

Abstract
Ultraviolet B and genotoxic drugs induce the expression of a vascular endothelial growth factor A (VEGF-A) splice variant (VEGF111) encoded by exons 1–4 and 8 in many cultured cells. Although not detected in a series of normal human and mouse tissue, VEGF111 expression is induced in MCF-7 xenografts in nude mice upon treatment by camptothecin. The skipping of exons that contain proteolytic cleavage sites and extracellular matrix–binding domains makes VEGF111 diffusible and resistant to proteolysis. Recombinant VEGF111 activates VEGF receptor 2 (VEGF-R2) and extracellularly regulated kinase 1/2 in human umbilical vascular endothelial cells and porcine aortic endothelial cells expressing VEGF-R2. The mitogenic and chemotactic activity and VEGF111's ability to promote vascular network formation during embyonic stem cell differentiation are similar to those of VEGF121 and 165. Tumors in nude mice formed by HEK293 cells expressing VEGF111 develop a more widespread network of numerous small vessels in the peritumoral tissue than those expressing other isoforms. Its potent angiogenic activity and remarkable resistance to proteolysis makes VEGF111 a potential adverse factor during chemotherapy but a beneficial therapeutic tool for ischemic diseases.
Introduction
VEGF-A is a potent angiogenic factor that stimulates vascular endothelial cell survival, proliferation, migration, and differentiation, alters their pattern of gene expression, and delays senescence (Dvorak, 2000). VEGF acts through binding to VEGF receptor 1 (VEGF-R1; flt-1) and 2 (VEGF-R2; KDR/flk-1), the latter apparently mediating most of the VEGF signaling, whereas the role of VEGF-R1 is more elusive (Waltenberger et al., 1994). Affinity binding of VEGF-A to VEGF-R2 is enhanced by neuropilin-1, a nonkinase coreceptor (Soker et al., 1998).
Both homozygous and heterozygous deletions of the VEGF-A gene in mice are embryonic lethal (Carmeliet et al., 1996; Ferrara et al., 1996), suggesting that a finely tuned regulation of VEGF expression is essential for development. Excessive and/or ectopic angiogenesis or its reverse, defective vascularization, is responsible for several pathologies. VEGF is a major initiator of tumor angiogenesis, a critical step in cancer growth and metastasis (Skobe et al., 1997). Overexpression of VEGF is also determinant in retinopathies (Caldwell et al., 2005) and many other diseases. A large effort is being produced to decrease the excessive production of VEGF or to tackle VEGF-induced angiogenesis with neutralizing antibodies, antagonists, function inhibitors, or inhibitory soluble receptors (Ferrara, 2005). In contrast, pathologies such as ischemia, chronic wounds, and erectile dysfunction could benefit from neovascularization driven by VEGF delivery (Lauer et al., 2002; Yoon et al., 2004; Burchardt et al., 2005).
VEGF is a disulfide-bonded dimeric glycoprotein with a molecular mass of 34–45 kD encoded by a gene that contains eight exons. The known isoforms of human VEGF generated by alternative splicing are encoded by exons 1–5 and a varying combination of the other exons (Jingjing et al., 1999; Whittle et al., 1999; Robinson and Stringer, 2001; Bates et al., 2002). The domains encoded by the first four exons contain the signal peptide, the sequence of recognition by VEGF-Rs, and a glycosylation site. The amino acid sequence encoded by exon 5 includes the main site of cleavage by plasmin (Claffey et al., 1995; Keyt et al., 1996) and matrix metalloproteinases (Lee et al., 2005). Exon 6 encodes the sequence that binds to heparin and a cell surface retention consensus sequence. The sequence encoded by exon 7, besides a moderate affinity for heparin and heparan sulfate proteoglycans, contains the binding site for neuropilin-1. VEGF121 (lacking exons 6 and 7) is freely diffusible, whereas VEGF189 (containing exons 1–6A, 7, and 8) is primarily associated with the cell surface and the ECM. VEGF165 (containing exons 1–5, 7, and 8) displays properties that are intermediate between VEGF189 and 121 (Park et al., 1993).
Cancers often arise as a consequence of the accumulation of genetic mutations induced by a wide variety of physical and chemical agents. UV irradiation is an environmental element with carcinogenic potential. UV-B (290–320 nm) induces the formation of cyclobutane pyrimidine dimers in the DNA, which are considered to play an essential role in skin cancer induction (Katiyar et al., 2001). Cutaneous alterations observed after a single exposure to UV-B include erythema and edema related to an altered expression of biological mediators, including angiogenic factors (Blaudschun et al., 2002). In this paper, we show that UV-B irradiation and genotoxic drugs induce the expression of a new VEGF splice variant, an isoform lacking the sequence encoded by exons 5–7. This isoform, which is biologically active and resistant to proteolysis, may be of therapeutic interest.
Results
UV-B and genotoxic agents induce the expression of a new VEGF isoform lacking exons 5–7
UV-B irradiation (30 mJ/cm2) of HaCat cells, MDA-MB-231 cells, and MCF-7 cells affected the expression of the mRNA encoding the various VEGF isoforms as evaluated by quantitative RT-PCR. Using primers P1 and P2 (Table I; Hajitou et al., 2001) chosen on exons 3 and 8, respectively, VEGF189, 165, and 121 mRNAs were detected in control and UV-exposed cells (Fig. 1 A, top left). Upon UV treatment, the mRNA levels of VEGF165 and, to a smaller extent, VEGF189 and 121 were decreased, whereas a faster migrating RT-PCR product (Fig. 1 A, arrows) was observed in MCF-7 cells and, at a lower level, in HaCat and MDA-MB-231 cells. This product was never observed in nonirradiated cells. Sequence analysis of this fast migrating product revealed the presence of sequences of exons 3, 4, and 8 and the absence of sequences encoded by exons 5–7. Sequencing of the full-length fast migrating VEGF cDNA variant amplified using primers P3 and P4 (GenBank/EMBL/DDBJ accession no. DQ229900; Table I) demonstrated that it was formed by exons 1–4 and 8. Skipping of the 30 bp encoded by exon 5 did not change the reading frame of the downstream sequence. According to the current nomenclature, this new splice variant was named VEGF111, as the sequence theoretically encodes a 111 amino acid–long human VEGF molecule after excision of the signal peptide. It is noteworthy that the total VEGF mRNA level, which can be quantitatively evaluated thanks to the synthetic RNA used as internal standard in each RT-PCR reaction tube, is only barely modified in the UV-treated cells. VEGF111 is formed mostly at the expense of the VEGF165 variant.
Table I. The amplified RNA, primer numbers, sequences, and length in base pairs of RT-PCR products.
| Primer | mRNA | Primers | Length | Std |
|---|---|---|---|---|
| bp | bp | |||
| P1 (forward) | VEGF | CCTGGTGGACATCTTCCAGGAGTA | 530, 479, 407, | 311 |
| P2 (reverse) | CTCACCGCCTCGGCTTGTCACA | 347, 275, 245a | ||
| P3 (forward) | VEGF (full length) | CACACGCGGCCGCCGAAACCATGAACTTTCTGCTGTC | 586, 454, 424b | NA |
| P4 (reverse) | ACACAGCTAGCTCACCGCCTCGGCTTGTCACA | |||
| P1 (forward) | VEGF111 specific | CCTGGTGGACATCTTCCAGGAGTA | 237 | NA |
| P5 (reverse) | CTCGGCTTGTCACATCTGCATTCA | |||
| P6 (forward) | 28S rRNA | GTTCACCCACTAATAGGGAACGTG | 212 | 269 |
| P7 (reverse) | GATTCTGACTTAGAGGCGTTCAGT | |||
| P8 (forward) | Bcl-X | ACCCCCAGTGCCATCAATGGCAA | 512 (Bcl-Xl) | NA |
| P9 (reverse) | CCCGTCAGGAACCAGCGGTTGA | 324 (Bcl-Xs) | ||
| P10 (forward) | CD31 | CAAGGCGATTGTAGCCACCTCCA | 206 | NA |
| P11 (reverse) | CCAACAACTCCCCTTGGTCCAGA |
Std, length in base pairs of amplification products of the synthetic standard RNA. NA, not available.
For humanVEGF206, 189, 165, 145, 121, and 111, respectively.
For human VEGF165, 121, and 111, respectively.
Figure 1.

