

Abstract
A solid tumor forms an organ-like entity comprised of neoplastic cells and non-transformed host stromal cells embedded in an extracellular matrix. Similar to normal tissues, blood vessels nourish cells residing in tumors. However, unlike normal blood vessels, tumor vasculature has abnormal organization, structure, and function. Tumor vessels are leaky and blood flow is heterogeneous and often compromised. Vascular hyperpermeability and the lack of functional lymphatic vessels inside tumors cause elevation of interstitial fluid pressure in solid tumors. Each of these abnormalities forms a physiological barrier to the delivery of therapeutic agents to tumors. Furthermore, elevated tumor interstitial fluid pressure increases fluid flow from the tumor margin into the peri-tumor area and may facilitate peri-tumor lymphatic hyperplasia and metastasis. Abnormal microcirculation in tumors also leads to a hostile microenvironment characterized by hypoxia and acidosis, which hinder the effectiveness of anti-tumor treatments such as radiation therapy and chemotherapy. In addition, host-tumor interactions regulate expression of pro- and anti-angiogenic factors and hence contribute to their imbalance and resulting pathophysiological characteristics of the tumor. Restoration of pro- and anti-angiogenic balance in tumors may “normalize” tumor vasculature and thus improve its function. Indeed, anti-angiogenic treatments directly targeting angiogenic signaling pathways as well as indirectly modulating angiogenesis show normalization of tumor vasculature and microenvironment at least transiently in both preclinical and clinical settings. Combination of cytotoxic therapy and anti-angiogenic treatment during the vascular normalization exhibits synergistic effect.
Keywords: Angiogenesis, lymphangiogensis, tumor, stromal cells, vascular endothelial growth factor, microenvironment, hypoxia, acidosis, interstitial fluid pressure, metastasis
Since solid tumors require angiogenesis for their growth and metastasis, anti-angiogenic therapy has been extensively studied in both preclinical and clinical settings (Folkman, 2000). While these anti-angiogenic drugs have been approved for cancer treatment, it appears that clinical application of anti-angiogenic therapy is more complex than originally thought (Jain et al., 2006). The mechanisms of action of anti-angiogenic therapy are yet to be sorted out. Therefore, it is important to understand the biology of the tumor vasculature – the target of anti-angiogenic therapy. Tumor vasculature is not a simple supply line of nutrients to tumors. It governs pathophysiology of solid tumors and thus, tumor growth, invasion, metastasis and response to various therapies. In this review, we will discuss structure and function of tumor vasculature, the resulting abnormal microenvironment, causes and consequences of these abnormalities, regulation of angiogenic factor expression by host-tumor interactions, and potential normalization of these abnormalities by anti-angiogenic therapy.
PATHOPHYSIOLOGICAL CHARACTERISTICS OF SOLID TUMORS
Abnormal Blood Vessel Architecture and Function in Tumors
The normal microvessels consist of arterioles, capillaries and venules, and form a well-organized, regulated and functional architecture (Figure 1A) (Jain, 2003). In contrast, tumor vessels are dilated, saccular, tortuous, and heterogeneous in their spatial distribution (Figure 1A) (Jain, 1988). Normal vasculature is characterized by dichotomous branching, but tumor vasculature is unorganized and has trifurcations and branches with uneven diameters. Vessel wall structure is also abnormal in tumors (Chang et al., 2000; di Tomaso et al., 2005; McDonald and Choyke, 2003). Large inter-endothelial junctions, increased numbers of fenestrations, vesicles and vesico-vacuolar channels, and a lack of normal basement membrane are often found in tumor vessels (Dvorak et al., 2002; Winkler et al., 2004). Perivascular cells have abnormal morphology and heterogeneous association with tumor vessels. The molecular mechanisms causing these abnormal vascular architectures are not well understood, but the imbalance of pro- and anti-angiogenic factors is considered to be a key contributor (Jain, 2005). Solid (mechanical) stress generated by proliferating tumor cells also compresses vessels in tumors (Padera et al., 2004; Roose et al., 2003). The combination of both molecular and mechanical factors may render the tumor vasculature abnormal.
Figure 1. Abnormal vasculature and microenvironment in tumors.

(A) Multiphoton laser-scanning microscopy image of normal blood vessels (left) and tumor vessels in LS174T human colon cancer xenografts (right) in mouse dorsal skin chambers. Blood vessels are contrast enhanced by FITC-dextran. Images are 550 μm across. (B) Microvascular permeability determined by intravital microscopy. Left, vascular permeability to dextran of 150,000 molecular weight in non-maligant (mature granulation) and neoplastic (VX2 carcinoma) tissues grown in the rabbit ear chamber. Right, vascular permeability to liposome with diameter between 86 and 90 nm in normal subcutaneous tissues (not detectable) and LS174T tumor tissues in the dorsal skin chambers. (C) Blood flow determined by intravital microscopy in normal pial vessels (maximum bead velocities, left) and tumor vessels in MCaIV murine breast cancer and U87 human glioma (RBC velocities, right) in mouse cranial windows. (D) Fluorescence microlymphangiography with FITC-dextran in mouse tail 28 days after the implantation of FSaII murine fobrosarcoma (left part of the image). The bar indicates 400 μm. (E) Mean interstitial pH and pO2 profiles in LS174T tumors in the dorsal skin chambers as one moves away from the nearest blood vessels. Tissue pO2 and pH were determined by phosphorescence quenching microscopy with a porphyrin probe and fluorescence ratio-imaging microscopy with BCECF, respectively. Open symbols, pH; closed symbols, pO2. Scale corresponds to distance from the vessel wall. (F) VEGF promoter activity (green), tissue pO2 (blue) and pH (red) in U87 tumors. Left, intravital microscopy image of GFP driven by VEGF promoter. The three parameters are determined along the yellow line. Center, this tumor is well oxygenated and there is no correlation between tissue pO2 and VEGF promoter activity. Right, on the other hand, the peak of VEGF promoter activity is observed in acidic pH region. A, courtesy of Dr. Edward Brown; B, data from (Gerlowski and Jain, 1986; Yuan et al., 1994a); C, adapted from (Yuan et al., 1994b); D, adapted from (Leu et al., 2000); E, adapted from (Helmlinger et al., 1997); F, adapted from (Fukumura et al., 2001).
Extravasation of molecules from the bloodstream occurs by diffusion, convection, and, to some extent, by transcytosis in an exchange vessel. Diffusion is considered to be the major form of transvascular transport in tumors (Jain, 1987). The diffusive permeability of a molecule depends on its size, shape, charge, and flexibility as well as the transvascular transport pathway. In agreement with the above-mentioned ultrastructural alterations in the tumor vessel wall, vascular permeability in solid tumors is generally higher than that in various normal tissues (Figure 1B).
