

Abstract
Using a cell-free content mixing assay containing rat liver endosomes and lysosomes in the presence of pig brain cytosol, we demonstrated that after incubation at 37°C, late endosome–lysosome hybrid organelles were formed, which could be isolated by density gradient centrifugation. ImmunoEM showed that the hybrids contained both an endocytosed marker and a lysosomal enzyme. Formation of the hybrid organelles appeared not to require vesicular transport between late endosomes and lysosomes but occurred as a result of direct fusion. Hybrid organelles with similar properties were isolated directly from rat liver homogenates and thus were not an artifact of cell-free incubations. Direct fusion between late endosomes and lysosomes was an N-ethylmaleimide–sensitive factor– dependent event and was inhibited by GDP-dissociation inhibitor, indicating a requirement for a rab protein. We suggest that in cells, delivery of endocytosed ligands to an organelle where proteolytic digestion occurs is mediated by direct fusion of late endosomes with lysosomes. The consequences of this fusion to the maintenance and function of lysosomes are discussed.
The earliest events in the endocytic pathway are relatively well established, both biochemically and microscopically. However, the later stages of the pathway to lysosomes, a route of major importance in most cells, are much less well characterized. The means by which material in a peripheral early endosome subsequently appears in a multivesicular late endosome deeper in the cell are still not completely understood. It may require both vesicular transport and/or maturation events, and may vary with cell type (for review see Gruenberg and Maxfield, 1995). However, agreement is emerging that content mixing with preexisting lysosomes occurs as a final step (Mullock et al., 1994; Futter et al., 1996; Mellman, 1996; Tjelle et al., 1996; Bright et al., 1997). To date, it is not clear whether such content mixing is the result of direct fusion (van Deurs et al., 1995; Futter et al., 1996; Griffiths, 1996; Bright et al., 1997), vesicular transport (Berg et al., 1995), or “kiss and run” events (Storrie and Desjardins, 1996). It is also unknown whether the common cytosolic fusion machinery (Rothman, 1994) involving N-ethylmaleimide (NEM)-sensitive factor (NSF),1 soluble NSF-attachment proteins (SNAPs), and a class of small GTP-binding proteins known as rabs (Pfeffer, 1994) is required for endosomal content transfer to lysosomes. However, it is clear that this general fusion machinery is used at earlier stages of the endocytic pathway in both mammalian and yeast cells (Rodriguez et al., 1994; Hicke et al., 1997).
Destruction of endocytosed labels by lysosomal enzymes has been a major difficulty for both biochemical and microscopic studies, together with the lack of morphologically recognizable lysosomes in many of the tissue culture cells studied. We have used a cell-free system from rat liver (Mullock et al., 1994), based on avidin ligand–loaded late endosomes and biotinylated ligand-containing lysosomes, to examine the biochemical aspects of late endosome–lysosome fusion. Receptor-mediated endocytosis was used to load these compartments in hepatocytes but not in other liver cell types, and cell-free incubations were kept short to minimize proteolytic damage. In the present study, we have shown that organelles containing the newly formed avidin–biotin complex can be separated from dense core lysosomes on the basis of their lower density. By immunoEM, we have independently demonstrated that these hybrid organelles contain endocytosed avidin together with lysosome-derived cathepsin L. Organelles of similar density containing endocytosed avidin and cathepsin L were isolated, using the same gradient, from the livers of rats previously injected with avidin ligand, indicating that the cell-free system was not generating an in vitro artifact. Using the cell-free system, we have shown that direct fusion between late endosomes and lysosomes required NSF and SNAPs and was inhibited by GDP dissociation inhibitor (GDI), demonstrating that it used the common NSF fusion machinery observed at many steps of vesicle-mediated intracellular membrane traffic.
Materials and Methods
Reagents
Avidin-asialofetuin (Av-ASF) and 125I-labeled biotinylated polymeric IgA (bpIgA) were as previously described (Mullock et al., 1994). Affinity purified goat anti–avidin (egg white) was obtained from Calbiochem (Nottingham, UK), and magnetic beads coated with rabbit anti–goat IgG were from Advanced Magnetics Inc. (Cambridge, MA). Wortmannin from Calbiochem was aliquoted and kept at −20°C as a 1 mM stock in DMSO. Nordihydroguaiaretic acid (NDGA) was from Affiniti Research Products (Nottingham, UK) and was made up as a 2 mM solution in ethanol. LY294002 was kindly provided by Dr. P. Shepherd (Department of Biochemistry, University College, London, UK), aliquoted, and kept at −20°C as a 10 mg/ml stock in DMSO. Recombinant Myc-tagged NSF was purified from cultures of Escherichia coli (strain from Dr. J. Rothman provided with permission by Dr. P. Woodman, Department of Biochemistry and Molecular Biology, University of Manchester, UK) by the procedure of Wilson and Rothman (1992). Recombinant His-tagged α- and γ-SNAPs were obtained from the same source and purified according to Whiteheart et al. (1993). Preparations of valosin-containing protein/p97 were gifts from Dr. P. Woodman and Dr. E. Smythe (Department of Biochemistry, University of Dundee, UK). Purified recombinant rab 7 was a gift from Dr. A. Wandinger-Ness (Northwestern University, Evanston, IL). A rabbit antiserum to the carboxy-terminal portion of rab 7 was raised against a glutathione S-transferase fusion protein encoded by pGEX1N (Smith and Johnson, 1988) containing the BamHI/PvuII fragment of dog rab 7 cDNA (sequence data available from GenBank/EMBL/DDBJ under accession number M 35522; the gift of Dr. M. Zerial, EMBL, Heidelberg, Germany) and was affinity purified on the same fusion protein. A plasmid containing NH2-terminal His-tagged bovine rab GDI cDNA, the gift of Dr. H. Davidson and Mr. D. McDonald (Department of Clinical Biochemistry, University of Cambridge) was expressed in E. coli BL21(DE3) and the recombinant GDI purified according to Ullrich et al. (1995). The rabbit polyclonal anti–rat MPR antiserum was as described previously (Reaves et al., 1996). The rabbit polyclonal anti–mouse cathepsin L antibody, which has been demonstrated to cross-react with rat fibroblast cathepsin L (Punnonen et al., 1994), was kindly provided by Dr. Michael Gottesman (National Cancer Institute, Bethesda, MD). Protein A conjugated to monodisperse 15-nm colloidal gold was purchased from the Department of Cell Biology, University of Utrecht. Polyclonal rabbit anti–goat Ig antibodies conjugated to 8-nm colloidal gold were purchased from Sigma (Poole, UK).
Content Mixing Assay
The method described by Mullock et al. (1994) was slightly modified. Late endosomes were prepared from the liver of a rat, which had received ∼10 nmol of Av-ASF i.v. 6 min before killing and were stored in 0.25 M sucrose containing 10 mM N-tris(hydroxymethyl)-methyl-2-aminoethane and 1 mM Mg2+, pH 7.4 (STM). Lysosomes loaded in vivo with 125I-labeled bpIgA were separated from endosomes by layering 14 ml of postmitochondrial supernatant over a step gradient composed of 4 ml of 45% Nycodenz and 21 ml of 20% Nycodenz, a simplification of the method described in Ellis et al. (1992). The content mixing assay was performed in pig brain cytosol prepared by extracting the tissue (stored in liquid N2) with STM containing 25 mM KCl. The suspension was centrifuged for 15 min at 103,000 g and the supernatant filtered through Biogel P6 columns (Bio-Rad Laboratories, Hercules, CA). The protein concentration was ∼10 mg/ml. Duplicate samples containing late endosomes from ∼50 mg liver and freshly prepared lysosomes from ∼80 mg liver were routinely incubated for 10 min at 37°C in 0.2 ml brain cytosol plus 1 mM ATP and 1 mM GTP in addition to an ATP-regenerating mixture of phosphocreatine and creatine kinase. 60 μg/ml biocytin was also present to block any formation of avidin-bpIgA outside a membrane-bounded compartment. After incubation, dilution and lysis were as previously described (Mullock et al., 1994). The mixtures were incubated with 2.5 μl goat anti–avidin at 4°C for 1–2 h before incubation with magnetic beads. Total immunoprecipitable radioactivity in the samples was measured by performing similar incubations in the absence of biocytin. NEM treatment and NSF depletion of cytosol were as described in Mullock et al. (1994).
