

Abstract
The evolutionarily conserved execution phase of apoptosis is defined by characteristic changes occurring during the final stages of death; specifically cell shrinkage, dynamic membrane blebbing, condensation of chromatin, and DNA fragmentation. Mechanisms underlying these hallmark features of apoptosis have previously been elusive, largely because the execution phase is a rapid event whose onset is asynchronous across a population of cells. In the present study, a model system is described for using the caspase inhibitor, z-VAD-FMK, to block apoptosis and generate a synchronous population of cells actively extruding and retracting membrane blebs. This model system allowed us to determine signaling mechanisms underlying this characteristic feature of apoptosis. A screen of kinase inhibitors performed on synchronized blebbing cells indicated that only myosin light chain kinase (MLCK) inhibitors decreased blebbing. Immunoprecipitation of myosin II demonstrated that myosin regulatory light chain (MLC) phosphorylation was increased in blebbing cells and that MLC phosphorylation was prevented by inhibitors of MLCK. MLC phosphorylation is also mediated by the small G protein, Rho. C3 transferase inhibited apoptotic membrane blebbing, supporting a role for a Rho family member in this process. Finally, blebbing was also inhibited by disruption of the actin cytoskeleton. Based on these results, a working model is proposed for how actin/myosin II interactions cause cell contraction and membrane blebbing. Our results provide the first evidence that MLC phosphorylation is critical for apoptotic membrane blebbing and also implicate Rho signaling in these active morphological changes. The model system described here should facilitate future studies of MLCK, Rho, and other signal transduction pathways activated during the execution phase of apoptosis.
Dynamic membrane blebbing, along with chromatin condensation and DNA laddering are three of the most commonly used criteria for distinguishing apoptosis from other physiological processes (Wyllie et al., 1980). Despite their importance, little is known about mechanisms underlying these conserved events. In most systems, the morphological changes that characterize apoptosis occur shortly before death during a rapid, evolutionarily conserved stage of invariant duration known as the execution phase (Earnshaw, 1995; Jacobson et al., 1997). During the execution phase, the caspase family of proteases is thought to be activated and to cleave specific substrates, rapidly leading to cell death (Chinnaiyan and Dixit, 1996; Nagata, 1997; Nicholson and Thornberry, 1997). The execution phase of apoptosis has resisted biochemical characterization because its onset is markedly asynchronous across a population of cells (Lazebnik et al., 1995; McCarthy et al., 1997; Mills et al., 1997; Messam and Pittman, 1998). Thus, a simple system for synchronizing cells in the execution phase of apoptosis would prove useful for elucidating key signal transduction pathways critical for controlling the biochemical and morphological changes occurring just before death. Recently, McCarthy et al. (1997) reported that inhibition of caspases during apoptosis in Rat-1 fibroblasts resulted in a population of cells that entered into and remained in the execution phase of apoptosis (measured by membrane blebbing), with the same time-course as dying cells but without the appearance of other features of apoptosis (e.g., DNA laddering and chromatin condensation). In the present study, a similar model is described that has allowed us to identify signaling pathways that regulate the dramatic membrane blebbing occurring during the execution phase of apoptosis.
The majority of studies examining the formation of membrane blebs have focused on the role of cytoskeletal proteins. Tumor cells lacking actin binding protein (ABP)1 bleb extensively under normal conditions (Cunningham et al., 1992); cleavage of two other proteins that bind actin, talin and α-actinin, correlate with peroxide-induced blebbing (Miyoshi et al., 1996), and a fourth actin-binding cytoskeletal protein, fodrin, is cleaved by caspases during apoptosis (Martin et al., 1995; Cryns et al., 1996; Nath et al., 1996; Vanags et al., 1996). Numerous studies have focused directly on the role of actin in these apoptotic membrane changes. F actin is necessary for blebbing and eventual “apoptotic body” formation (Cotter et al., 1992), and the concentration of F actin is correlated with bleb size (Cunningham, 1995). F actin is present at the base of blebs during apoptosis (Laster and MacKenzie, 1996; Pitzer et al., 1996; Vemuri et al., 1996), and several groups have proposed that actin is cleaved by caspases during apoptosis (Mashima et al., 1995; Kayalar et al., 1996; McCarthy et al., 1997; see also Song et al., 1997).
Although cytoskeletal proteins including actin seem to be involved in membrane blebbing during apoptosis, there is no direct evidence of a role for myosin as the motor behind these morphological changes (however, it is interesting that microinjection of catalytically active myosin light chain kinase [MLCK] induces membrane blebs; Fishkind et al., 1991). Through interactions with actin, the myosin family of motor proteins is involved in many forms of cell motility. Conventional nonmuscle myosin (myosin II) has been implicated in such basic cellular processes as cytokinesis, stress fiber pulling, maintenance of the cortical actin layer, and secretion of vesicles (for reviews see Grebecki, 1994; Maciver, 1996; Mitchison and Cramer, 1996). Myosin II contractile activity in smooth muscle and nonmuscle cells is stimulated through phosphorylation of myosin regulatory light chain (MLC) on serine 19 by MLCK (Kohama et al., 1996; Gallagher et al., 1997). This phosphorylation catalyzes the interaction of the myosin head with actin and subsequently allows the myosin ATPase to produce sliding force. Recent studies have shown the phosphorylation state of MLC also to be regulated by the small G protein, Rho. Rho is involved in cytoskeletal rearrangement, including cell contraction after lysophosphatidic acid or thrombin stimulation (Jalink et al., 1994; Tigyi et al., 1996; Gebbink et al., 1997), neurite retraction (Jalink et al., 1994; Tigyi et al., 1996; Gebbink et al., 1997), and actin stress fiber formation (Ridley and Hall, 1992; Chrzanowska-Wodnicka and Burridge, 1996). Rho also stimulates Rho kinase (ROK), which phosphorylates and inactivates MLC phosphatase (Noda et al., 1995; Kimura et al., 1996). In addition, ROK and MLCK phosphorylate MLC on the same serine residue (Amano et al., 1996), suggesting that ROK, like MLCK, increases myosin contractile activity.
In this report, we use the caspase inhibitor, z-VAD-FMK to block cell death late in the apoptotic process after initiation of blebbing. This results in a dramatically increased proportion of actively blebbing cells after serum removal. In contrast, this dynamic blebbing does not occur in apoptosis induced by the general kinase inhibitor, staurosporine (STS), suggesting that blebbing may be controlled by phosphorylation. We used the enriched population of blebbing cells after serum removal and z-Val-Ala-Asp-fluoromethylketone (z-VAD-FMK) treatment to characterize key phosphorylation events that occur during the execution phase of apoptosis. Using cellular and biochemical assays, we demonstrate essential roles for myosin II and phosphorylation of MLC in the formation of membrane blebs, and identify MLCK and Rho as key regulators of apoptotic blebbing. Thus, this report describes a model system for studying biochemical events that occur during the execution phase of apoptosis, and thus provides the first description of how MLCK and Rho signal transduction mechanisms regulate apoptotic membrane blebbing.
Materials and Methods
Materials
z-VAD-FMK was purchased from Kamiya Biomedical Co. (Seattle, WA); BaF(BOC-Asp-CH2F-OMe) was from Enzyme Systems Products (Livermore, CA); KT5926, KN-93, KT5823, H-89, bisindolylmaleimide 1, ML-9, and ML-7 were from Calbiochem-Novabiochem Corp. (San Diego, CA); Hoechst 33342 was from Molecular Probes (Eugene, OR); calyculin A was from Alamone Labs (Jerusalem, Israel); RPMI 1640, DME, and trypsin/ EDTA were obtained from GIBCO BRL (Gaithersburg, MD); horse serum was from ICN Pharmaceuticals Inc. (Costa Mesa, CA); FCS was purchased from Hyclone (Logan, UT); rat tail type 1 collagen was from Collaborative Research, Inc. (Bedford, MA); [32P]orthophosphate was from Dupont-NEN (Boston, MA); cytochalasin D, nocodazole, taxol, staurosporine, protein A–Sepharose, and all other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO).
Cell Culture
The PC6-3 subline of PC12 cells (Pittman et al., 1993) was cultured at 37°C with 5% CO2 in RPMI 1640 supplemented with 10% horse serum, 5% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin. COS-7 cells were cultured at 37°C with 5% CO2 in DME supplemented with 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin.
To remove serum, cells were trypsinized and washed three times in serum-free medium. Then 30,000 cells were plated per well in a 24-well tissue culture dish in serum-free medium with either 100 μM z-VAD-FMK, or an equivalent volume of the vehicle, DMSO. Control cells were washed and plated at the same density in serum-free N2 medium (5 μg/ml insulin, 30 nM selenium, 10 μg/ml transferrin, 20 nM progesterone, 100 μM putrescine, and 1 mg/ml BSA in RPMI 1640; Bottenstein et al., 1980), and then incubated with or without z-VAD-FMK. N2 medium prevents serum deprivation–induced cell death for up to 5 or 6 d (unpublished observations). STS (1 μM), and either 100 μM z-VAD-FMK or DMSO were added to cells plated in N2 medium. Cell viability was assessed by counting cells in randomly selected fields, in the presence of 0.0625% trypan blue. All experiments were also performed with the caspase inhibitor, BaF, with identical results.
