

Abstract
Phosphoinositide 3-kinase (PI3K) has been shown to regulate cell and organ size in Drosophila, but the role of PI3K in vertebrates in vivo is not well understood. To examine the role of PI3K in intact mammalian tissue, we have created and characterized transgenic mice expressing constitutively active or dominant-negative mutants of PI3K in the heart. Cardiac- specific expression of constitutively active PI3K resulted in mice with larger hearts, while dominant-negative PI3K resulted in mice with smaller hearts. The increase or decrease in heart size was associated with comparable increase or decrease in myocyte size. Cardiomyopathic changes, such as myocyte necrosis, apoptosis, interstitial fibrosis or contractile dysfunction, were not observed in either of the transgenic mice. Thus, the PI3K pathway is necessary and sufficient to promote organ growth in mammals.
Keywords: cell size/heart/hypertrophy/phosphoinositide 3-kinase/transgenic mice
Introduction
One of the least understood areas in biology is how the size of animals and their organs is determined (Conlon and Raff, 1999). In most animals, determination of the organ size is achieved by controlling the number of cells. However, in certain tissue such as cardiac or skeletal muscle, hypertrophy (increase in cell size without cell division) plays a major role in determining the organ mass. In particular, the cardiac myocyte withdraws from the cell cycle soon after birth and the postnatal growth of the heart, which is essential to meet the increasing demand for cardiac work to support animal growth, is achieved primarily by hypertrophy (Soonpaa and Field, 1998). Extensive studies over the last decade have yielded significant information on neurohumoral growth factors involved in cardiac hypertrophy utilizing cultured cardiac myocytes as a model system (Chien et al., 1991; Sadoshima and Izumo, 1997). However, the mechanism of cell-size regulation in cardiac myocytes in the intact heart is still not well understood (Sugden and Clerk, 1998).
Among various signaling pathways, growth hormone, insulin-like growth factor (IGF)-1 and their downstream effectors are particularly implicated in the regulation of body and organ size (Conlon and Raff, 1999). IGF-1 and their downstream effectors, such as insulin receptor substrate (IRS)-1 and p70 S6 kinase (p70S6K), play an important role in body size determination in mammals (DeChiara et al., 1990; Baker et al., 1993; Liu et al., 1993; Araki et al., 1994; Tamemoto et al., 1994; Shima et al., 1998; Withers et al., 1998). Recent genetic studies in Drosophila also revealed effectors of insulin-like protein play a critical role in the determination of organ size as well as body size (Leevers et al., 1996; Bohni et al., 1999; Weinkove et al., 1999).
Phosphoinositide 3-kinase (PI3K) lies downstream of many receptor tyrosine kinases including insulin and IGF-1 receptors. PI3Ks play crucial roles in many aspects of biological response, such as membrane trafficking, cytoskeletal organization, cell growth and apoptosis (Toker and Cantley, 1997; Rameh and Cantley, 1999). The genes encoding PI3Ks have been isolated from a wide range of tissues and organisms. They are divided into three major classes based on their amino acid sequence, the homology among their lipid-kinase domains and substrate specificity (Vanhaesebroeck et al., 1997; Fruman et al., 1998). Class I PI3Ks phosphorylate the 3-position of the inositol ring of phosphatidylinositol (PtdIns), PtdIns 4-phosphate and PtdIns 4,5-diphosphate to form PtdIns 3-phosphate, PtdIns 3,4-diphosphate and PtdIns 3,4,5-trisphosphate, respectively. Class I PI3Ks can be further divided into two subclasses. The class IA PI3Ks, which include at least three isozymes, α, β and δ, are heterodimers of the catalytic 110 kDa subunit (p110) and the regulatory subunit of 85 or 55 kDa (p85/p55). The interaction of p85/p55 and p110 via the inter-Src homology 2 (iSH2) domain of p85/p55 and the N-terminal 123 amino acids of p110 is critical for achieving the maximal activity of this class of PI3Ks in mammalian cells (Dhand et al., 1994; Klippel et al., 1994; Yu et al., 1998).
A serine/threonine kinase Akt, also known as protein kinase B, is the most well characterized target of PI3K (Alessi and Cohen, 1998; Downward, 1998; Chan et al., 1999). Akt is known to mediate cell survival signal by regulating several effectors such as Bad or procaspase-9 (Datta et al., 1997; Cardone et al., 1998). Another protein serine/threonine kinase downstream of PI3K is p70S6K, which is known to be a physiological kinase for the ribosomal S6 protein whose phosphorylation increases the rate of initiation of translation of mRNA by ribosomes (Chou and Blenis, 1995; Thomas and Hall, 1997).
Despite the wealth of knowledge of biochemical and biological effects of PI3K in cultured cells, little information is available regarding its physiological roles in the intact animal. Targeted deletion of the p110α catalytic subunit (Bi et al., 1999) or the p85α regulatory subunit (Fruman et al., 1999) in mice resulted in embryonic or perinatal lethality, respectively. This makes it difficult to examine the role of PI3K in the later developmental process. Interestingly, a previous study utilizing tissue-specific expression of constitutively active (ca) or dominant-negative (dn) PI3K in Drosophila suggested a cell-autonomous role of PI3K in the determination of organ size (Leevers et al., 1996). However, there is greater genetic diversity in vertebrates than in lower metazoans, and in many cases functional redundancy exists that is not revealed in less complex organisms (Miklos and Rubin, 1996). To examine the role of PI3K in organ size determination in mammals, we have made transgenic mice expressing constitutively active or dominant- negative mutants of PI3K specifically in the heart.
Results
Production of constitutively active PI3K (caPI3K) transgenic mice
iSH2p110 (Franke et al., 1997b) is a chimeric molecule that contains the iSH2 domain of p85 fused to the N-terminus of bovine p110α by a flexible glycine linker. iSH2p110 has been shown to function in vitro and in vivo as a constitutively active molecule (Hu et al., 1995; Martin et al., 1996). The α myosin heavy chain (MyHC) promoter drives transgenes exclusively in cardiac myocytes and has been used extensively in previous transgenic studies (Wakasaki et al., 1997; Fentzke et al., 1998; Kadambi and Kranias, 1998). In the atrium this promoter is expressed in both embryonic and adult myocytes. However, its expression in ventricular myocytes is observed mainly after birth (Ng et al., 1991; Palermo et al., 1996). The iSH2p110 gene together with the Myc epitope tag was cloned into the αMyHC promoter construct and transgenic mice were produced. Five independently derived founders for the caPI3K were produced from 16 F0 mice screened by Southern blot analysis. All of the founders transmitted the transgene to the F1 generation. To confirm the transgene expression, 15 mg of cardiac tissue lysates from transgenic mice or non-transgenic mice were immunoprecipitated with the anti-p110α antibody, and probed with either anti-p110α antibody or anti-Myc epitope tag antibody (Figure 1A). Four out of five lines expressed significant amounts of the transgene product and were named as lines A to D. Line A expressed significantly lower amounts of the transgene product than the other lines on western blot analysis (data not shown). Immunoprecipitation with anti-p110α antibody followed by western blotting using the same antibody weakly detected the endogenous p110α molecule in the non-transgenic heart (Figure 1A, lane 2, upper panel). Expression of the iSH2p110 transgene product was confirmed by western blotting using the anti-p110α antibody, as well as the anti-Myc epitope tag antibody (Figure 1A, lane 4, upper and lower panels). To confirm that transgene expression is specific to the heart, 5 mg of protein from several tissues were immunoprecipitated with anti-p110α antibody followed by western blotting using anti-Myc antibody. As shown in Figure 1B, the transgene product was expressed exclusively in the heart.
