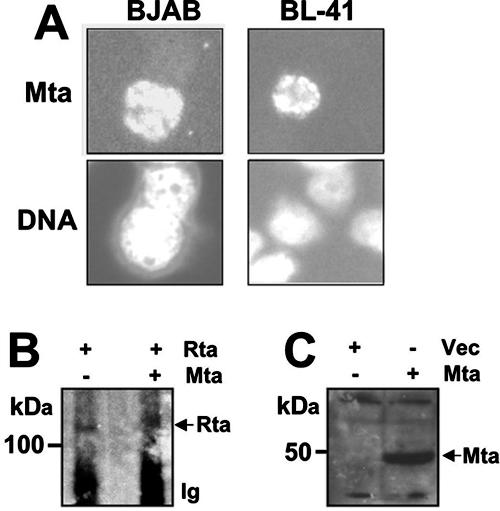
Mta is expressed in BJAB, BL-41, and 293 cells and does not stimulate expression of Rta from a plasmid in uninfected cells. (A) Mta is expressed in the nuclei of BJAB and BL-41 cells. BJAB and BL-41 cells were electroporated with 5 μg of the Mta expression vector, and immunofluorescence was determined as described in the legend to Fig. 2B. (B) Mta does not stimulate expression of Rta from a plasmid in uninfected cells. BL-41 cells were transfected with 5 μg each of expression plasmids for Rta and Mta, alone or together. Proteins immunoprecipitated by the Rta-specific antiserum were analyzed by Western blotting using the Rta antiserum as the primary probe. (C) Mta is expressed in 293 cells. 293 cells were transfected with 3.5 μg of the Mta expression vector or empty expression vector (Vec). Equal amounts of protein were separated by SDS-PAGE and analyzed by Western blotting using the anti-Mta serum.
Independent transactivation by Mta is promoter specific.
One potential mechanism of Mta-Rta synergy is that Rta stimulates an “intrinsic” transactivation function of Mta that is otherwise not observed when Mta is expressed independently. In Akata-31 and 293 cells, an endogenous protein might provide an Rta-like function for Mta to transactivate independently of Rta. Since Mta-Rta synergy was promoter specific (Fig. 3 and 4), we reasoned that our hypothesis would be supported if Rta-independent transactivation by Mta shared the same promoter specificity as Mta-Rta synergy.
Therefore, we tested Mta's promoter specificity for independent transactivation in transfected 293 cells. Figure 6A shows that Mta transactivated the full-length nut-1/PAN promoter and each of its deletion mutants with a preference that mirrored the relative differences in Mta-Rta synergy between each two promoters (i.e., as in Fig. 3D): nut-1 (−706) was activated more strongly by Mta than nut-1 (−1476), but deletion to −131 and −73 caused a gradual decrease in Mta-directed transactivation (Fig. 6A). However, the range of relative Mta transactivation among the four promoters was less pronounced than the range in Mta-Rta synergy (Fig. 3D).
FIG. 6.
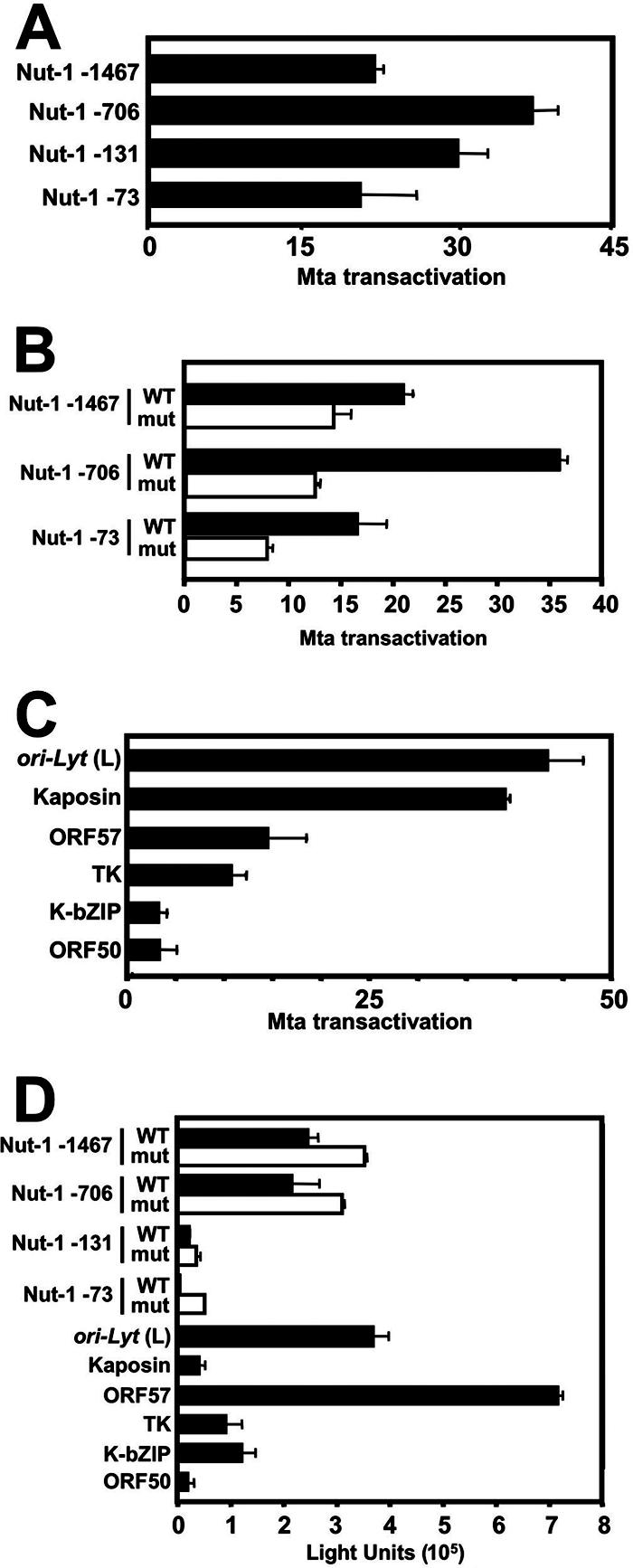
Mta transactivation is promoter specific. (A) Mta transactivation of the nut-1/PAN promoter deletions mirrors Mta-Rta synergy. 293 cells were cotransfected with the indicated reporter vectors and empty pcDNA3 vector or increasing amounts of pcDNA3-FLc57 plasmid. The greatest magnitude of transactivation by ORF57 relative to empty vector is graphed. The error bars indicate standard deviations. (B) Mutation of the RRE reduces but does not eliminate Mta transactivation of the nut-1/PAN promoter. Each of the indicated reporter vectors was tested for Mta transactivation in transfected 293 cells as described for panel A. (C) Promoter-specific transactivation by Mta mirrors promoter-specific synergy by Mta/Rta. Each of the indicated reporter vectors was tested for Mta transactivation in transfected 293 cells as described for panel A. (D) Promoter-specific Mta transactivation is independent of basal promoter activity. Shown are average light units of intrinsic luciferase activity for each of the indicated reporter vectors cotransfected with the empty pcDNA3 vector in 293 cells in the experiments shown in Fig. 6A, B, and C. mut, mutant.
In Fig. 3D, we confirmed our earlier observation (45) that the 13-bp RRE was required for Mta-Rta synergy. To test the effect of the RRE mutation on Rta-independent transactivation by Mta, we compared Mta transactivation of the WT and mutated versions of the full-length nut-1/PAN promoter and two of its truncated versions. As shown in Fig. 6B, the RRE mutation did not completely eliminate Mta transactivation in 293 cells but reduced it from about 33% to 67% in a deletion-specific manner.