Examination of Density of the Hybrid Organelles Formed by Fusion of Late Endosomes and Lysosomes
A 20-fold version (total volume, 4.8 ml) of the usual incubation mixture for endosome–lysosome fusion was incubated for 10 min at 37°C and then chilled and loaded over either a 0–35% Nycodenz gradient or a 1–22% Ficoll gradient (Ellis et al., 1992). After centrifugation in a vertical rotor (model VTi; Beckman Instruments, Fullerton, CA) for 1 h at 200,000 g, the gradient was collected in 1-ml fractions, and the distribution of total radiolabel was measured. To examine which fractions contained avidin-bpIgA complex, duplicate 250-μl samples were added to 25 μl 0.6 mg/ml biocytin and diluted, lysed, and immunoprecipitated as usual. A 250-μl blank sample from each tube received nonimmune goat IgG in place of antiavidin. Biotin-containing lysosomes were physically separated from avidin containing endosomes on the vertical gradient. Therefore, it was not possible to measure total immunoprecipitable radiolabel as in the assays of unfractionated incubation mixture. For the preparative separation of newly fused endosome–lysosome hybrids from dense core (electron-dense) lysosomes, 7 ml of the incubated fusion mixture was layered over a step gradient composed of 4 ml 45% Nycodenz, 14 ml of 20% Nycodenz, and 14 ml 20% Ficoll as described in Ellis et al. (1992). After centrifugation, the material at the Nycodenz/Ficoll interface contained the lysosome–endosome hybrids. Separation of in vivo–formed hybrids was achieved by loading the same gradient with 7 ml of postmitochondrial supernatant from a rat that had received an i.v. injection of Av-ASF 9 min before killing.
Removal of rab 7 from Late Endosomes with GDI and Effects of GDI on the Content Mixing Assay
Recombinant His-tagged GDI was assayed for its ability to solubilize rabs from membranes (Ullrich et al., 1993) by following the release of rab 7 from GDI-treated late endosome fractions. Late endosomes (300 μg protein) were prepared from rat liver and incubated with varying amounts of recombinant GDI in STM buffer containing 25 mM KCl in a total volume of 35 μl for 15 min at 37°C. The reaction mixtures were centrifuged at 245,000 g for 15 min, and both the pellet and the supernatant were assayed for rab 7, after SDS-PAGE and immunoblotting, by the enhanced chemiluminescence detection method (Amersham International, Buckinghamshire, UK), using the affinity purified rabbit antibodies to the carboxy-terminal portion of rab 7. Bands were quantitated using a scanner (model GT-9000; Seiko Epson Corp., Japan) with NIH Image 1.6 software. In a dose-dependent manner, incubation with recombinant GDI resulted in removal of rab 7 from the membrane fraction with concomitant recovery in the supernatant. Using 16 μM recombinant GDI, >90% solubilization was achieved.
In the content mixing assay, GDI function was assessed by adding various concentrations of purified recombinant GDI to aliquots of the standard incubation mixture and incubating for 10 min at 37°C. Release of rab 7 from membranes was assayed as above, although appearance in the supernatant could not be quantitatively analyzed because of the presence of rab 7 in pig brain cytosol. Calculation of the amount of GDI to cause 50% inhibition of content mixing or of removal of rab 7 from membranes took into account, respectively, a background value from an incubation at 4°C, or the remaining rab 7 associated with membranes in the presence of cytosol after treatment with 15 μM GDI. Attempts to overcome inhibition of content mixing by GDI were made in the presence of 2 μM GDI by the addition of 5 pmol rab 7 and 2 nmol of geranylgeranylpyrophosphate.
ImmunoEM
Organelles from gradient fractions were diluted at least fourfold with STM before pelleting at 90,000 g for 15 min for lysosomes and hybrids or at 174,000 g for 20 min for late endosomes in an ultracentrifuge (model TL-100; Beckman Instruments) and were prepared for ultrastructural immunocytochemistry as described by Griffiths (1993). Briefly, the material was fixed with 4% paraformaldehyde/0.1% glutaraldehyde in 250 mM Hepes, pH 7.2, at room temperature for 1 h, infused with 2.1 M sucrose in PBS overnight at 4°C, and frozen on aluminum stubs in liquid nitrogen. Frozen ultrathin sections were cut using an ultramicrotome equipped with a cryochamber attachment (Leica, Milton Keynes, UK), collected using 2.3 M sucrose in PBS or a methyl cellulose/sucrose mixture (Liou et al., 1996), mounted on formvar carbon–coated gold EM grids, and rinsed with PBS. The sections were transferred from PBS to 50 mM NH4Cl in PBS for 10 min at room temperature to quench unreacted aldehydes. Nonspecific interactions were blocked by transferring the grids to 2% gelatin in PBS for 10 min at room temperature and then 1% BSA in PBS for 10 min at room temperature. Double immunolabeling was performed as described previously (Reaves et al., 1996) using a sequential immunolabeling method (Slot et al., 1991). Sections were incubated for 30 min at room temperature with 5 μl of rabbit anti–cathepsin L diluted 1:100 in PBS containing 5% FCS and 0.1% BSA. The grids were washed with PBS/0.1% BSA (6× 3 min), and the rabbit antibodies were detected by incubating for 30 min at room temperature with PBS/0.1% BSA containing protein A conjugated to 15-nm colloidal gold. The sections were washed with PBS/0.1% BSA (2× 5 min) and PBS (4× 5 min), and the gold-labeled complex was stabilized using 1% glutaraldehyde in PBS (5 min). The above procedure was then repeated using a 1:100 dilution of the same goat anti–avidin antibody used for the immunoprecipitation assay followed by detection with 1:50 rabbit anti–goat IgG conjugated to 8-nm colloidal gold. Finally the sections were rinsed with distilled water (5 × 3 min) and contrasted by embedding them in freshly prepared 1.8% methyl cellulose/0.3% uranyl acetate (Tokuyasu, 1978). Grids were air dried and stored at room temperature before observation in a transmission electron microscope (model CM100; Philips Electron Optics, Mahwah, NJ). Micrographs were prepared according to conventional procedures.
Morphometry
Organelle diameters were measured using the mean caliper diameter method of Weibel (1979). Individual random sections through pellets were systematically scanned, and only organelles possessing two or more 8-nm and/or 15-nm gold conjugates were scored. The mean caliper diameter was recorded for each gold-labeled organelle, and the mean volume and surface area were derived assuming the organelles to be spherical. Between 31 and 48 organelles of each type were scored to define organelle diameter.
Quantitative analysis of the distribution of gold particles was performed on the electron microscope at a magnification of 15,500. Individual random sections through pellets were systematically scanned, and the presence of gold conjugates in organelles was scored in each experiment. Only organelles possessing two or more 8-nm and/or 15-nm gold conjugates were scored. The numbers of organelles possessing either single or double immunolabeling were expressed as percentages of the total number of organelles labeled. In each experiment, 200 gold-labeled organelles were scored to generate the data shown.
Where appropriate, the data were tested for statistical significance with analysis of variance using Super Anova software (version 1.11; Abacus Concepts Inc., Berkeley, CA). Only P values ⩽0.01 are shown. Unless otherwise stated, results are expressed as a mean ± SEM, with the number of experiments (n) shown in parentheses.
Results
A cell-free assay to determine endosome–lysosome content mixing was modified from that described previously (Mullock et al., 1994). The modified assay contained rat liver late endosomes preloaded with Av-ASF and rat liver lysosomes preloaded with 125I-bpIgA, together with pig brain cytosol plus an ATP-regenerating system and 1 mM GTP. A time course for content mixing is shown in Fig. 1 a. The assay showed a very reproducible fusion between late endosomes and lysosomes (6.8 ± 0.3% of total immunoprecipitable radiolabeled biotin complexed with avidin in 10 min over 55 experiments). Since lysosomes were freshly prepared for each experiment, all results are presented as a percentage of the standard fusion on the same occasion. Using brain cytosol, which lacks the enzymes to metabolize the glycogen in the lysosomal preparation, the energy dependence of the fusion reaction was much more obvious than that observed previously (Mullock et al., 1994); merely omitting ATP and GTP reduced the fusion by >75% (Fig. 1 b). Content mixing was dependent on the presence of cytosol and required incubation at 37°C (Fig. 1 b).