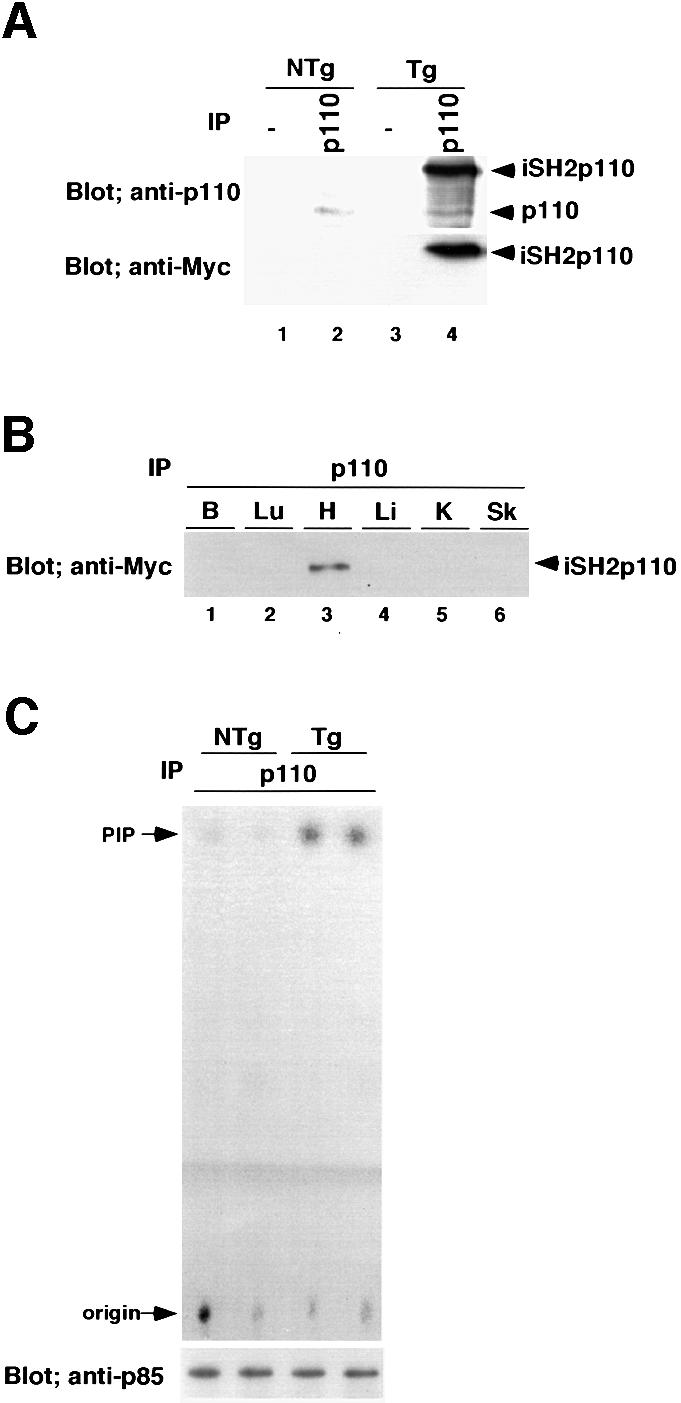
Fig. 1. Production of constitutively active PI3K (caPI3K) transgenic mice. (A) Expression of transgene in the heart. Cardiac tissue lysates (15 mg) from transgenic mice or non-transgenic mice were immunoprecipitated with anti-p110α-specific antibody, and probed with anti-p110α antibody or anti-Myc epitope tag antibody. (B) Tissue specificity of transgene expression. Protein (5 mg) from brain (B), lung (Lu), heart (H), liver (Li), kidney (K) or skeletal muscle (Sk) tissue was immunoprecipitated using anti-p110α antibody. The immunoprecipitated protein was blotted and probed with anti-Myc antibody. (C) PI3K activity in the heart. Heart tissue lysate (1 mg) was immunoprecipitated with anti-p110α-specific antibody and subjected to in vitro lipid kinase assay using phosphatidylinositol (PtdIns) as a substrate. Part of the immunoprecipitated enzyme was subjected to western blotting and probed with anti-p85 antibody to confirm that an equal amount of enzyme was used for the assay. PI3K activity in caPI3K transgenic mice increased 6.5-fold compared with non-transgenic mice. Data from two non-transgenic mice (NTg) and two transgenic mice (Tg) are shown. PIP, PtdIns 3-phosphate.
To confirm the activity of the transgene product, 1 mg of heart tissue lysate was immunoprecipitated with the anti-p110α antibody and was subjected to an in vitro lipid kinase assay using PtdIns as a substrate (Figure 1C). To confirm that an equal amount of enzyme was used for the assay, part of the immunoprecipitated enzyme was subjected to western blotting. The blot was probed with anti-p85 antibody, because we could not detect the endogenous p110 molecule when we used 1 mg of protein for immunoprecipitation (Figure 1C, lower panel). PI3K activity in caPI3K transgenic mice was increased 6.5-fold compared with that of non-transgenic mice (NTg, 1.00 ± 0.10 arbitrary units, n = 4; Tg, 6.54 ± 1.50 units, n = 4; p = 0.0104).
Production of dominant-negative PI3K (dnPI3K) transgenic mice
A catalytically inactive p110 molecule, when overexpressed in the cell, should compete with the endogenous p110 for interaction with the p85 regulatory subunit, thereby having an inhibitory effect on the function of the endogenous p110 molecule in vivo. Therefore, we made a truncated p110 mutant that has p85 binding domains, but lacks the kinase domain (p110Δkinase). The p110Δkinase gene, together with FLAG epitope tag, was cloned into the αMyHC promoter construct and transgenic mice were produced (Figure 2A). Four independently derived founders were produced from 39 mice screened by Southern blotting. Two of them transmitted the transgene to the progeny. To confirm the transgene expression, 5 mg of cardiac tissue lysates from dnPI3K transgenic mice or non-transgenic mice were immunoprecipitated with the anti-p85 antibody, and probed with anti-p110α antibody, anti-FLAG epitope tag antibody or anti-p85 antibody (Figure 2B). One of the two lines expressed the transgene. The amount of endogenous p110α that co-immunoprecipitated with p85 was 20% of that of non-transgenic mice (Figure 2B, compare lanes 2 and 4, upper panel), indicating that the majority of the endogenous p85 regulatory subunit was sequestered by the p110Δkinase molecule. The p110β protein was not detected in either dnPI3K mice or non-transgenic mice using the same blot (data not shown). To confirm that transgene expression is specific to the heart, 5 mg of protein from several tissues were immunoprecipitated with anti-p85 antibody followed by western blotting using anti-FLAG antibody. As shown in Figure 2B, the transgene product was expressed exclusively in the heart.
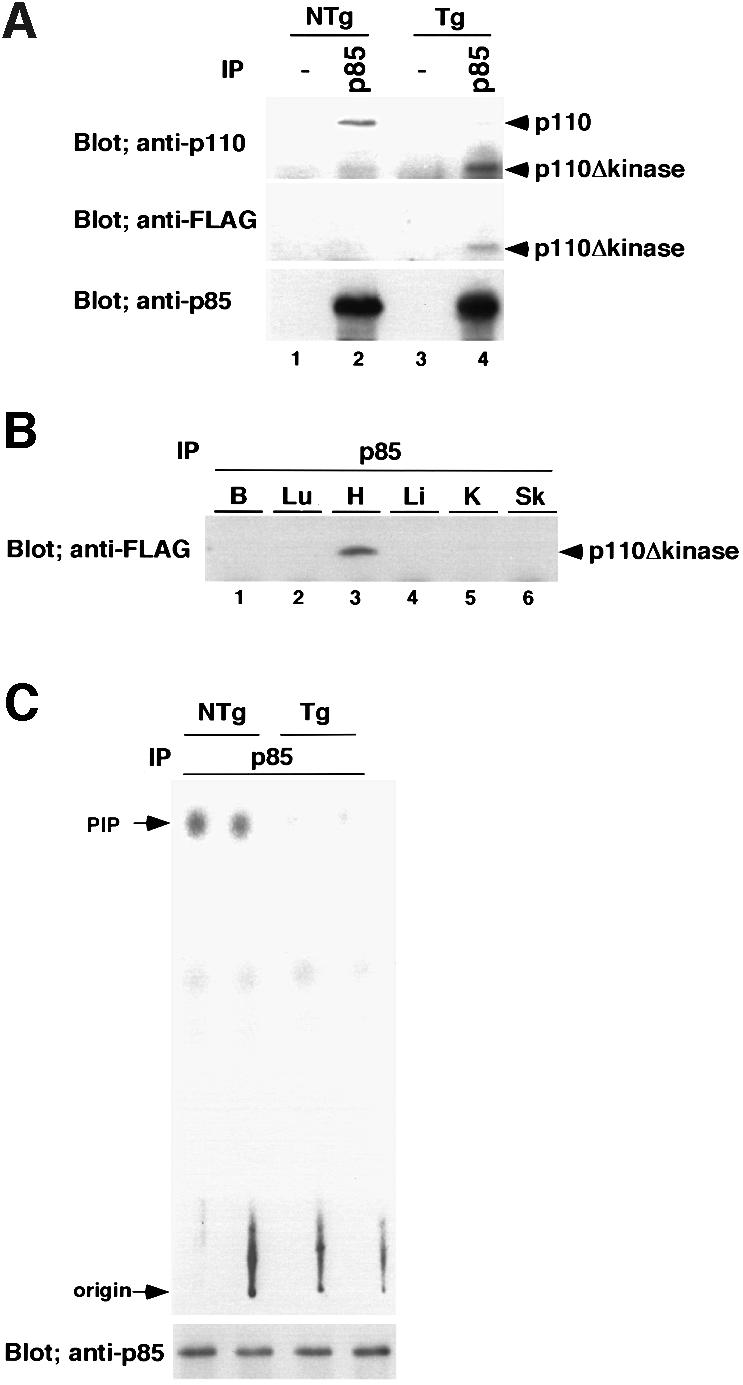
Fig. 2. Production of dominant-negative PI3K (dnPI3K) transgenic mice. (A) Expression of the transgene in the heart. Cardiac tissue lysates (5 mg) from dnPI3K transgenic mice or non-transgenic mice were immunoprecipitated with anti-p85 antibody, and probed with anti-p110α antibody, anti-FLAG epitope tag antibody or anti-p85 antibody. (B) Tissue specificity of transgene expression. Protein (5 mg) from brain (B), lung (Lu), heart (H), liver (Li), kidney (K) or skeletal muscle (Sk) tissue was immunoprecipitated using anti-p85 antibody. The immunoprecipitated protein was blotted and probed with anti-FLAG antibody. (C) PI3K activity in the heart tissue. Heart tissue lysate (1 mg) was immunoprecipitated with anti-p85-specific antibody and subjected to in vitro lipid kinase assay. Part of the immunoprecipitated enzyme was subjected to western blotting and probed with anti-p85 antibody to confirm that an equal amount of enzyme was used for the assay. PI3K activity of dnPI3K mice was decreased by 77% compared with that of non-transgenic mice. Data from two non-transgenic mice (NTg) and two transgenic mice (Tg) are shown.