Arterio-venous pressure difference and flow resistance govern blood flow in a vascular network. Flow resistance is a function of geometric (vascular architecture) and viscous (blood viscosity, rheology) resistances. Abnormalities in both vasculature and viscosity increase the resistance to blood flow in tumors (Jain, 1988). Focal leaks, which often exist in some of the tumor vessels, may also compromise the downstream blood flow. As a result, overall perfusion rates (blood flow rate per unit volume) in tumors are lower than in many normal tissues and the average RBC velocity in tumor vessels can be an order of magnitude lower than in normal vessels (Figure 1C). Unlike normal vessels, RBC velocity is independent of the diameter of tumor vessels. Furthermore, tumor blood flow is unevenly distributed, fluctuates with time and can even reverse its direction in some vessels – therefore regions with poor perfusion, or none at all, are commonly seen. The heterogeneity of tumor blood flow causes abnormal microenvironment in tumors and hinders the delivery and efficacy of therapeutic agents to tumors.
Interstitial Hypertension and Abnormal Lymphatics in Tumors
Both animal and human tumors exhibit interstitial hypertension while the interstitial fluid pressure (IFP) in normal tissues is around zero mmHg (Fukumura and Jain, 2007; Jain et al., 2007). The abnormal structure and function of blood and lymphatic vessels in tumors cause the IFP elevation. Tumor vessels lack perm selectivity due to the high vascular permeability. As a result, the hydrostatic and oncotic (colloid osmotic) pressures become almost equal between the intravascular and extravascular spaces (Boucher and Jain, 1992; Tong et al., 2004). In fact, tumor IFP equivalently increases or decreases in ~ 10 s after the modification of microvascular pressure (Netti et al., 1995). Reduced transmural pressure gradients decrease convection across tumor vessel walls. Furthermore, IFP is uniformly high throughout a tumor and drops precipitously in the tumor margin (Boucher et al., 1990). Fluid convection or bulk flow is negligible inside tumors due to the lack of interstitial pressure gradients. Thus, the uniformly elevated IFP compromises the delivery of therapeutic agents both across the blood vessel wall and interstitum in tumors. Furthermore, transmural coupling between IFP and microvascular pressure due to the high permeability of tumor vessels can abolish pressure difference between up and down stream of tumor blood vessels and leads to blood flow stasis in tumors without physically occluding the vessels. On the other hand, the interstitial fluid oozes out from the tumor periphery into the surrounding normal tissue, carrying angiogenic, lymphangiogenic growth factors and/or metastasizing tumor cells with it (Jain et al., 2007). This may facilitate tumor invasion and metastasis.
The normal lymphatic network maintains tissue interstitial fluid balance by draining excess fluid out the tissue. Proliferating tumor cells in a confined space create mechanical stress (solid stress) which compresses intra-tumor lymphatic vessels (Padera et al., 2004). Consequently, there are no functional lymphatic vessels inside solid tumors (Figure 1D) (Leu et al., 2000; Padera et al., 2002). Even if the structures with lymphatic endothelial marker are present in tumors, they do not transport fluid or macromolecules. The absence of functional lymphatics is another key contributor of elevated tumor IFP. Indeed, the placement of an “artificial lymphatics” could lower the IFP in tumors (DiResta et al., 2000). In contrast to the lack of functional intra-tumor lymphatics, functional lymphatic vessels are present in the tumor margin and the peri-tumoral tissue. These peripheral lymphatic vessels are hyperplastic, collect fluid, growth factors and cells exiting from tumors, and mediate metastases via the lymphatic system (Figure 2) (Hoshida et al., 2006; Padera et al., 2002).
Figure 2. Steps of lymphatic metastasis and the effect of anti-lymphangiogenic treatment.

Elevated tumor IFP increases interstitial fluid flow at the tumor margin (Jain et al., 2007). The exudates, which contain fluid, protein, and cells from tumors, are collected in the peritumoral lymphatic vessels. Although the mechanical signals that could trigger the lymphangiogenic switch are not known, hydrostatic pressure is likely to be such trigger (Boardman and Swartz, 2003). Furthermore, many tumors express lymphangiogenic factors such as VEGF-C (Alitalo et al., 2005). (A) FITC-dextran microlymphangiography of peritumor and normal lymphatics in mouse ears. Bar, 850 μm. MT, mock transduced. VEGF-C, VEGF-C overexpressing. Lymphatic vessels are hyperplastic in the peritumor region and ear base (further down stream) of the T241 fibrosarcomas. VEGF-C expression further increases lymphatic vessel diameter. Number of lymphatic vessels is also increased in the tumor periphery especially in the presence of excess VEGF-C (Isaka et al., 2004). Malformed lymphatic valves allow retrograde flow in these lymphatic vessels and facilitate transfer of metastasizing tumor cells, which can be visualized by intravital microscopy after the transfection of GFP expression vector (Hoshida et al., 2006). (B) Then GFP-positive tumor cells (green) are observed entering the cervical lymph node from afferent lymphatic (red, arrow) by MPLSM. Bar, 100 μm. VEGF-C overexpression significantly increases arrival of tumor cells to the lymph node. Macroscopic lymph node metastasis increases with the increase of tumor cell arrival to the lymph node (Hoshida et al., 2006). (C) The treatment with anti-VEGF receptor 3 antibody (mF4-31C1) significantly reduces peritumor and ear base lymphatic vessel size. (D) Tumor cells cannot reach to the cervical lymph node under the anti-VEGFR3 treatment. As a result, macroscopic lymph node is significantly reduced by anti-VEGFR3 treatment if it is given before the arrival of tumor cells to the lymph node (Hoshida et al., 2006). A–C, adapted or reproduced from (Hoshida et al., 2006).
Interstitial hypertension is a reflection of the global pathophysiology of solid tumors and may be used for diagnosis, prognosis and/or monitoring of treatment responses. In hyperplastic/dysplastic stages of a mouse model of spontaneous skin carcinogenesis, IFP is already elevated, accompanied by angiogenic vasculature that exhibits increased permeability and decreased perivascular cell coverage as well as partly compressed lymphatic vessels (Hagendoorn et al., 2006). Abnormal blood and lymphatic vascular functions with resultant interstitial hypertension in pre-malignant lesions can be used for early detection and may form a barrier to treatment similar to an established tumor. In the established tumors, the steep rise of IFP at the tumor periphery may be used to locate tumors during needle biopsy and improve diagnosis of patients (Jain et al., 1995). Furthermore, a study of cervical cancer has shown that elevated tumor IFP can predict poor outcome of radiation therapy (Jain, 2004). Thus, studies are warranted to monitor IFP in human tumors during various treatments (Willett et al., 2004).