Genotoxic agents induce the expression of VEGF111. The indicated cell lines were treated with 30 mJ/cm2 UV-B, 1 μM camptothecin, 5 mM l-mimosin, or 100 μg/ml mitomycin C for 24 h except otherwise indicated, and VEGF mRNA, VEGF111 mRNA, and 28S rRNA were measured by RT-PCR. (A) PAGE analysis of RT-PCR products of VEGF mRNA (top), VEGF111 mRNA (middle), and 28S rRNA (bottom). Arrows in the top panel indicate the VEGF111 amplification product. The drawings represent the exons (not to scale) encoding the corresponding VEGF isoforms. C, control cells; CPT, camptothecin treatment; M, 50-bp molecular weight markers; Mim, mimosin; MitC, mitomycin C; R, cells rinsed with red phenol–free medium but not irradiated; UV, UV-B irradiation; −, no cellular RNA. Asterisks indicate the RT-PCR products of synthetic RNA added to the test tubes to monitor reaction efficiency. (B–E) Dose-response analysis (B and D) and kinetics of induction (C and E) of the VEGF111 mRNA level in MCF-7 cells irradiated by UV-B (B and C) or treated with camptothecin (D and E). VEGF111 mRNA is given in percentages of the total VEGF isoform mRNA. Error bars represent SD.
As the junction between exons 4 and 8 is specific to VEGF111 mRNA, this isoform was specifically RT-PCR amplified by using a reverse primer (P5) sitting astride these exons, primer P1 being used as forward primer. A product of the expected size (237 bp) was detected using RNA from UV-B– irradiated cells but not from control cells (Fig. 1 A, middle left).
A dose-response analysis of the expression of VEGF111 mRNA upon treatment with UV-B up to 30 mJ/cm2 was performed in MCF-7 cells. Data indicated that the expression of VEGF111 progressively increased with the energy of the irradiation (Fig. 1 B). The level of VEGF111 mRNA reached ∼25% of the overall VEGF mRNA in the illustrated experiment but may vary between 25 and 40% from experiment to experiment. An energy of 30 mJ/cm2 was chosen in all subsequent experiments. A time-course relationship was also established in MCF-7 cells. VEGF111 mRNA was readily detected 12 h after irradiation, peaked at 24 h, and decreased thereafter (Fig. 1 C). A cDNA corresponding to the VEGF111 amplification product was induced upon UV irradiation, although to various levels, in several human and animal cells lines and in primary human keratinocytes and fibroblasts 24 h after treatment (Table II).
Table II. Induction of VEGF111 and Bcl-Xs/Bcl-Xl ratio by UV-B.
| Cells | VEGF111 | Bcl-Xs/Bcl-Xl |
|---|---|---|
| % | ||
| MCF-7 | 33 | 3.1 |
| HuH7 | 26 | ND |
| A2058 | 20 | 1.4 |
| MO59K | 16 | 1.4 |
| HCT116 | 15 | 1.8 |
| HT1080 | 14 | 0.7 |
| BT549 | 12 | 0.9 |
| AT5BIVA | 12 | ND |
| CHO | 11 | 2.3 |
| ES CGR8 | 8 | ND |
| HUVEC | 8 | ND |
| PAM212 | 8 | 1.4 |
| MEF | 7 | ND |
| HS578T | 7 | 3.9 |
| HaCat | 6 | 1.7 |
| HSF2 | 5 | ND |
| HSF1 | 4 | 4.0 |
| Human keratinocytes |
1–4 | ND |
| HEK293 | 3 | 0.6 |
| AT5BI | 2 | 3.3 |
| HeLa | 2 | 2.5 |
| MDA-MB-231 | 2 | 1.3 |
| COS-1 | 0 | 2.1 |
| NIH3T3 | 0 | 0.1 |
| CT-26 | 0 | ND |
| B16F10 | 0 | ND |
Percentage of VEGF111 (VEGF110 in rodent cell lines) mRNA over total VEGF mRNA and fold induction of the Bcl-X/Bcl-Xl ratio after UV-B treatment.
As UV-B is known to display genotoxic effects, the expression of VEGF111 was similarly investigated upon treatment with genotoxic agents inducing double-strand breaks, namely camptothecin, mimosin, and mitomycin C. 1 μM camptothecin, a DNA topoisomerase I poison, induced the expression of VEGF111 in HaCat and MCF-7 cells within 24 h to a level similar or even higher than that observed after UV irradiation (Fig. 1 A). A dose-response analysis indicated that VEGF111 induction progressively increased with the concentration of camptothecin in MCF-7 cells (Fig. 1 D). Time-course analysis showed that VEGF111 mRNA expression was already detected after 6 h and reached a plateau after 24 h (Fig. 1 E). MCF-7 cells treated with camptothecin for 24 h were washed and further incubated in fresh medium for up to 5 d. VEGF111 mRNA decreased rapidly after removal of the camptothecin, becoming undetectable after 24 h (unpublished data). 5 mM l-mimosin and 100 μg/ml mitomycin C, two other genotoxic agents, also induced the expression of VEGF111 mRNA (Fig. 1 A, top right). A similar induction of VEGF111 was observed in MCF-7 cells treated by UV or camptothecin in the absence of FCS in the medium. γ-Ray irradiation of some of these cell lines did not induce VEGF111 mRNA.
Mediums were conditioned for 24 h by MCF-7 cells either UV irradiated or treated with camptothecin for 6 h and were further washed to eliminate the compound. Naive MCF-7 cells incubated for 24 h with these conditioned mediums (CMs) did not express VEGF111 mRNA, suggesting that the induction of VEGF111 was not dependent on a paracrine mechanism (unpublished data).
Expression of VEGF111 in vivo
VEGF111 (or VEGF110, the equivalent of human VEGF111 in rodents) mRNA was measured by RT-PCR in a series of human and mice tissues as well as 6–18-d-old in toto mice embryos. No VEGF111/110 was detected in any of these tissues, whereas the traditional isoforms were easily detected, although at variable levels. Trials to induce VEGF110 in healthy BalbC or nude mice upon UV-B, γ-ray irradiation, or i.v. injection of the genotoxic agent camptothecin or its water-soluble analogue irinotecan were not successful.
Nude mice supplemented with oestrogens received a subcutaneous injection of MCF-7 cells (4 × 106 cells) mixed with 100 μl matrigel on both flanks as previously described (Sounni et al., 2002). After 3 wk, tumors had developed, and mice were treated with a daily intratumoral injection of 50 μl camptothecin (2 mg/ml) in one flank or the vehicle alone in the other flank for 1, 2, or 3 d and killed the day after the last injection. VEGF111 mRNA was detected in tumors injected with camptothecin but not or to a barely detectable level in the tumors injected with the vehicle in the controlateral flank (Fig. 2). No VEGF111 mRNA was detected in the tumors of noninjected mice while the other isoforms of VEGF were present.
Figure 2.
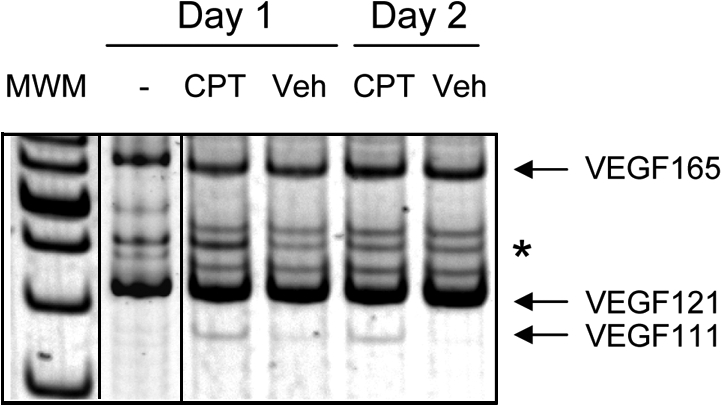
Expression of VEGF111 in MCF-7 tumors treated with camptothecin. Estrogen-supplemented nude mice received a subcutaneous injection of MCF-7 cells (4 × 106 cells) mixed with 100 μl matrigel in both flanks. After 3 wk, they received a daily intratumoral injection of 50 μl camptothecin (2 mg/ml) or the vehicle alone for 1 and 2 d. Tumors were collected 24 h after the last injection, and the VEGF mRNA expression was measured by RT-PCR. MWM, molecular weight marker; CPT, camptothecin treatment. The asterisk indicates the RT-PCR products of synthetic RNA added to the test tubes to monitor reaction efficiency.