The percentage of blebbing cells was determined for cells grown on collagen (50 μg/ml) coated 24-well tissue culture plates. Culturing cells on collagen provides a sharper distinction between normal wedge-shaped cells and round blebbing cells. An observer blinded to experimental condition counted all living cells and scored those with obvious membrane protrusions as blebbing. The effect of 5 μM KN-93, 30 μM H-89, 5 μM bisindolylmaleimide 1, 5 μM KT-5823, 4 μM KT-5926, 1 μM staurosporine, and 20 or 80 μM ML-9 on blebbing was determined by counting cells in the same fields before addition of drug, and 1 h after drug treatment. Six fields were counted per well, three wells per experiment, and experiments were performed three times.
The effects of cytoskeletal disruption on serum-deprived, z-VAD-FMK– treated cells were assessed by treating cells with cytochalasin D (0.5 μg/ml), taxol (100 μM), nocodazole (5 μM), and 2,3-butanedione monoxime (BDM; 15 mM). Drugs were added 24 h after serum removal and z-VAD-FMK administration, and cells were either counted 1 h later or, in the case of BDM experiments, cells were followed by time-lapse videomicroscopy.
DNA Laddering and Hoechst Staining
DNA laddering was performed as described previously (Pittman et al., 1993). Briefly, cells were either trypsinized or scraped directly into Eppendorf tubes (serum-free and staurosporine samples, respectively). Cell pellets were resuspended in 0.1 M EDTA, 1% SDS, 100 μg/ml proteinase K, and 0.2 M Tris, pH 8.5, and then incubated at 60°C for 2 h. After addition of 5 M potassium acetate, lysates were vortexed and placed on ice for 30 min. The supernatant obtained from centrifugation at 14,000 g for 15 min was precipitated with ice-cold ethanol, and DNA was electrophoresed.
For Hoechst staining after serum removal or staurosporine treatment, 300,000 cells were plated on collagen-coated, glass bottom 35-mm tissue culture dishes (Mat-Tek, Ashland, MA). After experimental treatments, cells were rinsed three times with PBS and then fixed for 15 min in 4% paraformaldehyde. Cells were rinsed with PBS three times, incubated with 5 μg/ml Hoechst 33342 in PBS for 15 min, rinsed once with PBS, coverslipped, and then allowed to dry for several hours.
Time-Lapse Videomicroscopy
Time-lapse videomicroscopy of cells was performed as described previously (Mills et al., 1995). Briefly, images were obtained with a video camera (AG-6050; Panasonic, Secaucus, NJ) attached to a Nikon Diaphot inverted microscope (Garden City, NJ), and recorded on either a JVC (BR-9000U; Pine Brook, NJ) or a Sony (SVT-5050; Park Ridge, NJ) time-lapse VHS VCR. Time-lapse images were then time-base corrected and digitized using Image 1 software (Universal Imaging, Inc., West Chester, PA). Culture conditions were identical to those in the incubator (5% CO2, 37°C), and cells could be recorded for at least 5 d without adverse effects. For time-lapse experiments, ∼200,000 cells were plated on 50 μg/ml collagen-coated, glass bottom 35-mm dishes. For assessment of drug effects, cells were transferred to the time-lapse chamber, and a field selected, and then recorded for at least 10 min (usually 30–60 min) before drug was added.
Phosphorylation of MLC
The procedure used was adapted from that described previously (Ludlowyke et al., 1989). Cells were labeled for 2 h in medium containing 100 μCi/ml [32P]orthophosphate, scraped into ice-cold PBS, and then pelleted 1 min at 12,000 g. The cell pellet was resuspended in ice-cold lysis buffer: 1% NP-40, 250 mM NaCl, 5 mM EGTA, 0.5 mM PMSF, 1 μg/ml leupeptin, 15 mM β-mercaptoethanol, 100 mM sodium pyrophosphate, 50 mM NaF, 20 mM Tris, pH 7.9. Lysates were homogenized through a 26-gauge needle until no longer viscous and then centrifuged at 14,000 g for 10 min. Cells lysates were treated with a 1:20 dilution of crude anti–myosin II antibody (obtained from Dr. R. Adelstein, National Institutes of Health, Bethesda, MD), and immune complexes isolated using protein A–Sepharose. Immunoprecipitates were then washed once in lysis buffer and once in PBS. For MLC separation and quantification, the method of Taylor and Stull (1988) was adapted as follows: immunoprecipitates were resuspended in 9 M urea, 2 mM DTT, 22 mM glycine, 20 mM sucrose, 1 mM EGTA, and 20 mM Tris, pH 8.8, and then heated to 90°C for 5 min. Samples were electrophoresed on a separating gel containing 10% acrylamide, 40% glycerol, 22 mM glycine, and 20 mM Tris, pH 8.8, after a stacking gel consisting of 4% acrylamide, 6 M urea, 22 mM glycine and 20 mM Tris, pH 8.8. Running buffer was 22 mM glycine and 20 mM Tris, pH 8.8. Gels were pre-electrophoresed for 1 h at 350 V with 1 mM DTT added to the upper chamber. Then samples were loaded and electrophoresed for 1.5 h at 450 V. Gels were dried and then exposed to PhosphorImager plates (Molecular Dynamics, Sunnyvale, CA). Bands were quantitated using ImageQuant software (Molecular Dynamics).
C3 Exoenzyme Experiments
The glutathione-S-transferase (GST) fusion protein of Clostridium botulinum C3 exoenzyme (construct obtained from Dr. A. Alberts, Imperial Cancer Research Fund, London, UK) was purified on glutathione Sepharose according to the manufacturer's instructions (Pharmacia Biotech, Uppsala, Sweden). Preliminary experiments were performed to determine optimal conditions for treating cells. Concentrations up to 25 μg/ml purified C3 in the presence or absence of 10 μg/ml lipofectamine as a vehicle for increasing cellular uptake were tested (Hirao et al., 1996). Concentrations of 10 μg/ml C3 transferase in 10 μg/ml lipofectamine or 25 μg/ml C3 transferase alone had marked biological effects on blebbing within 2 h. The C3/lipofectamine combination was used routinely in experiments. Counts of blebbing cells were made before and 2 h after addition of C3 and lipofectamine. Lipofectamine alone had no effect on blebbing.
Results
Serum Withdrawal and STS Induce Apoptosis, but Membrane Blebbing Occurs Only After Serum Withdrawal
Both serum removal and STS induce apoptosis in PC12 cells (Fig. 1). In each case, death is characterized by DNA laddering and chromatin condensation, and can be inhibited by the general inhibitors of caspases, z-VAD-FMK (Fig. 1) or BaF (data not shown). The time-course of death is faster in STS-treated cells; however, the most striking difference between serum deprivation and treatment with STS is the absence of membrane blebs in cells after treatment with STS, both with (Fig. 2 b) and without z-VAD-FMK (data not shown). The lack of membrane blebs in cells treated with STS suggests that the mechanism underlying apoptotic membrane blebbing may be a phosphorylation event.
Figure 1.

Characterization of apoptosis after removal of serum or exposure to STS. (a) PC12 cells were plated in either serum free (SF, □) or control N2 medium (C, ⋄), and plated with (+) or without 100 μM z-VAD-FMK. Viability was assessed by trypan blue exclusion. Serum removal from PC12 cells resulted in ∼50% death by 30 h; this death was inhibited by 100 μM z-VAD-FMK. Data represent the mean ± SEM for three experiments. (b) PC12 cells were subcultured into N2 medium and 1 μM staurosporine (STS) with (⋄) or without (□) 100 μM z-VAD-FMK. Treatment of PC12 cells with 1 μM STS resulted in ∼50% death by 6 h; this death was inhibited by 100 μM z-VAD-FMK. Data represent the mean ± SEM for three experiments. (c) DNA laddering caused by serum removal (SF) at 24 h, or staurosporine (STS) treatment at 12 h in the absence (−) or presence (+) of 100 μM z-VAD-FMK. Control cells (C) were in N2 medium with DMSO vehicle. 1-kb DNA ladders are shown for reference to the right of each gel. Note that z-VAD-FMK prevents DNA laddering. (d) Hoechst staining of apoptotic nuclei after serum removal (SF) at 24 h and staurosporine (STS) treatment at 12 h was prevented by 100 μM z-VAD-FMK (+). Control cells (CON) were in N2 medium with DMSO vehicle. Arrowheads indicate apoptotic nuclei. Bar, 40 μM.
Figure 2.