Figure 6C shows a similar analysis of Mta transactivation of the six additional KSHV promoters that we tested for Mta-Rta synergy (Fig. 4). The ori-Lyt (L) and kaposin promoters were strongly activated by Mta in 293 cells, to 44- and 39-fold, respectively. The ORF57 and TK promoters were moderately activated, to about 15-fold. The K-bZIP and ORF50 promoters were very weakly activated by Mta, to less than fivefold. Together, Fig. 6 demonstrates that the preference of independent promoter activation by Mta in 293 cells closely mirrored the specificity of Mta-Rta synergy in CV-1 cells. The RRE contributed to Mta transactivation in 293 cells but was not required.
Promoter-specific transactivation by Mta is independent of basal promoter activity.
All of the promoters tested in Fig. 6A to C were cloned into identical reporter vectors (pGL3 basic), each driving expression of the luciferase gene as a reporter molecule. It was conceivable that Mta transactivated the luciferase message posttranscriptionally and that Mta's promoter-specific effect was due to differences in intrinsic (basal) transcription controlled by each promoter. However, this was not the case: the intrinsic activity of each promoter (Fig. 6D) was independent of the magnitude of Mta transactivation shown in Fig. 6A to C. For example, the Nut-1 (−1467) and Nut-1 (−73) promoters were transactivated to nearly identical magnitudes by Mta (Fig. 6A), yet the basal activities of the two promoters differed by about eightfold. Similarly, the ORF57 promoter had the highest basal activity of all of the promoters tested, and the kaposin promoter had nearly the lowest basal activity, yet Mta activated the kaposin promoter to about a threefold-greater magnitude.
Mta enhances transcriptional initiation in 293 cells.
To further distinguish between transcriptional and posttranscriptional transactivation by Mta in 293 cells, we performed nuclear run-on transcription assays in cells transfected with the nut-1/PAN reporter in the absence or presence of increasing amounts of Mta. Run-on transcription assays are the classic method to distinguish transcriptional from posttranscriptional transactivation (85). These assays measure the relative number of RNA polymerase molecules actively transcribing a gene at a defined moment. This is achieved by separating cell nuclei from the remainder of the cell, which prevents active polymerase molecules from reinitiating transcription. By incubating the isolated nuclei in the presence of buffered nucleotides and DIG-11-UTP, only nascent transcripts incorporate DIG.
Total RNA from the nuclei was hybridized to excess transcript-specific DNA cross-linked to a nitrocellulose membrane (Fig. 7A). The label was then quantitated by detection of DIG. We quantitated the luciferase transcripts made from the transfected nut-1/PAN reporter and normalized those values with endogenous 7SK transcripts in duplicate transfections (Fig. 7A and B). As shown in Fig. 7C, nuclei from 293 cells transfected with the Mta expression vector showed a dose-responsive transcriptional transactivation up to 10-fold greater than that of nuclei from cells transfected with reporter plasmid and control empty vector. Figures 7A and B also demonstrate that the cellular 7SK gene was transactivated by ORF57/Mta when 4.5 μg of the Mta vector was transfected. This effect on endogenous 7SK transcription therefore reduced the apparent transactivation of the nut-1/PAN promoter by Mta in the normalized data (Fig. 7C), which otherwise would have been 5.5-fold by 4.5 μg of expression vector. These data suggested that Mta transactivation of the nut-1/PAN promoter in 293 cells occurred in part by enhancing transcriptional initiation, perhaps by a mechanism in which Mta functioned as a bona fide transcriptional transactivator. These experiments, however, do not exclude the possibility that Mta acted indirectly by hypothetically stimulating the abundance or activity of a cellular transcription factor that regulates the nut-1/PAN promoter.
FIG. 7.

ORF57/Mta is a transcriptional transactivator. 293 cells were cotransfected with the pGL3-nut-1/PAN (−1467) plasmid and the empty vector pcDNA3 (Vector) or the indicated amounts of pcDNA3-FLc57 plasmid. Total nuclei were isolated from each transfected-cell population and analyzed by run-on transcription assays as described in Materials and Methods. (A) Each signal for nut-1/PAN transcription is aligned vertically with the corresponding signal for 7SK. (B) Signals were quantitated by phosphorimager and graphed as relative units. (C) Each nut-1/PAN reporter signal was divided by the corresponding 7SK signal. Activation (n-fold) was calculated by dividing each of the resulting ORF57-stimulated values from that of the value from vector-transfected cells alone, which was normalized to 1. The error bars indicate standard deviations.
Promoter-specific Mta transactivation occurs at the levels of transcription and RNA stability.
In Fig. 6D, we showed that promoter-specific transactivation by Mta was independent of intrinsic promoter activity. However, a direct method for testing Mta's effect on reporter transcript stability is to compare transcript accumulation in the presence and absence of the transcriptional inhibitor ActD. Figure 8A shows that when luciferase was expressed from the nut-1/PAN promoter, its transcript was barely detectable when Mta was not cotransfected, regardless of ActD treatment (lanes 1 to 4). Figure 8A demonstrates that Mta transactivated the nut-1/PAN promoter in the absence of ActD (lanes 5 and 6) and maintained that level of luciferase transcript when ActD was added (lanes 7 and 8).
FIG. 8.

Promoter-specific transactivation by Mta functions at the levels of transcription and RNA stability. 293 cells were cotransfected in quadruplicate with the reporter plasmid pGL3-nut1 (−706) (A) or pCMV-Luc (B) and the expression vectors indicated at the top. Half of each set of transfections was treated with ActD, as indicated, and total RNA was analyzed by Northern blotting using the luciferase (lucif) gene or 7SK transcript (cellular control) as a probe. Each signal was quantitated by phosphorimager. Relative activity was calculated by normalizing each pair of luciferase signals to each other by comparison to the corresponding pairs of signals for the cellular 7SK transcript. Each normalized signal in panel A or B was then divided by the signals from lanes 1 and 2, which were set at 1. The numbers above each luciferase panel represent the average of the pair of resulting signals, with each corresponding standard deviation in parentheses.
Comparison of Fig. 8A to B supports a promoter-specific transcriptional and posttranscriptional role of Mta transactivation. When luciferase was expressed from the potent CMV promoter in the absence of Mta, the transcript was easily detectable when ActD was omitted from the culture but became virtually undetectable when ActD was added (Fig. 8B, compare lanes 1 and 2 to 3 and 4). Coexpression of Mta had no effect on luciferase transcript in the absence of ActD (Fig. 8B, compare lanes 5 and 6 to 1 and 2) but maintained luciferase transcript levels in the presence of ActD (Fig. 8B, compare lanes 7 and 8 to 5 and 6). If Mta were transactivating luciferase RNA only by enhancing its stability, then the levels of luciferase RNA would have increased proportionally regardless of the promoter (compare lanes 1 and 2 with 5 and 6 for both Fig. 8A and B). This was not the case, as Mta increased the luciferase transcript 11.8-fold for the nut-1/PAN promoter (Fig. 8A), but not the CMV promoter (Fig. 8B), in the absence of ActD. These data suggested that Mta was indeed transactivating the nut-1/PAN promoter, but not the CMV promoter. However, the data from Fig. 8 also demonstrated that once luciferase is expressed, whether as the result of Mta-mediated promoter transactivation (nut-1/PAN) (Fig. 8A) or by the intrinsic activity of the promoter (CMV) (Fig. 8B), Mta also enhanced the stability of the luciferase RNA (Fig. 8, lanes 7 and 8).
The Rta and Mta proteins bind directly to each other.
The least complicated mechanism to explain synergy between the Rta and Mta proteins is by direct interaction with each other. To test this hypothesis, we generated total protein extracts from BCBL-1 PEL cells that had been treated with TPA for 24 h. Equal amounts of extract were incubated with our antisera that were specific for Mta or Rta or with preimmune sera. Immunoprecipitated proteins were probed by Western blotting using the anti-Mta or anti-Rta antibodies that had been cross-linked to horseradish peroxidase (to eliminate background detection of the precipitated antibodies). As shown in Fig. 9A (left), the anti-Mta serum, but not the preimmune serum, coprecipitated Rta. As shown in Fig. 9A (right), the anti-Rta serum, but not the preimmune serum, coprecipitated Mta. These data prove that Mta and Rta interact in the same complex in extracts from infected cells (a similar result has been published elsewhere [61]).