Recombinant VEGF111 is glycosylated
Recombinant VEGF (reVEGF) 165, 121, and 111 were produced in serum-free medium of transfected HEK293 cells and thereafter were called reVEGFs (produced in eukaryotic cells). These cells were selected for their low intrinsic expression of VEGF. When evaluated in parallel cultures, the production of reVEGF by transfected HEK293 was always similar for VEGF121 and 165 and threefold lower for VEGF111. CM containing 20 ng reVEGF, as measured by ELISA, were analyzed by Western blotting in reducing conditions with or without treatment with PNGase F (Fig. 3 A). In the absence of enzymatic treatment, VEGF appeared as a single band at ∼18 kD in the CM of HEK293 expressing reVEGF111 and as a doublet at ∼17 and 21 kD and at 22 and 26 kD in the CM of HEK293 expressing reVEGF121 and reVEGF165, respectively. No signal was detected in CM from cells transfected with the empty plasmid (unpublished data). As VEGF contains one glycosylation site, the bands of lower mobility likely correspond to glycosylated forms of reVEGF121 and 165. They are indeed shifted to faster migrating forms at the expected size after deglycosylation with PNGase F. The deglycosylated reVEGF165 produced in HEK293 cells displayed a mobility identical to commercial reVEGF165 produced in bacteria. The reVEGF111 expressed as an 18-kD band was processed to the expected lower size (∼14 kD) upon PNGase F treatment, suggesting that the majority of reVEGF111 molecules are glycosylated in HEK293 cells.
Figure 3.

VEGF111 is biologically active in vitro. (A) Expression of reVEGF in HEK293 cells and deglycosylation. HEK293 cells were transformed with vectors enabling the expression of VEGF111, 121, and 165. CM containing 20 ng VEGF as measured by ELISA was analyzed by Western blotting before and after treatment with N-glycosidase F (PNGase). (B and C) Serum-starved overnight HUVECs, PAEC, PAEC/R1, or PAEC/R2 (expressing VEGF-R1 or -R2) were treated with CM of HEK293 cells expressing VEGF111, 121, or 165 or with commercially available reVEGF165 (cVEGF) produced in bacteria (10 ng/ml each) or with 10% FCS for 5 min. Untreated cells (−) or cells treated with CM of control HEK293 cells (CM) served as controls. Total and phosphorylated VEGF-R2 (B) and ERK1/2 (C) were measured by Western blotting. (D) HUVECs labeled with Fluo3-AM were treated with CM of HEK293 cells expressing 10 ng/ml of VEGF111, 121, or 165 or CM from control HEK293 cells (CM). The percentage of cells responding by a 20% increase in intracellular free calcium concentration was recorded. (E) HUVECs were treated with HEK293 CM containing 10 ng/ml VEGF111 (black circles), 121 (open triangles), or 165 (black triangles) or CM from control HEK293 cells (open circles). DNA was measured in triplicate wells harvested as a function of time. (F) A modified Boyden chamber assay was used to measure the effect of VEGF111 (black circles), 121 (open triangles), 165 (open squares), and control medium (dotted line) on the chemotactic migration of PAEC/R2 cells. Error bars represent SD.
reVEGF111 is biologically active in vitro
Biological effects of reVEGF111 were evaluated in vitro on endothelial cells, human umbilical vascular endothelial cells (HUVECs), and porcine aortic endothelial cells (PAECs) expressing VEGF-R1 (PAEC/R1) or VEGF-R2 (PAEC/R2) or transformed with empty vector (PAEC; Waltenberger et al., 1994). reVEGF121 and 165 were used as positive controls, and the CM of HEK293 cells transfected with the empty vector was used as a negative control. HUVEC, PAEC, PAEC/R1, or PAEC/R2 starved overnight were treated for 5 min with each of the three reVEGF isoforms and the control CM. reVEGF111 was able to induce VEGF-R2 phosphorylation in HUVECs and PAEC/R2 at a level similar to that induced by commercially available VEGF165, reVEGF165, and reVEGF121 (Fig. 3 B). Control medium had no effect. In contrast, we did not observe any phosphorylation of the VEGF-R1 in HUVECs or PAEC/R1. The phosphorylation of extracellularly regulated kinase 1/2 (ERK1/2) was induced in HUVECs, PAEC/R1, and PAEC/R2 but not in PAEC upon treatment with reVEGF111, similar to reVEGF121 or 165 (Fig. 3 C). Control CM had no effect on the ERK1/2 phosphorylation status. Together, these data indicate that VEGF111 signaling is mediated through VEGF-R1 and -R2.
The induction of intracellular calcium transients by reVEGF111 was investigated in HUVECs by single-cell real-time fluorescence microscopy. Although control medium induced calcium transients in only 15% of the cells, reVEGF111 induced calcium transients in 70% of them (i.e., to a level similar to that observed with reVEGF165 and 121; Fig. 3 D). reVEGF111 stimulated HUVEC proliferation to the same extent as reVEGF121 (twofold) and slightly less than reVEGF165 (2.5-fold; Fig. 3 E). Control CM induced a significantly lower cell proliferation rate.
PAEC/R2 cells and monocytes have been previously shown to migrate in response to a chemotactic gradient of VEGF165 (Waltenberger et al., 1994; Stadler et al., 2007). We tested VEGF111 for its ability to promote the migration of PAEC/R2 cells by using a modified Boyden chamber assay (Fig. 3 F). The strongest migratory response to VEGF111 was observed at concentrations ranging between 1 and 20 ng/ml with a peak at 10 ng/ml (20.8 ± 2.3 cells/high-power field; P = 0.006 vs. unstimulated migration). The peak migration of PAEC/R2 cells to VEGF111 was slightly lower than the peak migratory response to VEGF165 (28.7 ± 2.7 cell/high-power field peak migration; P = 0.09). In contrast, VEGF111-induced migration reached higher levels than that induced by VEGF121 (15.5 ± 1.7 cells/high-power field; P = 0.13). As tested by checkerboard analysis, the migration induced by VEGF111 occurred in the presence of a positive concentration gradient between the two compartments, indicating a true chemotactic activity without appreciable unspecific chemokinetic activity. It is well established that monocytes isolated from peripheral blood migrate in response to the activation of VEGF-R1, whereas the expression of VEGF-R2 is absent or below the detection limit (unpublished data). In our hands, VEGF111 showed no consistent effect on monocyte migration (unpublished data). These observations strongly suggest that VEGF111 mediates its chemotactic signal via VEGF-R2 rather than VEGF-R1.
VEGF111 induces an angiogenic phenotype in embryoid bodies
The proangiogenic activity of VEGF111 was tested in an embryoid bodies assay. Mouse embyonic stem cells were cultured for 6 d in the presence of the three reVEGF isoforms or the control medium. Embryoid bodies were fixed at day 6 and labeled by anti-CD31 antibodies to visualize vascular structures. The density of the capillary-like structures was increased in the samples treated with the three reVEGF isoforms as compared with the control (Fig. 4 A) and evaluated by a visual score (Fig. 4 B). reVEGF111 significantly increased the CD31 mRNA at day 4 (Fig. 4 C) to a level similar to that induced by reVEGF165 and 121.
Figure 4.

VEGF111 induces angiogenesis in embryoid bodies. Embryoid bodies were formed in the presence of CM from HEK293 cells expressing VEGF111, 121, or 165 or CM from control HEK293 cells (CM). (A) Representative microphotograph of embryoid bodies after immunofluorescent labeling of CD31. (B) Several microphotographs of six embryoid bodies from three independent experiments for each treatment were taken at random and analyzed by five investigators in double blind, and a score (from 0 to 3, where 0 indicates lack of vascular labeling and 3 indicates maximum labeling) was given to each sample. Statistical analysis was performed using a Chi square test. ***, P < 0.0001. (C) CD31 mRNA level was measured in embryoid bodies by RT-PCR. Statistical analysis was performed using a t test. *, P < 0.05; **, P < 0.01. (D and E) Embryoid bodies were differentiated in the presence of reVEGF111, 121, or 165. The angle between the plane of division of endothelial cells and the long axis of the vascular structures was analyzed after labeling of the vessels with anti-CD31 (green) and the mitotic cells with antiphosphohistone H3 (red) as illustrated by the cell in D for two sister cells having divided with a 90° angle. The angle of division plane was recorded for 43–63 cells in each group of embryoid bodies (n = 11). A scoring of the distribution of the angles is shown in E. Error bars represent SD.
To gain further insights into the mechanism of angiogenesis, the effect of the various reVEGF on the pattern of endothelial cell divisions in embryoid bodies was undertaken according to the procedure described by Zeng et al. (2007). Embryoid bodies were prepared and supplemented with 5 ng/ml reVEGF111, 121, or 165. At day 8, they were fixed and immunostained with anti-CD31 and antiphosphohistone H3. The angle between a line drawn along the long axis of the vessel and the division plane of mitotic cells was calculated for 43–63 cells in 11 embryoid bodies for each reVEGF. The majority of the division planes was oriented within 75–90° (i.e., close to perpendicular to the long axis of the vessel; Fig. 4, D and E).