METABOLIC ENVIRONMENT IN SOLID TUMORS
Causes and Consequences of Abnormal Metabolic Environment in Tumors
Hypoxia and acidosis are hallmarks of the abnormal metabolic environment in solid tumors (Figure 1E) (Harris, 2002; Helmlinger et al., 1997; Tatum et al., 2006). A key function of the vasculature is to provide adequate levels of nutrients and oxygen to the parenchymal cells and to remove waste products. Tumor vessels fail to do so adequately due to their abnormal structure and function. The imbalance of vascular network development and tumor cell proliferation results in the formation of hypovascular regions in tumors. Since tissue diffusion limit of oxygen is 100–200 μm (Krogh, 1922), the regions far from blood vessels become chronically hypoxic (Figure 1E). Furthermore, the existence of blood vessels does not guarantee tissue oxygenation in tumors. Blood flow in tumor vessels is often intermittent, and, thus, some regions of a tumor are starved for oxygen periodically, a phenomenon called “acute hypoxia” or “perfusion-limited hypoxia” (Brown and Giaccia, 1998; Dewhirst, 1998). In addition, high-resolution intravital microscopy and phosphorescence quenching microscopy revealed that there is no clear relationship between blood flow rate and oxygen tension (pO2) of individual tumor vessels (Helmlinger et al., 1997). Surprisingly, some perfused tumor vessels carry almost no oxygen. Prolonged transit time of the blood in tumors due to the heterogeneous, disorganized, dysfunctional vessel network in tumors and the imbalance of oxygen supply and consumption may cause tumor tissue hypoxia despite the presence of blood flow.
Low extracellular pH is another consequence of the abnormal microcirculation in the tumor. Lactic acid and carbonic acid are the known sources of H+ ions in tumors (Helmlinger et al., 2002; Pouyssegur et al., 2006). The former is the product of anaerobic glycolysis and the latter is formed from CO2 and H2O by carbonic anhydrase. Increased production of H+ ions and their reduced removal lower extracellular pH in tumors. The mean pH profiles also decreased in tumors with increasing distance from nearest blood vessels (Figure 1E). Interestingly, the mean pH profile exhibits a plateau phase between 100 μm and 170 μm away from blood vessel wall despite constant decrease in pO2 presumably because of lack of glucose availability and limited glycolysis. One would expect low extracellular pH and hypoxia to track each other and to co-localize with regions of poor blood flow. Surprisingly, optical microscope techniques that permit the simultaneous high-resolution mapping of multiple physiological parameters revealed that there is a lack of spatial correlation among these parameters (Helmlinger et al., 1997). As discussed above, some perfused tumor vessels may not be able to deliver adequate oxygen to the surrounding cells, although they may be able to carry away the waste products (e.g., lactic acid). Such discrepancy may cause the lack of concordance of the metabolic environment parameters in tumors (Helmlinger et al., 1997).
Both pO2 and pH are important determinants of tumor growth, metabolism, and response to a variety of therapies. Oxygen is an important component of radiation therapy (Brown, 1999). Hypoxia in solid tumors significantly reduces their radiation sensitivity. Tumor hypoxia is also associated with resistance to some chemotherapeutics such as bleomycin and neocarzinostatin (Brown, 1999). Likewise acidic extracellular pH hinders the cellular uptake of weak base drugs such as adriamycin, doxorubicin and mitoxantrone and thus, their efficacy (Vukovic and Tannock, 1997). Immune cells targeting tumor cells are not fully functional under hypoxic and/or acidic conditions and thus, allow tumors to evade the host immune response and cell based therapies. Furthermore, exposure to hypoxia and/or acidic pH renders tumor cells to be highly invasive and metastatic (Erler et al., 2006; Pennacchietti et al., 2003; Rofstad et al., 2006). The hostile metabolic environment in tumors selects tumor cells that are more malignant, aggressive and genetically unstable, and less susceptible to apoptosis, thus rendering them resistant to various therapies. Finally, both hypoxia and acidic pH can induce expression of angiogenic factors and thus, contribute to growth and metastasis of tumors (Fukumura, 2005).
Regulation of angiogenic gene expression by metabolic microenvironment
Hypoxia upregulates various angiogenic growth factors, including vascular endothelial growth factor (VEGF), angiopoietin (Ang) 2, platelet derived growth factor (PDGF), Placenta growth factor (PlGF), transforming growth factor α (TGFα), interleukin (IL)-8, and hepatocyte growth factor (HGF) (Harris, 2002). Of the various molecules involved in sensing and responding to hypoxia, hypoxia inducible factor 1α (HIF1α) is considered to be the master regulator of oxygen homeostasis (Semenza, 2003). This transcription factor is upregulated in a number of human tumors (Harris, 2002). HIF1α binds to the hypoxia responsive element (HRE) in the promoter hypoxia-responsive genes such as VEGF, PDGF and TGFα and induces their expression (Harris, 2002; Semenza, 2003). A few other factors, such as IL-8 and PlGF, are activated by HIF-independent mechanisms (Harris, 2002; Xu et al., 2004). Hypoxia may also play an important role in the angiogenic switch that is required for tumor growth and expansion.
Low extracellular pH causes stress-induced alteration of gene expression, including the upregulation of VEGF and IL-8 in tumor cells in vitro (Xu et al., 2002). Despite its importance, the effect of the low and heterogeneous interstitial pH on VEGF expression in vivo, especially in relationship with hypoxia remained unknown for many years due to the lack of appropriate techniques and animal models. The combination of fluorescence ratio imaging microscopy for pH measurements (Martin and Jain, 1993), phosphorescence quenching microscopy for pO2 measurements (Torres-Filho et al., 1994) and the transgenic technology for visualization of VEGF promoter activity (Fukumura et al., 1998) have allowed the coordinated study of pH, pO2, and VEGF expression in vivo (Figure 1F) (Fukumura et al., 2001). Detailed analysis indicated that in low pH or oxygenated regions, tissue pH, but not pO2, regulates VEGF promoter activity. Conversely, in hypoxic or neutral pH regions, tissue pO2 and not pH regulates VEGF expression (Fukumura et al., 2001). Tissue pO2 and pH appeared to regulate VEGF transcription in tumors independently. In fact, the analysis of the VEGF promoter region revealed that acidic pH induces VEGF expression via Ras-ERK1/2-AP1 pathway but not the HIF-HRE mediated pathway (Xu et al., 2002). Taken together these data suggest that two key microenvironmental parameters in solid tumors regulate angiogenic factors in a complementary manner.