To confirm that p110Δkinase functioned as a dominant-negative molecule, 1 mg of heart tissue lysate was immunoprecipitated with the anti-p85 antibody and was subjected to an in vitro lipid kinase assay (Figure 2C). To confirm that an equal amount of enzyme was used for the assay, part of the immunoprecipitated enzyme was subjected to western blotting and probed with anti-p85 antibody (Figure 2C, lower panel). PI3K activity of the heart from dnPI3K mice was decreased by 77% compared with that of non-transgenic mice (NTg, 1.00 ± 0.21 units, n = 4; Tg, 0.23 ± 0.03 units, n = 4; p = 0.0102). Taken together, we were able to achieve a significant increase (6.5-fold) or decrease (by 77%) in PI3K activity in the heart using caPI3K and dnPI3K transgenes, respectively.
Activation of potential downstream targets in PI3K transgenic mice
Next, we examined the activation of potential downstream targets of PI3K. Representative western blots are shown in the upper and middle panels of Figure 3 and the results of quantitative densitometry of 6–8 animals in each group are shown in the lower panels. Akt (or protein kinase B) is one of the best characterized targets of PI3K in cultured cells (Chan et al., 1999). Activation of Akt in the heart tissue was assessed by measuring the amount of Akt phosphorylated at Ser473 using a phosphospecific antibody (pAkt in Figure 3A, upper panel). In caPI3K mice, the amount of phosphorylated Akt was increased 2.2-fold, whereas the amount of unphosphorylated Akt (Akt in Figure 3A, middle panel) was decreased by 44%. In dnPI3K mice, the amount of phosphorylated Akt was decreased by 74%, whereas the unphosphorylated Akt was increased by 45%. The ratio of the phosphorylated Akt to the unphosphorylated Akt (pAkt/Akt in Figure 3A, lower panel) is increased 3.9-fold in caPI3K mice, but is decreased by 77% in dnPI3K mice compared with non-transgenic littermates. These results are concordant with the relative activities of PI3K in caPI3K and dnPI3K mice, thus suggesting that activation of Akt is regulated by PI3K in intact tissue.

Fig. 3. Activation of Akt and p70S6K, not ERK, was regulated by PI3K in the heart. (A) Activation of Akt in the heart tissue of PI3K transgenic mice. In caPI3K mice, the amount of phosphorylated Akt at Ser473 was increased, whereas the unphosphorylated form of Akt was decreased. In dnPI3K mice, the amount of phosphorylated Akt was decreased and the unphosphorylated Akt was increased. (B) Activation of ERK in the heart tissue of PI3K transgenic mice. The amount of phosphorylated ERK or total ERK2 was not different among non-transgenic, caPI3K and dnPI3K mice. (C) Activation of p70S6K in the heart tissue of PI3K transgenic mice. The ratio of the phosphorylated p70S6K to the total p70S6K (p-p70S6K/Σp70S6K) was 1.7-fold higher in caPI3K mice, but is decreased by 72% in dnPI3K mice compared with non-transgenic littermates. (D) Phosphorylation of ribosomal S6 protein in the heart tissue of PI3K transgenic mice. The amount of phosphorylated S6 was increased 1.8-fold in caPI3K mice and decreased by 36% in dnPI3K mice. Hearts from eight non-transgenic mice, six caPI3K mice and six dnPI3K mice were used for the assay of Akt and ERK. Hearts from eight non-transgenic mice, eight caPI3K mice and eight dnPI3K mice were used for the assay of p70S6K and S6 protein. *p<0.05 versus non-transgenic (NTg) mice.
We also examined the activation of extracellularly responsive kinase (ERK), which is one of the downstream targets of p21ras, by measuring the amount of ERK phosphorylated at Tyr204 (pERK in Figure 3B, upper panel). In both caPI3K and dnPI3K mice, there was no significant difference in the amounts of phosphorylated ERK or total amount of ERK2 (ERK2 in Figure 3B, middle panel), suggesting that ERK does not seem to be regulated by PI3K in the heart.
p70S6K phosphorylates 40S ribosomal protein S6, which results in increased protein synthesis. p70S6K has been shown to be downstream of PI3K (Chou and Blenis, 1995). We examined the activation of p70S6K by measuring the amount of p70S6K phosphorylated at Thr389 (p-p70S6K in Figure 3C, upper panel), which is known to correlate well with p70S6K activity (Weng et al., 1998). The amount of phosphorylated p70S6K was increased 2.2-fold in caPI3K mice, whereas it was decreased by 74% in the dnPI3K mouse heart. Western blotting using p70S6K antibody produces broad bands at ∼70 kDa due to the presence of p70S6K phosphorylated at multiple sites (Σp70S6K in Figure 3C, middle panel). The total amount of p70S6K was estimated by adding densitometric scores of these bands. The total amount of p70S6K was not significantly different among three animal groups (Σp70S6K in Figure 3C, lower panel). The ratio of the phosphorylated p70S6K to the total p70S6K (p-p70S6K/Σp70S6K in Figure 3C, lower panel) was 1.7-fold higher in caPI3K mice, but was decreased by 72% in dnPI3K mice compared with non-transgenic littermates. These results suggest that PI3K plays an important role in the regulation of p70S6K activity in the intact mammalian tissue.
To confirm the p70S6K activity in the heart, the amount of the phosphorylated form of ribosomal S6 protein was measured using antibody specific for S6 protein (Kimball et al., 1999). Loading of equal amounts of protein was confirmed by the Coomassie Blue staining of the membrane (data not shown). The amount of phosphorylated S6 was increased 1.8-fold in caPI3K mice, and it was decreased by 36% in dnPI3K mice (Figure 3D).
PI3K activity correlates with the size of the heart
Heart weight/body weight (HW/BW) ratio is a well established index of cardiac hypertrophy. HW/BW ratios of lines A, B, C and D were 4.18 ± 0.10 (n = 9), 4.93 ± 0.53 (n = 6), 4.94 ± 0.13 (n = 4) and 5.12 ± 0.31 (n = 5), respectively. HW/BW of non-transgenic mice was 4.18 ± 0.19 (n = 12). Lines B, C and D had a significant increase in HW/BW ratio (p <0.05). In all four lines of caPI3K mice, there was no premature death or sign of heart failure after 1 year of observation. Three- to four-month-old female mice from line B were used for the subsequent analysis. Male caPI3K mice showed the same degree of increase in HW/BW ratio as female mice. Body weight, lung weight and liver weight of the transgenic mice were not different from those of non-transgenic animals (Table I). However, as shown in Figure 4 (left panel), there was a proportional increase in the size of all chambers and the thickness of left ventricular walls, which resembles ‘physiological hypertrophy’ associated with normal growth of the animal.
Table I. Heart weight, lung weight and liver weight of PI3K transgenic mice.
| Non-transgenic | caPI3Ka | dnPI3Kb | |
|---|---|---|---|
| Number of animals | 6 | 6 | 7 |
| Body weight (g) | 22.9 ± 2.5 | 22.6 ± 1.3 | 22.5 ± 3.1 |
| Tibial length (mm) | 17.1 ± 0.2 | 17.1 ± 0.2 | 16.5 ± 0.5 |
| Heart weight (mg) | 94.4 ± 4.7 | 112.0 ± 10.3* | 78.5 ± 3.6* |
| Lung weight (mg) | 141.1 ± 21.4 | 146.8 ± 16.0 | 140.2 ± 13.4 |
| Liver weight (mg) | 1054 ± 91 | 1127 ± 155 | 1026 ± 130 |
| Heart weight/body weight (mg/g) | 4.18 ± 0.29 | 4.93 ± 0.53* | 3.53 ± 0.42* |
| Lung weight/body weight (mg/g) | 6.48 ± 0.47 | 6.17 ± 0.56 | 6.29 ± 0.80 |
| Liver weight/body weight (mg/g) | 46.7 ± 5.2 | 49.3 ± 3.1 | 45.8 ± 3.93 |
| Heart weight/tibial length (mg/mm) | 5.53 ± 0.30 | 6.55 ± 0.65* | 4.75 ± 0.18* |
| Lung weight/tibial length (mg/mm) | 8.60 ± 0.88 | 8.24 ± 1.19 | 8.50 ± 0.73 |
| Liver weight/tibial length (mg/mm) | 61.8 ± 5.3 | 65.9 ± 9.0 | 62.0 ± 6.4 |
Results are presented as mean ± SE.
acaPI3K, constitutively active PI3K transgenic.
bdnPI3K, dominant-negative PI3K transgenic.
*p <0.05 versus non-transgenic.

Fig. 4. Perturbation of PI3K activity alters heart size in transgenic mice. In caPI3K, there was a proportional increase in the size of all chambers and ventricular wall thickness. In dnPI3K mice, the heart was proportionally smaller than in non-transgenic mice. Bars represent 1 mm.
Transgenic mice that express dnPI3K did not show any premature death or sign of heart failure after 1 year of observation. Interestingly, the HW/BW ratio was decreased by 17% in dnPI3K mice (NTg, 4.18 ± 0.29, n = 6; Tg, 3.53 ± 0.42, n = 7; p = 0.008), even though there was no difference in body weight, lung weight, liver weight or tibial length compared with non-transgenic mice (Table I). Three- to four-month-old female mice were used for the detailed subsequent analysis. Male dnPI3K mice showed the same degree of decrease in the HW/BW ratio as female mice. All of the chambers of the transgenic heart were smaller compared with those of non-transgenic litter-mates, and the proportion of the chambers was maintained (Figure 4, right panel). The hearts of dnPI3K mice remained smaller without any sign of heart failure after >1 year’s observation.
We intercrossed caPI3K and dnPI3K mice. The HW/BW ratio of double transgenic mice having both caPI3K and dnPI3K transgenes was 4.83 ± 0.45 (n = 3), which is similar to that of caPI3K mice. This is consistent with the expected mechanism of how this caPI3K transgene functions, which should be independent of p85, while the dnPI3K transgene should work through sequestering p85.