Figure 1.

Characterization of content mixing between late endosomes and lysosomes. (a) Time course under standard incubation conditions, i.e., in the presence of cytosol, an ATP-regenerating system, 1 mM ATP, and 1 mM GTP. Results are expressed at each time point as the percentage of the total immunoprecipitable counts that had been formed within membranous compartments (i.e., which were immunoprecipitable in the presence of biocytin). (b) Requirements for the content mixing reaction. Results are expressed as percentages of the content mixing obtained on the same day under the standard conditions, i.e., in the presence of cytosol, an ATP-regenerating system, 1 mM ATP, and 1 mM GTP. 0.2 mM GMP-PNP or guanosine 5′-O-(3-thiotriphosphate (GTPγS) replaced GTP where indicated. incbn, incubation.
Late Endosomes Fuse with Lysosomes to Give an Organelle of Intermediate Density
Content mixing between late endosomes and lysosomes would be expected to produce a hybrid organelle of intermediate density if it occurred as a result of direct fusion or if a large fraction of content and membrane was transferred by vesicle movement. The formation of a hybrid organelle was investigated using isopycnic centrifugation on continuous Nycodenz and Ficoll gradients. Centrifugation on 0–35% Nycodenz gradients of the mixture of 125I-bpIgA– loaded lysosomes and Av-ASF–loaded late endosomes after the fusion incubation showed that the peak of radioactivity immunoprecipitable with antiavidin appeared reproducibly at a lower density than the peak of total counts, which remained at a lysosomal position (refractive indices at peaks 1.371 for immunoprecipitable counts, 1.377 for total counts). Centrifugation of the mixture on a 1–22% Ficoll gradient showed that ∼85% of the immunoprecipitable radioactivity was denser than the late endosomes. A step gradient designed to exploit these differences permitted separation of newly fused endosome–lysosome hybrids from both labeled dense core lysosomes and late endosomes (Fig. 2 a). The incubated endosome–lysosome mixture was loaded over a step gradient composed of a cushion of 45% Nycodenz, a layer of 20% Nycodenz, and a layer of 20% Ficoll. Late endosomes would be expected to remain above the 20% Ficoll, and the lysosomes containing radioactive bpIgA would be expected to sediment to the 45% Nycodenz cushion. The distribution of total radiolabel in Fig 2 a (open symbols) shows the expected peak on the 45% Nycodenz cushion, a small peak at the top of the Ficoll layer caused by degraded material, and in addition, a prominent peak at the Nycodenz/Ficoll interface. Fractions from each peak of total radioactivity were precipitated with antiavidin in the presence of excess biotin (Fig. 2 a, solid symbols). Negligible amounts of radioactivity were immunoprecipitable from the lysosomal peak and from the peak at the top of the gradient. Fractions from the Nycodenz/Ficoll interface, however, contained immunoprecipitable radioactivity whose distribution across the interface closely matched that of the total radioactivity. This peak was therefore enriched in the avidin-biotin–containing hybrid structures. Of the total radioactivity present at the Nycodenz/Ficoll interface, 7 ± 2% (4) was immunoprecipitable. This percentage was not altered by leaving out biocytin from the immunoprecipitation reaction, which suggests a lack of significant numbers of attached but unfused endosomes and lysosomes at the interface. The low percentage of immunoprecipitable material does not imply that the remainder of radioactivity at the interface is not in hybrid structures, just that it cannot be immunoprecipitated, probably because of proteolysis in the hybrid organelles (Mullock et al., 1994). Any single proteolytic cleavage occurring between the biotin and 125I, which are covalently attached to the pIgA, would be sufficient to reduce the amount of immunoprecipitable radioactivity. Immunoblotting experiments showed that small proportions of MPR and rab 7 were also found at the Nycodenz/Ficoll interface (Fig. 2 b). Immunoprecipitation from a gradient whose otherwise identical loaded material had been kept at 4°C as a control showed that only one tenth as much radioactivity was immunoprecipitable with no peak in its distribution (data not shown).
Figure 2.
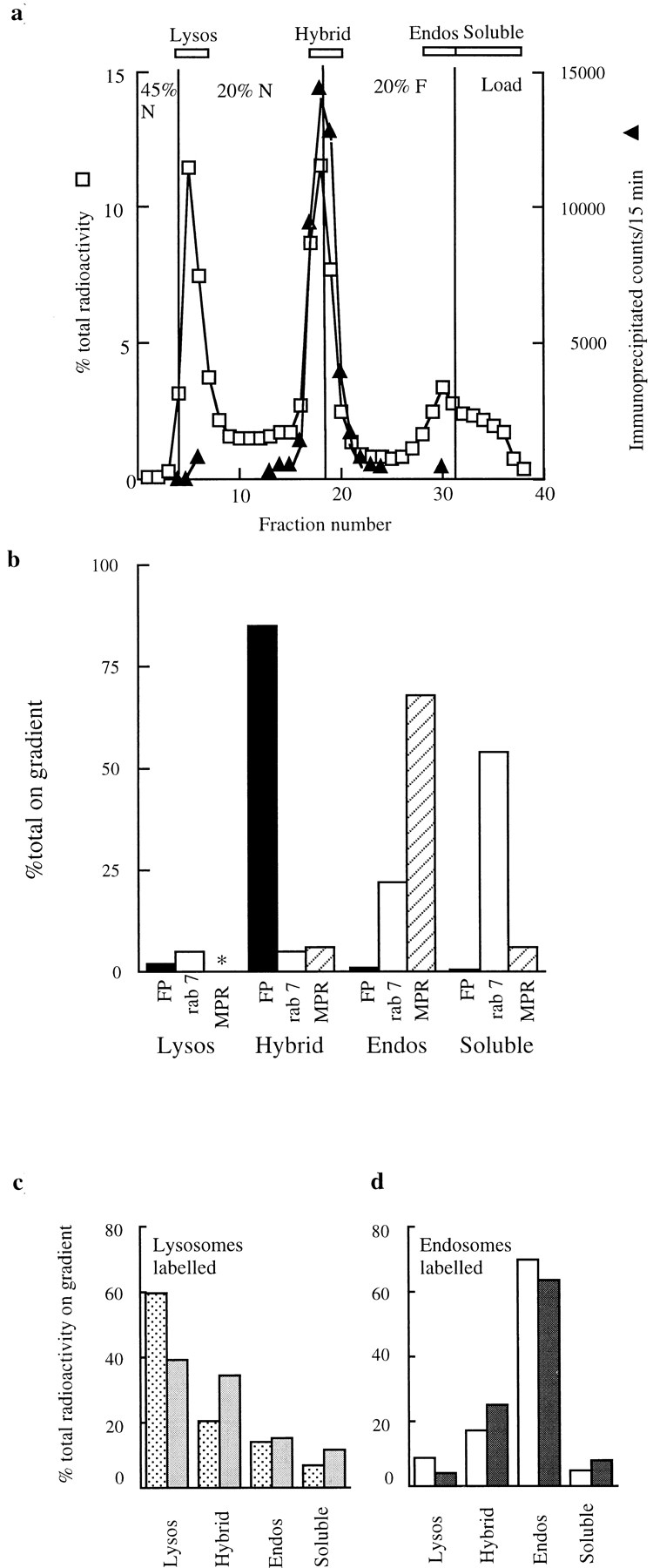
Separation of late endosome–lysosome hybrids on a step gradient. (a) Distribution of total and immunoprecipitable radiolabels after a standard content mixing assay containing 125I-bpIgA–loaded lysosomes and Av-ASF–loaded late endosomes incubated for 10 min at 37°C; □, percentage of total radioactivity on gradient; ▴, radioactivity in the peak fractions, immunoprecipitable with antiavidin. (Radioactive counts were routinely collected for 15 min and corrected for background.) The radioactivity at the light end of the step gradient peaking around fraction 30 was not precipitable with trichloroacetic acid, indicating that it represented soluble degradation products. (b) Distribution of immunoprecipitable radioactivity (FP, fusion product), immunoblotted rab 7, and immunoblotted MPR on the step gradient run under the same conditions as in a. The four regions of the gradient are shown at the top of a. *, no detectable immunoblotted MPR in the Lysos region. (c) Separate experiment showing distribution of total radioactivity before ( ) and after (
) the incubation of the content mixing assay (lysosomes loaded with 125I-bpIgA). (d) Distribution of total radioactivity before (□) and after (▪) incubation of 125I-Av-ASF–loaded late endosomes with unlabeled lysosomes under the conditions of the content mixing assay. Lysos, lysosomes; Endos, endosomes; N, Nycodenz; F, Ficoll.