ROLE OF HOST-TUMOR INTERACTION IN TUMOR ANGIOGENESIS
Involvement of host stromal cells in tumor angiogenesis
It is becoming increasingly apparent that the development and pathophysiology of a tumor cannot be explained simply by the genes in the tumor cells (Weinberg, 2006). We are beginning to understand that host stromal cells profoundly influence many steps of tumor progression, such as tumor cell proliferation, invasion, angiogenesis, metastasis, and even malignant transformation (Elenbaas and Weinberg, 2001; Fukumura et al., 1998; Li et al., 2003; Liotta and Kohn, 2001; Pollard, 2004; Ruiter et al., 2001; Tlsty, 2001). Interactions between the diverse cell types within a tumor, via both soluble factors and direct cell-to-cell contact, play an important role in the induction, selection, and expansion of the neoplastic cells. Successful tumor cells are those that have acquired the ability to co-opt their normal neighbors by inducing them to release abundant fluxes of growth-stimulating signals (Li et al., 2003; Tlsty, 2001; Weinberg, 2006).
Intravital observation of tumors grown in a GFP reporter mouse revealed that stromal fibroblasts express VEGF in tumors especially abundantly at the host-tumor interface (Fukumura et al., 1998) (Figure 3A). Furthermore, VEGF-expressing stromal cells co-localize with the vasculature and even surround tumor blood vessels deep inside the tumor (Brown et al., 2001b) (Figure 3A). These findings suggest that activated fibroblasts are involved in angiogenesis, fortification of blood vessels, and function of these vessels. In fact, co-implantation of fibroblasts enhanced the tumorigenicity of breast cancer cells in vivo (Noel et al., 1993). Carcinoma associated fibroblasts promote tumor growth and angiogenesis through secretion of SDF-1/CXCL12 (Orimo et al., 2005). In addition to fibroblasts, inflammatory cells recruited to tumors may also promote (rather than eliminate) angiogenesis and tumor cell growth (de Visser et al., 2006; Pollard, 2004). Some studies show the relative contribution of stromal cells and tumor cells in the expression of VEGF in tumors. For an example, teratomas derived from VEGF deficient embryonic stem cells (ES cells) show VEGF levels and angiogenic activity about one-half of those in the tumors derived from wild-type ES cells suggesting that host stromal cells can produce approximately half of the total VEGF in this tumor type (Tsuzuki et al., 2000). On the other hand, late stage orthotopic pancreatic tumors expressed significantly higher tumor cell-derived VEGF compared to early stage or ectopically-grown tumors (Tsuzuki et al., 2001). The ratio of tumor/host-derived VEGF and other growth factors may vary depending on tumor type, stage, and organ site.
Figure 3. Role of host-tumor interaction in angiogenesis and vessel function.

(A) Imaging of VEGF promoter activity in host stromal cells. MCaIV murine breast tumor is grown in the dorsal skin chamber of a transgenic mouse expressing GFP under the control of VEGF promoter. Left, activated fibroblasts exhibit strong VEGF promoter activity (green) at the host-tumor interface. Right, VEGF expressing host cells associate with blood vessels (red) inside tumors. Images are 733 μm across. (B) Microangiography of B16F10 murine melanoma grown in the cranial window (left) and dorsal skin chamber (right). Images are 3.75 mm across. (C) Vessel morphology in B16 melanomas grown in the dorsal skin chamber (DSC) and cranial window (CW) determined by intravital microscopy. (D) Pore cut-off size of tumor vessels in MCaIV tumors and murine hepatoma HCaI grown in the dorsal skin chamber (S.C.) and cranial window (Cranial) are determined by intravital microscopy after the injection of different size fluorescent nanoparticles. Filled and open circles indicate negative and positive extravasation, respectively. (E) Vascular permeability to BSA in normal blood vessels before (Pre) and after (Post) superfusion with VEGF. A, adapted from (Brown et al., 2001a); C adapted from (Kashiwagi et al., 2005); D, adapted from (Hobbs et al., 1998); E, adapted from (Monsky et al., 1999).
Regulation of angiogenesis and vessel functions by organ microenvironment
Angiogenesis and functions of resultant vessels differ significantly between the same tumors grown in different host organs (Fidler, 2001; Jain et al., 2002). For example, murine melanomas grown in a cranial window have higher vessel density and branching, and relatively smaller vessel size as compare to those in the same tumors grown in a dorsal skin chamber (Figure 3B, 2C) (Kashiwagi et al., 2005). A human glioma (HGL21) has fairly leaky vessels when grown subcutaneously in immunodeficient mice, but it exhibits blood-brain barrier properties in the cranial window (Jain, 1997). Furthermore, the vascular pore cut-off size (the maximum functional pore size for trans-vascular transport of macromolecules through the vessel wall) in various tumors decreased when the tumors were grown in the cranial window as compared to the dorsal skin chamber (Figure 3D) (Hobbs et al., 1998). Organ specific upregulation of angiogenic factors is one of the mechanisms causing differential angiogenic activity. In agreement with the higher angiogenic activity (Figure 3B, 3C), the murine melanomas exhibit higher tissue nitric oxide levels when grown in the cranium compared to those in the same tumors grown subcutaneously (Kashiwagi et al., 2005). Human renal cell carcinoma xenografts grown orthotopically in the kidneys of immunodeficient mice were highly vascularized and metastatic, and expressed 10- to 20-fold higher levels of bFGF mRNA than those from the same tumor grown subcutaneously (Singh et al., 1994). Human colon cancer and melanoma grown in the liver expressed lower levels of VEGF and IL-8 mRNA, respectively and had a lower vessel density than those in subcutaneous tissue (Fukumura et al., 1997; Gutman et al., 1995). The expression of endogenous anti-angiogenic factors is also regulated by organ specific host-tumor interaction. Human gall bladder primary tumors inhibit angiogenesis and growth of secondary tumors at a distant site in a TGFβ1–dependent mechanism. This result was observed when the primary tumors were grown in the gall bladder (orthotopic) but not in the subcutaneous space (ectopic) (Gohongi et al., 1999).