It is shown that there is a very tight correlation between heart weight and body weight in normal mice (Goodman et al., 1984; Ernsberger et al., 1996). Thus, perturbation of PI3K activity in the myocytes selectively modulated the size of the heart without changes in the size of other organs or overall body weight.
Cell size of isolated cardiac myocytes from PI3K transgenic mice
To examine whether the increase in organ size was due to cellular growth (increase in cell size) or proliferation (increase in cell number), we measured cell size and the number of nuclei in individual cardiac myocytes in isolated adult myocyte preparations (Figure 5A). The myocytes dissociated from caPI3K heart had a significant increase in the mean cell area compared with those from the non-transgenic heart (NTg, 2501 ± 49 µm2, data from five mice; Tg, 2974 ± 107 µm2, data from four mice; p = 0.004). Both the long axis length (NTg, 122.9 ± 1.2 µm, data from five mice; Tg, 127.3 ± 1.1 µm, data from four mice; p = 0.033) and the short axis length (NTg, 28.4 ± 0.7 µm, data from five mice; Tg, 32.1 ± 0.6 µm, data from four mice; p = 0.006) were increased in caPI3K mice. The ratio of the long axis to the short axis tended to be smaller in caPI3K mice, although it was not statistically significant (NTg, 4.63 ± 0.17, data from five mice; Tg, 4.26 ± 0.05, data from four mice; p = 0.098). In contrast, the mean cell area of the myocytes isolated from dnPI3K transgenic heart was significantly smaller than that from non-transgenic heart (NTg, 2501 ± 49 µm2, data from five mice; Tg, 2061 ± 51 µm2, data from four mice; p = 0.004), accompanied by the significant decrease in both the long axis length (NTg, 122.9 ± 1.2 µm, data from five mice; Tg, 108.6 ± 1.2 µm, data from four mice; p <0.001) and the short axis length (NTg, 28.4 ± 0.7 µm, data from five mice; Tg, 26.4 ± 0.8 µm, data from four mice; p = 0.036) of the cells. Figure 5B shows histogram analysis demonstrating that there were normal distributions of myocyte size isolated from transgenic and non-transgenic mice. Interestingly, the curve was shifted to the right (more bigger cells) for myocytes isolated from caPI3K mice, and it was shifted to the left for myocytes from dnPI3K mice.

Fig. 5. PI3K regulates cell size in transgenic heart. (A) The cardiac myocytes were enzymatically dissociated from the mice hearts. The mean cell areas of myocytes isolated from caPI3K mice hearts were significantly increased compared with non-transgenic controls. The mean cell areas of myocytes isolated from dnPI3K mice hearts were decreased. (B) Distribution of the cell size in the representative animals from non-transgenic, caPI3K and dnPI3K mice. The measurement of cell size was performed on 100 cells for each heart.
Cardiac myocytes withdraw from the cell cycle soon after birth in rodents (Yoshizumi et al., 1995). However, forced expression of cyclin D1 under αMyHC promoter has been shown to cause an increase in the number of multinucleated cells (Soonpaa et al., 1997). To determine whether PI3K affects the nuclear profile of cardiac myocytes, we counted the number of nuclei in isolated myocytes. Distributions of the number of nuclei in each myocyte were not different among caPI3K, dnPI3K and non-transgenic hearts (Table II).
Table II. Morphometric analysis of isolated cardiac myocytes.
| Number of nuclei (%) |
|||||||
|---|---|---|---|---|---|---|---|
| Mean cell area (µm2) | Long axis (µm) | Short axis (µm) | Long axis/short axis | 1 | 2 | >3 | |
| Non-transgenic (n = 5) | 2501 ± 49 | 122.9 ± 1.2 | 28.4 ± 0.7 | 4.63 ± 0.17 | 9.8 ± 3.0 | 85.9 ± 4.2 | 4.4 ± 1.4 |
| caPI3K (n = 4) | 2974 ± 107* | 127.3 ± 1.1* | 32.1 ± 0.6* | 4.26 ± 0.05 | 9.6 ± 2.4 | 85.1 ± 4.0 | 5.3 ± 2.2 |
| dnPI3K (n = 4) | 2061 ± 51* | 108.6 ± 1.2* | 26.4 ± 0.8* | 4.37 ± 0.08 | 11.6 ± 1.8 | 87.1 ± 2.0 | 1.3 ± 0.2 |
n, number of animals.
Mean values from each mouse were calculated using the measurements from 100 cells isolated from an individual mouse. Next, new mean values (± SE) for each experimental group were calculated based on the data from the mean values from the individual mouse, and are presented in the table.
*p <0.05 versus non-transgenic.
Absence of cardiomyopathic changes or apoptosis in PI3K transgenic mice
Upon microscopic observation, necrosis or myocyte disarray was not observed in caPI3K mice or dnPI3K mice (Figure 6A, upper panels). Masson trichrome stain showed no interstitial fibrosis in caPI3K mice or dnPI3K mice (Figure 6A, lower panels). Since apoptosis has been postulated to be a critical determinant of organ size by counterbalancing cell proliferation (Conlon and Raff, 1999), and PI3K has been implicated in cell survival in several systems (Chan et al., 1999), we used a DNA-laddering assay to detect the presence of apoptosis in the heart (Figure 6B). There was no evidence of DNA laddering in dnPI3K or caPI3K hearts even after a prolonged exposure of the gel (Figure 6B, lanes 2 and 3). In control experiments, this assay readily detected increased DNA fragmentation in heart tissue taken from mice injected with doxorubicin (Figure 6B, lane 4) and in the thymus of animals treated with dexamethasone (Figure 6B, lane 5). TUNEL staining of the heart sections showed no differences in TUNEL-positive cells in non-transgenic, caPI3K or dnPI3K hearts (data not shown). These results suggest that the regulation of organ size by PI3K does not involve apoptosis.


Fig. 6. Absence of cardiomyopathic changes or apoptosis in PI3K transgenic mice. (A) Histopathological analysis of heart tissue from PI3K transgenic mice. Myocardial necrosis, fibrosis or disarray was not observed in caPI3K or dnPI3K transgenic mice. Upper panels show sections stained with hematoxylin and eosin, and lower panels show sections stained with Masson trichrome. Bars represent 10 µm. (B) Absence of apoptosis in caPI3K transgenic or dnPI3K transgenic mice. Low molecular weight DNA was prepared from mouse tissues and end labeled using terminal deoxynucleotidyl transferase. No ‘DNA laddering’ was evident in the hearts from caPI3K mice or dnPI3K mice. Heart tissues from mice that received doxorubicin and thymus tissues from dexamethasone injected mice were used as positive controls.
Echocardiographic assessment of left ventricular function of PI3K transgenic mice
Contractile dysfunction leads to compensatory hypertrophy of the heart. Therefore, we assessed left ventricular (LV) dimensions and systolic function of caPI3K mice using M-mode echocardiography (Table III). There was a significant increase in wall thickness in both the anterior (NTg, 0.6 ± 0.0 mm, n = 8; Tg, 0.7 ± 0.0 mm, n = 4; p = 0.009) and the posterior (NTg, 0.6 ± 0.0 mm, n = 8; Tg, 0.7 ± 0.0 mm, n = 4; p <0.0001) LV walls. There was no difference in LV diastolic diameter or LV systolic diameter. Fractional shortening, an echocardiographic index of left ventricular systolic function, was not altered in these transgenic mice. Although there was a trend of decreased LV anterior wall thickness in dnPI3K mice (NTg, 0.6 ± 0.0 mm, n = 8; Tg, 0.5 ± 0.0 mm, n = 4; p = 0.120), there was no significant difference in LV posterior wall thickness, LV diastolic diameter, LV systolic diameter or fractional shortening compared with non-transgenic mice (Table III).
Table III. Echocardiographic data of PI3K transgenic mice.
| Non-transgenic | caPI3K | dnPI3K | |
|---|---|---|---|
| Number of animals | 8 | 4 | 4 |
| Body weight (g) | 22.3 ± 0.7 | 23.3 ± 0.5 | 21.0 ± 0.8 |
| Heart rate (b.p.m.) | 285 ± 23 | 269 ± 21 | 254 ± 5 |
| Diastolic anterior wall thickness (mm) | 0.6 ± 0.0 | 0.7 ± 0.0* | 0.5 ± 0.0 |
| Diastolic posterior wall thickness (mm) | 0.6 ± 0.0 | 0.7 ± 0.0* | 0.6 ± 0.0 |
| LV diastolic diameter (mm) | 3.4 ± 0.1 | 3.4 ± 0.1 | 3.6 ± 0.1 |
| LV systolic diameter (mm) | 1.2 ± 0.1 | 1.1 ± 0.1 | 1.5 ± 0.1 |
| Fractional shortening (%) | 65 ± 2 | 68 ± 3 | 58 ± 1 |
b.p.m., beats per minute; LV, left ventricle.
Results are presented as mean ± SE.
*p <0.05 versus non-transgenic.
ANF and βMyHC genes are differently regulated in PI3K transgenic mice
Myocardial hypertrophy is typically associated with transcriptional activation of several ‘fetal’ genes, such as atrial natriuretic factor (ANF) and βMyHC (Izumo et al., 1988; Chien et al., 1998). We analyzed the expression of αMyHC, βMyHC and ANF genes by northern hybridization analysis (Figure 7). In caPI3K mice, the expression of MyHC genes shifted from the α isoform to the β isoform, whereas ANF mRNA was not increased. The expression of MyHC isoforms was not different from non-transgenic mice, whereas ANF mRNA was markedly increased in dnPI3K mice. This discordant regulation of βMyHC and ANF genes suggests that PI3K differentially regulates the ‘fetal’ gene program (see Discussion).