Distribution of total label from labeled lysosomes before and after 10 min incubation at 37°C is shown from a separate experiment in Fig. 2 c. As expected, there was movement of radiolabel from the lysosomal to the hybrid region. In a complementary experiment in which the content mixing assay was carried out with 125I–Av-ASF–loaded endosomes and nonradiolabeled bpIgA-loaded lysosomes, redistribution of total label in the step gradient from the endosomal position in the 20% Ficoll to the hybrid position on the Nycodenz/Ficoll interface was similar (Fig. 2 d).
In control experiments, late endosome fractions were prepared from two rat livers, one of which had taken up Av-ASF for 6 min and the other 125I-bpIgA for 6 min. When these fractions were incubated together for 10 min at 37°C under the conditions of the standard content mixing assay, ∼40% of radioactivity was immunoprecipitable, showing that endosome–endosome fusions had occurred. However, when the mixture was fractionated on the step gradient described above, the Nycodenz/Ficoll interface appeared clear and no membrane could be sedimented from this interface. When lysosome fractions containing 125I-bpIgA were incubated for 10 min at 37°C under the conditions of the standard content mixing assay and then fractionated on the step gradient, <2% of total radioactivity and <2% of the marker enzyme N-acetyl glucosaminidase were observed at the Nycodenz/Ficoll interface. There is thus no evidence that organelles of a density resulting in their accumulation at the Nycodenz/Ficoll interface could arise from endosome–endosome or lysosome–lysosome fusions in the endosome–lysosome content mixing assays described above.
ImmunoEM was carried out on membrane-bound organelles isolated from the lysosomal position (45%/20% Nycodenz interface) and hybrid organelle position (Nycodenz/Ficoll interface) of the step gradient after centrifugation of endosome–lysosome content mixing assay material. When the content mixing assay had been incubated for 10 min at 37°C, the organelles recovered from the Nycodenz/ Ficoll interface were enriched in structures containing both endocytosed avidin and lysosome-derived cathepsin L compared to structures containing both labels in either endosome or lysosome fractions (Fig. 3). There was insufficient bpIgA in these structures to be detected by immunoEM with available antibodies.
Figure 3.

Quantitation of cathepsin L and avidin immunolabeled organelles of ultrathin frozen sections from the fractions illustrated in Fig. 4. Results are presented as a mean ± the range (lysosome fraction) or SEM (hybrid and endosome fractions). Cathepsin L+/avidin+ structures were significantly enriched in the hybrid fraction compared to the lysosome and endosome fractions, * P < 0.01. The cathepsin L+/avidin− structures in the lysosome fraction were electron dense, whereas similarly labeled structures in the endosome fraction were electron lucent, suggesting that the latter may be vesicles and organelles from the route of delivery of newly synthesized cathepsin to lysosomes.
Hybrid organelles formed after 10 min incubation of the standard content mixing assay and collected at the Nycodenz/Ficoll interface had a mean diameter of 957 ± 11 nm (derived surface area 0.96 μm2). After 5 min incubation, they were significantly smaller with a mean diameter of 850 ± 32 nm (derived surface area 0.75 μm2). Both values are significantly different from the mean diameter of the late endosomes, 341 ± 11 nm (derived surface area 0.12 μm2) and the mean diameter of lysosomes, 384 ± 11 nm (derived surface area 0.15 μm2), showing that multiple fusions must have occurred even after 5 min incubation. A single further fusion event with either a lysosome or endosome would be sufficient to explain the further increase in diameter and surface area observed after 10 min. There is no clear relationship between this data obtained by EM and the time course for content mixing measured biochemically (Fig. 1 a). The latter is most likely to measure fusions between endosomes and lysosomes or hybrids and lysosomes rather than hybrids and endosomes since we estimate that there was ∼100-fold excess of avidin-ASF over bpIgA in the endosome and lysosome fractions used in these experiments.
Representative micrographs demonstrating the morphology of the hybrid organelles and of lysosome and late endosome preparations are shown in Fig. 4 (8-nm gold particles label immunoreactive avidin and 15-nm gold particles label cathepsin L). Material from the lysosomal peak at the 45%/20% Nycodenz interface contained mostly electron-dense structures labeled with cathepsin L, but little avidin (Fig. 4 c). Quantitation of avidin- and cathepsin L–labeled structures in the late endosome preparation used in the fusion assay showed that many endosomes were heavily loaded with avidin but contained very little cathepsin L (Fig. 3). However, since a significant proportion of cathepsin L–positive, avidin-negative structures were also observed in the endosomal preparation, late endosomes were incubated in the content mixing assay in the absence of lysosomes at 37°C for 10 min. No membrane could be recovered from the Nycodenz/Ficoll interface of a step gradient after such an incubation (data not shown).
Figure 4.
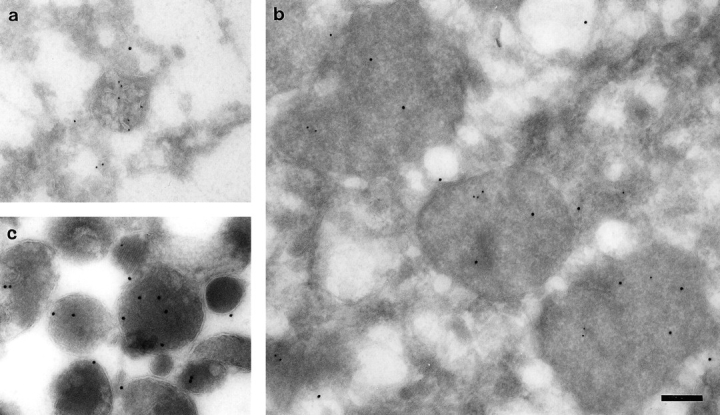
Immunoelectron microscopy identifying avidin (8-nm gold) and cathepsin L (15-nm gold) immunoreactivity in organelles from late endosome, lysosome, and hybrid fractions. (a) A late endosome peak fraction from the preparative 1–22% Ficoll gradient showing an endosomal structure containing Av-ASF. (b) Hybrid organelles formed after the content mixing assay incubation accumulated at the 20% Nycodenz/20% Ficoll interface of the step gradient (see Fig. 2). These membrane-bounded organelles from the interface peak fractions were immunolabeled with antibodies to both cathepsin L and avidin. (c) Electron-dense lysosomes that could be immunolabeled with anti–cathepsin L antibodies accumulated at the 45%/20% Nycodenz interface of the step gradient after the content mixing assay. Bar, 200 nm.
In a separate experiment, it was found that hybrid structures containing avidin and cathepsin L could be recovered at the Nycodenz/Ficoll interface after centrifugation of postmitochondrial supernatant from a rat injected with Av-ASF 9 min before (data not shown). This demonstrated that hybrid endosome–lysosome structures were not simply an artifact of the cell-free system.