FIG. 9.
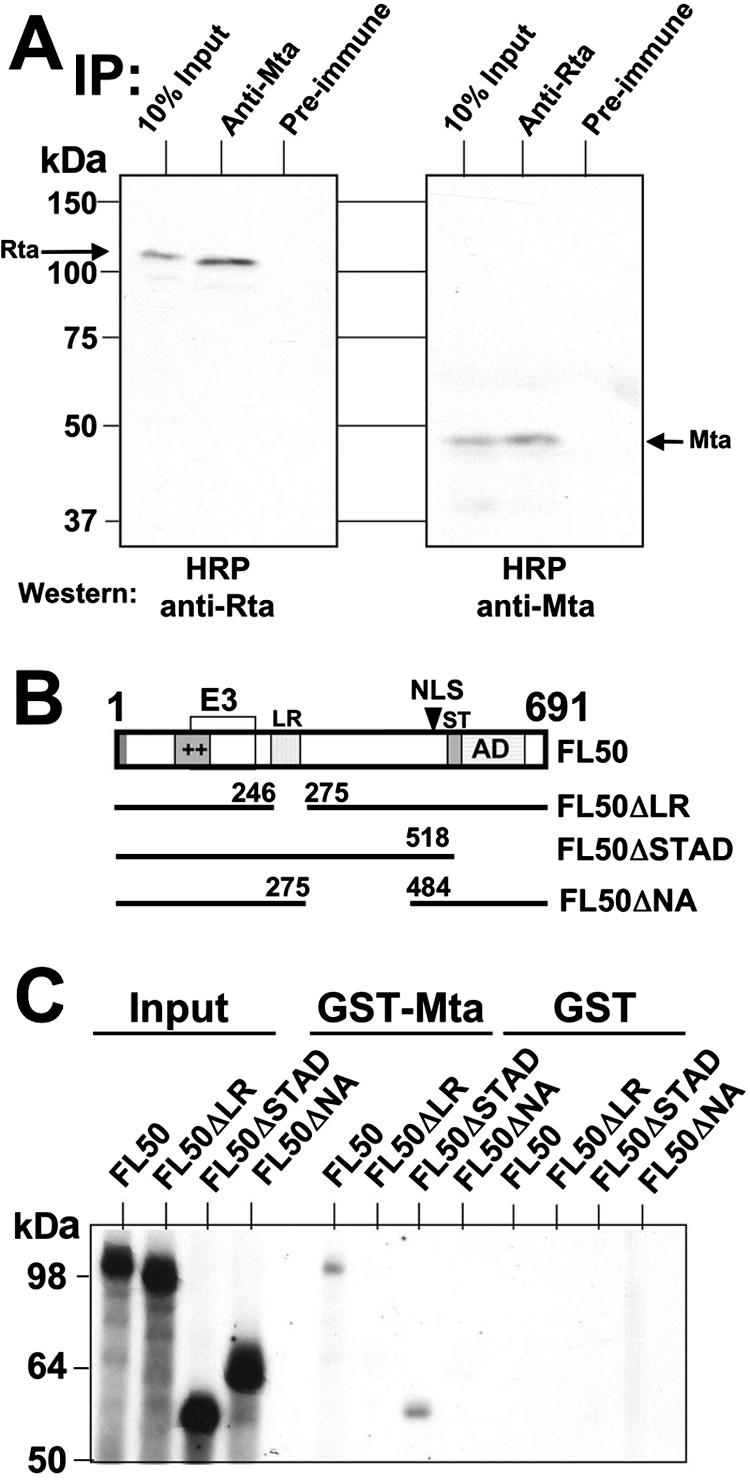
ORF50/Rta and ORF57/Mta proteins interact physically. (A) Coimmunoprecipitations. Total protein extracts from induced PEL cells were incubated with the indicated antisera, as described in Materials and Methods. Immunoprecipitated (IP) proteins were displayed by SDS-PAGE and analyzed by Western blotting using horseradish peroxidase (HRP)-conjugated anti-Rta or anti-Mta sera, as indicated. (B) Schematic of ORF50/Rta protein and deletions. The box shows structural and functional protein domains of the 691-aa ORF50/Rta protein. The bars below represent each of the deletion mutants used in Fig. 7C. ++, basic amino acids; E3, E3 ubiquitin ligase activity (109); LR, leucine rich (10); NLS, nuclear localization signal; AD, activation domain (56, 98). (C) GST pull downs. GST-Mta or GST moiety alone was immobilized on glutathione-agarose beads and incubated with the indicated 35S-labeled Rta proteins (from programmed RRL). Following washes, the beads were boiled in 2× Laemmli buffer, and bound proteins were electrophoresed on a 10% denaturing polyacrylamide gel. The fixed, amplified, and dried gels were analyzed by autoradiography.
To confirm the Mta-Rta interaction in vitro, and to determine the amino acid requirements for Rta to bind to Mta, we employed a GST pull-down approach using Mta generated as an N-terminal fusion to the GST moiety. RRLs were programmed with plasmids expressing full-length Rta, or three deletion mutants (Fig. 9B), in the presence of [35S]methionine. The migration of each of the input Rta proteins is demonstrated on the left side of Fig. 9C. The right side of Fig. 9C shows that full-length Rta bound to GST-Mta in vitro, confirming the coimmunoprecipitation (Fig. 9A). Only the C-terminally truncated mutant of Rta, called ORF50ΔSTAD, interacted with Mta. Deletion of the leucine-rich repeats (ΔLR) of Rta, or of Rta aa 275 to 484 (ΔNA), eliminated interaction with Mta. None of the Rta proteins bound to the GST moiety expressed alone (right side of Fig. 9C). Our data suggested that two individual regions of Rta were required for interactions with Mta.
Mta is a DNA-binding protein.
The data described above demonstrated that Mta/Rta synergy and Rta-independent Mta transactivation were promoter specific. The simplest mechanism by which Mta achieved promoter specificity was by binding DNA in a sequence-specific fashion. To determine whether Mta bound to DNA directly, we generated Mta as an N-terminal fusion to a six-histidine epitope. The full-length protein was expressed and purified to near homogeneity in E. coli (shown by GelCode Blue stain in Fig. 10A). Using EMSAs, the His6-Mta was tested for binding to a 99-bp DNA from the nut-1 promoter. The DNA included the 13-bp RRE, the TATA box, and the transcriptional start site of nut-1/PAN (−73 to +26) (Fig. 3A shows the probe location). As shown in Fig. 10B, His6-Mta formed at least four complexes with the DNA at the lowest concentration of protein tested. As we increased the concentration of His6-Mta, there was a dose-responsive increase in the intensity of the protein-DNA complexes and a concomitant appearance of slower-migrating complexes. As a negative control, we repeated the EMSAs but substituted His6-RBP-Jk for Mta, which did not result in formation of DNA-protein complexes (Fig. 10B). These data suggested that Mta protein bound directly to the nut-1/PAN promoter in the absence of Rta.
FIG. 10.

ORF57/Mta is a DNA-binding protein. (A) Expression and purification of His6-Mta in E. coli. His6-Mta protein was expressed and purified from E. coli, as described in Materials and Methods. Equal volumes of flowthrough (FT) and the indicated fractions were analyzed by SDS-PAGE, followed by visualization using Gel-Code Blue stain. (B) His6-Mta binds to nut-1/PAN promoter DNA in vitro. A DNA spanning bp −73 to +26 of the nut-1/PAN promoter (Fig. 3A) was end labeled with 32P and incubated with DNA-binding buffer alone or increasing amounts of His6-Mta or His6-RPB-Jk protein, as indicated. Nondenaturing PAGE was used to analyze each reaction, the gel was dried, and signals were visualized by autoradiography. (C) His6-Mta binds to the nut-1/PAN promoter in vivo. ChIP assays were performed as described in Materials and Methods using the indicated antisera and chromatin from uninduced (−TPA) or induced (+TPA) BC-3 cells. The nut-1/PAN promoter was detected and quantitated by real-time PCR; end products of the amplification were analyzed by agarose gel electrophoresis and visualized by staining them with ethidium bromide. − or +, omission or addition, respectively, of indicated proteins. The number of plus signs indicates the relative amount of protein added.