VEGF111 induces vascularization in vivo
Nude mice (six per group) were injected with matrigel containing empty plasmid–transfected (control) HEK293 in one flank and HEK293 expressing VEGF111, 121, or 165 in the controlateral flank. In addition, three mice were injected with control cells in both flanks. Tumors were observed at the site of injection in nearly all mice: 21/21 in control HEK293, six/six in HEK293/111, three/six in HEK293/121, and six/six in HEK293/165. High levels of each VEGF mRNA splice variant were found in their respective tumors but not in control tumors (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200703052/DC1). No VEGF121 mRNA was found in the remnant plugs of matrigel in the three unresponsive mice. Circulating human VEGF, which was measured by ELISA, was detected in the six mice injected with HEK293/111 (386 ± 122 pg/ml), in the three responsive mice injected with HEK293/121 (411 ± 131 pg/ml), and in the six mice injected with HEK293/165 (436 ± 279 pg/ml). None was found in mice injected with control HEK293 in both flanks.
The tumors displayed different macroscopic aspects according to the VEGF isoform expressed by HEK293 cells. The tumor mass and its peritumoral vascularization visualized through the skin are delineated by dotted lines in Fig. 5 A. In the control and HEK293/121, the tumors remained restricted to the site of injection, appearing as a white mass in the control and bluish in the 121 tumors. In mice injected with HEK293/111 and HEK293/165, the tumors appeared largely surrounded by a highly vascularized zone as judged from the bluish color of the skin. The cyanotic skin surface was measured individually, and the mean was 133 ± 57 mm2 for the VEGF121 tumors, 258 ± 115 mm2 for the VEGF165 tumors, and significantly larger, 397 ± 102 mm2, for the VEGF111 tumors. After dissection (Fig. 5 B), the control tumors and its surrounding tissues were indeed poorly vascularized. A dense network of vessels had developed in the peritumoral tissue in HEK293/111 mice (six/six), whereas the tumor themselves were poorly vascularized. In contrast, the 165 tumors (six/six mice) and, to a lesser extent, the 121 tumors (three/three mice) where highly vascularized, as shown by their reddish appearance, whereas the peritumoral tissue was poorly vascularized.
Figure 5.

VEGF111 induces angiogenesis in vivo. Mice were injected with a mixture of matrigel and HEK293 cells expressing reVEGF111, 121, or 165 or were transfected with empty vector (Cont) and killed after 3 wk. (A) Photographs of one representative mouse of each group before dissection. Dotted lines delimit the surface of the tumor and the peritumoral vascular network visible through the skin. (B) Photographs of one representative mouse of each group after dissection showing the tumor and the peritumoral tissue. Arrows indicate the lateral thoracic veins. (C) Histopathological analysis of the tumor and peritumoral tissues. Sections were stained with anti-αSMA antibodies and hematoxylin-eosin. (D) Expression of CD31 mRNA in the tumors: the mRNA was measured by RT-PCR in control HEK295 tumors (n = 21) and tumors grown from HEK293/111 (n = 6), HEK293/121 (n = 3), and HEK293/165 (n = 6). The data were corrected by the signals obtained for the 28S rRNA. (E) Number of vessels per millimeter squared of peritumoral tissue. The vessels were counted on paraffin sections of control mice (20 sections) and 111 (six sections), 121 (three sections), and 165 (six sections) mice. (F) Surface of the αSMA-positive vessels in the peritumoral tissue. Dots represent individual values, and horizontal bars indicate the mean value. *, P < 0.01; **, P < 0.0001 (versus control tumor). Statistical analysis was performed using the t test. Error bars represent SD.
These observations were confirmed by histological analyses (Fig. 5 C). Sections through the tumor and the peritumoral tissue were stained for α smooth muscle actin (αSMA). The control and the VEGF111 and 121 tumors contained a moderate number of vessels delineated by αSMA-positive cells, whereas the VEGF165 tumors showed large kystic ectasia containing red blood cells and tumoral cells. The peritumoral tissue was poorly vascularized in control HEK293 tumors, whereas it contained a very large number of vessels in VEGF111 tumors. Fewer but larger size vessels were observed in VEGF121 and 165 peritumoral tissue. This observation was confirmed by quantifying the number and size of αSMA-positive vessels per millimeter2 of peritumoral tissue. The vessel density within the tissue surrounding the tumor (Fig. 5 E) was actually significantly higher in VEGF111 mice (P < 0.01) than in control, VEGF121, or VEGF165 mice that contained a similar low number of vessels. The mean size of these peritumoral vessels (Fig. 5 F) was significantly higher in VEGF121 and VEGF165 mice (2,042 ± 2,537 μm2 and 4,505 ± 9,060 μm2, respectively), whereas it was similar in control and VEGF111 mice (740 ± 1,073 μm2 and 1,042 ± 1,710 μm2, respectively). The level of CD31 mRNA in the tumors expressing each isoform of VEGF was significantly higher than in the control tumor (Fig. 5 D).
The lateral thoracic vein and afferent vessels draining the tumor were significantly enlarged in mice bearing tumors expressing the three VEGF isoforms as compared with control tumors. Moreover, this enlargement was significantly higher in mice bearing VEGF111 tumors than the two other isoforms (Fig. 5 B and Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200703052/DC1).
As an element of interpretation of the in vivo data, clearance of the various reVEGF isoforms was determined after an i.v. injection. The half-life was 74 min for VEGF111, 73 min for VEGF121, and 55 min for VEGF165 (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200703052/DC1). Altogether, these results demonstrate that the VEGF111 isoform displays potent angiogenic activity both in vitro and in vivo.
reVEGF111 is resistant to proteolytic degradation and remains biologically active
The main site of cleavage of VEGF by plasmin has been identified as Arg110-Ala111 (Keyt et al., 1996) in the sequence encoded by exon 5, suggesting that VEGF111 should be resistant to plasmin. Upon incubation of the reVEGF isoforms with purified plasmin at increasing concentrations for 4 h, both reVEGF121 and 165 were progressively degraded in fragments with an apparent molecular size of 14–18 kD under reducing conditions, which is consistent with the expected size of glycosylated and nonglycosylated monomers of VEGF110. In contrast, reVEGF111 was completely resistant to plasmin (Fig. 6 A). Similar results were obtained after incubation with fluids collected from human nonhealing wounds known to be rich in proteases, including plasmin. reVEGF121 and 165 were almost completely degraded already after a 4-h incubation, whereas reVEGF111 remained intact even after 24 h (Fig. 6 B). HUVECs were incubated with control CM and CM containing each of the three VEGF isoforms, pretreated or not pretreated with plasmin or fluids from chronic ulcer. As expected, [3H]thymidine incorporation was stimulated in cells treated with reVEGF isoforms (Fig. 6 C). Pretreatment of reVEGF165 and 121 with plasmin (0.32 U) induced a slight but significant decrease in proliferative activity (Fig. 6 C), whereas pretreatment with wound fluid completely abolished it (Fig. 6 D). In contrast, neither plasmin nor chronic wound fluid affected [3H]thymidine incorporation induced by reVEGF111.
Figure 6.

VEGF111 is resistant to degradation by plasmin and proteases from nonhealing chronic wound fluid. (A and B) CM from HEK293 containing 20 ng reVEGF165, 121, and 111 were treated at 37°C with the indicated concentrations of plasmin for 4 h (A) or 10 μl of fluid collected from a chronic wound for the indicated times (B). The products were analyzed by Western blotting. (C and D) Cultures of HUVECs were treated with HEK293 cell CM containing 2 ng/ml VEGF111, 121, or 165, CM from control HEK293 (CM), or 2 ng/ml of commercial reVEGF165 (cVEGF) produced in bacteria either untreated or pretreated with 0.32 U plasmin for 4 h (C) or fluids collected from a chronic ulcer for 24 h (D). After 48 h, the cells were incubated with [3H]thymidine for 18 h, and TCA-precipitable radioactivity was measured. Statistics were performed using the analysis of variance test followed by the Tukey-Kramer test. *, P < 0.05; **, P < 0.001. Error bars represent SD.
VEGF111 is not induced by hypoxia, hypoglycemia, apoptosis, or reactive oxygen species but seems to be related to DNA damage
Hypoxia, a recognized mechanism able to stimulate VEGF expression (Stein et al., 1995), was induced by culturing MCF-7 cells under 1% O2 or mimicked by treatment with cobalt ions. Although both treatments induced an overall VEGF mRNA overexpression, about twofold in agreement with published observations (Maity et al., 2000), they failed to induce a detectable level of VEGF111 (unpublished data). Hypoglycemia, which is also known to increase VEGF expression, was induced by culturing MCF-7 cells in glucose-free DME compared with DME supplemented with 4.5 g/liter glucose. Again, VEGF111 mRNA was not detected, whereas the overall VEGF expression was stimulated by twofold.