Content Mixing Is Unlikely to Be Vesicle Mediated
The rapid appearance of a distinct peak of content-mixed lysosome–endosome hybrids suggested direct fusion between the two organelles, rather than transfer of material from one to the other by small vesicles. This suggestion was supported by two further lines of evidence. Firstly, transfer vesicles usually have coats, which have to be removed before fusion with the receptor organelle. Such coat loss is often prevented by substitution of nonhydrolyzable GTP analogues for GTP, and hence content mixing is inhibited (Rothman, 1994). However, endosome–lysosome content mixing was actually stimulated by nonhydrolyzable GTP analogues (Fig. 1). Secondly, it is usually possible to generate transfer vesicles by a preincubation of the donor compartment and thereby produce more content mixing when the acceptor compartment is added. However, in the case of late endosomes, 5 min preincubation with cytosol and an energy regenerating system reduced the amount of content mixing subsequently observed after addition of lysosomes (Fig. 5), to less than 60% of the standard fusion. This reduction was cytosol and energy dependent since in the absence of cytosol and energy at least 20 min preincubation of endosomes was required to reduce the subsequent content mixing below 60%. The effect of preincubation with cytosol and energy was not prevented by protease inhibitors or the protein kinase inhibitors staurosporine or tyrphostin 25 (data not shown). Replacing GTP with its nonhydrolyzable analogue 5′-guanylylimidodiphosphate (GMP-PNP) increased the inhibition still further (Fig. 5).
Figure 5.

Content mixing after preincubation of late endosomes for 5 min. Late endosomes were preincubated (preinc) at 37°C for 5 min in cytosol with an ATP-regenerating system (E) and 1 mM GTP, with omissions as shown or with 200 μM GMP-PNP replacing GTP. As a control in STM, late endosomes were preincubated at 37°C for 5 min in STM alone.
Fusion Is NSF and SNAP Dependent
Although lysosomes and late endosomes appeared to fuse directly, they showed several of the same requirements as fusion events involving transfer vesicles at other sites in membrane traffic pathways. NEM treatment of cytosol inhibited content mixing in a dose-dependent manner (Fig. 6 a). NEM treatment of endosomes and lysosomes also inhibited content mixing, and when cytosolic NSF was depleted by incubation without ATP, content mixing was reduced still further (Fig. 6 b). Addition of recombinant NSF relieved this inhibition (Fig. 6 b) in a dose-dependent manner (Fig. 6 c). Pretreatment of the NSF with NEM destroyed its activity (data not shown). The lysosome and endosome fractions contained sufficient NSF to support content mixing fully in the presence of cytosol in which NSF was depleted by incubation without ATP (Fig. 6 b). The involvement of α- and γ-SNAP proteins was shown by treating late endosomes with 1 M KCl for 15 min on ice. Content mixing, reduced to 68 ± 4% (5) of standard fusion by such KCl treatment, was restored to 86 ± 5% (3) by addition of recombinant SNAPs (∼50 pmol α-SNAP and 15 pmol γ-SNAP per assay).
Figure 6.
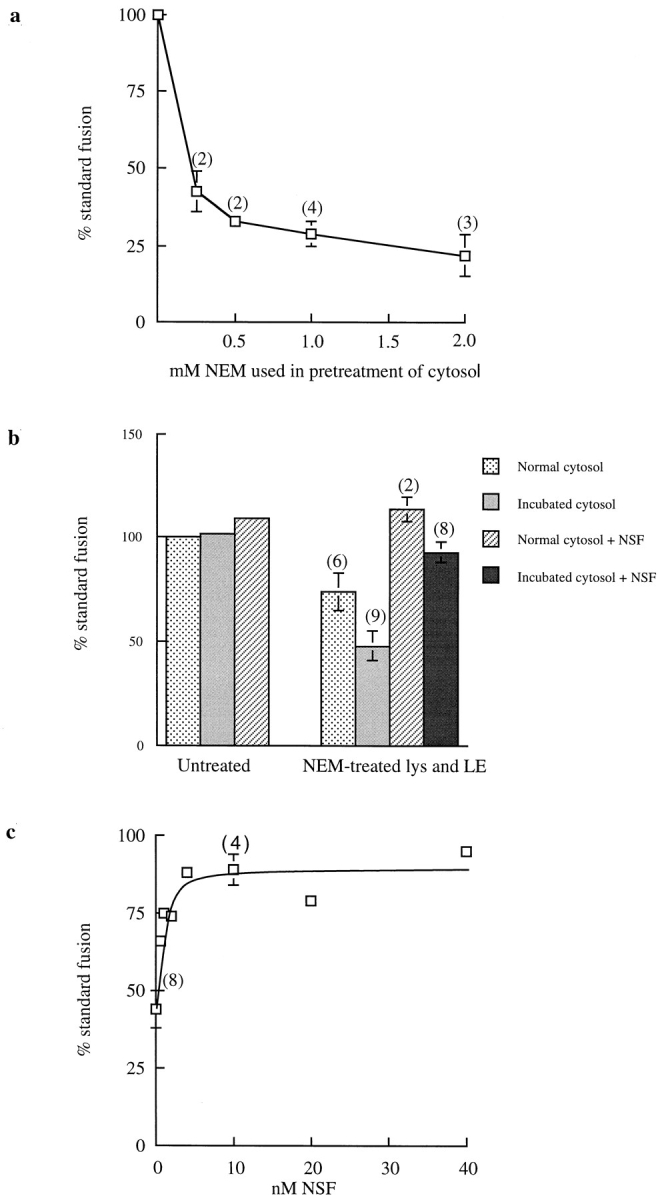
Content mixing requires NSF. (a) Dose-dependent inhibition of content mixing by NEM treatment of cytosol. Cytosol was pretreated with NEM at 4°C for 15 min before quenching with DTT. (b) Destruction of NSF and replacement with recombinant NSF. Cytosol was incubated at 37°C for 15 min in the absence of ATP to destroy NSF; late endosomes (LE) and lysosomes (lys) were separately treated with 2 mM NEM at 4°C for 15 min before quenching with DTT. Recombinant NSF was added to 10 nM. (c) Dose dependence of recombinant NSF addition. Cytosol was NSF-depleted by incubation and late endosomes and lysosomes were NEM treated as in b.
Content Mixing Is Inhibited by GDI
The presence of recombinant GDI inhibited the content mixing reaction in a dose-dependent manner (Fig. 7 a). Such incubation with GDI was shown to remove rab 7 from membranes with the same dose response (Fig. 7, b and c). Content mixing was inhibited 50% by 5 μM GDI (calculated from Fig. 7 a) and 50% of rab 7 was removed from membranes in the assay by 5 ± 0.5 (3) μM GDI. Addition of rab 7 and geranylgeranyl pyrophosphate to the incubation mixture did not prevent GDI inhibition of content mixing, suggesting that a different rab protein might be required for late endosome–lysosome fusion.
Figure 7.

Inhibition of content mixing by GDI. (a) Content mixing in the presence of GDI. (b) Removal of rab 7 from the membranes in a representative content mixing assay by GDI. (c) Quantitation of the data shown in b.
Phosphatidylinositol 3-Kinase Inhibitors Prevented Content Mixing
Low concentrations of wortmannin and another very specific phosphatidylinositol 3-kinase (PI3-K) inhibitor, LY294002, partially inhibited content mixing (Table I). Since wortmannin has also been reported to inhibit phospholipases (for references see Reaves et al., 1996), the membrane-permeable phospholipase A2 inhibitor, NDGA, was used for comparison, but this compound had no effect on content mixing. 3-methyladenine, a known inhibitor of autophagy and late endosome to lysosome transport, which is also a PI3-K inhibitor (Blommaart et al., 1997), also reduced content mixing. Although chloroquine and primaquine had a similar inhibitory effect, bafilomycin, an inhibitor of vacuolar proton pumping ATPase, had little or no effect even when present during a preincubation period (Table I). There was no effect on the content mixing assay of the protein phosphatase inhibitors, okadaic acid (1.5 μM), vanadate (100 μM), or tyrphostin 25 (100 μM), nor of 20 μM taxol, 5 mM EGTA, or 10 μM cytochalasin D (data not shown).
Table I.
The Effects of Potential Inhibitors on Late Endosome–Lysosome Content Mixing
| Inhibitor | Percent normal fusion | |
|---|---|---|
| Wortmannin 100 nM | 65 ± 3 (2) | |
| LY294002 100 μM | 44 | |
| NDGA 20 μM | 96 | |
| 3-methyladenine 1 mM | 82 | |
| 10 mM | 50 | |
| Chloroquine 250 μM | 93 | |
| 1 mM | 76 | |
| Primaquine 250 μM | 66 ± 2 (2) | |
| * Bafilomycin 1 μM | 85 |
Inhibitors were added at the stated concentrations to the standard cell-free assay.