To determine whether Mta associated with the nut-1/PAN promoter in infected cells, we performed ChIP experiments. Chromatin from mock-induced or TPA-induced BC-3 cells was cross-linked with formaldehyde 40 h post-TPA treatment. Extracts were incubated with our anti-Mta serum, positive control anti-Rta serum, or negative control preimmune serum or purified rabbit IgG. We analyzed the ChIP-assayed DNA using quantitative real-time PCR with primers specific for a 178-bp fragment of the nut-1/PAN promoter spanning the TATA box and 13-bp RRE (positions −141 to +37) as described previously (64). Figure 10C shows the results of amplification of the ChIP-assayed DNA. Equal amounts of chromatin from mock-induced and TPA-induced BC-3 cells were used as input for the ChIP (“input” lanes). However, the anti-Mta serum precipitated the nut-1/PAN promoter only following TPA induction, but not in mock-induced cells. Since the rabbit IgG control showed only weak background bands, this suggested that Mta bound to the nut-1/PAN promoter in infected cells during reactivation of KSHV from latency. Note that the lanes for the anti-Rta serum (Fig. 10C) surpassed the linear range of the PCR; quantitation is shown in Table 2.
TABLE 2.
Mta is enriched on the Nut-1/PAN promoter during reactivation
| Target DNA | Enrichment with immunoprecipitating antibodya:
|
|||
|---|---|---|---|---|
| α-Mta | α-Rta | Preimmune | IgG | |
| Nut-1/PAN promoter | 27.4 | 6.3 | 1.0 | 1.4 |
| K6 ORF | 2.9 | 1.6 | 1.0 | 4.5 |
ChIPs, as described in Materials and Methods, are represented as enrichment (n-fold) of Mta or Rta DNA binding in the presence of TPA.
The PCRs were quantitated in real time by detecting amplification products using fluorescent, self-quenching molecular beacons. As shown in Table 2, the background level of chromatin that was immunoprecipitated using the negative control, preimmune serum, was set at 1.0× for normalization of the other ChIPs. Mta was enriched 27.4-fold on the nut-1/PAN promoter in infected cells following TPA addition. In comparison, at the same time point following reactivation, Rta was enriched 6.3-fold on the nut-1/PAN promoter. The negative control antibody, total rabbit IgG, yielded a background value of 1.4-fold.
As a negative control for gene-specific DNA binding, we used the same ChIP-assayed DNA but substituted PCR primers and a corresponding molecular beacon that was designed to specifically detect the DNA encoding the KSHV K6 ORF. As shown in Table 2, Mta and Rta were enriched on the K6 ORF by 2.9- and 1.6-fold, respectively. These values were below the value for enrichment using the negative control IgG, which was 4.5-fold. Together, these data suggest that both Mta and Rta bound to the nut-1/PAN promoter in infected cells within the same TATA-proximal span of 178 bp. It is unclear whether direct binding of Mta to Rta contributes to our ability to ChIP Mta on the nut-1/PAN promoter in vivo.
A putative A/T hook domain contributes to DNA binding and transactivation by Mta.
Analysis of Mta's primary sequence revealed a basic domain that was enriched in arginine (Fig. 11A). The software program MOTIF (http://www.genome.jp/) revealed a putative A/T hook domain within the R domain of Mta (aa 119 to 131) (Fig. 11B). A/T hooks are DNA-binding domains found in a great variety of cellular proteins that act as accessory proteins for transcription factors (1, 4). To determine whether the putative A/T hook contributed to Mta DNA binding, we deleted the domain in the full-length His6 fusion of Mta (called MtaΔA/T) and performed EMSA with the nut-1/PAN (−73 to +26) DNA. As shown in Fig. 11C, both WT and mutant Mta formed multiple complexes with the PAN DNA; however, three of the DNA-protein complexes formed only with WT Mta but not MtaΔA/T.
FIG. 11.

A lesion in Mta's putative A/T hook domain alters its association with promoter DNA and reduces Mta transactivation. (A) Mta primary structure map; a schematic of the ORF57/Mta protein. The numbers refer to amino acid positions. Asterisks, putative nuclear export signals; arrowheads, RXP tri-peptides; SR, serine-arginine dipeptide-rich domain; R, arginine-rich domain; L, leucine repeat; HCC, amino acids conserved in herpesviral Mta homologs (93). (B) Mta contains a putative A/T hook DNA-binding domain. Shown is an alignment of the putative AT hook motif of Mta with a sequence logo (19) created by comparison of selected A/T hook motifs contained in the protein block IPB000637B (38). The height of each amino acid in the logo represents the relative frequency of that residue at the indicated position. The lowercase r at position 8 in the Mta sequence is the only amino acid not commonly found in A/T hooks. (C) Deletion of the putative A/T hook alters the association of Mta with promoter DNA. Purified His6-Mta WT or MtaΔA/T protein was preincubated in buffer alone or with the unlabeled −73 to +26 nut-1/PAN promoter DNA (Fig. 3A) at two concentrations. Labeled probe was then added, and EMSA results were analyzed as described in the legend to Fig. 10B. (D) Deletion of the putative A/T hook reduces Mta-mediated transactivation of the nut-1/PAN promoter. The indicated amounts of expression vectors for the indicated proteins were cotransfected with the pGL3-nut1 (−706) reporter plasmid into 293 cells, and luciferase assays were performed as described in the legend to Fig. 6A. (E) Proteins expressed from the indicated vectors were visualized by immunofluorescence of transfected 293 cells as for Fig. 5A.
To test the functional significance of the Mta DNA-binding mutation, we asked whether the mutant Mta could transactivate the nut-1/PAN promoter/reporter in transfected 293 cells. As shown in Fig. 11D, transactivation by the MtaΔA/T mutant was reduced by at least 60% at all input plasmid amounts compared to WT Mta. These data supported a role for Mta DNA binding in transactivation of the nut-1/PAN promoter.
The A/T hook overlaps a portion of Mta that has been suggested to contribute to Mta's nuclear localization (59). We found that the MtaΔA/T DNA-binding mutant was well expressed and correctly localized to the nuclei of transfected 293 cells (Fig. 11E).
DISCUSSION
In this study, we have demonstrated that the KSHV ORF57/Mta protein cooperates with ORF50/Rta to reactivate the virus from latency in PEL models of infection (Fig. 1). Three independent strategies confirmed that ectopic expression of Rta alone was sufficient to reactivate the virus, but Mta could not induce KSHV reactivation in the absence of Rta expression (Fig. 1). Since Mta cannot reactivate the virus independently, it presumably does not activate the ORF50 promoter when ectopically expressed independently in infected cells. This hypothesis was supported by the inability of Mta to significantly activate the ORF50 promoter under any of our experimental conditions (Fig. 4 and 6C). Mta has been previously reported to activate the ORF50 promoter in cooperation with Rta (61); the small magnitude of Mta's effect in that publication agreed with our data.