Beside genotoxicity, UV-B and camptothecin induce nongenomic effects as the production of reactive oxygen species (ROS) and apoptosis (Hattori-Nakakuki et al., 1994). To decipher whether genomic effect, cell death, or oxidative stress triggers the expression of VEGF111 mRNA, MCF-7 cells were treated by puromycin and H2O2 as inducers of apoptosis and ROS, respectively. The Bcl-Xs/Bcl-Xl mRNA ratio monitored in parallel progressively increased with H2O2 concentrations to reach a concentration of 250 μM, a level similar to that observed after treatment with UV-B (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200703052/DC1). However the level of VEGF111 mRNA remained 20–30-fold lower in H2O2-treated cells than in the UV-B–treated cells, suggesting that apoptosis and ROS do not trigger the splicing mechanism leading to VEGF111 expression. Similarly, puromycin barely induced VEGF111 expression while increasing the Bcl-Xs/Bcl-Xl mRNA ratio to a level similar to that observed in UV-B. Cells treated with ROS inducers FeSO4 (10 μM) or Na2Fe(CN)5NO (80 μM) also failed to express VEGF111 (unpublished data). Together, these data strongly suggest that induction of VEGF111 expression by genotoxic agents is not related to apoptosis or to oxidative stress. They also indicate that the regulation of pre-mRNA splicing that results in the expression of VEGF111 and Bcl-Xs are different processes. Accordingly, the Bcl-Xs/Bcl-Xl mRNA ratios measured in the various cell types after UV irradiation did not correlate with the expression of VEGF111 (Table II).
Preliminary data suggest that VEGF111 expression could depend on ataxia telangiectasia mutated/ATR (ataxia telangiectasia and Rad3 related) and p53 known to be activated by genotoxic agents. Further work is progressing to decipher the signaling mechanisms involved in this alternative splicing.
Discussion
VEGF111 is a new splice variant of VEGF-A lacking exons 5–7. Its expression is induced in several types of cultured cells by genotoxic agents such as UV-B, camptothecin, l-mimosin, and mitomycin C. UV-B triggers the formation of pyrimidine dimers in DNA, whereas camptothecin, l-mimosin, and mitomycin C induce double-strand breaks. Surprisingly, γ irradiation, which also leads to double-strand breaks, does not induce VEGF111 expression.
Relationship between structure and biological properties of VEGF111
The VEGF mRNA variants described up to now contain exons 1–5 and a combination of exons 6a, 6b, 7, and 8 (Robinson and Stringer, 2001; Bates et al., 2002). The VEGF111 encoded by exons 1–4 and 8 contain the sequences responsible for binding to VEGF-R1 and VEGF-R2, which were located in exons 3 and 4, respectively, whereas binding sites for NRP-1, heparin, and ECM components encoded by exons 6 and 7 are lacking. Lack of the ECM-binding domain of the protein confers to VEGF111 a larger bioavailability, which might explain some of our findings in the in vivo experiments that are discussed later. No specific function has been ascribed for the short amino acid sequence encoded by exon 8. However, its role may be underestimated because VEGF165b containing an alternative exon 8 has antiangiogenic properties (Bates et al., 2002). It has also been suggested that exon 8 stabilizes VEGF binding to NRP-1 (von Wronski et al., 2006).
Although recombinant human VEGF121 and 165 produced in HEK293 cells were only partly glycosylated, VEGF111 was fully glycosylated, as shown by the difference in the electrophoretic pattern before and after treatment with PNGase. This suggests that Asn74 is more accessible to the glycosylation machinery in VEGF111. Although VEGF glycosylation appears to have no major effect on its biological activity, it seems required for efficient secretion (Peretz et al., 1992; Claffey et al., 1995). The new splice variant VEGF111 is indeed efficiently secreted and is as active as the other known VEGF isoforms.
Houck et al. (1991) reported that VEGF165 and 121, the shortest previously described isoform encoded by exons 1–5 and 8, as well as VEGF110, a proteolytic fragment obtained after partial digestion by a plasmin-containing sequence encoded by exons 1–4 and a partial sequence of exon 5, have similar endothelial cell mitogenic activity. In agreement with these observations, we found that the mitogenic activity of VEGF111 on endothelial cells was similar to that of VEGF121 and slightly less efficient than VEGF165. The lack of sequences encoded by exons 5–7 in VEGF111 and exons 6–7 in VEGF121 might affect the positioning of the receptor-binding interfaces in the dimeric molecules. Moreover, the absence of the NRP-1–binding site in these two isoforms could also be responsible for their slightly reduced mitogenic activity (Soker et al., 1997). The phosphorylation level of the VEGF-R2 induced by VEGF111 was found to be similar to that induced by VEGF121 and slightly lower than the activation produced by VEGF165.
The main sites of cleavage of VEGF by plasmin and matrix metalloproteinases were found in the sequence encoded by exon 5 (Claffey et al., 1995; Keyt et al., 1996; Lee et al., 2005). As expected on the basis of the lack of exon 5, VEGF111 is not sensitive to degradation by plasmin in conditions that cleave VEGF121 and165 isoforms primarily between residues 110 and 111, producing VEGF110 homodimers. Accordingly, the mitogenic potential of VEGF111 is unaffected by plasmin treatment, whereas that of VEGF121 and 165 is reduced. Fluid collected from a nonhealing chronic wound was used as a source of proteolytic enzymes found in cutaneous impaired healing conditions. Incubation of VEGF165 and 121 results in their complete proteolysis and a total loss of their mitogenic activity, whereas VEGF111 is completely resistant to degradation and retains its full biological properties. A similar degradation of VEGF165 by fluids from chronic ulcers was reported by Lauer et al. (2000). It was prevented by mutating the Arg110 and Ala111 plasmin cleavage site (Lauer et al., 2002), further suggesting that the cleavage of VEGF within exon 5 unmasks proteolytic sites that are cryptic otherwise.
VEGF111, phosphorylated VEGF-R2, and ERK1/2 induced calcium transient and stimulated HUVEC proliferation similarly to the 121 and 165 isoforms. Although we have no direct evidence of the binding of VEGF111 to VEGF-R1, two arguments are in favor of VEGF111 signaling via this receptor. The first is the induction of ERK1/2 phosphorylation by VEGF111 in the PAEC/R1 cells similarly to the VEGF121 and 165 isoforms. The second comes from the results of the oriented endothelial cell division in the embryoid bodies assay. According to Zeng et al. (2007), the preferential orientation perpendicular to the long axis of the vessel, as we observed in embryoid bodies differentiated by VEGF111, 121, and 165, is determined by Flt-1 because embyonic stem Flt-1−/− loses this behavior. A controversial argument is the lack of induction by VEGF111 of monocyte chemotactic migration. Further work is needed to clarify the issue.
Although VEGF111 displays biological activities in vitro close to those of VEGF121 and 165, the three isoforms elicited a different pattern of the neovasculature in and around the tumors produced by the injection of transfected HEK293 with their respective cDNA. VEGF111-expressing tumors were macroscopically pale and exhibited moderate intratumoral vascularization in high contrast with VEGF165-expressing tumors that were highly vascularized and contained large blood lacunae. No or little inflammatory infiltrate was observed in any type of tumor. A most striking feature was the presence in the peritumoral tissue of an extensively developed network of small size vessels around VEGF111-expressing tumors, whereas the vessels in the peritumoral area of VEGF121 and 165 tumors were less numerous but of larger size. These findings are in agreement with those of Lee et al. (2005), who reported that tissues adjacent to xenograft tumors expressing a protease-resistant mouse VEGF (VEGFΔ108–118, corresponding to VEGF164 but lacking most of the sequence encoded by exon 5) had smaller vessels at higher density than tumors expressing wild-type VEGF164. The development of a high density vascular network in the adjacent tissue of VEGF111-expressing tumors may be correlated with its reduced potential for ECM binding. However, the diffusible VEGF121 that shares a similar feature and has a similar half-life did not induce the same vascular organization. The widespread peritumoral vascular network in VEGF111 mice may also contribute to enlargement of the draining thoracic vein.
As the HEK293 cells transfected with the cDNA of either isoform proliferate and migrate at the same rate, at least in vitro, and the mRNA level of each isoform is similar in the three types of tumors (Fig. S1), the differences observed in the tumoral and peritumoral vascular network are likely linked to the diffusible properties and/or possibly to the resistance to proteolysis of the various isoforms. The mechanisms that regulate these different patterns require additional investigations.
VEGF111, a promising therapeutic agent?