In the case of bafilomycin, additional experiments were carried out in which the components of the standard assay were preincubated for 30 min at 4°C with 1 and 2 μM bafilomycin before incubation at 37°C. Similar results were obtained, with 90 and 85% normal fusion observed, respectively.
Discussion
The biochemical results, complemented by immunoEM, described in this paper indicate that late endosomes can fuse directly and completely with lysosomes. The immunoprecipitable product formed from mixing avidin in late endosomes and biotin in lysosomes was shown to be located in a hybrid lysosome–endosome, which was less dense than lysosomes and denser than late endosomes and which could be separated from both. Late endosomes, rather than peripheral early endosomes, were the species involved. The endosomes were prepared on Ficoll gradients, which separate early and late endosomes, the late endosomes peaking at a higher density than early endosomes (Mullock et al., 1994). Late endosomes were routinely taken from this denser section of the gradient. These late endosomes showed a higher content mixing ability than material taken from the lighter end of the same gradient, where any residual labeled early endosomes would be found. In any case, after 6 min in vivo labeling, as used here to load the endosomes, most labeled asialoglycoprotein has left peripheral early endosomes (Mullock et al., 1994).
It is conceivable that rapid vesicular transfer of material between late endosomes and lysosomes could produce a compartment of intermediate density. However, no evidence for such transfer could be found. On the contrary, attempts to increase content mixing by preincubation of late endosomes under conditions that might facilitate vesicle formation (i.e., with cytosol and an energy-regenerating system) inhibited content mixing when lysosomes were subsequently added. Substitution of nonhydrolyzable GTP analogues for GTP, which inhibit vesicular transfer in many systems by preventing detachment of vesicular coats (Rothman, 1994), actually increased endosome–lysosome content mixing.
ImmunoEM showed clearly that the late endosome fraction contained, as expected, organelles heavily loaded with avidin, but these contained very little of the lysosomal enzyme cathepsin L. After the content mixing incubation of avidin-loaded late endosomes with lysosomes, the lysosomal fraction, which had passed through 20% Nycodenz, contained cathepsin L but little avidin. The fraction that was retained above the 20% Nycodenz layer at the Nycodenz/Ficoll interface was enriched in organelles containing both cathepsin L and avidin, as would be expected for the fraction in which immunoprecipitable radiolabel was shown to be concentrated by the biochemical assay. The formation of these structures was not simply an in vitro phenomenon since similar organelles, containing both avidin and cathepsin L, were visible in material from the Nycodenz/ Ficoll interface, when the step gradient had been loaded with postmitochondrial supernatant from a rat injected with Av-ASF. We conclude that in hepatocytes, late endosomes fuse completely with preexisting dense lysosomes, giving a hybrid organelle of intermediate density. Recent reports show that similar direct fusions without vesicular transfer can also occur between endosomes and autophagic vacuoles (Liou et al., 1997), and between lysosomes and the plasma membrane (Rodriguez et al., 1997), phagosomes (Funato et al., 1997), and other lysosomes (Ward et al., 1997).
In spite of the fact that content mixing between endosomes and lysosomes did not appear to be mediated by small vesicles, the process required some of the same proteins as vesicle-mediated transfer between Golgi compartments (Rothman, 1994). Thus, NSF was shown to be necessary by removal and replacement with recombinant protein. The inhibition of content mixing due to NEM treatment of cytosol could not, however, be relieved by addition of recombinant NSF even at 40 nM (data not shown), suggesting that a further NEM-sensitive cytosolic component was required. This was not α-SNAP, which can be NEM sensitive (Rodriguez et al., 1994), since addition of recombinant α-SNAP with NSF did not relieve the inhibition (data not shown). In further experiments that are not shown, we were unsuccessful in attempts to recover the fusion reaction by the addition of the p97/valosin-containing protein shown to be required for homologous fusions (Acharya et al., 1995; Rabouille et al., 1995) or thioredoxin shown to be required for vacuole inheritance in Saccharomyces cerevisiae (Xu and Wickner, 1996). Other non-NSF NEM-sensitive cytosolic factors have been described in transport systems between endosomes and TGN (Goda and Pfeffer, 1991) and in endosomal vesicle fusion (Rodriguez et al., 1994). In addition to NSF, SNAPs were required for fusion. Our data are consistent with some NSF as well as SNAPs being found associated with the organelle membranes before fusion, as has also been observed for membrane fractions isolated from earlier in the endocytic pathway (Steel et al., 1996). Demonstration of the need for SNAPs was difficult in that extraction procedures more thorough than incubation of the endosomes in 1 M KCl on ice for 15 min produced irreversible damage to the subsequent content mixing reaction, as did attempts to remove SNAPs from lysosomes or cytosol. However, Wilson et al. (1992) showed that such a KCl extraction stripped SNAPs from Golgi membranes. Removal of SNAPs from the late endosome membranes was therefore probably responsible for the decrease in content mixing activity, which could be partly restored by addition of recombinant SNAPs to the mixture. SNAPs have previously been shown to be essential for early endocytic vesicle fusion (Rodriguez et al., 1994), but in this work also, complete removal of SNAPs could not be achieved.
Transfer between compartments mediated by transport vesicles usually involves the participation of a rab protein in its GTP-bound form (Pfeffer, 1994). Most rab proteins can be removed from membranes by incubation with GDI (Pfeffer et al., 1995). Content mixing between late endosomes and lysosomes was inhibited by GDI in a dose- dependent manner, indicating that a rab protein was also involved in this fusion. Moreover, the activation of the late endosome–lysosome content mixing assay by nonhydrolyzable GTP analogues also suggests the involvement of a rab protein since rab proteins are active in their GTP-bound form. The rab proteins known to be associated with late endosomes are rab 7 and rab 9 (Zerial and Stenmark, 1993). Rab 9 has been shown to be involved in trafficking between late endosomes and the TGN (Lombardi et al., 1993). A rab 7 mutant has been observed in association with lysosomal membranes (Meresse et al., 1995), but Feng et al. (1995) concluded that rab 7 acts in the regulation of early to late endosome transport, a result reinforced by the data of Papini et al. (1997). Therefore, it is not surprising that addition of rab 7 and geranylgeranyl pyrophosphate did not relieve the GDI inhibition of late endosome–lysosome content mixing. It may be argued that the rab 7 was not properly incorporated into membranes under these conditions, but Lombardi et al. (1993) used such conditions to successfully reincorporate functional rab 9 into late endosomal membranes. It is possible that a novel rab-type protein is required for fusion of late endosomes with lysosomes. Small GTP-binding proteins have been reported on lysosomal membranes (Sai and Ohkuma, 1992), but the precise rab family members have not been identified.
A variety of factors have been shown to modulate membrane traffic through the endocytic pathway. The protein phosphatase inhibitor okadaic acid inhibits early endosome fusion (Woodman et al., 1992), but neither this nor other protein phosphatase inhibitors had an effect on endosome–lysosome fusion. Similarly, the polymerized microtubule stabilizing agent taxol, which stimulates the fusion of endocytic carrier vesicles with late endosomes indicating a microtubule requirement (Aniento et al., 1993), had no effect on endosome–lysosome fusion. A previous study of the transport of fluid phase markers through the endocytic pathway suggested that vacuolar ATPase activity was required for delivery to lysosomes (van Weert et al., 1995). Although the membrane-permeant weak bases chloroquine and primaquine, which reduce intraorganelle acidity, inhibited endosome–lysosome fusion, the lack of effect of the vacuolar ATPase inhibitor bafilomycin suggested that this inhibition was unrelated to alteration of organelle acidity and that vacuolar ATPase activity was not required for fusion. Actin microfilaments have been suggested to increase delivery to the degradative endocytic compartment in a mouse hepatoma cell line (Durrbach et al., 1996) and to facilitate endosome fusion with preexisting lysosomes in HEp-2 cells (van Deurs et al., 1995), but no effect of cytochalasin D on endosome–lysosome fusion was observed in our cell-free system. In contrast, inhibitors of PI3-K, which have dramatic morphological effects on late endocytic compartments (Reaves et al., 1996), did cause inhibition of endosome–lysosome fusion. It is conceivable that the inhibitory action of the PI3-K inhibitor wortmannin in the late endosome–lysosome content mixing assay was due to its effect on a rab protein since preliminary data showed that substitution of nonhydrolyzable GMP-PNP for GTP prevented wortmannin inhibition. Li et al. (1995) showed that addition of a constitutively activated rab 5 mutant (i.e., a mutant permanently in the GTP-bound form) reversed wortmannin inhibition of early endosome fusion in a cell-free system. The effects of the PI3-K inhibitors and weak bases and the non–NSF- dependent effects of NEM all suggest that factors in addition to the NSF/SNAP fusion machinery are required for endosome–lysosome fusion.