Furthermore, the response of the blood vessels to a given stimuli may also vary depending on the host organ site and host-tumor interaction. With the presence of a blood-brain-barrier, a significantly higher amount of VEGF was required to induce vascular hyperpermeability in normal vessels in the cranial window than in the dorsal skin chamber (Figure 3E) (Monsky et al., 1999). On the other hand, the cranial environment is more angiogenic and forms new vessels faster than the subcutaneous tissue in response to a given angiogenic factor such as VEGF and bFGF (Dellian et al., 1996). These differences are presumably due to differences in the phenotype of vascular endothelial cells, which is defined by their origin, by cell-cell and cell-matrix interactions, and by the local microenvironment. These findings indicate that VEGF level alone may not be a sufficient predictor of angiogenesis or vascular permeability in the tumors growing in different organs. Indeed, the vascular permeability of LS174T human colon cancer grown in the liver versus subcutaneous space was inversely correlated with the expression levels of VEGF at these sites, while angiogenesis paralleled the VEGF levels (Fukumura et al., 1997). Conversely, higher VEGF expression and permeability but lower angiogenesis were observed in ZR75 human breast cancers grown in the mammary fat pad (primary site) compared to those grown in the cranial window (metastatic site) (Monsky et al., 2002). Knowledge of organ-dependent profiles of gene expression and protein level as well as responsiveness to these factors, in stromal cells and tumor cells from different organ microenvironments, will provide new insight into tumor biology and should allow us to understand why a given tumor behaves differently in different organs.
NORMALIZATION OF TUMOR VASCULATURE AND MICROENVIRONMENT BY ANTI-ANGIOGENIC THERAPIES
Normalization by targeting VEGF signaling
As discussed earlier excess production of pro-angiogenic molecules and/or diminished production of anti-angiogenic molecules may cause the abnormalities in vessels and microenvironment in tumors resulting in insufficient drug delivery and therapeutic efficacy (Jain, 2005). If one can restore the balance of pro- and anti-angiogenic factors, the vasculature might revert back to a more “normal” state (Jain, 2001; Jain, 2005). Targeting angiogenic signaling such as provided by VEGF, which is overexpressed in the majority of solid tumors, can be a potential strategy to reverse some of the abnormalities. Indeed, neutralizing antibodies against VEGF and its receptor 2 as well as VEGF receptor tyrosine kinase inhibitors have been shown to improve tumor vasculature and microenvironment in various tumor models. Bevacizumab (anti-VEGF antibody) and DC101 (anti-VEGFR2 antibody) prune some tumor vessels and remodel the remaining vasculature in human colon cancer and glioma xenografts as well as murine breast cancers so that it more closely resembles the normal vasculature (Figure 4A) (Tong et al., 2004; Winkler et al., 2004; Yuan et al., 1996). The anti-VEGF treatments reduce the size and length as well as permeability of these abnormally dilated and tortuous vessels (Figure 4B) (Tong et al., 2004; Winkler et al., 2004; Yuan et al., 1996). The “normalized” vasculature during the anti-VEGFR2 treatment has greater coverage of perivascular cells (Figure 4C) and a more normal thickness of basement membrane in breast cancers, squamous cell carcinomas and gliomas (Tong et al., 2004; Vosseler et al., 2005; Winkler et al., 2004). These changes in tumor vasculature are accompanied by normalization of the tumor microenvironment. Bevacizumab, DC101 and SU11657 (small molecule inhibitor of VEGFRs and PDGFR) decrease tumor IFP in breast, colon cancers and gliomas (Huber et al., 2005; Lee et al., 2000; Tong et al., 2004). Decreased IFP restores pressure gradient across blood vessel wall as well as tumor interstitum and thus, increases drug penetration in tumors (Jain et al., 2007; Tong et al., 2004; Wildiers et al., 2003). Both Bevacizumab and DC101 improve tumor tissue oxygenation in human glioma xenografts (Figure 4D) (Lee et al., 2000; Winkler et al., 2004). As a result, the efficacy of radiation treatments is significantly improved when combined with anti-VEGF treatments (Figure 4E) (Kozin et al., 2001; Lee et al., 2000; Winkler et al., 2004). However, anti-VEGF treatment-induced vascular normalization may be transient. Eight days after the start of anti-VEGFR2 treatment, excess reduction of vessel density and reappearance of abnormal feature of tumor vasculature such as lack of perivascular cell coverage and hypoxia are observed in a glioma model, although vessel diameter and permeability remained low (Figure 4B, C, D) (Winkler et al., 2004). The combination of anti-VEGFR2 treatment and radiation therapy delayed tumor growth synergistically only when ionizing radiation was given during this “normalization window” (Figure 4E) (Winkler et al., 2004).
Figure 4. Normalization of tumor vasculature and microenvironment by anti-VEGFR2 treatment.

(A) Normal microvessels (left) and U87 tumor vessels pre (center) and 2 days after anti-VEGFR2 antibody (DC101) treatment (right) in the mouse brain visualized by MPLSM through cranial window. (B) U87 tumor vessel morphology and function during anti-VEGFR2 treatment determined by intravital microscopy. (C) Immunohistochemical analysis of NG2-positive perivascular cell (red) coverage of U87 tumor vessels (green) during anti-VEGFR2 treatment. (D) Hypoxic fraction of U87 tumor tissues determined by a redox marker (pimonidazole) staining. (E) U87 tumor growth delay by anti-VEGFR2 (DC101), radiation (RT), and the combination (RT1-RT5) treatments compared to control tumor growth (C). Inset shows schedules of the combined treatments. Fractionate radiation (8Gy × 3 days) is given starting at 9 days before (RT1), 2 days before (RT2), 1 day after (RT3), 4 days after (RT4), or 7 days after (RT5) the start of DC101 treatment. A–E, adapted or reproduced from (Winkler et al., 2004).
Normalization by indirect anti-angiogenic therapy
Similar normalization has been observed following indirect anti-angiogenic treatments, which target upstream signaling of angiogenic factors or modulate response to angiogenic factors and are readily available in clinic. The blockade of human epidermal growth factor receptor (HER)-2 signaling by a neutralizing antibody trastuzumab (Herceptin) downregulates angiogenic factors such as VEGF, TGFα, Ang1, and PAI-1, and also induces anti-angiogenic factor thrombospondin-1, thus, Herceptin acts as an anti-angiogenic cocktail against HER2 expressing tumors (Izumi et al., 2002b). Indeed, Herceptin normalizes the vasculature of HER2 overexpressing human breast cancer xenografats (Figure 5A) (Izumi et al., 2002b). Whereas vessels in the control antibody-treated tumors are dilated and leaky, those in the Herceptin-treated tumors have diameters and vascular permeability closer to those of normal vessels (Figure 5B) (Izumi et al., 2002b). It is noteworthy that although Herceptin significantly inhibits VEGF expression in tumor cells, the overall VEGF expression in the tumor tissue does not change, due to compensation by the host stromal cells. Thus, a combination of anti-VEGF and Herceptin treatments may have superior anti-angiogenic effects. These findings also suggest that host stromal cells may compensate for the loss of critical growth factors during anti-tumor treatment, and will thus provide a survival advantage that allows repopulation of treatment-resistant tumor cells.