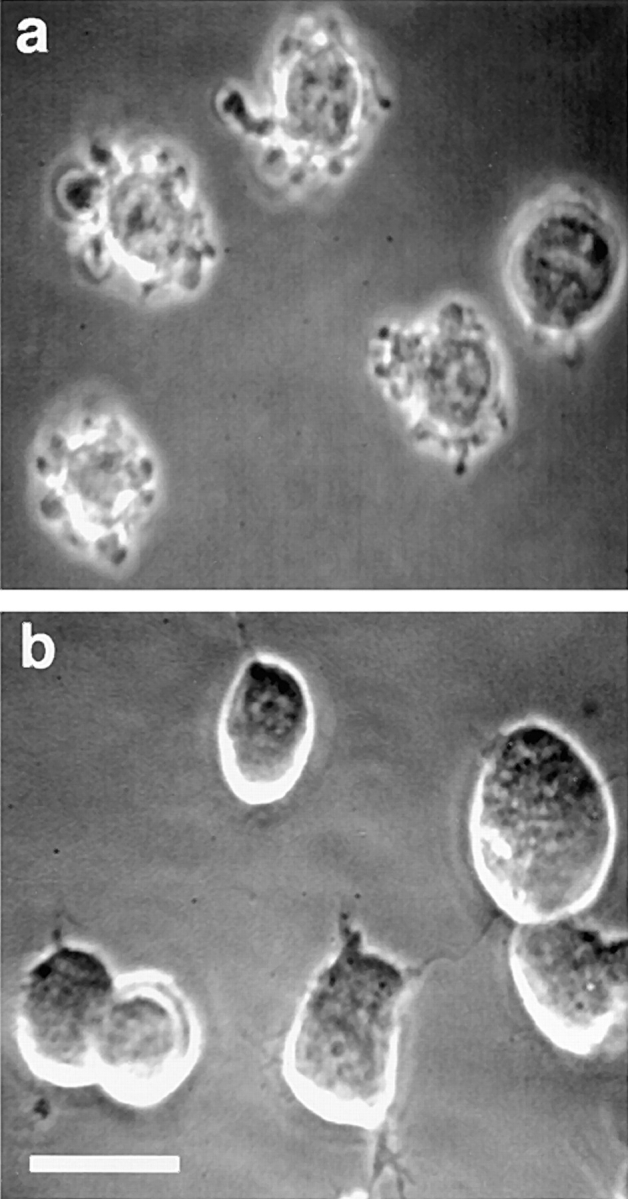
Dynamic membrane blebbing is present in cells treated with z-VAD-FMK after serum removal, but is absent in cells treated with STS and z-VAD-FMK. (a) Representative field of serum-free z-VAD-FMK–treated cells at 24 h. (b) Representative field of STS- and z-VAD-FMK–treated cells at 12 h. Note that some cells form neurite-like projections after STS treatment. Phase rings were purposely misaligned slightly to decrease phase “halo” and emphasize surface morphology. Bar, 30 μM.
An additional striking observation in these initial experiments is the greatly increased percentage of cells dynamically extruding and retracting membrane blebs in serum-deprived cultures after treatment with z-VAD-FMK (Fig. 2 a), compared to serum-deprived cells in the absence of z-VAD-FMK (data not shown). To determine the increased number of blebbing cells in serum-deprived, z-VAD-FMK–treated cultures, cells were counted and scored for blebbing at time zero and at 24 h (Fig. 3 a). At time zero, low percentages of cells with blebs are present in all groups, which probably reflects the normal background death in these cultures. After 24 h in serum-free medium without z-VAD-FMK, only 10% of cells are blebbing, presumably because these cells undergo the asynchronous death characteristic of apoptosis. Consequently, only a small percentage of these cells are blebbing at any one time (Fig. 3 a). Control cells maintained for 24 h in serum-free medium supplemented with N2 components have a low background death and very few cells are blebbing (Fig. 3 a). Inhibition of caspases in the absence of an apoptotic stimulus does not cause blebbing by itself, as only a small percentage of z-VAD-FMK–treated cells are blebbing in medium containing N2 components at 24 h (Fig. 3 a). The small increase in blebbing in the N2 z-VAD-FMK condition compared to N2 alone at 24 h is consistent with inhibition of normal background death by z-VAD-FMK. After serum removal and treatment with z-VAD-FMK, there is a marked, nearly fivefold increase in the fraction of cells blebbing at 24 h (Fig. 3 a), supporting a previous report that z-VAD-FMK blocks death after onset of blebbing (McCarthy et al., 1997). This raised the possibility of using the increased number of cells stopped early in the execution phase of apoptosis to investigate the mechanisms underlying dynamic membrane blebbing.
Figure 3.
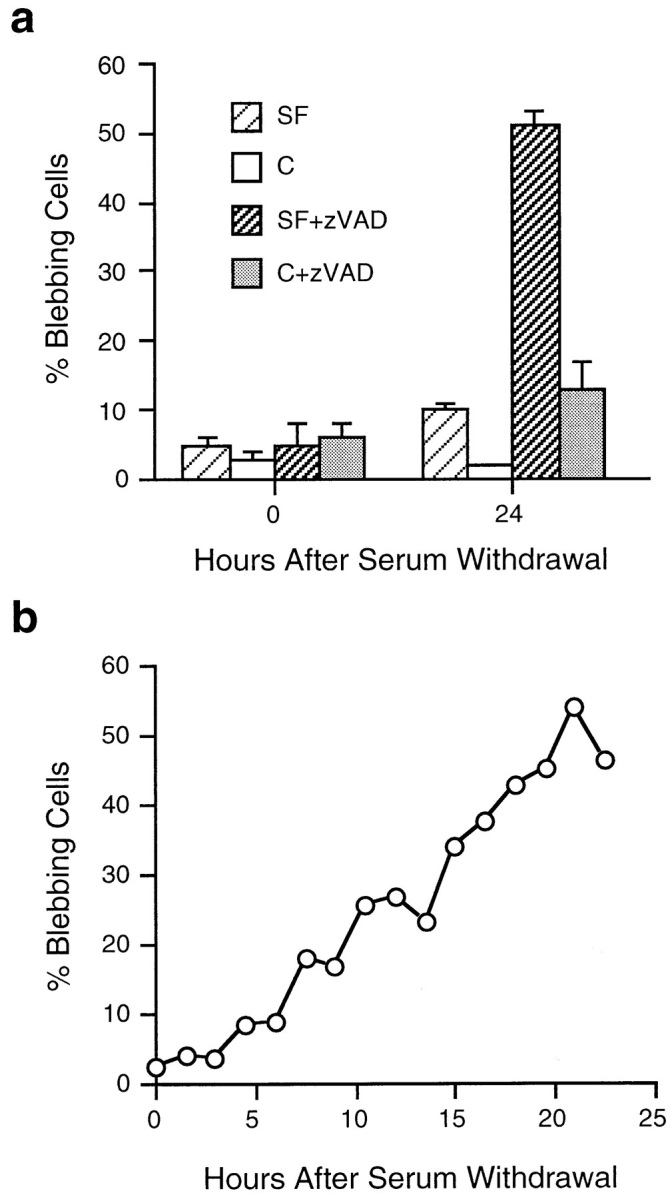
The number of blebbing cells increase over time after serum removal in the presence of z-VAD-FMK. (a) Blebbing cells were scored at 0 and 24 h after serum removal and initiation of z-VAD-FMK treatment. The percentage of blebbing cells was calculated by dividing the number of blebbing cells by the total number of living cells per field. At 24 h, ∼50% of serum-deprived, z-VAD-FMK–treated cells were blebbing (SF + z-VAD, dark striped bars). SF, serum free; C, control N2 medium plus DMSO; and C + z-VAD N2 medium plus 100 μM z-VAD-FMK. Data represent the mean ± SEM for three experiments. (b) The percentage of blebbing cells was calculated every 1.5 h from a time-lapse video recording of a field of cells treated with 100 μM z-VAD-FMK after removal of serum. The results depicted are representative of three separate experiments.
The time-course of entry into blebbing was determined using long term time-lapse videomicroscopy (Fig. 3 b) of cultures of PC12 cells after serum removal and z-VAD-FMK treatment. The number of actively blebbing cells increasing over time seems to reflect normal, asynchronous entry into the execution phase of apoptosis (McCarthy et al., 1997). The time-course for accumulation of blebbing cells is similar to that for cell death seen in cells in serum-free medium such that ∼50% of the cells are dead (Fig. 1 a) or blebbing (Fig. 3 b) by 24 h. A similar asynchronous entry into blebbing is seen in serum-deprived, caspase-inhibited COS-7 cells, although the time-course is longer, and the proportion of blebbing cells is not as large (data not shown). In both cases, the morphology of blebbing in z-VAD-FMK–treated, serum-deprived cells is indistinguishable from that seen in the execution phase of apoptosis, except that, once these cells start to bleb, they continue to do so for hours to days rather than dying within the hour as serum-deprived cells without z-VAD-FMK do. Thus, treatment of apoptotic cultures with caspase inhibitors causes accumulation of blebbing cells not only in the Rat-1 fibroblast cell line as previously shown (McCarthy et al., 1997) but also in adrenal-derived PC12 cells, and kidney-derived COS-7 cells.
MLCK Activity Regulates Apoptotic Membrane Blebbing
As all other agents used to induce apoptosis in our laboratory (e.g., growth factor withdrawal, UV irradiation, H2O2, ceramide, and menadione) cause membrane blebbing, the absence of this feature of apoptosis in STS-treated cells was intriguing. Two hypotheses could explain this observation: (1) STS initiates apoptosis downstream of blebbing; or (2) STS initiates apoptosis upstream of blebbing but prevents blebbing by concomitantly inhibiting a necessary downstream kinase. To differentiate between these alternatives, cultures of PC12 and COS-7 cells were deprived of serum, and treated with z-VAD-FMK until a substantial portion of cells were actively blebbing (24 h of serum deprivation for PC12 cells and 48 h for COS-7 cells). Then STS was added and cells were followed using time-lapse videomicroscopy. Both PC12 and COS-7 cells stop blebbing within minutes after addition of STS (Fig. 4) but remain viable for hours. In other experiments, blebbing of UV-irradiated PC12 cells was also inhibited by STS treatment (data not shown), suggesting that the effect of STS is not limited to trophic factor withdrawal induced apoptotic blebbing.