When analyzed across a population of infected cells, Rta is necessary and sufficient to induce reactivation. However, our single-cell reactivation data suggest that a productive lytic cycle will not necessarily be completed in every infected cell that expresses Rta. Instead, we hypothesize that a “commitment point” downstream of Rta expression must be passed for viral DNA replication to occur and progeny virions to be produced. One such “commitment factor” for KSHV reactivation appears to be Mta. In Fig. 1, we show that Mta cooperates with Rta in inducing lytic reactivation when the two proteins are coexpressed ectopically. In Fig. 2D, we show that greater than 80% of Mta-expressing cells coexpress the true late protein K8.1 at 72 h post-TPA induction. In contrast, less than 20% of Rta-positive cells have progressed to K8.1 expression at that time point. In Fig. 2C, we show that the population of Mta-expressing cells “branches away” from the population of Rta-expressing cells by 48 h post-TPA addition and begins to plateau in number while Rta-expressing cells continue to increase linearly. We hypothesize that the Mta-expressing population declines as those cells are eliminated due to complete viral reactivation and lysis. Genetic studies have clearly demonstrated that Mta is required for productive viral reactivation, especially for optimal expression of DNA replication factors (37, 58). Among the promoters we tested, we show in Fig. 3 and 4 that Mta-Rta synergy is greatest on the two ori-Lyt-associated promoters (as well as nut-1/PAN). Transcription from ori-Lyt (L) is required for successful origin function and KSHV lytic replication (101, 102). Therefore, Mta appears to satisfy the definition of a commitment factor for KSHV reactivation, whose expression seems to determine whether reactivation proceeds to full viral productive replication.
Although KSHV Mta clearly has a robust posttranscriptional transactivation function (34, 37, 45, 58-60, 62), our data show that Mta is also a DNA-binding protein that can activate transcription independently of Rta in a cell- and promoter-specific manner (Table 1 and Fig. 6). Similarly, Mta-Rta synergy was also cell and promoter specific. However, the cell line specificity of Mta transactivation did not correspond to that of Mta-Rta synergy (Table 1). Among the lymphocyte lines that we tested, there was no apparent correspondence between a particular B-cell lineage and the effects of Mta: for example, in Burkitt's lymphomas, only Akata-31 cells supported Rta-independent transcriptional activation by Mta and only BL-41 cells supported robust Mta-Rta synergy. Since Mta is not expressed independently of Rta in reactivating cells (56), it is difficult to determine whether Mta activates any KSHV promoters independently of Rta in infected cells. Multiple myeloma/plasma cell lymphoma lines, which are closest phenotypically to PELs (42, 47), supported a CV-1-like response to Mta and Rta: Rta, but not Mta, activated transcription independently, and Mta and Rta synergized (Table 1).
It is unknown why 293 and Akata-31 cells support Rta-independent transactivation by Mta. 293 cells are human embryonic kidney cells that are transformed by the adenovirus E1a and E1b genes (53). In experiments (not shown), coexpression of E1a with Mta did not convert Mta to a transcriptional transactivator in cells in which Mta is normally transcriptionally silent. Therefore, 293 cells are ideal for identifying transcriptional mechanisms used by Mta independently of a requirement for coexpression of Rta.
In Fig. 9, we demonstrate that the Mta and Rta proteins physically interact by coimmunoprecipitation and GST pull-down assays. This interaction satisfies the simplest explanation for their ability to cooperate in transactivation. Slightly more than 10% of total Mta and Rta in PEL extracts is coimmunoprecipitated (Fig. 9A). These data suggest that the Mta-Rta interaction is limited, with the majority of the two proteins engaged in heterologous interactions during reactivation. Such conditions would be ideal for the Mta-Rta interaction to regulate the progression of reactivation. The domains of Rta that were required for directly binding to Mta were the LR and Rta central region (aa 275 to 484) (Fig. 9B and C). Both domains are required for correct multimerization of Rta (10), but it is unclear whether Mta binds to or regulates a specific homomultimeric configuration of Rta.
We showed that Mta bound the nut-1/PAN promoter DNA in vitro (Fig. 10B and 11C) and in vivo (Fig. 10C and Table 2), and the promoter specificity of Rta-independent transactivation by Mta (Fig. 6) mirrored the specificity of Mta-Rta synergy (Fig. 3 and 4). The only exception to this correspondence was that mutations of the RRE ablated Mta-Rta synergy (Fig. 3D) and reduced, but did not eliminate, Mta transactivation (Fig. 6B). Therefore, the mechanism of Mta-Rta synergy appears to be specified by direct interactions of Mta with promoter DNA and with Rta. The contribution of Rta to promoter-specific synergy might be determined by its affinity for DNA binding, as the preferred promoters for synergy all contain the high-affinity RRE (90). Coexpression of Rta with Mta might stimulate the promoter-specific “latent” transcriptional-transactivation function of Mta. This transcriptional-transactivation function of Mta appears to be active independently of Rta in 293 or Akata-31 cells but inactive in most other cells (Table 1). However, our data do not exclude the possibility that Rta-independent transactivation by Mta in 293 and Akata-31 cells reflects Mta's hypothetical ability to increase the abundance or activity of a cellular transcription factor that regulates the nut-1/PAN promoter.
Deletion of the putative A/T hook between aa 119 and 131 of Mta (Fig. 11B) altered the pattern of protein-DNA complexes formed by Mta (Fig. 11C) and severely reduced Mta-mediated transactivation of the nut-1/PAN promoter (Fig. 11D). Twelve of the 13 aa in Mta's putative A/T hook are positionally conserved with a subset of A/T hook proteins that are annotated in the Protein Blocks database (38). Although Mta lacks the highly conserved glycine at position 8 of the A/T hook (Fig. 11B) (4), recent publications have shown that the glycine is dispensable for DNA binding of heterologous A/T hook proteins (68, 97). Deletion of the A/T-hook did not completely ablate Mta's functions (Fig. 11), agreeing with the observation that A/T hooks cooperate in cis with other protein domains to mediate DNA binding (52) and supporting a role for A/T hook-independent (i.e., posttranscriptional) mechanisms of Mta transactivation.
The association of Mta with promoter DNA (Fig. 10 and 11) and stimulation of transcriptional initiation by Mta (Fig. 7) support a mechanism in which Mta specifies genes for transactivation at the earliest step in RNA biogenesis, transcriptional initiation. The observation that Mta also transactivates genes posttranscriptionally in our studies (Fig. 8) (45) and others (34, 37, 45, 58-60, 62) suggests that Mta might subsequently associate with the nascent RNA transcript in the nucleus. Indeed, a recent publication suggested that nuclear-cytoplasmic shuttling of Mta was not required for posttranscriptional activation (72).
A prime mechanism that might enable coupled transcriptional and posttranscriptional transactivation by Mta is interaction with the CTD of cellular RNA polymerase II (78). The best-studied homolog of KSHV Mta, the HSV ICP27 protein, interacts with the CTD (21, 111) and transactivates using transcriptional and posttranscriptional mechanisms (20, 21, 23, 29, 39, 66, 67, 70, 74, 82, 84, 95, 110, 111). However, it is unclear how a putative interaction of KSHV Mta with the RNA polymerase II CTD could explain Mta's promoter specificity. Other Mta homologs within the Herpesviridae, including the human CMV UL69 protein, varicella-zoster virus 4 protein, bovine herpesvirus type 1 ICP27 protein, and equine herpesvirus type 1 ICP27 protein, all demonstrate transcriptional mechanisms of transactivation (36, 69, 77, 88, 106). The EICP27 protein physically interacts with and synergistically cooperates with the immediate-early EICP0 protein in transactivating viral promoters (2). EICP27 also binds directly to the TATA box-binding protein (3). It must be stressed, however, that the amino acid identity among KSHV Mta and its Herpesviridae homologs is relatively low, so understanding KSHV Mta's mechanism of cooperation with Rta has the potential to establish new paradigms for control of herpesviral reactivation and contributions to pathogenesis.
Our data suggest that selective cooperation of the KSHV Mta and Rta proteins, in a promoter- and transcript-specific fashion, might regulate alternative sublytic or productive gene expression programs. Such differences in viral lytic gene expression pathways would be expected to determine significant pathophysiologic consequences for KSHV infection. According to our data, single-cell expression of Mta identifies those cells that successfully produce viral progeny (Fig. 2D). Since Mta expression requires prior expression of Rta, every Rta-expressing cell has the potential to proceed down the lytic cascade. However, our data suggest that the progression from Rta single positive to Rta/Mta double positive is a key step in viral pathogenesis.
Acknowledgments
We thank Harvey Ozer, Abraham Pinter, Paul Farrell, Yan Yuan, Hua Zhu, and S. J. Gao for cell lines, plasmids, and bacmid. We thank Richard Pine for helpful discussions regarding nuclear run-on assays.