VEGF111 mRNA is absent from all healthy human or murine tissues that we tested. We also failed to detect it in mouse skin after UV-B irradiation. This is not surprising, as its expression in UV-B–treated human skin fibroblasts and keratinocytes in vitro is very low. In contrast, it was induced in MCF-7 tumors upon treatment with camptothecin. The potential induction of VEGF111 by chemotherapy might be an adverse side effect that could contribute to the acquisition of drug resistance. The finding that VEGF111 expression by xenografts of HEK293 cells surpasses VEGF121 and 165 in inducing the formation of a large array of vessels around the tumor supports this hypothesis, which needs further experimental and clinical investigations to be firmly established.
Defective vascularization and ischemia is a significant medical problem in many disciplines, including cardiovascular diseases and wound healing. VEGF-A plays a pivotal role in the angiogenic process during tissue repair. In chronic wounds, although increased VEGF mRNA was detected in the epidermis (Brown et al., 1992), the proteolytic environment and mainly serine proteases such as plasmin lead to VEGF degradation (Lauer et al., 2000; and unpublished data) and impaired vascularization. Mutation of the plasmin cleavage site Arg110/Ala111 in VEGF165 (Lauer et al., 2002) generated a proteolysis-resistant variant that induced an improved healing response in the db/db mouse (Roth et al., 2006). The potent angiogenic properties of VEGF111 and its remarkable resistance to proteolysis make it an interesting alternative candidate for therapeutic use in ischemic diseases. This is presently under investigation.
Materials and methods
Cell culture
Cells were cultured in DME containing 10% FCS (Cambrex), 2 mM glutamine, 50 μg/ml ascorbic acid, penicillin, and streptomycin except otherwise indicated. MO59K was cultured in DME/Nut Mix F-12 (Invitrogen) with 10% FCS, 2 mM glutamine, nonessential amino acids (Invitrogen), penicillin, and streptomycin; HEK293 cells were cultured in DME containing 10% FCS, nonessential amino acids, penicillin, streptomycin, 2 mg/liter fungizone, and 40 mg/liter gentamycin. PAECs were cultured in Ham F-12 (Invitrogen) with 10% FCS, gentamycin, penicillin, streptomycin, and fungizone, and HCT116 cells were cultured in MacKoy's medium (Cambrex) containing 10% FCS, penicillin, and streptomycin. Primary human keratinocytes were cultured in KGM-2 (Cambrex), and HUVECs were cultured in MCDB-131 medium (Invitrogen) complemented with 20% FCS, 2 mM glutamine, 5.8 U/ml heparin (Sigma-Aldrich), penicillin, and streptomycin on a coat of 0.2% gelatin. All cultures were kept at 37°C under 5% CO2 except otherwise indicated. Monocytes were isolated from buffy coats obtained from healthy donors (Transfusion Center, Liège, Belgium) as described previously (Stadler et al., 2007).
Chemicals
reVEGF165, plasmin, camptothecin, mimosin, FeSO4, and Na2Fe(CN)5NO were obtained from Sigma-Aldrich. CoCl2 was obtained from Merck, mitomycin C was purchased from Kyowa Hakko Kogyo, hygromycin was obtained from Invitrogen, and irinotecan was purchased from Aventis.
Antibodies, Western blotting, immunohistochemistry, and ELISA
The following antibodies were used: anti-ERK1/2 (rabbit polyclonal), antiphospho-ERK1/2 (monoclonal), and FITC-conjugated anti-αSMA were obtained from Sigma-Aldrich; anti–VEGF-R2 (rabbit polyclonal) and anti-VEGF (rabbit polyclonal) were purchased from Santa Cruz Biotechnology, Inc.; antiphospho–VEGF-R2 (rabbit polyclonal) was purchased from EMD; polyclonal rabbit antiphosphohistone H3 (Ser10) was obtained from Millipore; rat anti–mouse CD31 was purchased from BD Biosciences; sheep anti-FITC antibodies conjugated to HRP were purchased from Roche; secondary antibodies conjugated with biotin, HRP, or streptavidin/FITC were obtained from Dako; and FITC-labeled donkey anti–rat antibodies and rhodamine-labeled donkey anti–rabbit antibodies were purchased from Jackson ImmunoResearch Laboratories.
Proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride transfer membranes (PerkinElmer) by electroblotting. Membranes were blocked by nonfat dry milk (3% in PBS-Tween buffer) and probed with primary antibodies revealed by HRP-conjugated secondary antibodies. Signals were detected by chemiluminescence using an ECL Western Blotting Analysis System (Thermo Fisher Scientific) and x-ray film exposure and were quantified using a Fluor-S MultiImager (Bio-Rad Laboratories).
Tissue samples were fixed in formaldehyde and embedded in paraffin, and sections of 5 μm were successively reacted with anti-αSMA antibodies conjugated with FITC and anti-FITC conjugated with HRP and were stained with DAB, hematoxylin, and eosin. The samples were mounted with Aqua PolyMount (Polysciences Inc.) and visualized at room temperature using an inverted fluorescent microscope (Axiovert 25; Carl Zeiss, Inc.) with a 63× NA 1.25 objective magnification, camera (Axiocam; Carl Zeiss, Inc.), and KS400 acquisition software (Carl Zeiss, Inc.). Human VEGF was measured in the CM of cultured cells or in blood samples by ELISA (R&D Systems) as described by the manufacturer.
RNA purification
Total RNA was purified from cell culture using a High Pure RNA Isolation kit (Roche) and from early mouse embryos (days 6–9) using a High Pure RNA Tissue kit (Roche) after grinding in lysis solution with a Dounce homogenizer. Tissues from older mouse embryos and adult mouse or human tissues were crushed in liquid N2 (Dismembrator; Braun Biotech International), and RNA was collected by cesium chloride floatation (Chirgwin et al., 1979) or by using the High Pure RNA Tissue kit.
RT-PCR amplification
RT-PCR amplifications were performed using the GeneAmp PCR System 2400 or 9800 (PerkinElmer) and GeneAmp Thermostable rTth Reverse Transcriptase RNA PCR kit (PerkinElmer), 10 ng of total RNA, and the different pairs of primers (5 pmol each; see Table I). For amplification of the VEGF isoforms mRNA and 28S ribosomal RNA (rRNA), a known copy number of a synthetic standard RNA was included in each sample to monitor the reaction efficiency (Hajitou et al., 2001; Lambert et al., 2001). Bcl-X and CD31 mRNA were measured using the same conditions as the 28S rRNA. For Bcl-X mRNA amplification, the primers chosen on exons 1 and 3 amplified both Bcl-Xs and Bcl-Xl isoforms. For specific detection of the VEGF111 isoform, mRNA was reverse transcribed using oligodT (Eurogentec) and Superscript II (Invitrogen) as described by the manufacturer. Reverse transcription at 42°C for 50 min was followed by denaturation of the enzyme at 70°C for 15 min. 40 ng VEGF111 cDNA was amplified by PCR, the primers P1 and P5 (Table I), and Taq polymerase (Takara). Conditions for PCR amplification were 94°C for 15 s, 60°C for 20 s, and 72°C for 10 s.
Irradiation with UV-B light and γ rays
15 × 103 cells/cm2 were seeded for 24 h. Culture medium was replaced by a thin layer of phenol red–free DME, and cells were irradiated in open dishes with UV (30 mJ/cm2 except otherwise indicated) using two lamps (TL 20W/12; Philips) in the hood. The UV light spectrum was 90% UV-B and 10% UV-A, whereas no UV-C was detected as measured by a radiometer (UVX; UVP Inc.). After irradiation, the phenol red–free DME was replaced by fresh culture medium. Mice were irradiated by using the same equipment after a short anesthesia. Cells in DME containing 25 mM Hepes, pH 7.25, and mice were γ irradiated in an exactor (Gammacell 40; MDS Analytical Technologies) at 1.15 Gy/min.
Characterization of the VEGF111 splice variant mRNA
VEGF mRNA from UV-irradiated HaCat cells were RT-PCR amplified using the P1 and P2 primers (Table I). The VEGF111-specific product was extracted from the acrylamide gel and sequenced using a Thermo-sequenase radiolabeled terminator cycle sequence kit (GE Healthcare). Full-length VEGF111 cDNA was obtained by RT-PCR amplification using primers P3 and P4 (Table I) and was sequenced.
Production of reVEGF
RNA purified from UV-irradiated HaCat cells was reverse transcribed using SuperScriptII and an oligodT primer. The complete coding sequences of VEGF111, 121, and 165 were amplified with Pwo DNA polymerase (Roche) using P3 (having an NotI restriction site sequence at its 5′ end) and P4 (having an NheI restriction site sequence at its 5′ end). After restriction with NotI–NheI and purification of the cDNA of interest, the PCR products were ligated (Ligation kit version II; Takara) between the NotI and NheI sites of a pCEP4 vector (Invitrogen) containing a home-modified multiple cloning site (Colige et al., 2005). Plasmids were amplified in XL10-Gold ultracompetent bacteria (Stratagene) and prepared using the Plasmid Miniprep kit (Bio-Rad Laboratories). HEK293 cells were transfected by 1–2 μg plasmid using FuGene 6 (Roche), and transformed cells were selected by 100 mg/liter hygromycin for 2–3 wk.