Figure 4.
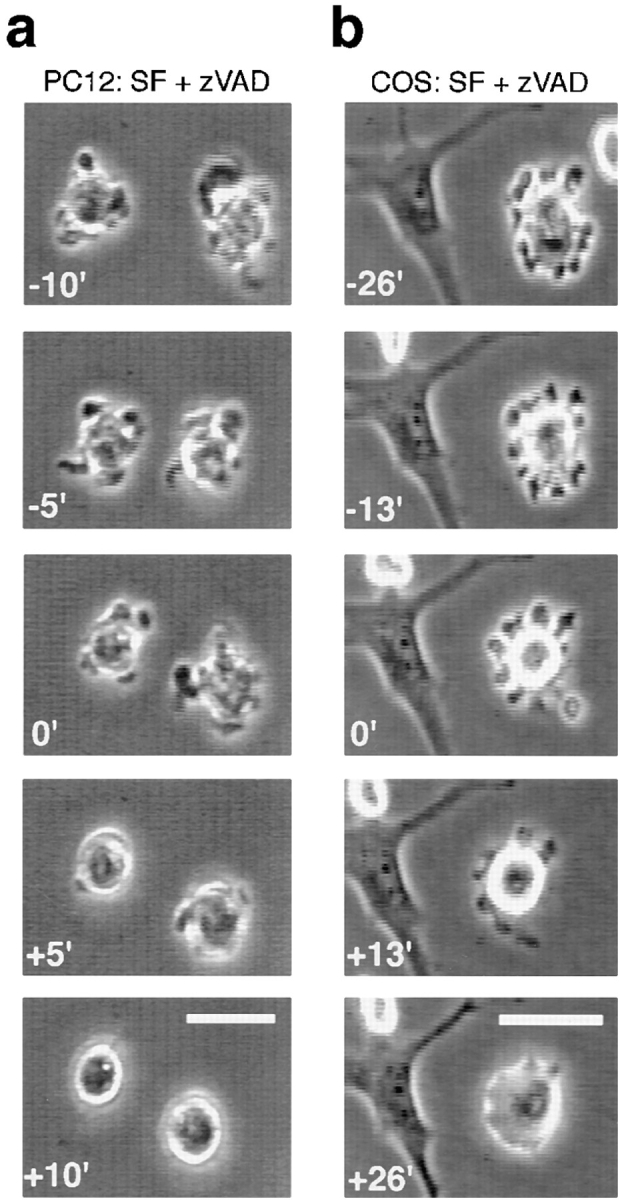
Staurosporine inhibits apoptotic blebbing in PC12 and COS-7 cells. Serum removal and z-VAD-FMK treatment were performed as described in the legend to Fig. 1. Cells were recorded for at least 10 min before addition of 1 μM STS. (a) Time-lapse images of a representative field of PC12 cells 24 h after removal of serum and treatment with 100 μM z-VAD-FMK. 5-min intervals are shown before and after addition of 1 μM STS (added at time zero). Note that 10 min after adding STS, blebbing ceased. (b) Time-lapse images of a representative field of COS-7 cells 48 h after removal of serum and treatment with 100 μM z-VAD-FMK. 13-min intervals are shown before and after addition of 1 μM STS (added at time zero). Approximately 26 min after STS addition, blebbing ceased. Bars: (a) 25 μM; (b) 50 μM.
To determine which kinase(s) STS inhibits to block blebbing, a panel of more specific inhibitors was screened. PC12 cells, deprived of serum and treated with z-VAD-FMK for 24 h, were treated for 1 h with either STS, KT5926 (an inhibitor of MLCK), KN-93 (an inhibitor of CAMKII), KT5823 (an inhibitor of protein kinase G), bisindolylmaleimide 1 (an inhibitor of protein kinase C) or H-89 (an inhibitor of protein kinase A). Only kinase inhibitors that inhibit MLCK (i.e., STS and KT5926) have a statistically significant effect on blebbing—STS decreases blebbing by 89%, and KT5926 by 39% (Fig. 5 a). The PKA inhibitor, H89, induces rapid death of cells at a concentration similar to that used in other studies (30 μM; Chijiwa et al., 1990) and has no effect on blebbing at a slightly lower concentration (7.5 μM).
Figure 5.
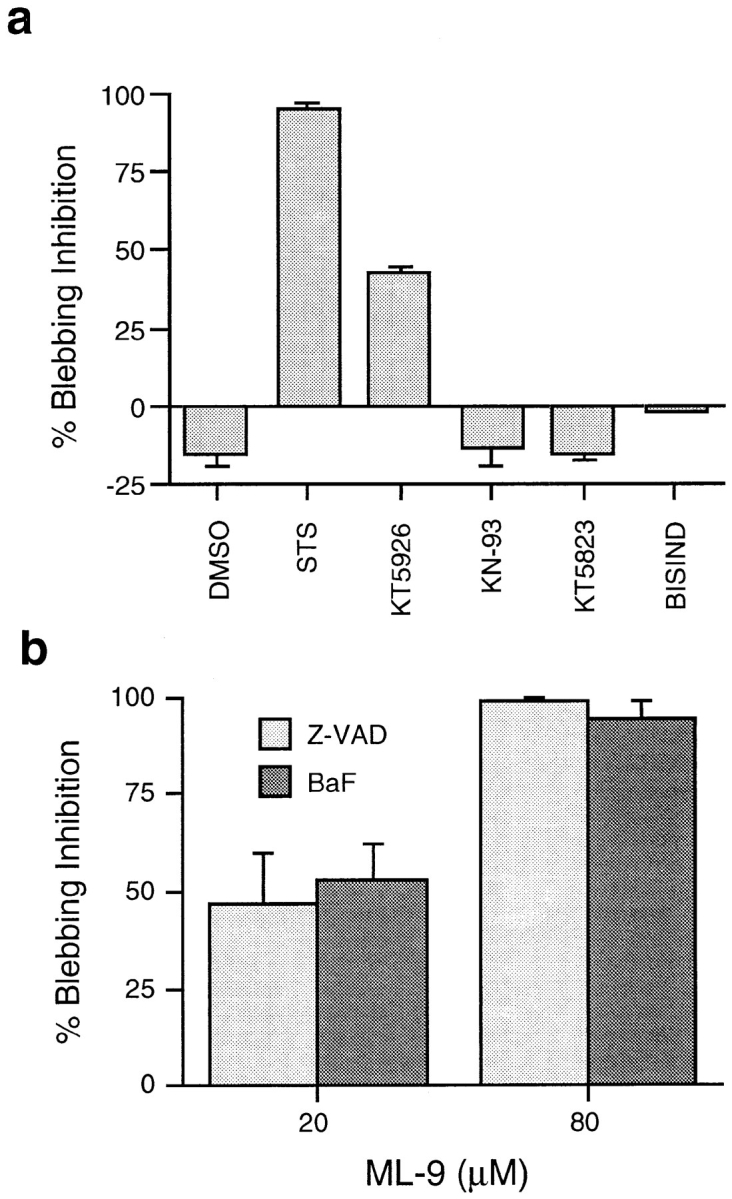
Inhibitors of MLCK stop apoptotic membrane blebbing. (a) Addition of either kinase inhibitor (1 μM STS, 4 μM KT-5926, 5 μM KN-93, 5 μM KT-5823, or 5 μM bisindolylmaleimide 1) or equivalent volume of DMSO 24 h after serum deprivation and z-VAD-FMK treatment. Cells were counted immediately before addition of kinase inhibitor, and the same fields counted 1 h after addition of inhibitor. Percent inhibition of blebbing was calculated by comparing the portion of cells blebbing before and after treatment. Data represent the mean ± SEM for three experiments. (b) The MLCK inhibitor, ML-9, inhibits blebbing of cells deprived of serum and treated with 100 μM z-VAD-FMK or 100 μM BaF (a general caspase inhibitor) in a dose-dependent manner. Cells were counted as above, in the presence of 20 or 80 μM ML-9. Data represent the mean ± SEM for three experiments.
In addition to inhibiting MLCK, KT5926 has activity against CAMKII (Hashimoto et al., 1991). To ensure that decreased blebbing is the result of MLCK inhibition, ML-9 and ML-7 (MLCK inhibitors that do not inhibit CAMKII) were used (Saitoh et al., 1986, 1987). ML-9 at 20 μM inhibits blebbing of serum-deprived cells treated with either z-VAD-FMK by 47%, or BaF by 53% within 1 h (Fig. 5 b). At 80 μM, ML-9 inhibits blebbing in z-VAD-FMK by 99%, and in BaF by 94% within 1 h (Fig. 5 b). At the higher concentration, ML-9 causes cells to lift off the tissue culture plate. When ML-9 is used at an intermediate concentration (40 μM), it rapidly inhibits blebbing after serum removal not only in the presence (Fig. 6 a) of z-VAD-FMK, but also in the absence (Fig. 6 b) of z-VAD-FMK, without causing cells to lift off the tissue culture dish. Similar results were seen with 10 μM ML-7 (data not shown). This suggests that MLCK regulates apoptotic membrane blebbing, and the morphological changes that occur in serum-deprived, z-VAD-FMK cells are mechanistically the same as those seen in dying, serum-deprived cells.