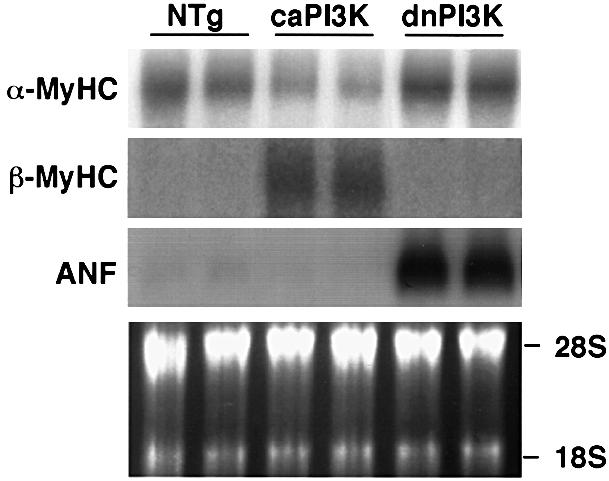
Fig. 7. PI3K differentially regulates ‘fetal’ gene expression in the transgenic heart. Expression of ANF and βMyHC gene was analyzed by northern hybridization analysis. In caPI3K mice, the expression of MyHC mRNA shifted from the α isoform to the β isoform, whereas ANF mRNA was not increased. In dnPI3K mice, the expression of MyHC isoforms was not different from non-transgenic mice, whereas ANF mRNA was markedly increased. Data from two animals from each group are shown.
Discussion
In order to determine the role of PI3K in the adult heart, we have perturbed the activity of PI3K specifically in cardiac myocytes by expressing constitutively active or dominant-negative forms of PI3K in transgenic mice. The caPI3K transgene caused a larger heart associated with an increase in myocyte size, whereas dnPI3K expression resulted in a smaller heart associated with smaller myocytes. The proportion of chamber size, architecture of myocardium or cardiac function was not disturbed by the modulation of PI3K activity, suggesting the specific role of PI3K in organ size determination. Thus, our study represents the first example to show that PI3K is necessary and sufficient to promote organ growth in mammals, similar to the demonstrated role of PI3K in Drosophila (Leevers et al., 1996).
The availability of the highly cardiac-specific αMyHC promoter prompted the creation of many transgenic mice that overexpress ‘molecules of interest’ in the heart (Izumo and Shioi, 1998; Kadambi and Kranias, 1998). However, it is possible that an overexpressed molecule could affect a biological process in which the endogenous counterpart is not normally involved. To circumvent this potential problem, we created a pair of transgenic lines that express ‘gain-of-function’ and ‘loss-of-function’ mutants of the p110 catalytic subunit of PI3K, and carefully compared the resultant phenotypes caused by the respective mutant transgenes. The fact that the dnPI3K transgene had exactly the opposite effects on the cardiac phenotype, compared with those caused by the caPI3K transgene, leads us to conclude that the phenotypic changes observed in these transgenic hearts are most likely due to specific modulations of PI3K activity.
The size of an organ is determined by the coordinate regulation of cell growth, proliferation and death (Conlon and Raff, 1999). It is very difficult, if not impossible, to count accurately the cell number in a compact organ, such as the heart. However, we speculate that PI3K regulated heart size mainly through the regulation of cell size in our transgenic mice, because (i) an 18% increase of the HW/BW ratio was associated with a comparable (19%) increase in the mean cell size in caPI3K mice, and a 17% decrease of the HW/BW ratio was associated with a similar degree (18%) of decrease in cell size in dnPI3K mice; (ii) distribution of the number of nuclei in each myocyte was not different among caPI3K, dnPI3K and non-transgenic hearts; and (iii) no apoptosis was observed in either transgenic mice using a DNA-laddering assay or TUNEL analysis. However, we cannot exclude the potential role of PI3K in the regulation of cell number in mitotically competent organs, because most of the increase in heart size occurs postnatally when the myocytes are post-mitotic (Soonpaa et al., 1996; Soonpaa and Field, 1998), and because the promoter used to generate transgenic mice in this study is active mainly after birth in the ventricles (Ng et al., 1991; Palermo et al., 1996). Modulation of PI3K activity in Drosophila wing alters both cell size and number in the organ (Leevers et al., 1996).
What are the downstream targets of PI3K that are involved in the regulation of organ size? The available information does not provide us with the definitive answer to this question. However, Akt, a well characterized downstream target of PI3K (Alessi and Cohen, 1998; Downward, 1998), is likely to be one of the major mediators of this process. Akt is necessary and sufficient for phosphorylation and subsequent inactivation of 4E-BP1, a repressor of mRNA translation (Gingras et al., 1998; Dufner et al., 1999; Takata et al., 1999). Akt can also activate p70S6K in some contexts (Burgering and Coffer, 1995), although activation of p70S6K might not be solely dependent on Akt (Conus et al., 1998; Dufner et al., 1999). Recent genetic experiments in Drosophila show that Akt regulates organ growth (Verdu et al., 1999). Our results showed that PI3K is necessary and sufficient for the activation of Akt in in situ heart.
Another potential candidate is p70S6K, which is upregulated in caPI3K hearts and downregulated in dnPI3K hearts. The amount of phosphorylated S6 protein was correlated with the activation of p70S6K. It was shown previously that the inhibition of the p70S6K pathway by rapamycin at nanomolar concentrations selectively suppresses an increase in protein synthesis of cultured neonatal myocytes in response to growth factors (Sadoshima and Izumo, 1995; Boluyt et al., 1997). Interestingly, rapamycin did not inhibit other phenotypic changes associated with myocyte hypertrophy, such as re-activation of fetal genes and sarcomere organization (Sadoshima and Izumo, 1995). This raises the possibility that p70S6K may selectively regulate cell size via controlling the rate of protein synthesis. It was recently observed that gene disruption of p70S6K resulted in smaller body size in mice (Shima et al., 1998). In Drosophila, deficiency of the S6K gene is associated with a reduction in body size associated with smaller cells (Montagne et al., 1999).
PI3K is shown to promote cell survival in several cell systems (Franke et al., 1997a). We did not detect evidence of cardiomyocyte apoptosis in dnPI3K mice. This may be because the residual PI3K activity is sufficient to prevent apoptosis, or because other survival pathways compensate for the anti-apoptotic function of PI3K. Interestingly, no significant apoptosis was reported in p110 knockout mice (Bi et al., 1999) or in Drosophila expressing a kinase-deficient p110 transgene (Leevers et al., 1996).
ERK is known to be activated by most, if not all, hypertrophic factors for the cardiac myocytes (Bogoyevitch et al., 1994; Foncea et al., 1997). However, the activity of ERK was not changed by modulation of PI3K activity in transgenic animals. This is consistent with the report in vitro which showed that a constitutively active PI3K failed to activate ERK (Klippel et al., 1996). It is also reported that ERK is only affected by PI3K under restrictive conditions (Duckworth and Cantley, 1997).
ANF and βMyHC mRNA are usually co-regulated in the ventricle during embryonic development and in cardiac hypertrophy induced by mechanical overload (Chien et al., 1998; Izumo et al., 1988). However, in our transgenic mice, expression of these two genes was independently regulated and did not correlate with the increase in heart size. No cardiac dysfunction was observed in either transgenic mice. IGF-1, which is known to activate PI3K activity in cardiac myocytes (Foncea et al., 1997), decreases ANF gene expression in intact heart (Donath et al., 1997), as well as in cultured myocytes (Eppenberger-Eberhardt et al., 1997). The PI3K pathway is implicated in the differentiation of skeletal myoblasts (Coolican et al., 1997; Jiang et al., 1999) and smooth muscle cells (Hayashi et al., 1998). It was recently shown that a member of the Forkhead family of transcription factors is directly phosphorylated by Akt (Brunet et al., 1999). It is likely that PI3K regulates the activity of a selective number of transcription factors that regulate the differentiation of cardiac muscle cells. Additional work is necessary to determine the effectors of PI3K-dependent regulation of ANF and βMyHC gene expression in the heart.
In Drosophila, overexpression of constitutively active or dominant-negative mutants of PI3K in the eye causes an increase or decrease in organ size through cell size regulation (Leevers et al., 1996), which is highly reminiscent of our findings in the mouse heart. Disruption of the Drosophila IRS-1 homolog (an upstream regulator of PI3K) results in a small fly due to a decrease in body and organ size (Bohni et al., 1999). Drosophila p70S6K determines body size exclusively through cell size regulation (Montagne et al., 1999). In the mouse, dwarfism occurs in knockout mice of IGFs or their receptors (DeChiara et al., 1990; Baker et al., 1993; Liu et al., 1993), IRS-1 (Araki et al., 1994; Tamemoto et al., 1994), IRS-2 (Withers et al., 1998) or p70S6K (Shima et al., 1998). Our findings, together with these previous reports, suggest that the conserved PI3K pathway plays a critical role in the determination of organ size in insects and mammals.