Work in the Lukac laboratory was supported by the American Cancer Society, American Foundation for AIDS Research, NJ Commission on Cancer Research, Ruth Estrin Goldberg Memorial for Cancer Research, and American Heart Association.
Footnotes
Published ahead of print on 3 October 2007.
REFERENCES
- 1.Agresti, A., and M. E. Bianchi. 2003. HMGB proteins and gene expression. Curr. Opin. Genet. Dev. 13:170-178. [DOI] [PubMed] [Google Scholar]
- 2.Albrecht, R. A., S. K. Kim, and D. J. O'Callaghan. 2005. The EICP27 protein of equine herpesvirus 1 is recruited to viral promoters by its interaction with the immediate-early protein. Virology 333:74-87. [DOI] [PubMed] [Google Scholar]
- 3.Albrecht, R. A., S. K. Kim, Y. Zhang, Y. Zhao, and D. J. O'Callaghan. 2004. The equine herpesvirus 1 EICP27 protein enhances gene expression via an interaction with TATA box-binding protein. Virology 324:311-326. [DOI] [PubMed] [Google Scholar]
- 4.Aravind, L., and D. Landsman. 1998. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 26:4413-4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.AuCoin, D. P., K. S. Colletti, Y. Xu, S. A. Cei, and G. S. Pari. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) contains two functional lytic origins of DNA replication. J. Virol. 76:7890-7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bello, L., A. Davison, M. Glenn, A. Whitehouse, N. Rethmeier, T. Schulz, and J. Clements. 1999. The human herpesvirus-8 ORF57 gene and its properties. J. Gen. Virol. 80:3207-3215. [DOI] [PubMed] [Google Scholar]
- 7.Boneschi, V., L. Brambilla, E. Berti, S. Ferrucci, M. Corbellino, C. Parravicini, and S. Fossati. 2001. Human herpesvirus 8 DNA in the skin and blood of patients with Mediterranean Kaposi's sarcoma: clinical correlations. Dermatology 203:19-23. [DOI] [PubMed] [Google Scholar]
- 8.Bowser, B. S., S. M. DeWire, and B. Damania. 2002. Transcriptional regulation of the K1 gene product of Kaposi's sarcoma-associated herpesvirus. J. Virol. 76:12574-12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown, C. R., M. S. Nakamura, J. D. Mosca, G. S. Hayward, S. E. Straus, and L. P. Perera. 1995. Herpes simplex virus trans-regulatory protein ICP27 stabilizes and binds to 3′ ends of labile mRNA. J. Virol. 69:7187-7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bu, W., K. D. Carroll, D. Palmeri, and D. M. Lukac. 2007. The Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 81:5788-5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell, T. B., M. Borok, L. Gwanzura, S. MaWhinney, I. E. White, B. Ndemera, I. Gudza, L. Fitzpatrick, and R. T. Schooley. 2000. Relationship of human herpesvirus 8 peripheral blood virus load and Kaposi's sarcoma clinical stage. AIDS 14:2109-2116. [DOI] [PubMed] [Google Scholar]
- 12.Carroll, K. D., W. Bu, D. Palmeri, S. Spadavecchia, S. J. Lynch, S. A. Marras, S. Tyagi, and D. M. Lukac. 2006. Kaposi's sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J. Virol. 80:9697-9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll, K. D., F. Khadim, S. Spadavecchia, D. Palmeri, and D. M. Lukac. 2007. Direct interactions of KSHV/HHV-8 ORF50/Rta protein with the cellular protein Octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 81:8451-8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cesarman, E., and E. Mesri. 2000. Viral G protein-coupled receptor and Kaposi's sarcoma: a model of paracrine neoplasia? J. Exp. Med. 191:417-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang, P. J., D. Shedd, L. Gradoville, M. S. Cho, L. W. Chen, J. Chang, and G. Miller. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K-12 genes by binding to related response elements. J. Virol. 76:3168-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang, P. J., D. Shedd, and G. Miller. 2005. Two subclasses of Kaposi's sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J. Virol. 79:8750-8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen, J., K. Ueka, S. Sakakibara, T. Okuno, and K. Yamanishi. 2000. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor gene. J. Virol. 74:8623-8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Countryman, J., and G. Miller. 1985. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc. Natl. Acad. Sci. USA 82:4085-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crooks, G. E., G. Hon, J. M. Chandonia, and S. E. Brenner. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curtin, K. D., and D. M. Knipe. 1993. Altered properties of the herpes simplex virus ICP8 DNA-binding protein in cells infected with ICP27 mutant viruses. Virology 196:1-14. [DOI] [PubMed] [Google Scholar]
- 21.Dai-Ju, J. Q., L. Li, L. A. Johnson, and R. M. Sandri-Goldin. 2006. ICP27 interacts with the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it undergoes proteasomal degradation during infection. J. Virol. 80:3567-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Damania, B., J. H. Jeong, B. S. Bowser, S. M. DeWire, M. R. Staudt, and D. P. Dittmer. 2004. Comparison of the Rta/Orf50 transactivator proteins of gamma-2-herpesviruses. J. Virol. 78:5491-5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Bruyn Kops, A., S. L. Uprichard, M. Chen, and D. M. Knipe. 1998. Comparison of the intranuclear distributions of herpes simplex virus proteins involved in various viral functions. Virology 252:162-178. [DOI] [PubMed] [Google Scholar]
- 24.Deng, H., J. T. Chu, M. B. Rettig, O. Martinez-Maza, and R. Sun. 2002. Rta of the human herpesvirus 8/Kaposi sarcoma-associated herpesvirus up-regulates human interleukin-6 gene expression. Blood 100:1919-1921. [DOI] [PubMed] [Google Scholar]
- 25.Deng, H., M. J. Song, J. T. Chu, and R. Sun. 2002. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 76:8252-8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dourmishev, L. A., A. L. Dourmishev, D. Palmeri, R. A. Schwartz, and D. M. Lukac. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol Rev. 67:175-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engels, E. A., R. J. Biggar, V. A. Marshall, M. A. Walters, C. J. Gamache, D. Whitby, and J. J. Goedert. 2003. Detection and quantification of Kaposi's sarcoma-associated herpesvirus to predict AIDS-associated Kaposi's sarcoma. AIDS 17:1847-1851. [DOI] [PubMed] [Google Scholar]
- 28.Ensoli, B., M. Sturzl, and P. Monini. 2000. Cytokine-mediated growth promotion of Kaposi's sarcoma and primary effusion lymphoma. Semin. Cancer Biol. 10:367-381. [DOI] [PubMed] [Google Scholar]
- 29.Everett, R. D., G. Sourvinos, C. Leiper, J. B. Clements, and A. Orr. 2004. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J. Virol. 78:1903-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fakhari, F. D., and D. P. Dittmer. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213-6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fixman, E. D., G. S. Hayward, and S. D. Hayward. 1992. trans-acting requirements for replication of Epstein-Barr virus ori-Lyt. J. Virol. 66:5030-5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gradoville, L., J. Gerlach, E. Grogan, D. Shedd, S. Nikiforow, C. Metroka, and G. Miller. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gruffat, H., J. Batisse, D. Pich, B. Neuhierl, E. Manet, W. Hammerschmidt, and A. Sergeant. 2002. Epstein-Barr virus mRNA export factor EB2 is essential for production of infectious virus. J. Virol. 76:9635-9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta, A. K., V. Ruvolo, C. Patterson, and S. Swaminathan. 2000. The human herpesvirus 8 homolog of Epstein-Barr virus SM protein (KS-SM) is a posttranscriptional activator of gene expression. J. Virol. 74:1038-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gwack, Y., H. Nakamura, S. H. Lee, J. Souvlis, J. T. Yustein, S. Gygi, H. J. Kung, and J. U. Jung. 2003. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol. Cell. Biol. 23:8282-8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamel, F., and C. Simard. 2003. Mapping of the bovine herpesvirus 1 glycoprotein C promoter region and its specific transactivation by the viral BICP27 gene product. Arch. Virol. 148:137-152. [DOI] [PubMed] [Google Scholar]