Cell multiplication
15,000 HUVECs were seeded in gelatin-coated multiwells in the presence of MCDB-131 supplemented with 20% FCS. After 3 h, the medium was replaced by fresh medium with or without VEGF and renewed every 2 d. Cells were collected at various times, and the DNA was measured by fluorimetry using a SpectraMax Gemini XS apparatus (MDS Analytical Technologies) after labeling with bis-benzimide. In some experiments, 1 μM [3H]thymidine (2.5 Ci/mol; PerkinElmer) was added to cell cultures at day 2, and the TCA-precipitable radioactivity was measured after 18 h.
Cell migration
Chemotactic migration assays of PAEC/R2 and human monocytes were performed in a modified Boyden chamber assay as previously described (Waltenberger et al., 1994; Stadler et al., 2007).
Intracellular calcium measurement
Measurements of intracellular free calcium transients induced by VEGF111, 121, and 165 in HUVECs were performed as previously described (Mineur et al., 2005).
N-deglycosylation of VEGF
CM of HEK293 cells expressing VEGF165, 121, or 111 or from control cells containing the empty plasmid were treated with PNGase F (N-glycosidase F; New England Biolabs, Inc.) as described by the manufacturer. The electrophoretic pattern of the various VEGF isoforms before and after enzymatic treatment was determined by SDS-PAGE and Western blotting.
In vitro angiogenesis
Embryoid bodies were formed as previously described (Deroanne et al., 2002). In brief, undifferentiated embryonic stem CGR8 cells were aggregated for 4 d in a 20-μl drop of DME supplemented with 10% FCS, 0.1 mM nonessential amino acids, 0.1 mM β-mercaptoethanol, and 5 ng/ml of the various reVEGF isoforms and were further kept in culture on gelatin-coated coverslips for 6 d in the same medium (n = 6 for each isoform). For immunohistochemistry, embryoid bodies were fixed in methanol and incubated with rat anti–mouse CD31 antibodies, biotin-conjugated anti–rat IgG, and streptavidin/FITC. The CD31 mRNA was measured by RT-PCR. A similar procedure was used (n = 11 embryoid bodies for each isoform) to determine the orientation plane of mitotic endothelial cells as described previously (Zeng et al., 2007) by a double immunostaining using anti-CD31 as above, antiphosphohistone H3, and rhodamine-labeled secondary antibodies. Samples were mounted in Aqua PolyMount. Immunostaining was observed at room temperature by inverted confocal microscopy (TSP2; Leica) with an objective magnification of 10× NA 0.3 and confocal acquisition software (Leica) or by an inverted microscope (Axiovert 25; Carl Zeiss, Inc.).
In vivo angiogenesis
HEK293 cells transfected with the empty vector or expressing human reVEGF111, 121, or 165 were mixed (2 × 106 cells) with 200 μl matrigel depleted in growth factors (Becton Dickinson) and injected subcutaneously in the flanks of nude mice (6-wk-old Swiss Nu/Nu). After 3 wk, mice were killed according to the ethical policy of our institute. Blood was collected by heart puncture. Tumors and the surrounding tissues were inspected and dissected. Half of each specimen was used for morphological analysis, and the other half was used for RNA extraction.
Experimental chemotherapy
Nude mice supplemented with oestrogens (SE-121 17 β-Estradiol 1.7 mg/pellet 60-d release; Innovative Research of America) received a subcutaneous injection of 4× 106 MCF-7 cells mixed with 100 μl matrigel on both flanks according to a previously described procedure (Sounni et al., 2002). After 3 wk, mice were treated by a daily dose of 50 mg/Kg irinotecan or 10 mg/kg camptothecin, an intratumoral injection of 50 μl camptothecin (2 mg/ml), or the vehicle alone for 1, 2, and 3 d and were killed 24 h after the last injection. The tumors as well as various organs and tissues were collected. VEGF111 mRNA was detected by RT-PCR.
Online supplemental material
Fig. S1 illustrates the expression of VEGF mRNA in tumors obtained after injection in nude mice of HEK293 cells expressing the various VEGF isoforms. Fig. S2 shows the diameter of the lateral thoracic vein in mice bearing tumors obtained after injection of HEK293 cells expressing the various VEGF isoforms. Fig. S3 gives the blood concentration of the various VEGF isoforms as a function of time after injection in mice. Fig. S4 illustrates the expression of VEGF111 mRNA and the Bcl-Xs/Bcl-Xl ratio in MCF-7 cells treated with UV-B, puromycin, or H2O2. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200703052/DC1.
Supplementary Material
Acknowledgments
HaCat and MO59K cells were gifts from N. Fusenig (German Cancer Research Center, Heidelberg, Germany) and J. Turner (Cross Cancer Institute, Edmonton, Canada), respectively. The technical assistance of M.-J. Nix, G. Rega, A. Hoffman, A. Heyeres, and Fabrice Olivier was greatly appreciated. We thank G. Poncin and M.P. Defresne for their help in cytology analyses.
This work was supported, in part, by a grant from the Centre Anticancéreux of the University of Liège, the Belgian Fonds National de la Recherche Scientifique, the Fonds de la Recherche Scientifique Médicale, and the Foundation Against Cancer.
Abbreviations used in this paper: αSMA, α smooth muscle actin; CM, conditioned medium; ERK1/2, extracellularly regulated kinase 1/2; HUVEC, human umbilical vascular endothelial cell; PAEC, porcine aortic endothelial cell; reVEGF, recombinant VEGF; ROS, reactive oxygen species; rRNA, ribosomal RNA; VEGF-R, VEGF receptor.
References
- Bates, D.O., T.G. Cui, J.M. Doughty, M. Winkler, M. Sugiono, J.D. Shields, D. Peat, D. Gillatt, and S.J. Harper. 2002. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 62:4123–4131. [PubMed] [Google Scholar]
- Blaudschun, R., C. Sunderkotter, P. Brenneisen, R. Hinrichs, T. Peters, L. Schneider, Z. Razi-Wolf, N. Hunzelmann, and K. Scharffetter-Kochanek. 2002. Vascular endothelial growth factor causally contributes to the angiogenic response upon ultraviolet B irradiation in vivo. Br. J. Dermatol. 146:581–587. [DOI] [PubMed] [Google Scholar]
- Brown, L.F., K.T. Yeo, B. Berse, T.K. Yeo, D.R. Senger, H.F. Dvorak, and L. van de Water. 1992. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J. Exp. Med. 176:1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchardt, M., T. Burchardt, A.G. Anastasiadis, R. Buttyan, A. de la Taille, A. Shabsigh, J. Frank, and R. Shabsigh. 2005. Application of angiogenic factors for therapy of erectile dysfunction: protein and DNA transfer of VEGF 165 into the rat penis. Urology. 66:665–670. [DOI] [PubMed] [Google Scholar]
- Caldwell, R.B., M. Bartoli, M.A. Behzadian, A.E. El-Remessy, M. Al-Shabrawey, D.H. Platt, G.I. Liou, and R.W. Caldwell. 2005. Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr. Drug Targets. 6:511–524. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., V. Ferreira, G. Breier, S. Pollefeyt, L. Kieckens, M. Gertsenstein, M. Fahrig, A. Vandenhoeck, K. Harpal, C. Eberhardt, et al. 1996. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 380:435–439. [DOI] [PubMed] [Google Scholar]
- Chirgwin, J.M., A.E. Przybyla, R.J. MacDonald, and W.J. Rutter. 1979. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 18:5294–5299. [DOI] [PubMed] [Google Scholar]
- Claffey, K.P., D.R. Senger, and B.M. Spiegelman. 1995. Structural requirements for dimerization, glycosylation, secretion, and biological function of VPF/VEGF. Biochim. Biophys. Acta. 1246:1–9. [DOI] [PubMed] [Google Scholar]
- Colige, A., F. Ruggiero, I. Vandenberghe, J. Dubail, F. Kesteloot, J. Van Beeumen, A. Beschin, L. Brys, C.M. Lapiere, and B. Nusgens. 2005. Domains and maturation processes that regulate the activity of ADAMTS-2, a metalloproteinase cleaving the aminopropeptide of fibrillar procollagens types I-III and V. J. Biol. Chem. 280:34397–34408. [DOI] [PubMed] [Google Scholar]