Materials and methods
Mutants of PI3K used in this study
iSH2p110 is a kind gift from T.Franke. To make p110Δkinase, nucleotides 1–1731 (amino acids 1–577) of p110α (Klippel et al., 1994) were cloned by PCR using the mouse p110 cDNA as a template. p110Δkinase sense (5′-CGGGATCCACCATGGACTACAAGGACGACGATGACAAGCCTCCACGACCATCTTCGGGT-3′) primer included a BamHI restriction site, ATG, FLAG epitope tag sequence and nucleotides 4–24 of the coding sequence of p110. p110Δkinase antisense (5′-TGCTCTAGATCAGCTCAGCACGCATCTGGAATTCCACTTGACAGACAGAAG) primer included nucleotides 1705–1731, the CAAX box of H-ras, stop codon and an XbaI restriction site. PCR product was cloned into pcDNA3 (Invitrogen). Expression of the mutant protein was confirmed by transient transfection into COS7 cells followed by western blotting using M2 anti-FLAG antibody (Eastman Kodak).
Generation of transgenic mice
The cDNA insert for iSH2p110 or p110Δkinase gene was cloned into SalI-digested αMyHC promoter construct (clone 26, a generous gift from J.Robbins; Gulick et al., 1991). This vector contains a 5.8 kb BamHI–MaeIII fragment of the murine αMyHC gene that includes the promoter and exons 1–3 from the 5′ untranslated region of the gene, as well as the human growth hormone polyadenylation site. The bacterial sequence was removed by digestion with NotI and injected into the male pronucleus of fertilized single-cell FVB/N embryos. Transgenic founders were identified by Southern blot analysis of tail DNA using the human growth hormone poly(A) as a probe. All aspects of animal care and experimentation performed in this study were approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center.
Protein preparation
The hearts were removed after cervical dislocation and were immediately frozen in liquid nitrogen. The heart lysates were obtained by homogenization in ice-cold buffer [1% NP-40, 10% glycerol, 137 mM NaCl, 20 mM Tris–HCl pH 7.4, 4 µg/ml aprotinin, 4 µg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride (PMSF), 4 µg/ml pepstatin, 20 mM NaF, 1 mM sodium pyrophosphate, 1 mM orthovanadate]. The lysates were kept on ice for 15 min and cleared by centrifugation at 15 000 g for 20 min at 4°C. Protein concentration was determined by the Bradford method (Bio-Rad).
Analysis of transgene expression by western blotting
Protein (5–15 mg) was mixed with protein A–Sepharose for 1 h at 4°C, which was subsequently removed by centrifugation. Each aliquot was then mixed with anti-p85 (5 µl; Upstate Biotechnology) or anti-p110α (5 µg; Santa Cruz) antibodies overnight at 4°C. Protein A–Sepharose was added for an additional 1 h at 4°C, and the beads were collected by centrifugation. The beads were washed four times with 1 ml of lysis buffer and resuspended in 50 µl of 2× SDS/gel loading buffer (1× buffer contains 62.5 mmol/l Tris–HCl pH 6.8, 1% SDS, 10% glycerol, 0.005% Bromophenol blue and 5% β-mercaptoethanol). Immunoprecipitates were subjected to SDS–PAGE, and proteins were transferred onto polyvinylidene difluoride membranes (Immobilon-P, Millipore). The blots were probed with anti-p110α (1 µg/ml), anti-Myc (1 µg/ml; Santa Cruz) or anti-FLAG (3 µg/ml), followed by horseradish peroxidase-conjugated protein A (1:10 000; Jackson) or anti-mouse IgG (1:10 000; Jackson). The protein probed was then visualized by the enhanced chemiluminescence system (Amersham).
Lipid kinase assay
Lipid kinase assays were performed essentially as described (Serunian et al., 1991). Briefly, 10 µl of a 1 mg/ml mixture of phosphatidylinositol and phosphatidylserine (3:2) (sonicated in 20 mM HEPES pH 7.0 and 0.1 mM EGTA) were added to immunoprecipitated enzyme from 1 mg of lysate (30 µl volume), followed by the addition of 10 µl of 5× ATP/MgCl2 mixture (100 µM ATP, 25 mM MgCl2, 50 mM HEPES pH 7.0) containing 10 µCi of [γ-32P]ATP. The kinase reaction was performed at room temperature for 10 min, and the reaction was stopped with 60 µl of 2 M HCl. Lipids were extracted by the addition of 160 µl of chloroform–methanol (1:1), and the organic phase was collected and analyzed by TLC (Merk). The TLC solvent was a mixture of propanol and acetic acid (2 M) (65:35). After drying, TLC plates were subjected to autoradiography.
Activation of Akt, ERK, p70S6K and S6
The amount of phosphorylated Akt was examined by western blotting using the phosphospecific Akt antibody (Ser473; New England Biolabs). Membranes were stripped and then blotted with an anti-Akt antibody raised against peptide including unphosphorylated Ser473 (New England Biolabs). The amount of phosphorylated ERK was evaluated by the phosphospecific ERK antibodies (Tyr204; Santa Cruz) and the anti-ERK2 antibody (Transduction Lab). The activation of 70 kDa ribosomal S6 kinase (p70S6K) was examined by phosphospecific p70S6K antibody (Thr389; New England Biolabs) and the anti-p70S6K antibody (Santa Cruz). The phosphorylation of ribosomal S6 protein was examined using antibody specific for the phosphorylated form of S6 (gift from M.Birnbaum).
Histological analysis
Mice were given 500 U of heparin i.p. 15 min before anesthesia. The mice were anesthetized by i.p. injection of ketamine (Parke-Davis; 50 mg/kg) and xylazine (Loyd Laboratories; 10 mg/kg). The thorax was opened and the LV was punctured by a 25 G needle. The heart was arrested in diastole by the injection of 0.15 ml of cadmium chloride (100 mM) (Li et al., 1997). The inferior vena cava was cut to allow drainage, and mice were perfused first by 10 ml of phosphate-buffered saline and then 30 ml of 3.7% buffered formaldehyde at a speed of 2 ml/min. Mouse tissues were dehydrated and embedded in paraffin and 6 µm sections were cut. Sections were stained with hematoxylin and eosin or Masson trichrome. Photomicrographs were obtained using Zeiss Axiophot microscopes.
Morphometric analysis of isolated cardiac myocytes
The cardiac myocytes were enzymatically dissociated from the mouse heart according to the previously published protocol with minor modifications (Wolska and Solaro, 1996). The hearts from 10- to 12-week-old female transgenic or non-transgenic mice were retrogradely perfused and enzymatically dissociated using 0.3% collagenase. The dissociated myocytes were plated on laminin (10 µg/ml) coated dishes. After 1 h of plating, unattached cells were removed by changing the media. Photographs were taken under a phase microscope. The cell area was measured by tracing the edge of the cell and determining the area inside the outline. The long axis and short axis of the cells were determined by measuring from the two points furthest apart on the edge of the cell and from the two points closest together on the edge of the cell. Morphometric analysis was performed using IPLab software (Scanalytics, Inc.). Some of the dissociated myocytes were also smeared onto positively charged slides, fixed with 3.7% buffered formaldehyde and stained with hematoxylin. The number of nuclei in each myocyte was counted under a light microscope. The measurement of cell size and nuclei number was performed on 100 cells for each heart. The mean values from each heart were used for the statistical analysis.
DNA-laddering assay
Preparation of the low molecular weight DNA was performed according to the method of Frisch and Francis (1994). An aliquot (40 mg) of tissues was homogenized using 1 ml of extraction buffer (0.5% Triton X-100, 5 mM Tris pH 7.5, 20 mM EDTA) and the lysate was incubated for 20 min on ice. The soluble fraction containing low molecular weight DNA was obtained by centrifugation at 15 000 g for 10 min, followed by phenol–chloroform extraction, ethanol precipitated, and resuspended in Tris–EDTA pH 8.0. RNA was digested by 20 µg/ml RNase A for 1 h at 37°C, phenol–chloroform extracted twice, ethanol precipitated, and resuspended in 50 µl of water. A 20 µl aliquot of extracted DNA was end labeled using 25 U of terminal deoxynucleotidyl transferase and 50 µCi of [α-32P]ddATP (Amersham) in a reaction volume of 50 µl for 1 h at 37°C. A 20 µl aliquot of labeled products was electrophoresed on 2% agarose gel, dried and subjected to autoradiography. Thymus tissue from a mouse that received 150 µg of dexamethasone was used as a positive control for DNA laddering (Ahmed and Sriranganathan, 1994). Heart tissues from mice that received i.p. injection of doxorubicin (15 mg/kg) 1 h before they were killed were also used as another positive control (Wang et al., 1998).
Echocardiography
Echocardiographic examination of mice was performed as described (Manning et al., 1994). Briefly, mice were anesthetized with an i.p. injection of ketamine HCl (50 mg/kg) and xylazine (10 mg/kg). Echocardiography was performed in prone decubitus position with a Hewlett-Packard Sonos 1500 sector scanner equipped with a 7.5 MHz phased-array transducer. Two-dimensionally guided M-mode tracings were recorded on strip-chart paper at a paper speed of 100 mm/s. Anterior and posterior wall thickness and LV internal dimensions were measured according to the leading-edge method of the American Society of Echocardiography (Sahn et al., 1978). All measurements were performed with an off-line analysis system (Cardiac Workstation, Freeland Systems) by one observer who was blinded to the genotype of the animals.
Northern hybridization analysis
Total RNA was purified from mouse tissues using the TRIzol Reagent (Life Technologies). A 20 µg aliquot of total RNA was electrophoresed in 1.2% denaturing formaldehyde agarose gels and blotted onto Hybond N (Amersham). The radiolabeled probes were hybridized at 42°C in a hybridization solution (50% deionized formamide, 6× SSC, 5× Denhardt’s solution, 0.5% SDS, 200 µg/ml denatured salmon sperm DNA). Blots were washed twice at room temperature in 2× SSC/1% SDS for 5 min, and twice at 50°C in 2× SSC for 30 min. The probe for mouse ANF was cloned by RT–PCR using mouse heart cDNAs as a template with the following primers (sense, 5′-ATGGGCTCCTTCTCCATCAC; antisense, 5′-TTATCTTCGGTACCGGAAGCTG). RatβMyHC 3′ untranslated region cDNA and mouse αMyHC 3′ untranslated region cDNA were kind gifts from M.Buckingham.