- 37.Han, Z., and S. Swaminathan. 2006. Kaposi's sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J. Virol. 80:5251-5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henikoff, J. G., E. A. Greene, S. Pietrokovski, and S. Henikoff. 2000. Increased coverage of protein families with the blocks database servers. Nucleic Acids Res. 28:228-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jean, S., K. M. LeVan, B. Song, M. Levine, and D. M. Knipe. 2001. Herpes simplex virus 1 ICP27 is required for transcription of two viral late (gamma 2) genes in infected cells. Virology 283:273-284. [DOI] [PubMed] [Google Scholar]
- 40.Jenkins, P., U. Binne, and P. Farrell. 2000. Histone acetylation and reactivation of Epstein-Barr virus from latency. J. Virol. 74:710-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jenner, R., M. Alba, C. Boshoff, and P. Kellam. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75:891-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jenner, R. G., K. Maillard, N. Cattini, R. A. Weiss, C. Boshoff, R. Wooster, and P. Kellam. 2003. Kaposi's sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Natl. Acad. Sci. USA 100:10399-10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones, J. L., D. L. Hanson, M. S. Dworkin, and H. W. Jaffe. 2000. Incidence and trends in Kaposi's sarcoma in the era of effective antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 24:270-274. [DOI] [PubMed] [Google Scholar]
- 44.Kedes, D. H., and D. Ganem. 1997. Sensitivity of Kaposi's sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J. Clin. Investig. 99:2082-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirshner, J. R., D. M. Lukac, J. Chang, and D. Ganem. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J. Virol. 74:3586-3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirshner, J. R., K. Staskus, A. Haase, M. Lagunoff, and D. Ganem. 1999. Expression of the open reading frame 74 (G-protein-coupled receptor) gene of Kaposi's sarcoma (KS)-associated herpesvirus; implications for KS pathogenesis. J. Virol. 73:6006-6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klein, U., A. Gloghini, G. Gaidano, A. Chadburn, E. Cesarman, R. Dalla-Favera, and A. Carbone. 2003. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood 101:4115-4121. [DOI] [PubMed] [Google Scholar]
- 48.Liang, Y., J. Chang, S. Lynch, D. M. Lukac, and D. Ganem. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jk, the target of the Notch signaling pathway. Genes Dev. 16:1977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang, Y., and D. Ganem. 2004. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi's sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 78:6818-6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liao, W., Y. Tang, Y. L. Kuo, B. Y. Liu, C. J. Xu, and C. Z. Giam. 2003. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 transcriptional activator Rta is an oligomeric DNA-binding protein that interacts with tandem arrays of phased A/T-trinucleotide motifs. J. Virol. 77:9399-9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 52.Llano, M., M. Vanegas, N. Hutchins, D. Thompson, S. Delgado, and E. M. Poeschla. 2006. Identification and characterization of the chromatin-binding domains of the HIV-1 integrase interactor LEDGF/p75. J. Mol. Biol. 360:760-773. [DOI] [PubMed] [Google Scholar]
- 53.Louis, N., C. Evelegh, and F. L. Graham. 1997. Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line. Virology 233:423-429. [DOI] [PubMed] [Google Scholar]
- 54.Lu, M., J. Suen, C. Frias, R. Pfeiffer, M. H. Tsai, E. Chuang, and S. L. Zeichner. 2004. Dissection of the Kaposi's sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J. Virol. 78:13637-13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lukac, D., L. Garibyan, J. Kirshner, D. Palmeri, and D. Ganem. 2001. DNA binding by the Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 75:6786-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 58.Majerciak, V., N. Pripuzova, J. P. McCoy, S. J. Gao, and Z. M. Zheng. 2007. Targeted disruption of Kaposi's sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, K8α, and K8.1 and the production of infectious virus. J. Virol. 81:1062-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Majerciak, V., K. Yamanegi, S. H. Nie, and Z. M. Zheng. 2006. Structural and functional analyses of Kaposi sarcoma-associated herpesvirus ORF57 nuclear localization signals in living cells. J. Biol. Chem. 281:28365-28378. [DOI] [PubMed] [Google Scholar]
- 60.Majerciak, V., K. Yamanegi, and Z. M. Zheng. 2006. Gene structure and expression of kaposi's sarcoma-associated herpesvirus ORF56, ORF57, ORF58, and ORF59. J. Virol. 80:11968-11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malik, P., D. J. Blackbourn, M. F. Cheng, G. S. Hayward, and J. B. Clements. 2004. Functional co-operation between the Kaposi's sarcoma-associated herpesvirus ORF57 and ORF50 regulatory proteins. J. Gen. Virol. 85:2155-2166. [DOI] [PubMed] [Google Scholar]
- 62.Malik, P., D. J. Blackbourn, and J. B. Clements. 2004. The evolutionarily conserved Kaposi's sarcoma-associated herpesvirus ORF57 protein interacts with REF protein and acts as an RNA export factor. J. Biol. Chem. 279:33001-33011. [DOI] [PubMed] [Google Scholar]
- 63.Malik, P., and J. B. Clements. 2004. Protein kinase CK2 phosphorylation regulates the interaction of Kaposi's sarcoma-associated herpesvirus regulatory protein ORF57 with its multifunctional partner hnRNP K. Nucleic Acids Res. 32:5553-5569. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Marras, S. A., F. R. Kramer, and S. Tyagi. 2003. Genotyping SNPs with molecular beacons. Methods Mol. Biol. 212:111-128. [DOI] [PubMed] [Google Scholar]
- 65.Martin, D. F., B. D. Kuppermann, R. A. Wolitz, A. G. Palestine, H. Li, C. A. Robinson, et al. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N. Engl. J. Med. 340:1063-1070. [DOI] [PubMed] [Google Scholar]
- 66.McCarthy, A. M., L. McMahan, and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP27 deletion mutants exhibit altered patterns of transcription and are DNA deficient. J. Virol. 63:18-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGregor, F., A. Phelan, J. Dunlop, and J. B. Clements. 1996. Regulation of herpes simplex virus poly(A) site usage and the action of immediate-early protein IE63 in the early-late switch. J. Virol. 70:1931-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Metcalf, C. E., and D. A. Wassarman. 2006. DNA binding properties of TAF1 isoforms with two AT-hooks. J. Biol. Chem. 281:30015-30023. [DOI] [PubMed] [Google Scholar]
- 69.Moriuchi, H., M. Moriuchi, H. A. Smith, and J. I. Cohen. 1994. Varicella-zoster virus open reading frame 4 protein is functionally distinct from and does not complement its herpes simplex virus type 1 homolog, ICP27. J. Virol. 68:1987-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mullen, M. A., S. Gerstberger, D. M. Ciufo, J. D. Mosca, and G. S. Hayward. 1995. Evaluation of colocalization interactions between the IE110, IE175, and IE63 transactivator proteins of herpes simplex virus within subcellular punctate structures. J. Virol. 69:476-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murphy, S., F. Altruda, E. Ullu, M. Tripodi, L. Silengo, and M. Melli. 1984. DNA sequences complementary to human 7 SK RNA show structural similarities to the short mobile elements of the mammalian genome. J. Mol. Biol. 177:575-590. [DOI] [PubMed] [Google Scholar]
- 72.Nekorchuk, M., Z. Han, T. T. Hsieh, and S. Swaminathan. 2007. Kaposi's sarcoma-associated herpesvirus ORF57 protein enhances mRNA accumulation independently of effects on nuclear RNA export. J. Virol. 81:9990-9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nishimura, K., K. Ueda, E. Guwanan, S. Sakakibara, E. Do, E. Osaki, K. Yada, T. Okuno, and K. Yamanishi. 2004. A posttranscriptional regulator of Kaposi's sarcoma-associated herpesvirus interacts with RNA-binding protein PCBP1 and controls gene expression through the IRES. Virology 325:364-378. [DOI] [PubMed] [Google Scholar]