Figure 6.

ML-9 inhibits apoptotic blebbing in PC12 cells. (a) Time-lapse images of a representative field of 24-h serum- deprived, z-VAD-FMK–treated cells at 4-min intervals before and after addition of 40 μM ML-9 at time zero. Similar results are seen with 10 μM ML-7 (data not shown). (b) Time-lapse images of a field of cells after serum removal in the absence of z-VAD-FMK before and after addition of 40 μM ML-9 at time zero. Note the field in b was selected to include several blebbing cells. Due to asynchrony (see Results, and Fig. 3), only ∼10% of PC12 cells after serum removal are blebbing at one time, so it is not typical to find several blebbing cells in the same field in the absence of z-VAD-FMK. Bar, 25 μM.
Populations of Blebbing Cells Have Increased Levels of MLC Phosphorylation
Results obtained with inhibitors of MLCK implicate regulation of MLC as a critical component of apoptotic blebbing. To demonstrate that phosphorylation of MLC increases in a population enriched for actively blebbing cells, levels of MLC phosphorylation were directly assessed by immunoprecipitating 32P-labeled PC12 cells with antibodies against myosin II. To increase the percentage of actively blebbing cells, PC12 cells in serum-free medium were treated with z-VAD-FMK for 24 h. Increased MLC phosphorylation is observed in these cells, compared to control, non-blebbing cells (Fig. 7 a). The relatively low signal, in both control and serum-deprived, z-VAD-FMK–treated cultures (Fig. 7 a), can be proportionally enhanced by treatment with the phosphatase inhibitor, calyculin A, for 15 min (Fig. 7 a; Deery and Heath, 1993). Therefore, calyculin A–treated cells were used for the remainder of the immunoprecipitation experiments. In three independent experiments, three- to fivefold higher levels of MLC phosphorylation were seen in cultures enriched for blebbing cells (i.e., serum-free, z-VAD-FMK–treated) compared to serum-free cells without z-VAD-FMK. MLC phosphorylation in z-VAD-FMK–treated control cells is similar to phosphorylation levels of serum-deprived or control cells in the absence of z-VAD-FMK (Fig. 7 b); therefore, z-VAD-FMK itself has no effect on MLC phosphorylation. The MLCK inhibitor, ML-9, not only inhibits blebbing (Figs. 5 and 6), but also decreases MLC phosphorylation (Fig. 7 c).
Figure 7.

Phosphorylation of MLC is increased in blebbing cells and blocked by ML-9 treatment. Serum removal and z-VAD-FMK treatment were performed as described in Fig. 1. 32P-Labeled cell lysates were immunoprecipitated with anti–human platelet myosin II antibody, electrophoresed on a urea gel, and then analyzed on a Molecular Dynamics PhosphorImager. (a) Pre-treatment with 200 nM of the phosphatase inhibitor, calyculin A (CalA), increases MLC phosphorylation in both serum free (SF) and control (C) cells. Cells were pretreated with calyculin A for the rest of the experiments. A nonspecific rabbit IgG was used as a control, all other immunoprecipitations were performed with the anti–human platelet myosin II antibody. (b) Neither serum removal nor z-VAD-FMK alone increase phosphorylation of myosin light chain; however, cells treated with z-VAD-FMK after serum removal for 24 h (i.e., blebbing) have increased phosphorylation of MLC. (c) Addition of 40 μM ML-9 to cells deprived of serum and treated with 100 μM z-VAD-FMK decreases the phosphorylation of MLC. The gels in b and c were electrophoresed longer to visualize the mono- and di-phosphorylated forms of MLC. Bars shown to the right of image indicate positions of purified proteins run next to immunoprecipitates on the gel. The essential MLC (E-MLC), and the slower migrating (top) regulatory MLC (R-MLC) are not phosphorylated. The two lower R-MLC bands indicate the mono- and di-phosphorylated forms of MLC.
Involvement of Rho and Cytoskeletal Proteins in Membrane Blebbing
Two lines of evidence support a role for the small G protein Rho in MLC phosphorylation. First, Rho activates ROK, which phosphorylates MLC on serine 19, the same residue phosphorylated by MLCK (Amano et al., 1996). Second, ROK phosphorylates and deactivates a MLC phosphatase subunit, causing increased MLC phosphorylation levels in cells with rapid phosphate turnover (Noda et al., 1995; Kimura et al., 1996). To test the role of Rho in blebbing, serum-deprived, z-VAD-FMK–treated cells were incubated for 2 h with Clostridium botulinum toxin C3 transferase, an enzyme that ADP ribosylates and inactivates Rho (Sekine et al., 1989; Aktories et al., 1990; Paterson et al., 1990). C3 treatment for 2 h decreases the percentage of blebbing cells by ∼60% (Fig. 8). Time-lapse videomicroscopy shows that overnight treatment with C3 completely inhibits blebbing without cell loss (data not shown).
Figure 8.

The Rho inhibitor C3 transferase decreases blebbing. 10 μg/ml C3 and 10 μg/ml lipofectamine were added to cultures deprived of serum and treated with z-VAD-FMK for 24 h. Percent blebbing cells was calculated by dividing the number of blebbing cells by the number of total cells after 2 h of treatment with either vehicle (DMSO) or C3 transferase (C3). Data are mean ± SEM from three independent experiments.
Several agents that alter the cytoskeleton, were tested for immediate effects on blebbing. Incubation of serum-deprived, z-VAD-FMK–treated cells with nocodazole, a microtubule destabilizer, modestly increases the already considerable fraction of blebbing cells (Table I). Taxol, a microtubule stabilizer, has little effect during the short time frame of the experiment, whereas cytochalasin D, a destabilizer of actin filaments, greatly inhibits blebbing in serum-deprived, z-VAD-FMK–treated cells within an hour (Table I). Inhibition of myosin ATPase function with BDM does not affect the number of cells with blebs when viewed in static cultures (Table I). However, time-lapse videomicroscopy reveals that BDM dramatically slows the blebbing process, in fact, cells are often “frozen” with blebs extended (Table I; see Discussion). Thus, inactivation of myosin function with BDM appears to inhibit both protrusion and retraction of blebs.
Table I.
Immediate Effects on Blebbing
| Treatment | Response | Description | ||
|---|---|---|---|---|
| Cytochalasin D (actin depolymerizer) | ↓ ↓ ↓ | A decrease in blebbing was seen 10 min after addition of cytochalasin D, and this inhibition was seen for the length of the experiment (1 h). | ||
| Taxol (microtubule stabilizer) | — | No change was observed in blebbing over the time-course of the experiment (1 h). | ||
| Nocodazole (microtubule destablizer) | ↑ | No initial changes are apparent in amount of blebbing cells, although by 1 h there is an increase in number of blebbing cells. | ||
| BDM (inhibits myosin ATPase) | ↓ ↓ | No changes are apparent by counting static cultures. Observation of cells via time-lapse however, shows a striking decrease in the rate of protrusions and retractions (cells appear “frozen” in place with blebs extended). |
24 h after serum removal and z-VAD-FMK treatment, either cytochalasin D (0.5 μg/ml), taxol (100 μM), nocodazole (5 μM), or BDM (15 mM) was added to cultures. 1 h later, the number of blebbing cells was counted. Time-lapse videomicroscopy was also used to determine effects of BDM on blebbing.
Discussion
By increasing the fraction of cells undergoing morphological changes characteristic of the execution phase of apoptosis, we were able to demonstrate that MLCK activity is critical for dynamic membrane blebbing, and moreover, that an increased level of myosin II regulatory light chain phosphorylation is present in enriched populations of blebbing cells. Our data also implicate the small G protein, Rho, in cellular blebbing. Finally, evidence is presented that several cytoskeletal proteins play an important role in normal apoptotic blebbing. Thus, we extend the previously known motile events requiring myosin II by demonstrating for the first time, an essential role for this key cellular motor in membrane blebbing—a form of cell motility present not only during apoptosis but also during cell division. The model system described here, as well as the broader implications of MLCK and Rho signaling during the execution phase should have considerable impact on future studies characterizing signaling events during the execution phase of apoptosis.
Apoptosis is a physiological process, defined by characteristic and evolutionarily conserved morphological changes including DNA laddering, chromatin condensation, membrane blebbing, cell shrinkage, and apoptotic body formation. Morphological events such as these distinguish apoptosis from other cellular processes; therefore, it is likely that these conserved features of apoptosis are critical aspects of the apoptotic process in vivo. Recently, there has been considerable interest in identifying mechanisms underlying these late processes (McCarthy et al., 1997; Rudel and Bokoch, 1997); however, the pathways that lead to these downstream events have been difficult to analyze biochemically because they occur asynchronously in a population of cells, with only ∼10% of cells in the execution phase at one time (Earnshaw, 1995; Mills et al., 1997; Messam and Pittman, 1998). Using z-VAD-FMK to inhibit death and synchronize cells early in the execution phase (i.e., before DNA laddering and chromatin condensation; McCarthy et al., 1997) has allowed us to characterize one of the hallmark features of apoptosis, membrane blebbing. This system should prove useful for continued characterization of regulatory mechanisms involved in blebbing, and for defining other signaling pathways activated during the execution phase of apoptosis.