In the present study we have demonstrated the direct fusion of preexisting dense lysosomes with late endosomes. By morphological examination of NRK cells that had endocytosed colloidal gold–albumin conjugates, we have previously obtained evidence that dense-core lysosomes fuse with late endosomes in living cells to form hybrid compartments (Bright et al., 1997). In both that and other studies (van Deurs et al., 1995; Futter et al., 1996), fine striations were sometimes observed between bound endosomes and lysosomes. These may contribute to the binding involved in the interaction of late endosomes with lysosomes. The formation of a hybrid organelle as a result of lysosome– endosome fusion, which has now been observed both in a cell-free system and in vivo (Bright et al., 1997), suggests the presence of a membrane recovery system to avoid the build up of swollen hybrid structures within the cell. This is likely to be a very dynamic process allowing rapid retrieval of membrane proteins present in endosomes but not lysosomes and ensuring that individual hybrid organelles exist only for a short period of time unless further fusion events take place. It is also of interest that in the present cell-free system, a nonhydrolyzable GTP analogue added to the preincubation of late endosomes caused inhibition of subsequent content mixing between endosomes and lysosomes, suggesting that vesicles were being formed from late endosomes but were not destined for fusion with lysosomes. There is accumulating evidence that coat proteins exist on endosomes and lysosomes and could play a role in membrane retrieval (Seaman et al., 1993; Whitney et al., 1995; Aniento et al., 1996; Stoorvogel et al., 1996; Traub et al., 1996). A diagrammatic representation of the processes involved in endosome–lysosome fusion and subsequent recovery from the hybrid organelle is shown in Fig. 8. In vivo, the endosome–lysosome hybrid almost certainly recondenses to a dense lysosome by a combination of membrane retrieval and digestive processes (Bright et al., 1997). The ability to separate the hybrids from both late endosomes and dense lysosomes should enable us to examine the condensation process, as well as to search for the proteins involved in controlling the fusion reaction.
Figure 8.

Diagrammatic representation of the direct fusion of late endosomes with lysosomes to form a hybrid organelle of intermediate density. Binding of late endosomes to lysosomes may be mediated by filamentous structures that have been observed by electron microscopy. The formation of the hybrid organelle requires NSF and SNAPs and is inhibited by GDI, although the exact step at which they function and their order of interaction are unknown. Previous experiments with NRK cells (Bright et al., 1997) suggest that dense-core lysosomes may reform from hybrid organelles. This requires recondensation of contents and probably retrieval of membrane to an unknown location, possibly to an earlier site in the endocytic pathway. cat L, cathepsin L-positive structures.
Acknowledgments
We thank Margaret Robinson, Howard Davidson, Rainer Duden, Barbara Reaves, and Gudrun Ihrke, all of the Department of Clinical Biochemistry, University of Cambridge, for much valuable discussion.
Abbreviations used in this paper
- Av-ASF
avidin-asialofetuin
- bpIgA
biotinylated polymeric IgA
- GDI
GDP dissociation inhibitor
- GMP-PNP
5′-guanylylimidodiphosphate
- MPR
cation-independent mannose 6-phosphate receptor
- NDGA
nordihydroguaiaretic acid
- NEM
N-ethylmaleimide
- NSF
NEM-sensitive factor
- PI3-K
phosphatidylinositol 3-kinase
- SNAP
soluble NSF attachment protein
Footnotes
This work was funded by the Medical Research Council and Smith Kline Beecham.
Received for publication 2 May 1997 and in revised form 15 December 1997.
B.M. Mullock and N.A. Bright contributed equally to this work.
Address all correspondence to Paul Luzio, Department of Clinical Biochemistry, University of Cambridge, Addenbrooke's Hospital, Hills Road, Cambridge, CB2 2QR, UK. Tel.: +44-1223-336780. Fax: +44-1223-330598. E-mail: jpl10@cam.ac.uk
References
- Acharya U, Jacobs R, Peters J, Watson N, Farquhar MG, Malhotra V. The formation of Golgi stacks from vesiculated Golgi membranes requires two distinct fusion events. Cell. 1995;82:895–904. doi: 10.1016/0092-8674(95)90269-4. [DOI] [PubMed] [Google Scholar]
- Aniento F, Emans N, Griffiths G, Gruenberg J. Cytoplasmic dynein–dependent vesicular transport from early to late endosomes. J Cell Biol. 1993;123:1373–1387. doi: 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniento F, Gu F, Parton RG, Gruenberg J. An endosomal β-COP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol. 1996;133:29–41. doi: 10.1083/jcb.133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T, Gjoen T, Bakke O. Physiological functions of endosomal proteolysis. Biochem J. 1995;307:313–326. doi: 10.1042/bj3070313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blommaart EF, Krause U, Schellens JPM, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- Bright NA, Reaves BJ, Mullock BM, Luzio JP. Dense core lysosomes can fuse with late endosomes and are reformed from the resultant hybrid organelles. J Cell Sci. 1997;110:2027–2040. doi: 10.1242/jcs.110.17.2027. [DOI] [PubMed] [Google Scholar]
- Durrbach A, Louvard D, Coudrier E. Actin filaments facilitate two steps of endocytosis. J Cell Sci. 1996;109:457–465. doi: 10.1242/jcs.109.2.457. [DOI] [PubMed] [Google Scholar]
- Ellis, J.A., M.R. Jackman, J.H. Perez, B.M. Mullock, and J.P. Luzio. 1992. Membrane pathways in polarised epithelial cells. In Protein Targeting: A Practical Approach. A.I. Magee and T. Wileman, editors. Oxford University Press, London. 25–57.
- Feng Y, Press B, Wandinger-Ness A. Rab 7: an important regulator of late endocytic membrane traffic. J Cell Biol. 1995;131:1435–1452. doi: 10.1083/jcb.131.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Beron W, Yang CZ, Mukhopadhyay A, Stahl PD. Reconstitution of phagosome-lysosome fusion in streptolysin O-permeabilized cells. J Biol Chem. 1997;272:16147–16151. doi: 10.1074/jbc.272.26.16147. [DOI] [PubMed] [Google Scholar]
- Futter CE, Pearse A, Hewlett LJ, Hopkins CR. Multivesicular endosomes containing internalised EGF–EGF receptor complexes mature then fuse directly with lysosomes. J Cell Biol. 1996;132:1011–1023. doi: 10.1083/jcb.132.6.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Pfeffer SR. Identification of a novel, N-ethylmaleimide– sensitive cytosolic factor required for vesicular transport from endosomes to the trans-Golgi network in vitro. J Cell Biol. 1991;112:823–831. doi: 10.1083/jcb.112.5.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, G. 1993. Fine structure immunocytochemistry. Springer-Verlag, Berlin. 459 pp.