- Deroanne, C.F., K. Bonjean, S. Servotte, L. Devy, A. Colige, N. Clausse, S. Blacher, E. Verdin, J.M. Foidart, B.V. Nusgens, and V. Castronovo. 2002. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene. 21:427–436. [DOI] [PubMed] [Google Scholar]
- Dvorak, H.F. 2000. VPF/VEGF and the angiogenic response. Semin. Perinatol. 24:75–78. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. 2005. VEGF as a therapeutic target in cancer. Oncology. 69:11–16. [DOI] [PubMed] [Google Scholar]
- Ferrara, N., K. Carver-Moore, H. Chen, M. Dowd, L. Lu, K.S. O'Shea, L. Powell-Braxton, K.J. Hillan, and M.W. Moore. 1996. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 380:439–442. [DOI] [PubMed] [Google Scholar]
- Hajitou, A., N.E. Sounni, L. Devy, C. Grignet-Debrus, J.M. Lewalle, H. Li, C.F. Deroanne, H. Lu, A. Colige, and B.V. Nusgens, et al. 2001. Down-regulation of vascular endothelial growth factor by tissue inhibitor of metalloproteinase-2: effect on in vivo mammary tumor growth and angiogenesis. Cancer Res. 61:3450–3457. [PubMed] [Google Scholar]
- Hattori-Nakakuki, Y., C. Nishigori, K. Okamoto, S. Imamura, H. Hiai, and S. Toyokuni. 1994. Formation of 8-hydroxy-2'-deoxyguanosine in epidermis of hairless mice exposed to near-UV. Biochem. Biophys. Res. Commun. 201:1132–1139. [DOI] [PubMed] [Google Scholar]
- Houck, K.A., N. Ferrara, J. Winer, G. Cachianes, B. Li, and D.W. Leung. 1991. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol. 5:1806–1814. [DOI] [PubMed] [Google Scholar]
- Jingjing, L., Y. Xue, N. Agarwal, and R.S. Roque. 1999. Human Muller cells express VEGF183, a novel spliced variant of vascular endothelial growth factor. Invest. Ophthalmol. Vis. Sci. 40:752–759. [PubMed] [Google Scholar]
- Katiyar, S.K., B.M. Bergamo, P.K. Vyalil, and C.A. Elmets. 2001. Green tea polyphenols: DNA photodamage and photoimmunology. J. Photochem. Photobiol. B. 65:109–114. [DOI] [PubMed] [Google Scholar]
- Keyt, B.A., L.T. Berleau, H.V. Nguyen, H. Chen, H. Heinsohn, R. Vandlen, and N. Ferrara. 1996. The carboxyl-terminal domain (111-165) of vascular endothelial growth factor is critical for its mitogenic potency. J. Biol. Chem. 271:7788–7795. [DOI] [PubMed] [Google Scholar]
- Lambert, C.A., A.C. Colige, C. Munaut, C.M. Lapiere, and B.V. Nusgens. 2001. Distinct pathways in the over-expression of matrix metalloproteinases in human fibroblasts by relaxation of mechanical tension. Matrix Biol. 20:397–408. [DOI] [PubMed] [Google Scholar]
- Lauer, G., S. Sollberg, M. Cole, I. Flamme, J. Sturzebecher, K. Mann, T. Krieg, and S.A. Eming. 2000. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J. Invest. Dermatol. 115:12–18. [DOI] [PubMed] [Google Scholar]
- Lauer, G., S. Sollberg, M. Cole, T. Krieg, and S.A. Eming. 2002. Generation of a novel proteolysis resistant vascular endothelial growth factor165 variant by a site-directed mutation at the plasmin sensitive cleavage site. FEBS Lett. 531:309–313. [DOI] [PubMed] [Google Scholar]
- Lee, S., S.M. Jilani, G.V. Nikolova, D. Carpizo, and M.L. Iruela-Arispe. 2005. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J. Cell Biol. 169:681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity, A., N. Pore, J. Lee, D. Solomon, and D.M. O'Rourke. 2000. Epidermal growth factor receptor transcriptionally up-regulates vascular endothelial growth factor expression in human glioblastoma cells via a pathway involving phosphatidylinositol 3′-kinase and distinct from that induced by hypoxia. Cancer Res. 60:5879–5886. [PubMed] [Google Scholar]
- Mineur, P., A. Guignandon, C.A. Lambert, M. Amblard, C.M. Lapiere, and B.V. Nusgens. 2005. RGDS and DGEA-induced [Ca2+]i signalling in human dermal fibroblasts. Biochim. Biophys. Acta. 1746:28–37. [DOI] [PubMed] [Google Scholar]
- Park, J.E., G.A. Keller, and N. Ferrara. 1993. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell. 4:1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz, D., H. Gitay-Goren, M. Safran, N. Kimmel, D. Gospodarowicz, and G. Neufeld. 1992. Glycosylation of vascular endothelial growth factor is not required for its mitogenic activity. Biochem. Biophys. Res. Commun. 182:1340–1347. [DOI] [PubMed] [Google Scholar]
- Robinson, C.J., and S.E. Stringer. 2001. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 114:853–865. [DOI] [PubMed] [Google Scholar]
- Roth, D., M. Piekarek, M. Paulsson, H. Christ, T. Krieg, W. Bloch, J.M. Davidson, and S.A. Eming. 2006. Plasmin modulates vascular endothelial growth factor-A-mediated angiogenesis during wound repair. Am. J. Pathol. 168:670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobe, M., P. Rockwell, N. Goldstein, S. Vosseler, and N.E. Fusenig. 1997. Halting angiogenesis suppresses carcinoma cell invasion. Nat. Med. 3:1222–1227. [DOI] [PubMed] [Google Scholar]
- Soker, S., S. Gollamudi-Payne, H. Fidder, H. Charmahelli, and M. Klagsbrun. 1997. Inhibition of vascular endothelial growth factor (VEGF)-induced endothelial cell proliferation by a peptide corresponding to the exon 7- encoded domain of VEGF165. J. Biol. Chem. 272:31582–31588. [DOI] [PubMed] [Google Scholar]
- Soker, S., S. Takashima, H.Q. Miao, G. Neufeld, and M. Klagsbrun. 1998. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform- specific receptor for vascular endothelial growth factor. Cell. 92:735–745. [DOI] [PubMed] [Google Scholar]
- Sounni, N.E., L. Devy, A. Hajitou, F. Frankenne, C. Munaut, C. Gilles, C. Deroanne, E.W. Thompson, J.M. Foidart, and A. Noel. 2002. MT1-MMP expression promotes tumor growth and angiogenesis through an up- regulation of vascular endothelial growth factor expression. FASEB J. 16:555–564. [DOI] [PubMed] [Google Scholar]
- Stadler, N., J. Eggermann, S. Voo, A. Kranz, and J. Waltenberger. 2007. Smoking-induced monocyte dysfunction is reversed by vitamin C supplementation in vivo. Arterioscler. Thromb. Vasc. Biol. 27:120–126. [DOI] [PubMed] [Google Scholar]
- Stein, I., M. Neeman, D. Shweiki, A. Itin, and E. Keshet. 1995. Stabilization of vascular endothelial growth factor mRNA by hypoxia and hypoglycemia and coregulation with other ischemia-induced genes. Mol. Cell. Biol. 15:5363–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Wronski, M.A., N. Raju, R. Pillai, N.J. Bogdan, E.R. Marinelli, P. Nanjappan, K. Ramalingam, T. Arunachalam, S. Eaton, K.E. Linder, et al. 2006. Tuftsin binds neuropilin-1 through a sequence similar to that encoded by exon 8 of vascular endothelial growth factor. J. Biol. Chem. 281:5702–5710. [DOI] [PubMed] [Google Scholar]
- Waltenberger, J., L. Claesson-Welsh, A. Siegbahn, M. Shibuya, and C.H. Heldin. 1994. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 269:26988–26995. [PubMed] [Google Scholar]
- Whittle, C., K. Gillespie, R. Harrison, P.W. Mathieson, and S. Harper. 1999. Heterogeneous vascular endothelial growth factor (VEGF) isoform mRNA and receptor mRNA expression in human glomeruli, and the identification of VEGF148 mRNA, a novel truncated splice variant. Clin. Sci. (Lond.). 97:303–312. [PubMed] [Google Scholar]
- Yoon, Y.S., I.A. Johnson, J.S. Park, L. Diaz, and D.W. Losordo. 2004. Therapeutic myocardial angiogenesis with vascular endothelial growth factors. Mol. Cell. Biochem. 264:63–74. [DOI] [PubMed] [Google Scholar]
- Zeng, G., S.M. Taylor, J.R. McColm, N.C. Kappas, J.B. Kearney, L.H. Williams, M.E. Hartnett, and V.L. Bautch. 2007. Orientation of endothelial cell division is regulated by VEGF signaling during blood vessel formation. Blood. 109:1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}