A summary of our data in the context of the execution phase of apoptosis is provided in Fig. 9. An apoptotic stimulus (such as serum removal) activates several pathways that lead to a characteristic apoptotic death (i.e., DNA laddering, chromatin condensation, and membrane blebbing). Using caspase inhibitors to block cell death, a synchronous population of cells “trapped” early in the execution phase of apoptosis is generated, after initiation of blebbing, but before other characteristic changes such as chromatin condensation and DNA laddering (McCarthy et al., 1997). This system was used to demonstrate that MLC phosphorylation is crucial for membrane blebbing, as inhibition of MLCK (with STS, KT5926, ML-9, and ML-7) decreases blebbing, and phosphorylation of MLC is markedly enhanced in blebbing cells. Also, the small GTPase, Rho, which increases phosphorylation of MLC, was shown to be important for apoptotic membrane blebbing, as Rho inactivation also inhibits blebbing. Therefore, the data described in this paper provide starting points to begin elucidating upstream and downstream signaling events from MLCK and Rho during the execution phase of apoptosis.
Figure 9.

Signal transduction pathways regulating membrane blebbing during the execution phase of apoptosis. An apoptotic stimulus, such as serum removal, triggers pathways leading to DNA laddering, chromatin condensation, membrane blebbing, and the eventual death of the cell. Using z-VAD-FMK to inhibit caspases, cell death is blocked, along with the characteristic features, DNA laddering and chromatin condensation (McCarthy et al., 1997). Apoptotic membrane blebbing still occurs; therefore, blebbing is probably initiated before caspase activation. For blebbing to occur, the myosin regulatory light chain (MLC) must be phosphorylated, as demonstrated by inhibitors of myosin light chain kinase (MLCK) blocking blebbing and the increased phosphorylation state of MLC in serum-deprived, z-VAD-FMK–treated cells. This phosphorylation can be inhibited by inhibition of MLCK. Also, the small G protein Rho regulates blebbing, as inactivation of Rho with C3 transferase inhibits bleb protrusion. ROK, Rho kinase; MP, myosin phosphatase.
Signal transduction pathways implicated in regulation of apoptosis such as those involving extracellular signal-regulated kinase 1/2 (Xia et al., 1995; Cuvillier et al., 1996), stress-activated protein kinases (Johnson et al., 1996; Verheij et al., 1996; Zanke et al., 1996; Ichijo et al., 1997), phosphotidylinositol-3 kinase (Dudek et al., 1997; Kauffmann-Zeh et al., 1997), and AKT/PKB kinase (Dudek et al., 1997; Kauffmann-Zeh et al., 1997) have been identified; however, these are activated early in the apoptotic process, long before cells enter the execution phase (Park et al., 1996). Being able to accumulate a large number of synchronized cells late in the apoptotic process allowed us to demonstrate a critical role for MLC phosphorylation in maintaining membrane blebbing. Phosphorylation of its regulatory light chain allows myosin II to bind actin and ultimately results in force generation for many types of cell motility. The phosphorylation state of MLC is mediated by MLCK (Kohama et al., 1996), a myosin-specific phosphatase (Alessi et al., 1992; Hubbard and Cohen, 1993), and ROK. ROK has a dual role, both directly by phosphorylation and activation of MLC and indirectly by inhibiting the MLC phosphatase (Noda et al., 1995; Amano et al., 1996; Kimura et al., 1996). In addition to regulating MLC phosphorylation, Rho is involved in rearrangement of the cytoskeleton, including cell contraction after lysophosphatidic acid or thrombin stimulation (Jalink et al., 1994; Tigyi et al., 1996; Gebbink et al., 1997), neurite retraction (Jalink et al., 1994; Tigyi et al., 1996; Gebbink et al., 1997), and actin stress fiber formation (Ridley and Hall, 1992; Chrzanowska-Wodnicka and Burridge, 1996). Therefore, the involvement of Rho signaling in the morphological changes during apoptosis is not surprising.
The involvement of both MLCK and Rho signaling in the phosphorylation state of MLC may at first seem unnecessary, but this overlap in function may play an important regulatory role. For instance, MLCK could work synergistically with ROK. In this scenario, MLCK-stimulated phosphorylation of MLC may be increased by ROK-mediated inhibition of the myosin phosphatase. Here, the primary effect of ROK is the inhibition of myosin phosphatase, and the MLC phosphorylation by ROK may play a secondary role. Cells may need such a two-pronged approach on MLC phosphorylation to sustain the uninterrupted increase in cellular contractility that characterizes apoptotic blebbing. Another possibility is that, unlike MLCK (which requires calcium/calmodulin for activation), ROK function is not calcium-dependent. Thus, ROK phosphorylation of MLC may provide a calcium-independent mechanism for activation of myosin II.
The implication of myosin II in the mechanism of membrane blebbing, allows us to propose a working model of how early execution phase morphological changes occur in a cell. It has long been thought that myosin contraction of the peripheral actin ring produces centripetal force (Grebecki, 1994), which acts to compress the cytoplasm in the center of the cell. If structural proteins (such as ABP and fodrin) that link the cell membrane to the cortical actin layer are cleaved, then the centripetal force pushing the actin ring inward would cause extrusion of the cytoplasm in areas of weak actin/membrane linkage, resulting in bleb formation. To support this model for blebbing force production, we show that myosin II is necessary for blebbing.
Also supporting the model is the observation in the present study and in other reports (Keller and Zimmerman, 1986; Cotter et al., 1992; Ghibelli et al., 1995) that destabilization of actin filaments with cytochalasin greatly inhibits blebbing (cytochalasins can induce membrane bubbling in some non-apoptotic cells, but the morphological characteristics of this process are very different from those of apoptotic blebbing; Prentki et al., 1979, Miranda et al., 1974). Actin and fodrin are substrates for caspases (Martin et al., 1995; Cryns et al., 1996; Kayalar et al., 1996; Nath et al., 1996; Vanags et al., 1996; McCarthy et al., 1997); therefore, it has been speculated that caspase activation leading to actin and fodrin degradation, might lead to blebbing (Martin et al., 1995; Kayalar et al., 1996). However, data in this report and that of McCarthy et al. (1997) suggest that in these systems, blebbing is upstream of caspase inhibition. If this is true and our proposed working model is correct, then non-caspase–induced weaknesses in the cortical actin–membrane linkage must be occurring. Likely agents to perform this function would be calpains, which are known to cleave fodrin and other cytoskeletal proteins (Miyoshi et al., 1996; Nath et al., 1996). Inhibition of calpains prevents cleavage of the cytoskeletal proteins, α-actinin and talin, and also inhibits hepatocyte blebbing (Miyoshi et al., 1996). Preliminary data from our lab show that cells do not bleb if calpain inhibitors are added at the time of z-VAD-FMK treatment and serum removal (Stone, N.L., unpublished observations), which is consistent with the possibility that calpains are activated upstream of caspases and cleave key cytoskeletal proteins.
Destabilization of microtubules also may play an important role in the modulation of apoptotic blebbing. Treatment with nocodazole increases blebbing in our system and in others (Table I; Keller et al., 1985; Keller and Zimmerman, 1986). Consistent with this observation are studies showing that microtubules disassemble in the late stages of apoptosis (Bonfoco et al., 1996; Mills et al., 1998). Additional studies showing that microtubule disassembly increases cellular contractility (Danowski, 1989; Kolodney and Elson, 1993), and that this increase in contractility is mediated by an increase in MLC phosphorylation (Kolodney and Elson, 1995) support our working model of force generation for membrane blebbing.
Apoptotic membrane blebbing is a combination of dynamic protrusion and retraction events. MLCK inhibitors decrease the fraction of cells with blebs in static cell– counting experiments and lead to a loss of blebs in time-lapse videomicroscopy studies. Thus, inhibition of myosin II activity seems to prevent further protrusion of blebs, whereas retraction of blebs is not affected by inhibition of MLCK. However, both protrusion and retraction of blebs are slowed or stopped by the low affinity inhibitor of the myosin ATPase, BDM. BDM inhibits unconventional myosins as well as myosin II. Therefore, inhibition of bleb retraction by BDM suggests that a form of unconventional myosin may be responsible for retracting blebs. Interestingly, similar observations with BDM and MLCK inhibitors on growth cone protrusion and retraction have been observed recently (Ruchhoeff and Harris, 1997).
In addition to regulating blebbing, myosin and Rho activity may be involved in mediating other functions during the execution phase of apoptosis. The first morphological sign of apoptosis in vivo is a pulling up and away (rounding up) from extracellular matrix attachments. Cell contraction is mediated by Rho in cells that don't have strong adhesions to extracellular matrix, like PC12 cells (Jalink et al., 1994; Tigyi et al., 1996). In epithelial cell monolayers, individual apoptotic cells contract, using an actin-dependent mechanism to pull on neighboring cells (Peralta-Soler et al., 1996). Because intercellular attachments are maintained, this contraction performs the important function of automatically closing gaps that would be formed by death of a cell. Thus, actin-based contraction (likely involving Rho/ myosin regulation) could serve crucial in vivo functions, by preventing gaps in epithelia or mechanically recruiting neighboring cells to phagocytose apoptotic cells.