Statistical analysis
Results are presented as mean ± SE. The difference between the groups was compared using the two-tailed unpaired Student’s t-test; p <0.05 was considered as significant.
Acknowledgments
Acknowledgements
We thank J.Robbins, T.Franke and M.Buckingham for cDNA clones, M.Birnbaum for S6 antibody, L.Zhou and K.Converso for the assistance in echocardiography, and D.Fruman and H.Aoki for helpful discussions. This work was supported in part by GM 41890 and SCOR Grant on Atheroscrelosis to L.C.C., and NIH grant AG 44976 to S.I.
References
- Ahmed S.A. and Sriranganathan,N. (1994) Differential effects of dexamethasone on the thymus and spleen: alterations in programmed cell death, lymphocyte subsets and activation of T cells. Immunopharmacology, 28, 55–66. [DOI] [PubMed] [Google Scholar]
- Alessi D.R. and Cohen,P. (1998) Mechanism of activation and function of protein kinase B. Curr. Opin. Genet. Dev., 8, 55–62. [DOI] [PubMed] [Google Scholar]
- Araki E., Lipes,M.A., Patti,M.E., Bruning,J.C., Haag,B.,III, Johnson,R.S. and Kahn,C.R. (1994) Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature, 372, 186–190. [DOI] [PubMed] [Google Scholar]
- Baker J., Liu,J.P., Robertson,E.J. and Efstratiadis,A. (1993) Role of insulin-like growth factors in embryonic and postnatal growth. Cell, 75, 73–82. [PubMed] [Google Scholar]
- Bi L., Okabe,I., Bernard,D.J., Wynshaw-Boris,A. and Nussbaum,R.L. (1999) Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110α subunit of phosphoinositide 3-kinase. J. Biol. Chem., 274, 10963–10968. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch M.A., Glennon,P.E., Andersson,M.B., Clerk,A., Lazou,A., Marshall,C.J., Parker,P.J. and Sugden,P.H. (1994) Endothelin-1 and fibroblast growth factors stimulate the mitogen-activated protein kinase signaling cascade in cardiac myocytes. The potential role of the cascade in the integration of two signaling pathways leading to myocyte hypertrophy. J. Biol. Chem., 269, 1110–1119. [PubMed] [Google Scholar]
- Bohni R., Riesgo-Escovar,J., Oldham,S., Brogiolo,W., Stocker,H., Andruss,B.F., Beckingham,K. and Hafen,E. (1999) Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell, 97, 865–875. [DOI] [PubMed] [Google Scholar]
- Boluyt M.O., Zheng,J.S., Younes,A., Long,X., O’Neill,L., Silverman,H., Lakatta,E.G. and Crow,M.T. (1997) Rapamycin inhibits α1-adrenergic receptor-stimulated cardiac myocyte hypertrophy but not activation of hypertrophy-associated genes. Evidence for involvement of p70 S6 kinase. Circ. Res., 81, 176–186. [DOI] [PubMed] [Google Scholar]
- Brunet A. et al. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell, 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Burgering B.M. and Coffer,P.J. (1995) Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature, 376, 599–602. [DOI] [PubMed] [Google Scholar]
- Cardone M.H., Roy,N., Stennicke,H.R., Salvesen,G.S., Franke,T.F., Stanbridge,E., Frisch,S. and Reed,J.C. (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science, 282, 1318–1321. [DOI] [PubMed] [Google Scholar]
- Chan T.O., Rittenhouse,S.E. and Tsichlis,P.N. (1999) Akt/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem., 68, 965–1014. [DOI] [PubMed] [Google Scholar]
- Chien K.R., Knowlton,K.U., Zhu,H. and Chien,S. (1991) Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. FASEB J., 5, 3037–3046. [DOI] [PubMed] [Google Scholar]
- Chien K.R., Grace,A.A. and Hunter,J.J. (1998) Molecular biology of cardiac hypertrophy and heart failure. In Chein,K.R. (ed.), Molecular Basis of Cardiovascular Disease W.B.Saunders Co., Philadelphia, PA, pp. 211–250. [Google Scholar]
- Chou M.M. and Blenis,J. (1995) The 70 kDa S6 kinase: regulation of a kinase with multiple roles in mitogenic signalling. Curr. Opin. Cell Biol., 7, 806–814. [DOI] [PubMed] [Google Scholar]
- Conlon I. and Raff,M. (1999) Size control in animal development. Cell, 96, 235–244. [DOI] [PubMed] [Google Scholar]
- Conus N.M., Hemmings,B.A. and Pearson,R.B. (1998) Differential regulation by calcium reveals distinct signaling requirements for the activation of Akt and p70S6k. J. Biol. Chem., 273, 4776–4782. [DOI] [PubMed] [Google Scholar]
- Coolican S.A., Samuel,D.S., Ewton,D.Z., McWade,F.J. and Florini,J.R. (1997) The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J. Biol. Chem., 272, 6653–6662. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Dudek,H., Tao,X., Masters,S., Fu,H., Gotoh,Y. and Greenberg,M.E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell, 91, 231–241. [DOI] [PubMed] [Google Scholar]
- DeChiara T.M., Efstratiadis,A. and Robertson,E.J. (1990) A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature, 345, 78–80. [DOI] [PubMed] [Google Scholar]
- Dhand R. et al. (1994) PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J., 13, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath M.Y., Gosteli-Peter,M.A., Hauri,C., Froesch,E.R. and Zapf,J. (1997) Insulin-like growth factor-I stimulates myofibrillar genes and modulates atrial natriuretic factor mRNA in rat heart. Eur. J. Endocrinol., 137, 309–315. [DOI] [PubMed] [Google Scholar]
- Downward J. (1998) Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol., 10, 262–267. [DOI] [PubMed] [Google Scholar]
- Duckworth B.C. and Cantley,L.C. (1997) Conditional inhibition of the mitogen-activated protein kinase cascade by wortmannin. Dependence on signal strength. J. Biol. Chem., 272, 27665–27670. [DOI] [PubMed] [Google Scholar]
- Dufner A., Andjelkovic,M., Burgering,B.M., Hemmings,B.A. and Thomas,G. (1999) Protein kinase B localization and activation differentially affect S6 kinase 1 activity and eukaryotic translation initiation factor 4E-binding protein 1 phosphorylation. Mol. Cell. Biol., 19, 4525–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppenberger-Eberhardt M., Aigner,S., Donath,M.Y., Kurer,V., Walther,P., Zuppinger,C., Schaub,M.C. and Eppenberger,H.M. (1997) IGF-I and bFGF differentially influence atrial natriuretic factor and α-smooth muscle actin expression in cultured atrial compared to ventricular adult rat cardiomyocytes. J. Mol. Cell. Cardiol., 29, 2027–2039. [DOI] [PubMed] [Google Scholar]
- Ernsberger P., Koletsky,R.J., Baskin,J.S. and Collins,L.A. (1996) Consequences of weight cycling in obese spontaneously hypertensive rats. Am. J. Physiol., 270, R864–R872. [DOI] [PubMed] [Google Scholar]
- Fentzke R.C., Korcarz,C.E., Lang,R.M., Lin,H. and Leiden,J.M. (1998) Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J. Clin. Invest., 101, 2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foncea R., Andersson,M., Ketterman,A., Blakesley,V., Sapag-Hagar,M., Sugden,P.H., LeRoith,D. and Lavandero,S. (1997) Insulin-like growth factor-I rapidly activates multiple signal transduction pathways in cultured rat cardiac myocytes. J. Biol. Chem., 272, 19115–19124. [DOI] [PubMed] [Google Scholar]
- Franke T.F., Kaplan,D.R. and Cantley,L.C. (1997a) PI3K: downstream AKTion blocks apoptosis. Cell, 88, 435–437. [DOI] [PubMed] [Google Scholar]
- Franke T.F., Kaplan,D.R., Cantley,L.C. and Toker,A. (1997b) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science, 275, 665–668. [DOI] [PubMed] [Google Scholar]
- Frisch S.M. and Francis,H. (1994) Disruption of epithelial cell–matrix interactions induces apoptosis. J. Cell Biol., 124, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D.A., Meyers,R.E. and Cantley,L.C. (1998) Phosphoinositide kinases. Annu. Rev. Biochem., 67, 481–507. [DOI] [PubMed] [Google Scholar]
- Fruman D.A., Snapper,S.B., Yballe,C.M., Davidson,L., Yu,J.Y., Alt,F.W. and Cantley,L.C. (1999) Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science, 283, 393–397. [DOI] [PubMed] [Google Scholar]
- Gingras A.C., Kennedy,S.G., O’Leary,M.A., Sonenberg,N. and Hay,N. (1998) 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev., 12, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M.N., Lowell,B., Belur,E. and Ruderman,N.B. (1984) Sites of protein conservation and loss during starvation: influence of adiposity. Am. J. Physiol., 246, E383–E390. [DOI] [PubMed] [Google Scholar]
- Gulick J., Subramaniam,A., Neumann,J. and Robbins,J. (1991) Isolation and characterization of the mouse cardiac myosin heavy chain genes. J. Biol. Chem., 266, 9180–9185. [PubMed] [Google Scholar]
- Hayashi K., Saga,H., Chimori,Y., Kimura,K., Yamanaka,Y. and Sobue,K. (1998) Differentiated phenotype of smooth muscle cells depends on signaling pathways through insulin-like growth factors and phosphatidylinositol 3-kinase. J. Biol. Chem., 273, 28860–28867. [DOI] [PubMed] [Google Scholar]