- Griffiths G. On vesicles and membrane compartments. Protoplasma. 1996;195:37–58. [Google Scholar]
- Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- Hicke L, Zanolari B, Pypaert M, Rohrer J, Riezman H. Transport through the yeast endocytic pathway occurs through morphologically distinct compartments and requires an active secretory pathway and Sec18p/N-ethylmaleimide-sensitive fusion protein. Mol Biol Cell. 1997;8:13–31. doi: 10.1091/mbc.8.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, d'Souza-Schorey C, Barbieri MA, Roberts RL, Klippel A, Williams LT, Stahl PD. Evidence for phosphatidylinositol 3-kinase as a regulator of endocytosis via activation of Rab 5. Proc Natl Acad Sci USA. 1995;92:10207–10211. doi: 10.1073/pnas.92.22.10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou W, Geuze HJ, Slot JW. Improving structural integrity of cryosections for immunogold labelling. Histochem Cell Biol. 1996;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- Liou W, Geuze HJ, Geelen MJH, Slot JW. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J Cell Biol. 1997;136:61–70. doi: 10.1083/jcb.136.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi D, Soldati T, Riederer MA, Goda Y, Zerial M, Pfeffer SR. Rab 9 functions in transport between late endosomes and the trans Golgi network. EMBO (Eur Mol Biol Organ) J. 1993;12:677–682. doi: 10.1002/j.1460-2075.1993.tb05701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- Meresse S, Gorval J, Chavrier P. The rab 7 GTPase resides on a vesicular compartment connected to lysosomes. J Cell Sci. 1995;108:3349–3358. doi: 10.1242/jcs.108.11.3349. [DOI] [PubMed] [Google Scholar]
- Mullock BM, Perez JH, Kuwana T, Gray SR, Luzio JP. Lysosomes can fuse with a late endosomal compartment in a cell-free system from rat liver. J Cell Biol. 1994;126:1173–1182. doi: 10.1083/jcb.126.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papini E, Satin B, Bucci C, de Bernard M, Telford JL, Manetti R, Rappuoli R, Zerial M, Montecucco C. The small GTP binding protein rab7 is essential for cellular vacuolation induced by Helicobacter pylori cytotoxin. EMBO (Eur Mol Biol Organ) J. 1997;16:15–24. doi: 10.1093/emboj/16.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR. Rab GTPases: master regulators of membrane trafficking. Curr Opin Cell Biol. 1994;6:522–526. doi: 10.1016/0955-0674(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR, Dirac-Svejstrup AB, Soldati T. Rab GDP dissociation inhibitor: putting Rab GTPases in the right place. J Biol Chem. 1995;270:17057–17059. doi: 10.1074/jbc.270.29.17057. [DOI] [PubMed] [Google Scholar]
- Punnonen E-L, Marjomaki VS, Reunanen H. 3-methyladenine inhibits transport from late endosomes to lysosomes in cultured rat and mouse fibroblasts. Eur. J Cell Biol. 1994;65:14–25. [PubMed] [Google Scholar]
- Rabouille C, Levine TP, Peters J, Warren G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic fragments. Cell. 1995;82:905–914. doi: 10.1016/0092-8674(95)90270-8. [DOI] [PubMed] [Google Scholar]
- Reaves BJ, Bright NA, Mullock BM, Luzio JP. The effect of wortmannin on the localisation of lysosomal type I integral membrane glycoproteins suggests a role for phosphoinositide 3-kinase activity in regulating membrane traffic late in the endocytic pathway. J Cell Sci. 1996;109:749–762. doi: 10.1242/jcs.109.4.749. [DOI] [PubMed] [Google Scholar]
- Rodriguez L, Stirling CJ, Woodman PG. Multiple N-ethylmaleimide-sensitive components are required for endosomal vesicle fusion. Mol Biol Cell. 1994;5:773–783. doi: 10.1091/mbc.5.7.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Sai Y, Ohkuma S. Small GTP-binding proteins on rat liver lysosomal membranes. Cell Struct Funct. 1992;17:363–369. doi: 10.1247/csf.17.363. [DOI] [PubMed] [Google Scholar]
- Seaman MNJ, Ball CL, Robinson MS. Targeting and mistargeting of plasma membrane adaptors in vitro. J Cell Biol. 1993;123:1093–1105. doi: 10.1083/jcb.123.5.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. Immunolocalization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Steel GJ, Tagaya M, Woodman PG. Association of the fusion protein NSF with clathrin-coated vesicle membranes. EMBO (Eur Mol Biol Organ) J. 1996;15:745–752. [PMC free article] [PubMed] [Google Scholar]
- Stoorvogel W, Oorschot V, Geuze HJ. A novel class of clathrin-coated vesicles budding from endosomes. J Cell Biol. 1996;132:21–33. doi: 10.1083/jcb.132.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storrie B, Desjardins M. The biogenesis of lysosomes: is it a kiss and run, continuous fusion and fission process? . BioEssays. 1996;18:895–903. doi: 10.1002/bies.950181108. [DOI] [PubMed] [Google Scholar]
- Tjelle TE, Brech A, Juvet LK, Griffiths G, Berg T. Isolation and characterization of early endosomes, late endosomes and terminal lysosomes: their role in protein degradation. J Cell Sci. 1996;109:2905–2914. doi: 10.1242/jcs.109.12.2905. [DOI] [PubMed] [Google Scholar]
- Tokuyasu KY. A study of positive staining of ultrathin frozen sections. J Ultrastruct Res. 1978;63:287–307. doi: 10.1016/s0022-5320(78)80053-7. [DOI] [PubMed] [Google Scholar]
- Traub LM, Bannykh SI, Rodel JE, Aridor M, Balch WE, Kornfeld S. AP-2–containing clathrin coats assemble on mature lysosomes. J Cell Biol. 1996;135:1801–1814. doi: 10.1083/jcb.135.6.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Stenmark H, Alexandrov K, Huber LA, Kaibuchi K, Sasaki T, Takei Y, Zerial M. Rab GDP dissociation inhibitor as a general regulator for the membrane association of rab proteins. J Biol Chem. 1993;268:18143–18150. [PubMed] [Google Scholar]
- Ullrich O, Horiuchi H, Alexandrov K, Zerial M. Use of Rab-GDP dissociation inhibitor for solubilisation and delivery of Rab proteins to biological membranes in streptolysin O-permeabilised cells. Methods Enzymol. 1995;257:243–253. doi: 10.1016/s0076-6879(95)57029-2. [DOI] [PubMed] [Google Scholar]
- van Deurs B, Holm PK, Kayser L, Sandvig K. Delivery to lysosomes in the human carcinoma cell line HEp-2 involves an actin filament- facilitated fusion between mature endosomes and preexisting lysosomes. Eur J Cell Biol. 1995;66:309–323. [PubMed] [Google Scholar]
- van Weert AWM, Dunn KW, Geuze HJ, Maxfield FR, Stoorvogel W. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J Cell Biol. 1995;130:821–834. doi: 10.1083/jcb.130.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DM, Leslie JD, Kaplan J. Homotypic lysosome fusion in macrophages: analysis using an in vitro assay. J Cell Biol. 1997;139:665–673. doi: 10.1083/jcb.139.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel, E.W. 1979. Stereological Methods. Vol. 1. Practical Methods for Biological Morphometry. Academic Press, London. 415 pp.
- Whiteheart SW, Griff IC, Brunner M, Clary DO, Mayer T, Bushrow SA, Rothman JE. SNAP family of NSF attachment proteins includes a brain specific isoform. Nature. 1993;362:353–355. doi: 10.1038/362353a0. [DOI] [PubMed] [Google Scholar]
- Whitney JA, Gomez M, Sheff D, Kreis TE, Mellman I. Cytoplasmic coat proteins involved in endosome function. Cell. 1995;83:703–713. doi: 10.1016/0092-8674(95)90183-3. [DOI] [PubMed] [Google Scholar]
- Wilson DE, Rothman JE. Expression and purification of recombinant N-ethylmaleimide-sensitive fusion protein from Escherichia coli. Methods Enzymol. 1992;219:309–318. doi: 10.1016/0076-6879(92)19031-z. [DOI] [PubMed] [Google Scholar]
- Wilson DW, Whiteheart SW, Wiedmann M, Brunner M, Rothman JE. A multisubunit particle implicated in membrane fusion. J Cell Biol. 1992;117:531–538. doi: 10.1083/jcb.117.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman PG, Mundy DI, Warren G. Cell-free fusion of endocytic vesicles is regulated by phosphorylation. J Cell Biol. 1992;116:331–338. doi: 10.1083/jcb.116.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wickner W. Thioredoxin is required for vacuole inheritance in Saccharomyces cerevisiae. . J Cell Biol. 1996;132:787–794. doi: 10.1083/jcb.132.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M, Stenmark H. Rab GTPases in vesicular transport. Curr Opin Cell Biol. 1993;5:613–620. doi: 10.1016/0955-0674(93)90130-i. [DOI] [PubMed] [Google Scholar]