This is the first report to define a potential mechanism for myosin II–mediated force generation during apoptotic membrane blebbing and to propose a working model of how blebbing might occur. Synchronizing cells early in the final phase of apoptosis will facilitate future characterization of myosin and Rho in bleb protrusion, as well as providing a powerful system for studying upstream and downstream signaling pathways involved in other conserved events in the execution phase of apoptosis. Thus, this study advances our understanding of the signaling events that occur during apoptosis, and describes the use of a model system that will allow other features of the execution phase of apoptosis to be understood mechanistically.
Acknowledgments
We thank Drs. Jean and Joseph Sanger (University of Pennsylvania, Philadelphia, Pennsylvania), and Dr. L. Sweeney (University of Pennsylvania) for helpful discussions; Dr. J. Meinkoth (University of Pennsylvania) for critical reading and discussions of the manuscript; Dr. R. Adelstein for kindly providing myosin II antibodies, and Dr. A. Alberts for generously providing the C3 transferase construct.
This work was supported by National Institutes of Health grant NS 32465 (to R.N. Pittman) and MSTP 5-T32-6M07170 (to J.C. Mills).
Abbreviations used in this paper
- ABP
actin binding protein
- BDM
2,3-butanedione monoxime
- MLC
myosin regulatory light chain
- MLCK
myosin light chain kinase
- ROK
Rho kinase
- STS
staurosporine
- z-VAD-FMK
z-Val-Ala-Asp-fluoromethylketone
Footnotes
J.C. Mills and N.L. Stone contributed equally to this work.
Address all correspondence to Nicole L. Stone, University of Pennsylvania School of Medicine, 155 John Morgan Building, 3620 Hamilton Walk, Philadelphia, PA 19104-6084. Tel.: (215) 898-7099. Fax: (215) 573-2236. E-mail: burkhardt@pharm.med.upenn.edu
J.C. Mills' present address is Department of Pathology, Washington University School of Medicine, 550 South Euclid Avenue, Box 8118, St. Louis, MO 63110.
References
- Aktories K, Mohr C, Koch G. Clostridium botulinum C3 ADP- ribosyltransferase. Curr Top Micro Immunol. 1992;175:115–131. doi: 10.1007/978-3-642-76966-5_6. [DOI] [PubMed] [Google Scholar]
- Alessi D, Macdougall LK, Sola MM, Ikebe M, Cohen P. The control of protein phospatase-1 by targeting subunits. The major myosin phosphatase in avian smooth muscle is a novel form of protein phosphatase-1. Eur J Biochem. 1992;210:1023–1035. doi: 10.1111/j.1432-1033.1992.tb17508.x. [DOI] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Leist M, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Cytoskeletal breakdown and apoptosis elicited by NO donors in cerebellar granule cells require NMDA receptor activation. J Neurochem. 1996;67:2484–2493. doi: 10.1046/j.1471-4159.1996.67062484.x. [DOI] [PubMed] [Google Scholar]
- Bottenstein JE, Skaper SD, Varon SS, Sato GH. Selective survival of neurons from chick embryo sensory ganglionic dissociates utilizing serum-free supplemented medium. Exp Cell Res. 1980;125:183–190. doi: 10.1016/0014-4827(80)90202-5. [DOI] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Chinnaiyan AM, Dixit VM. The cell-death machine. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter TG, Lennon SV, Glynn JM, Green DR. Microfilament-disrupting agents prevent the formation of apoptotic bodies in tumor cells undergoing apoptosis. Cancer Res. 1992;52:997–1005. [PubMed] [Google Scholar]
- Cryns, V.L., Bergeron L., Zhu H., Li H., and Yuan J. 1996. Specific cleavage of α-fodrin during Fas- and tumor necrosis factor-induced apoptosis is mediated by an interleukin-1β-converting enzyme/Ced-3 protease distinct from the poly(ADP-ribose) polymerase protease. J. Biol. Chem. 271:31277–31282. [DOI] [PubMed]
- Cunningham CC. Actin polymerization and intracellular solvent flow in cell surface blebbing. J Cell Biol. 1995;129:1589–1599. doi: 10.1083/jcb.129.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers R, Stossel TP. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325–327. doi: 10.1126/science.1549777. [DOI] [PubMed] [Google Scholar]
- Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind JS, Spiegel S. Suppression of ceramide-mediated programmed cell death by spingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- Danowski BA. Fibroblast contractility and actin organization are stimulated by microtubule inhibitors. J Cell Science. 1989;93:255–266. doi: 10.1242/jcs.93.2.255. [DOI] [PubMed] [Google Scholar]
- Deery WJ, Heath JP. Phagocytosis induced by thyrotropin in cultured thyroid cells is associated with myosin light chain dephosphorylation and stress fiber disruption. J Cell Biol. 1993;122:21–37. doi: 10.1083/jcb.122.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC. Nuclear changes in apoptosis. Curr Opin Cell Biol. 1995;7:337–343. doi: 10.1016/0955-0674(95)80088-3. [DOI] [PubMed] [Google Scholar]
- Fishkind DJ, Cao L, Wang Y. Microinjection of the catalytic fragment of myosin light chain kinase into dividing cells:Effects on mitosis and cytokinesis. J Cell Biol. 1991;114:967–975. doi: 10.1083/jcb.114.5.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PJ, Herring BP, Stull JT. Myosin light chain kinases. J Muscle Res Cell Motil. 1997;18:1–16. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- Gebbink MFBG, Kranenburg O, Poland M, van Horck FPG, Houssa B, Moolenaar WH. Identification of a novel, putative Rho-specific GDP/GTP exchange factor and a RhoA-binding protein: Control of neuronal morphology. J Cell Biol. 1997;137:1603–1613. doi: 10.1083/jcb.137.7.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghibelli L, Nosseri C, Coppola S, Maresca V, Dini L. The increase in H2O2-induced apoptosis by ADP-ribosylation inhibitors is related to cell blebbing. Exper Cell Res. 1995;221:470–477. doi: 10.1006/excr.1995.1398. [DOI] [PubMed] [Google Scholar]
- Grebecki A. Membrane and cytoskeleton flow in motile cells with emphasis on the contribution of free-living amoebae. Int Rev Cytol. 1994;148:37–80. [Google Scholar]
- Hashimoto Y, Nakayama T, Teramoto T, Kato H, Watanabe T, Kinoshita M, Tsukamoto K, Tokunaga K, Kurokawa K, Nakanishi S, et al. Potent and preferential inhibition of Ca2+/calmodulin-dependent protein kinase II by K252a and its derivative, KT5926. Biochem Biophys Res Commun. 1991;181:423–429. doi: 10.1016/s0006-291x(05)81436-6. [DOI] [PubMed] [Google Scholar]
- Hirao M, Sato N, Kondo T, Yonemura S, Monden M, Sasaki T, Takai Y, Tsukita S. Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and Rho-dependent signaling pathway. J Cell Biol. 1996;135:37–51. doi: 10.1083/jcb.135.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard MJ, Cohen P. On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem Sci. 1993;18:172–177. doi: 10.1016/0968-0004(93)90109-z. [DOI] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, Ten P, Dijke, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Jalink K, van Corven EJ, Hengeveld T, Morii N, Narumiya S, Moolinaar WH. Inhibition of lysophosphatide- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein Rho. J Cell Biol. 1994;126:801–810. doi: 10.1083/jcb.126.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NL, Gardner AM, Diener KM, Lange-Carter CA, Gleavy J, Jarpe MB, Minden A, Karin M, Zon LI, Johnson GL. Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J Biol Chem. 1996;271:3229–3237. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signaling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Kayalar C, Ord T, Testa MP, Zhong L-T, Bredesen DE. Cleavage of actin by interleukin 1β-converting enzyme to reverse DNaseI inhibition. Proc Natl Acad Sci USA. 1996;93:2234–2238. doi: 10.1073/pnas.93.5.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller HU, Zimmermann A. Shape changes and chemotaxis of Walker 256 carcinosarcoma cells in response to colchicine, vinblastine, nocodazole, and taxol. Invasion and Metastasis. 1986;6:33–43. [PubMed] [Google Scholar]
- Keller HU, Zimmermann A, Cottier H. Phorbol myristate acetate (PMA) suppresses polarization and locomotion and alters F-actin content of Walker carcinosarcoma cells. Int J Cancer. 1985;36:495–501. doi: 10.1002/ijc.2910360414. [DOI] [PubMed] [Google Scholar]