- Hu Q., Klippel,A., Muslin,A.J., Fantl,W.J. and Williams,L.T. (1995) Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science, 268, 100–102. [DOI] [PubMed] [Google Scholar]
- Izumo S. and Shioi,T. (1998) Cardiac transgenic and gene-targeted mice as models of cardiac hypertrophy and failure: a problem of (new) riches. J. Card. Fail., 4, 263–270. [DOI] [PubMed] [Google Scholar]
- Izumo S., Nadal-Ginard,B. and Mahdavi,V. (1988) Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc. Natl Acad. Sci. USA, 85, 339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B.H., Aoki,M., Zheng,J.Z., Li,J. and Vogt,P.K. (1999) Myogenic signaling of phosphatidylinositol 3-kinase requires the serine-threonine kinase Akt/protein kinase B. Proc. Natl Acad. Sci. USA, 96, 2077–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadambi V.J. and Kranias,E.G. (1998) Genetically engineered mice: model systems for left ventricular failure. J. Card. Fail., 4, 349–361. [DOI] [PubMed] [Google Scholar]
- Kimball S.R., Shantz,L.M., Horetsky,R.L. and Jefferson,L.S. (1999) Leucine regulates translation of specific mRNAs in L6 myoblasts through mTOR-mediated changes in availability of eIF4E and phosphorylation of ribosomal protein S6. J. Biol. Chem., 274, 11647–11652. [DOI] [PubMed] [Google Scholar]
- Klippel A., Escobedo,J.A., Hirano,M. and Williams,L.T. (1994) The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity. Mol. Cell. Biol., 14, 2675–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A., Reinhard,C., Kavanaugh,W.M., Apell,G., Escobedo,M.A. and Williams,L.T. (1996) Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol. Cell. Biol., 16, 4117–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leevers S.J., Weinkove,D., MacDougall,L.K., Hafen,E. and Waterfield,M.D. (1996) The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J., 15, 6584–6594. [PMC free article] [PubMed] [Google Scholar]
- Li Q., Li,B., Wang,X., Leri,A., Jana,K.P., Liu,Y., Kajstura,J., Baserga,R. and Anversa,P. (1997) Overexpression of insulin-like growth factor-1 in mice protects from myocyte death after infarction, attenuating ventricular dilation, wall stress, and cardiac hypertrophy. J. Clin. Invest., 100, 1991–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.P., Baker,J., Perkins,A.S., Robertson,E.J. and Efstratiadis,A. (1993) Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell, 75, 59–72. [PubMed] [Google Scholar]
- Manning W.J., Wei,J.Y., Katz,S.E., Litwin,S.E. and Douglas,P.S. (1994) In vivo assessment of LV mass in mice using high-frequency cardiac ultrasound: necropsy validation. Am. J. Physiol., 266, H1672–H1675. [DOI] [PubMed] [Google Scholar]
- Martin S.S., Haruta,T., Morris,A.J., Klippel,A., Williams,L.T. and Olefsky,J.M. (1996) Activated phosphatidylinositol 3-kinase is sufficient to mediate actin rearrangement and GLUT4 translocation in 3T3-L1 adipocytes. J. Biol. Chem., 271, 17605–17608. [DOI] [PubMed] [Google Scholar]
- Miklos G.L. and Rubin,G.M. (1996) The role of the genome project in determining gene function: insights from model organisms. Cell, 86, 521–529. [DOI] [PubMed] [Google Scholar]
- Montagne J., Stewart,M.J., Stocker,H., Hafen,E., Kozuma,S.C. and Thomas,G. (1999) Drosophila S6 kinase: a regulator of cell size. Science, 285, 2126–2129. [DOI] [PubMed] [Google Scholar]
- Ng W.A., Grupp,I.L., Subramaniam,A. and Robbins,J. (1991) Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circ. Res., 68, 1742–1750. [DOI] [PubMed] [Google Scholar]
- Palermo J., Gulick,J., Colbert,M., Fewell,J. and Robbins,J. (1996) Transgenic remodeling of the contractile apparatus in the mammalian heart. Circ. Res., 78, 504–509. [DOI] [PubMed] [Google Scholar]
- Rameh L.E. and Cantley,L.C. (1999) The role of phosphoinositide 3-kinase lipid products in cell function. J. Biol. Chem., 274, 8347–8350. [DOI] [PubMed] [Google Scholar]
- Sadoshima J. and Izumo,S. (1995) Rapamycin selectively inhibits angiotensin II-induced increase in protein synthesis in cardiac myocytes in vitro. Potential role of 70-kD S6 kinase in angiotensin II-induced cardiac hypertrophy. Circ. Res., 77, 1040–1052. [DOI] [PubMed] [Google Scholar]
- Sadoshima J. and Izumo,S. (1997) The cellular and molecular response of cardiac myocytes to mechanical stress. Annu. Rev. Physiol., 59, 551–571. [DOI] [PubMed] [Google Scholar]
- Sahn D.J., DeMaria,A., Kisslo,J. and Weyman,A. (1978) Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation, 58, 1072–1083. [DOI] [PubMed] [Google Scholar]
- Serunian L.A., Auger,K.R. and Cantley,L.C. (1991) Identification and quantification of polyphosphoinositides produced in response to platelet-derived growth factor stimulation. Methods Enzymol., 198, 78–87. [DOI] [PubMed] [Google Scholar]
- Shima H., Pende,M., Chen,Y., Fumagalli,S., Thomas,G. and Kozma,S.C. (1998) Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J., 17, 6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soonpaa M.H. and Field,L.J. (1998) Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ. Res., 83, 15–26. [DOI] [PubMed] [Google Scholar]
- Soonpaa M.H., Kim,K.K., Pajak,L., Franklin,M. and Field,L.J. (1996) Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol., 271, H2183–H2189. [DOI] [PubMed] [Google Scholar]
- Soonpaa M.H., Koh,G.Y., Pajak,L., Jing,S., Wang,H., Franklin,M.T., Kim,K.K. and Field,L.J. (1997) Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J. Clin. Invest., 99, 2644–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden P.H. and Clerk,A. (1998) Cellular mechanisms of cardiac hypertrophy. J. Mol. Med., 76, 725–746. [DOI] [PubMed] [Google Scholar]
- Takata M. et al. (1999) Requirement for Akt (protein kinase B) in insulin-induced activation of glycogen synthase and phosphorylation of 4E-BP1 (PHAS-1). J. Biol. Chem., 274, 20611–20618. [DOI] [PubMed] [Google Scholar]
- Tamemoto H. et al. (1994) Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature, 372, 182–186. [DOI] [PubMed] [Google Scholar]
- Thomas G. and Hall,M.N. (1997) TOR signalling and control of cell growth. Curr. Opin. Cell Biol., 9, 782–787. [DOI] [PubMed] [Google Scholar]
- Toker A. and Cantley,L.C. (1997) Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature, 387, 673–676. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B., Leevers,S.J., Panayotou,G. and Waterfield,M.D. (1997) Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem. Sci., 22, 267–272. [DOI] [PubMed] [Google Scholar]
- Verdu J., Buratovich,M.A., Wilder,E.L. and Birnbaum,M.J. (1999) Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nature Cell Biol., 1, 500–506. [DOI] [PubMed] [Google Scholar]
- Wakasaki H., Koya,D., Schoen,F.J., Jirousek,M.R., Ways,D.K., Hoit, B.D., Walsh,R.A. and King,G.L. (1997) Targeted overexpression of protein kinase C β2 isoform in myocardium causes cardiomyopathy. Proc. Natl Acad. Sci. USA, 94, 9320–9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Ma,W., Markovich,R., Chen,J.W. and Wang,P.H. (1998) Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor I. Circ. Res., 83, 516–522. [DOI] [PubMed] [Google Scholar]
- Weinkove D., Neufeld,T.P., Twardzik,T., Waterfield,M.D. and Leevers,S.J. (1999) Regulation of imaginal disc cell size, cell number and organ size by Drosophila class I(A) phosphoinositide 3-kinase and its adaptor. Curr. Biol., 9, 1019–1029. [DOI] [PubMed] [Google Scholar]
- Weng Q.P., Kozlowski,M., Belham,C., Zhang,A., Comb,M.J. and Avruch,J. (1998) Regulation of the p70 S6 kinase by phosphorylation in vivo. Analysis using site-specific anti-phosphopeptide antibodies. J. Biol. Chem., 273, 16621–16629. [DOI] [PubMed] [Google Scholar]
- Withers D.J. et al. (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature, 391, 900–904. [DOI] [PubMed] [Google Scholar]
- Wolska B.M. and Solaro,R.J. (1996) Method for isolation of adult mouse cardiac myocytes for studies of contraction and microfluorimetry. Am. J. Physiol., 271, H1250–H1255. [DOI] [PubMed] [Google Scholar]
- Yoshizumi M. et al. (1995) Disappearance of cyclin A correlates with permanent withdrawal of cardiomyocytes from the cell cycle in human and rat hearts. J. Clin. Invest., 95, 2275–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Zhang,Y., McIlroy,J., Rordorf-Nikolic,T., Orr,G.A. and Backer,J.M. (1998) Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110α catalytic subunit by the p85 regulatory subunit. Mol. Cell. Biol., 18, 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]