Figure 5. Tumor vascular normalization by indirect anti-angiogenesis.
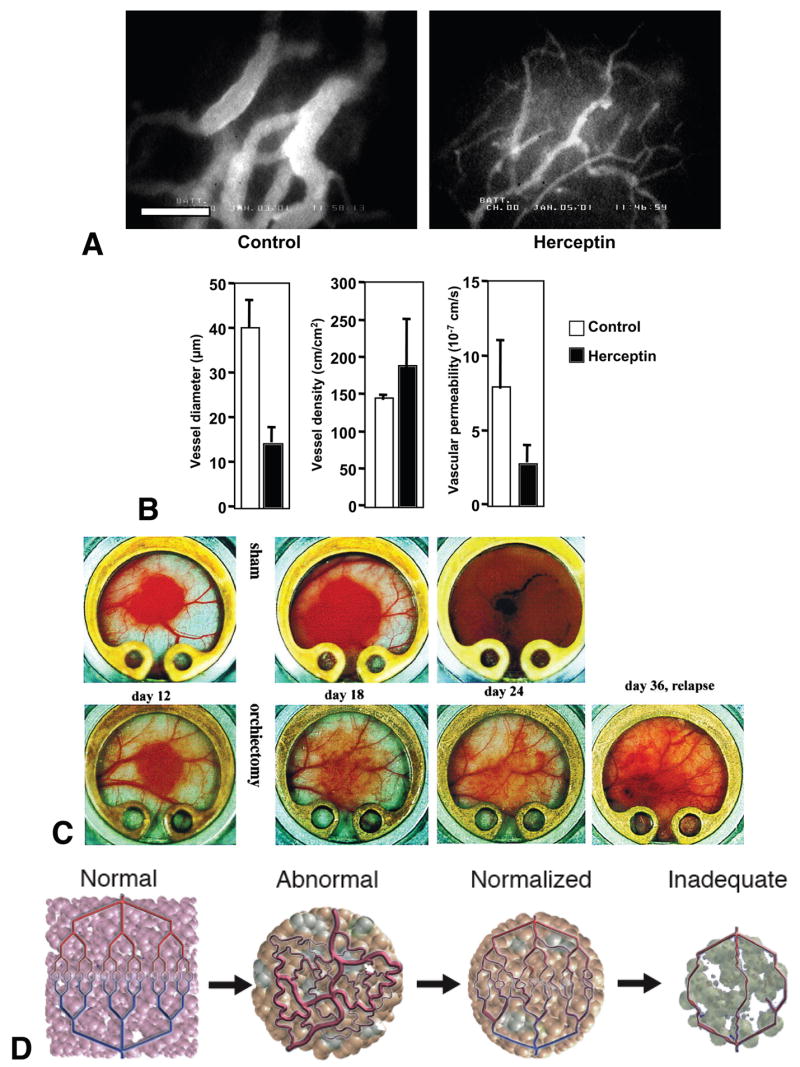
(A) Microangiography in control and Herceptin treated MDA-MB-361HK tumors grown in the mouse cranial window. The bar represents 100 μm. (B) MDA-MB-361HK tumor vessel morphology and function during Herceptin treatment determined by intravital microscopy. (C) Macroscopic images of androgen-dependent Shionogi tumors grown in the mouse dorsal skin chamber. Castration or sham operation is performed 15 days after the tumor implantation. (D) Changes in tumor vasculature during the course of anti-angiogenic therapy. Compared to the well-organized structure of normal vessels (Normal, far left), tumor vasculature is structurally and functionally abnormal (Abnormal, middle left). Anti-angiogenic therapies initially alleviate these abnormalities (Normalized, middle right). However, sustained or excessive anti-angiogenic treatments result in significant vessel reduction and thus, inadequate delivery of drugs or oxygen (Inadequate, far right). A, B adapted from (Izumi et al., 2002a); C, reproduced from (Jain et al., 1998); D, adapted from (Jain, 2001).
Several conventional therapies also have indirect anti-angiogenic effects. For example, sex steroid hormone upregulates expression of angiogenic factors such as VEGF in hormone-sensitive tumor cells and induces angiogenesis and tumor growth (Jain et al., 1998). In addition, a recent report showed that non-neoplastic stromal cells directly mediate hormone-dependent angiogenesis via recruitment of bone marrow-derived cells (Gupta et al., 2007). Thus, anti-hormone therapy acts as an anti-angiogenic therapy and potentially normalizes tumor vasculature. Indeed, hormone depletion downregulates VEGF expression in an androgen-dependent tumor, and decreases vessel diameter, density and permeability (Figure 5C) (Jain et al., 1998). Furthermore, tissue pO2 level increases after the hormone ablation in this tumor suggesting normalization of tumor vasculature (Hansen-Algenstaedt et al., 2000). However, anti-angiogenic/normalization effect of anti-hormone therapy is not permanent. A second wave of angiogenesis is fueled by hormone-independent cells that repopulate the tumor and express VEGF in the absence of androgen causing tumor relapse (Figure 5C).
Likewise, thalidomide, which inhibits VEGF- and bFGF-induced angiogenesis, lowers IFP and increases tumor perfusion, oxygenation and radiation sensitivity of murine fibrosarcomas (Ansiaux et al., 2005). Thalidomide-induced tumor reoxygenation and radiosensitization also occurs in a relatively narrow time window (2–3 days after the start of thalidomide treatment). In addition to these indirect anti-angiogenic therapies, certain doses and schedules of cytotoxic therapies such as chemotherapy and radiation therapy have been shown to exhibit anti-angiogenic activity via the direct cytotoxic effect on proliferating vascular endothelial cells (Garcia-Barros et al., 2003; Kerbel and Kamen, 2004). These treatments might also normalize the tumor vasculature via upregulating TSP-1 and thus improve their own uptake and efficacy in tumors.