- Kimura K, Iho M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kohama K, Ye LH, Hayakawa K, Okagaki T. Myosin light chain kinase: an actin-binding protein that regulates an ATP-dependent interaction with myosin. Trends Pharmacol Sci. 1996;17:284–287. doi: 10.1016/0165-6147(96)10033-x. [DOI] [PubMed] [Google Scholar]
- Kolodney MS, Elson EL. Correlation of myosin light chain phosphorylation with isometric contraction of fibroblasts. J BiolChem. 1993;268:23850–23855. [PubMed] [Google Scholar]
- Kolodney MS, Elson EL. Contraction due to microtubule disruption is associated with increased phosphorylation of myosin regulatory light chain. Proc Natl Acad Sci USA. 1995;92:10252–10256. doi: 10.1073/pnas.92.22.10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laster SM, Mackenzie JM., Jr Bleb formation and F-actin distribution during mitosis and tumor necrosis factor-induced apoptosis. Microscopy Res Tech. 1996;34:272–280. doi: 10.1002/(SICI)1097-0029(19960615)34:3<272::AID-JEMT10>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Lazebnik, Y.A., A. Takahashi, G.G. Poirier, S.H. Kaufman, and W.C. Earnshaw. 1995. Characterization of the execution phase of apoptosis in vitro using extracts from condemned-phase cells. J. Cell Sci. Suppl. 19:41–49. [DOI] [PubMed]
- Ludowyke RI, Peleg I, Beaven MA, Adelstein RS. Antigen- induced secretion of histamine and the phosphorylation of myosin by protein kinase C in rat basophilic leukemia cells. J Biol Chem. 1989;264:12492–12501. [PubMed] [Google Scholar]
- Maciver SK. Myosin II function in nonmuscle cells. Bioessays. 1996;18:179–182. doi: 10.1002/bies.950180304. [DOI] [PubMed] [Google Scholar]
- Martin SJ, O'Brien GA, Nishioka WK, McGahon AJ, Mahboubi A, Saido TC, Green DR. Proteolysis of fodrin (non-erythroid spectrin) during apoptosis. J Biol Chem. 1995;270:6425–6428. doi: 10.1074/jbc.270.12.6425. [DOI] [PubMed] [Google Scholar]
- Mashima T, Naito M, Fujita N, Noguchi K, Tsuruo T. Identification of actin as a substrate of ICE and an ICE-like protease and involvement of an ICE-like protease but not ICE in VP-16-induced U937 apoptosis. Biochem Biophys Res Comm. 1995;217:1185–1192. doi: 10.1006/bbrc.1995.2894. [DOI] [PubMed] [Google Scholar]
- McCarthy NJ, Whyte MKB, Gilbert CS, Evan GI. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messam, C.A., and R.N. Pittman. 1998. Asynchrony and commitment to die during apoptosis. Exp. Cell Res. In Press. [DOI] [PubMed]
- Mills J C, Wang S L, Erecinska M, Pittman R N. Use of cultured neurons and neuronal cell lines to study morphological, biochemical, and molecular changes occurring in cell death. Methods Cell Biol. 1995;46:217–242. doi: 10.1016/s0091-679x(08)61931-7. [DOI] [PubMed] [Google Scholar]
- Mills JC, Kim LH, Pittman RN. Differentiation to an NGF- dependent state and apoptosis following NGF removal both occur asynchronously in cultures of PC12 cells. Exp Cell Res. 1997;231:337–345. doi: 10.1006/excr.1997.3474. [DOI] [PubMed] [Google Scholar]
- Mills, J.C., V.M.-Y. Lee, and R.N. Pittman. 1998. Activation of a PP2A-like phosphatase and dephosphorylation of τ protein characterize onset of the execution phase of apoptosis. J. Cell Sci. In press. [DOI] [PubMed]
- Miranda A, Godman GC, Deitch AD, Tanenbaum SW. Action of cytochalasin D on cells of established lines. J Cell Biol. 1974;61:481–500. doi: 10.1083/jcb.61.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Umeshita K, Sakon M, Imajoh-Ohmi S, Fujitani K, Gotoh M, Oiki E, Kambayashi J, Monden M. Calpain activation in plasma membrane bleb formation during tert-butyl hydroperoxide-induced rat hepatocyte injury. Gastroenterology. 1996;110:1897–1904. doi: 10.1053/gast.1996.v110.pm8964416. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Nath R, Raser KJ, Stafford D, Hajimohammadreza I, Posner A, Allen H, Talanian RV, Yuen P, Gilbertsen RB, Wang KK. Non-erythroid α-spectrin breakdown by calpain and interleukin 1 β-converting-enzyme-like protease(s) in apoptotic cells:contributory roles of both protease families in neuronal apoptosis. Biochem J. 1996;319:683–690. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Noda M, Yasuda-Fukazawa C, Moriishi K, Kato T, Okuda T, Kurokawa K, Takuwa Y. Involvement of Rho in GTPγS-induced enhancement of phosphorylation of 20 kDa myosin light chain in vascular smooth muscle cells: inhibition of phosphatase activity. FEBS (Fed Eur Biochem Sci) Lett. 1995;367:246–250. doi: 10.1016/0014-5793(95)00573-r. [DOI] [PubMed] [Google Scholar]
- Park DS, Stefanis L, Yan CYI, Farinelli SE, Greene LA. Ordering the cell death pathway. J Biol Chem. 1996;271:21898–21905. doi: 10.1074/jbc.271.36.21898. [DOI] [PubMed] [Google Scholar]
- Paterson HF, Self A, Garrett MD, Just I, Aktories K, Hall A. Microinjection of recombinant p21 Rho induces rapid changes in cell morphology. J Cell Biol. 1990;111:1001–1007. doi: 10.1083/jcb.111.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta-Soler A, Mullin JM, Knudsen KA, Marano CW. Tissue remodeling during tumor necrosis factor-induced apoptosis in LLC-PK1 renal epithelial cells. Am J Physiol. 1996;270:869–879. doi: 10.1152/ajprenal.1996.270.5.F869. [DOI] [PubMed] [Google Scholar]
- Pittman RN, Wang SL, DiBenedetto AJ, Mills JC. A system for characterizing cellular and molecular events in programmed neuronal cell death. J Neurosci. 1993;13:3669–3680. doi: 10.1523/JNEUROSCI.13-09-03669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitzer F, Dantes A, Fuchs T, Baumeister W, Amsterdam A. Removal of proteosomes from the nucleus and their accumulation in apoptotic blebs during programmed cell death. FEBS (Fed Eur Biochem Sci) Lett. 1996;394:47–50. doi: 10.1016/0014-5793(96)00920-9. [DOI] [PubMed] [Google Scholar]
- Prentki M, Chaponnier C, Jeanrenaud B, Gabbiani G. Actin microfilaments, cell shape, and secretory processes in isolated rat hepatocytes. J Cell Biol. 1979;81:592–607. doi: 10.1083/jcb.81.3.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein Rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ruchhoeff ML, Harris WA. Myosin functions in Xenopus retinal ganglion cell growth cone motility in vivo. J Neurobiol. 1997;32:567–578. [PubMed] [Google Scholar]
- Rudel T, Bokoch GM. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science. 1997;276:1571–1574. doi: 10.1126/science.276.5318.1571. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Naka M, Hidaka H. The modulatory role of myosin light chain phosphorylation in human platelet activation. Biochem Biophys Res Commun. 1986;140:280–287. doi: 10.1016/0006-291x(86)91087-9. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Ishikawa T, Matsushima S, Naka M, Hidaka H. Selective inhibition of catalytic activity of smooth muscle myosin light chain kinase. J Biol Chem. 1987;262:7796–7801. [PubMed] [Google Scholar]
- Sekine A, Fujiwara M, Narumiya S. Asparagine residue in the Rho gene product is the modification site for botulinum ADP-ribosyltransferase. J Biol Chem. 1989;264:8602–8605. [PubMed] [Google Scholar]
- Song, Q., T. Wei, S. Lees-Miller, E. Alnemri, D. Watters, and M.F. Lavin. 1997. Resistance of actin to cleavage during apoptosis. Proc. Natl. Acad. Sci. USA. 94:157–162. [DOI] [PMC free article] [PubMed]
- Taylor DA, Stull JT. Calcium dependence of myosin light chain phosphorylation in smooth muscle cells. J Biol Chem. 1988;263:14456–14462. [PubMed] [Google Scholar]
- Tigyi G, Fischer DJ, Sebok A, Yang C, Dyer DL, Miledi R. Lysophosphatidic acid-induced neurite retraction in PC12 cells: control by phosphoinositide-Ca2+ signaling and Rho. J Neurochem. 1996;66:537–548. doi: 10.1046/j.1471-4159.1996.66020537.x. [DOI] [PubMed] [Google Scholar]
- Vanags DM, Porn-Ares MI, Coppola S, Burgess DH, Orrenius S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J Biol Chem. 1996;271:31075–31085. doi: 10.1074/jbc.271.49.31075. [DOI] [PubMed] [Google Scholar]
- Vemuri GS, Zhang J, Huang R, Keen JH, Rittenhouse SE. Thrombin stimulates wortmannin-inhibitable phosphoinositide 3-kinase and membrane blebbing in CHRF-288 cells. Biochem J. 1996;314:805–810. doi: 10.1042/bj3140805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedman A, Fuks Z, Kolesnick RN. Requirement for ceramide-initiated SAPK/JNK signaling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Zanke BW, Boudreau K, Rubie E, Winnett E, Tibbles LA, Zon L, Kyriakis J, Liu F-F, Woodgett JR. The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr Biol. 1996;6:606–613. doi: 10.1016/s0960-9822(02)00547-x. [DOI] [PubMed] [Google Scholar]