- 74.Panagiotidis, C., E. Lium, and S. Silverstein. 1997. Physical and functional interactions between herpes simplex virus immediate-early proteins ICP4 and ICP27. J. Virol. 71:1547-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paulose-Murphy, M., N.-K. Ha, C. Xiang, Y. Chen, L. Gillim, R. Yarchoan, P. Meltzer, M. Bittner, J. Trent, and S. Zeichner. 2001. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 75:4843-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pellet, C., S. Chevret, C. Frances, S. Euvrard, M. Hurault, C. Legendre, S. Dalac, D. Farge, C. Antoine, C. Hiesse, M. N. Peraldi, P. Lang, D. Samuel, Y. Calmus, F. Agbalika, P. Morel, F. Calvo, and C. Lebbe. 2002. Prognostic value of quantitative Kaposi sarcoma-associated herpesvirus load in posttransplantation Kaposi sarcoma. J. Infect. Dis. 186:110-113. [DOI] [PubMed] [Google Scholar]
- 77.Perera, L. P., S. Kaushal, P. R. Kinchington, J. D. Mosca, G. S. Hayward, and S. E. Straus. 1994. Varicella-zoster virus open reading frame 4 encodes a transcriptional activator that is functionally distinct from that of herpes simplex virus homology ICP27. J. Virol. 68:2468-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Phatnani, H. P., and A. L. Greenleaf. 2006. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20:2922-2936. [DOI] [PubMed] [Google Scholar]
- 79.Phelan, A., and J. Clements. 1998. Posttranscriptional regulation in herpes simplex virus. Semin. Virol. 8:309-318. [Google Scholar]
- 80.Preston, C. M. 1979. Control of herpes simplex virus type 1 mRNA synthesis in cells infected with wild-type virus or the temperature-sensitive mutant tsK. J. Virol. 29:275-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Robles, R., D. Lugo, L. Gee, and M. A. Jacobson. 1999. Effect of antiviral drugs used to treat cytomegalovirus end-organ disease on subsequent course of previously diagnosed Kaposi's sarcoma in patients with AIDS. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 20:34-38. [DOI] [PubMed] [Google Scholar]
- 82.Sacks, W. R., C. C. Greene, D. P. Aschman, and P. A. Schaffer. 1985. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J. Virol. 55:796-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sakakibara, S., K. Ueda, J. Chen, T. Okuno, and K. Yamanishi. 2001. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 75:6894-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Samaniego, L. A., A. L. Webb, and N. A. DeLuca. 1995. Functional interactions between herpes simplex virus immediate-early proteins during infection: gene expression as a consequence of ICP27 and different domains of ICP4. J. Virol. 69:5705-5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sambrook, J., and D. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 86.Sarid, R., O. Flore, R. A. Bohenzky, Y. Chang, and P. S. Moore. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sirianni, M. C., L. Vincenzi, S. Topino, A. Giovannetti, F. Mazzetta, F. Libi, D. Scaramuzzi, M. Andreoni, E. Pinter, S. Baccarini, G. Rezza, P. Monini, and B. Ensoli. 2002. NK cell activity controls human herpesvirus 8 latent infection and is restored upon highly active antiretroviral therapy in AIDS patients with regressing Kaposi's sarcoma. Eur. J. Immunol. 32:2711-2720. [DOI] [PubMed] [Google Scholar]
- 88.Smith, R. H., Y. Zhao, and D. J. O'Callaghan. 1993. The equine herpesvirus 1 (EHV-1) UL3 gene, an ICP27 homolog, is necessary for full activation of gene expression directed by an EHV-1 late promoter. J. Virol. 67:1105-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Song, M., H. Brown, T.-T. Wu, and R. Sun. 2001. Transcription activation of polyadenylated nuclear RNA by Rta in human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:3129-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song, M. J., H. Deng, and R. Sun. 2003. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 77:9451-9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Song, M. J., X. Li, H. J. Brown, and R. Sun. 2002. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 76:5000-5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Swaminathan, S. 2005. Post-transcriptional gene regulation by gamma herpesviruses. J. Cell Biochem. 95:698-711. [DOI] [PubMed] [Google Scholar]
- 94.Ueda, K., K. Ishikawa, K. Nishimura, S. Sakakibara, E. Do, and K. Yamanishi. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) replication and transcription factor activates the K9 (vIRF) gene through two distinct cis elements by a non-DNA-binding mechanism. J. Virol. 76:12044-12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Uprichard, S. L., and D. M. Knipe. 1996. Herpes simplex ICP27 mutant viruses exhibit reduced expression of specific DNA replication genes. J. Virol. 70:1969-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vieira, J., and P. M. O'Hearn. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225-240. [DOI] [PubMed] [Google Scholar]
- 97.Walters, M. S., K. T. Hall, and A. Whitehouse. 2004. The herpesvirus saimiri open reading frame (ORF) 50 (Rta) protein contains an AT hook required for binding to the ORF 50 response element in delayed-early promoters. J. Virol. 78:4936-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang, S., S. Liu, M. Wu, Y. Geng, and C. Wood. 2001. Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50 gene product contains a potent C-terminal activation domain which activates gene expression via a specific target sequence. Arch. Virol. 146:1415-1426. [DOI] [PubMed] [Google Scholar]
- 99.Wang, S. E., F. Y. Wu, M. Fujimuro, J. Zong, S. D. Hayward, and G. S. Hayward. 2003. Role of CCAAT/enhancer-binding protein alpha (C/EBPα) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 77:600-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang, S. E., F. Y. Wu, Y. Yu, and G. S. Hayward. 2003. CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 77:9590-9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang, Y., H. Li, M. Y. Chan, F. X. Zhu, D. M. Lukac, and Y. Yuan. 2004. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 78:8615-8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang, Y., Q. Tang, G. G. Maul, and Y. Yuan. 2006. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: dual role of replication and transcription activator. J. Virol. 80:12171-12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Watson, R. J., and J. B. Clements. 1980. A herpes simplex virus type 1 function continuously required for early and late virus RNA synthesis. Nature 285:329-330. [DOI] [PubMed] [Google Scholar]
- 104.Whitby, D., M. R. Howard, M. Tenant-Flowers, N. S. Brink, A. Copas, C. Boshoff, T. Hatzioannou, F. E. Suggett, D. M. Aldam, A. S. Denton, et al. 1995. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi's sarcoma. Lancet 346:799-802. [DOI] [PubMed] [Google Scholar]
- 105.Wingender, E., X. Chen, R. Hehl, H. Karas, I. Liebich, V. Matys, T. Meinhardt, M. Prub, I. Reuter, and F. Schacherer. 2000. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 28:316-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Winkler, M., S. A. Rice, and T. Stamminger. 1994. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J. Virol. 68:3943-3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wu, L., R. Renne, D. Ganem, and B. Forghani. 2000. Human herpesvirus 8 glycoprotein K8.1: expression, post-translational modification and localization analyzed by monoclonal antibody. J. Clin. Virol. 17:127-136. [DOI] [PubMed] [Google Scholar]
- 108.Xu, Y., D. P. AuCoin, A. R. Huete, S. A. Cei, L. J. Hanson, and G. S. Pari. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79:3479-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu, Y., S. E. Wang, and G. S. Hayward. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59-70. [DOI] [PubMed] [Google Scholar]
- 110.Zhong, L., and G. S. Hayward. 1997. Assembly of complete, functionally active herpes simplex virus DNA replication compartments and recruitment of associated viral and cellular proteins in transient cotransfection assays. J. Virol. 71:3146-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhou, C., and D. M. Knipe. 2002. Association of herpes simplex virus type 1 ICP8 and ICP27 proteins with cellular RNA polymerase II holoenzyme. J. Virol. 76:5893-5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhou, F. C., Y. J. Zhang, J. H. Deng, X. P. Wang, H. Y. Pan, E. Hettler, and S. J. Gao. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]