Clinical evidence of vascular normalization
Data obtained in recent clinical trials with anti-VEGF agents also support vascular normalization mirroring the preclinical findings discussed above. IFP measured by a wick-in-needle technique showed a 70% drop in rectal carcinomas 12 days after infusion of bevacizumab in patients (Willett et al., 2004; Willett et al., 2005). At the same time, tumor blood perfusion determined by functional CT as well as microvessel density in the biopsy specimen decreased by 30% and 50%, respectively while PET scans showed no change in the tumor FDG uptake (Willett et al., 2004; Willett et al., 2005). These data suggest that these tumor vessels are normalized by bevacizumab treatment and the resulting network is more efficient. In recurrent glioblastoma patients, tumor and vascular response to AZD2171 (a tyrosine kinase inhibitor for VEGF receptors) treatment was monitored using various MRI techniques (Batchelor et al., 2007). Relative tumor vessel size significantly decreased at 1 and 28 days after the start of AZD2171 treatment, while it returned to the abnormally large size at day 56. Tumor volume was also decreased up to 28 days but regrowth was observed after that. On the other hand, vascular permeability to gadolium remained decreased from day 1 through 112. As a result, parameters reflecting vasogenic edema were also decreased. This allowed reduction of steroid usage in these patients. It should be noted that most glioma cells express PDGFR, which is also a target of AZD2171. Furthermore, a subset of tumor cells express VEGF receptors. Potential direct cytotoxic effects against tumor cells should also be taken into account to evaluate the effect of anti-angiogenic therapy for tumor treatment. However, in this trial, there was no correlation between the tumor response and receptor levels.
Collectively, these data indicate that anti-angiogenic therapies can normalize tumor vasculature and microenvironment at least transiently in both preclinical and clinical settings. Timing, duration and extent of the normalization may depend on the agents used, type of tumors and the site of their growth. Therefore, it is critical to determine the timing and extent of vascular normalization using clinically available anti-angiogenic agents in various orthotopic tumors in conjunction with clinical trials with the same agents and tumor types. Finding and validating reliable surrogate markers is also urgently needed not only for clinical translation of the vascular normalization strategy but also for anti-angiogenic treatment in general. Kinetics of the number of viable circulating endothelial cells may be used to monitor response to anti-angiogenic treatments and disease progression (Batchelor et al., 2007; Duda et al., 2007; Willett et al., 2004; Willett et al., 2005). While plasma VEGF and PlGF levels are elevated and soluble VEGFR2 (sVEGFR2) level is low during the anti-VEGF treatments, the increase in plasma bFGF, stromal derived factor 1α and sVEGFR2 correlate with the escape form anti-VEGF therapy (Batchelor et al., 2007; Willett et al., 2005). Finally, understanding of cellular and molecular mechanisms of vascular normalization process is an important step for the application and improvement of this strategy. Perivascular cell recruitment was improved via Tie2 signaling and basement membrane was normalized by balancing its synthesis and degradation during the vascular normalization induced by anti-VEGFR2 treatment (Winkler et al., 2004). Modification of these pathways may extend the duration and magnitude of normalization window and make the normalization strategy more useful (Figure 5D).
Acknowledgments
This review is based on the following previous review articles: Jain RK (2005): Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 307:58–62; Fukumura D (2005): Role of microenvironment on gene expression, angiogenesis and microvascular functions in tumors. In Meadows GG (ed): “Integration/Interaction of Oncologic Growth.” Dordrecht: Springer Science + Business Media B.V., pp 23–36; Fukumura D and Jain RK (2007): Tumor microenvironment abnormalities: Causes, consequences, and strategies to normalize. Journal of Cellular Biochemistry (in press). We thank Dr. Lance L. Munn (Harvard Medical School) for his insightful suggestions and Dr. Edward Brown (University of Rochester) for MPLSM images. The work summarized here has been supported by continuous support from the National Cancer Institute since 1980 including P01-CA-80124 (R.K. Jain and D. Fukumura), R01-CA85140 (R.K. Jain) and R01-CA96915 (D. Fukumura), R01-CA115767-01 (R.K. Jain), and U01-CA084301 (R.K. Jain).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alitalo K, et al. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–953. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- Ansiaux R, et al. Thalidomide Radiosensitizes Tumors through Early Changes in the Tumor Microenvironment. Clin Cancer Res. 2005;11:743–750. [PubMed] [Google Scholar]
- Batchelor TT, et al. AZD2171, a Pan-VEGF Receptor Tyrosine Kinase Inhibitor, Normalizes Tumor Vasculature and Alleviates Edema in Glioblastoma Patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman KC, Swartz MA. Interstitial flow as a guide for lymphangiogenesis. Circ Res. 2003;92:801–8. doi: 10.1161/01.RES.0000065621.69843.49. [DOI] [PubMed] [Google Scholar]
- Boucher Y, et al. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Research. 1990;50:4478–84. [PubMed] [Google Scholar]
- Boucher Y, Jain RK. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: implications for vascular collapse. Cancer Research. 1992;52:5110–4. [PubMed] [Google Scholar]
- Brown EB, et al. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nature Medicine. 2001a;7:1069–1069. doi: 10.1038/89997. [DOI] [PubMed] [Google Scholar]
- Brown EB, et al. In vivo measurement of gene expression, angiogenesis, and physiological function in tumors using multiphoton laser scanning microscopy. Nature Medicine. 2001b;7:864–868. doi: 10.1038/89997. [DOI] [PubMed] [Google Scholar]
- Brown JM. The hypoxic cell: a target for selective cancer therapy- Eighteenth Bruce F. Cain Memorial Award lecture. Cancer Research. 1999;59:5863–70. [PubMed] [Google Scholar]
- Brown JM, Giaccia AJ. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Research. 1998;58:1408–1416. [PubMed] [Google Scholar]
- Chang YS, et al. Mosaic blood vessels in tumors: Frequency of cancer cells in contact with flowing blood. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:14608–14613. doi: 10.1073/pnas.97.26.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser KE, et al. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- Dellian M, et al. Quantitation and physiological characterization of angiogenic vessels in mice: Effect of basic fibroblast growth factor, vascular endothelial growth factor/vascular permeability factor, and host microenvironment. Am J Pathol. 1996;149:59–72. [PMC free article] [PubMed] [Google Scholar]
- Dewhirst MW. Concepts of oxygen transport at the microcirculatory level. Seminars in Radiation Oncology. 1998;8:143–150. doi: 10.1016/s1053-4296(98)80040-4. [DOI] [PubMed] [Google Scholar]
- di Tomaso E, et al. Mosaic Tumor Vessels: Cellular Basis and Ultrastructure of Focal Regions Lacking Endothelial Cell Markers. Cancer Res. 2005;65:5740–5749. doi: 10.1158/0008-5472.CAN-04-4552. [DOI] [PubMed] [Google Scholar]
- DiResta GR, et al. “Artificial lymphatic system”: A new approach to reduce interstitial hypertension and increase blood flow, pH and pO(2) in solid tumors. Annals of Biomedical Engineering. 2000;28:543–555. doi: 10.1114/1.295. [DOI] [PubMed] [Google Scholar]
- Duda DG, et al. A protocol for phenotypic detection and enumeration of circulating endothelial cells and circulating progenitor cells in human blood. Nat Protocols. 2007;2:805–810. doi: 10.1038/nprot.2007.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF, et al. Tumor Architecture and Targeted Delivery. In: Abrams PG, Fritzberg AR, editors. Radioimmunotherapy of Cancer. Marcel Dekker, Inc; New York: 2002. pp. 107–135. [Google Scholar]
- Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Experimental Cell Research. 2001;264:169–184. doi: 10.1006/excr.2000.5133. [DOI] [PubMed] [Google Scholar]
- Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. Angiogenic heterogenity: regulation of neoplastic angiogenesis by the organ microenvironment. Journal of the National Cancer Institute. 2001;93:1040–1041. doi: 10.1093/jnci/93.14.1040. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis. In: Holland JF, et al., editors. Cancer Medicine. B. C. Decker Inc.; Ontario, Canada: 2000. pp. 132–152. [Google Scholar]
- Fukumura D. Role of microenvironment on gene expression, angiogenesis and microvascular functions in tumors. In: Meadows GG, editor. Integration/Interaction of Oncologic Growth. Vol. 15. Springer Science + Business Media B.V.; Dordrecht: 2005. pp. 23–36. [Google Scholar]
- Fukumura D, Jain RK. Tumor microenvironment abnormalities: Causes, consequences, and strategies to normalize. Journal of Cellular Biochemistry. 2007 doi: 10.1002/jcb.21187. in press. [DOI] [PubMed] [Google Scholar]
- Fukumura D, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- Fukumura D, et al. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Research. 2001;61:6020–6024. [PubMed] [Google Scholar]
- Fukumura D, et al. Effect of host microenvironment on the microcirculation of human colon adenocarcinoma. American Journal of Pathology. 1997;151:679–688. [PMC free article] [PubMed] [Google Scholar]
- Garcia-Barros M, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–9. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- Gerlowski LE, Jain RK. Microvascular permeability of normal and neoplastic tissues. Microvascular Research. 1986;31:288–305. doi: 10.1016/0026-2862(86)90018-x. [DOI] [PubMed] [Google Scholar]
- Gohongi T, et al. Tumor-host interactions in the gallbladder suppress distal angiogenesis and tumor growth: Involvement of transforming growth factor b1. Nature Medicine. 1999;5:1203–1208. doi: 10.1038/13524. [DOI] [PubMed] [Google Scholar]
- Gupta PB, et al. Systemic Stromal Effects of Estrogen Promote the Growth of Estrogen Receptor-Negative Cancers. Cancer Res. 2007;67:2062–2071. doi: 10.1158/0008-5472.CAN-06-3895. [DOI] [PubMed] [Google Scholar]
- Gutman M, et al. Regulation of interleukin-8 expression in human melanoma cells by the organ environment. Cancer Research. 1995;55:2470–2475. [PubMed] [Google Scholar]
- Hagendoorn J, et al. Onset of abnormal blood and lymphatic vessel function and interstitial hypertension in early stages of carcinogenesis. Cancer Res. 2006;66:3360–4. doi: 10.1158/0008-5472.CAN-05-2655. [DOI] [PubMed] [Google Scholar]
- Hansen-Algenstaedt N, et al. Tumor oxygenation in hormone-dependent tumors during vascular endothelial growth factor receptor-2 blockage, hormone ablation, and chemotherapy. Cancer Research. 2000;60:4556–4560. [PubMed] [Google Scholar]
- Harris AL. Hypoxia: a key regulatory factor in tumour growth. Nature Reviews Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Helmlinger G, et al. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clinical Cancer Research. 2002;8:1284–1291. [PubMed] [Google Scholar]
- Helmlinger G, et al. Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nature Medicine. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- Hobbs SK, et al. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshida T, et al. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. Cancer Res. 2006;66:8065–8075. doi: 10.1158/0008-5472.CAN-06-1392. [DOI] [PubMed] [Google Scholar]
- Huber PE, et al. Trimodal Cancer Treatment: Beneficial Effects of Combined Antiangiogenesis, Radiation, and Chemotherapy. Cancer Res. 2005;65:3643–3655. doi: 10.1158/0008-5472.CAN-04-1668. [DOI] [PubMed] [Google Scholar]
- Isaka N, et al. Peritumor lymphatics induced by vascular endothelial growth factor-C exhibit abnormal function. Cancer Res. 2004;64:4400–4. doi: 10.1158/0008-5472.CAN-04-0752. [DOI] [PubMed] [Google Scholar]
- Izumi Y, et al. Tumor biology - Herceptin acts as an anti-angiogenic cocktail. Nature. 2002a;416:279–280. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- Izumi Y, et al. Herceptin acts as an anti-angiogenic cocktail. Nature. 2002b;416:279–280. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- Jain RK. Transport of molecules across tumor vasculature. Cancer and Metastasis Reviews. 1987;6:559–93. doi: 10.1007/BF00047468. [DOI] [PubMed] [Google Scholar]
- Jain RK. Determinants of tumor blood flow: a review. Cancer Research. 1988;48:2641–58. [PubMed] [Google Scholar]
- Jain RK. The Eugene M. Landis Award Lecture. Delivery of molecular and cellular medicine to solid tumors. Microcirculation. 1997;4:1–23. doi: 10.3109/10739689709148314. [DOI] [PubMed] [Google Scholar]
- Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nature Medicine. 2001;7:987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- Jain RK. Molecular regulation of vessel maturation. Nature Medicine. 2003 doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- Jain RK. Vascular and Interstitial Biology of Tumors. In: Abeleff M, et al., editors. Clinical Oncology. Elsevier; Philadelphia: 2004. pp. 153–172. [Google Scholar]
- Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Jain RK, et al. Method for locating tumors prior to needle biopsy. No. 5,396,897. USA patent. 1995
- Jain RK, et al. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nature Clinical Practice Oncology. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- Jain RK, et al. Dissecting tumor pathophysiology using intravital microscopy. Nature Reviews Cancer. 2002;2:266–276. doi: 10.1038/nrc778. [DOI] [PubMed] [Google Scholar]
- Jain RK, et al. Endothelial cell death, angiogenesis, and microvascular function after castration in an androgen-dependent tumor: Role of vascular endothelial growth factor. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10820–10825. doi: 10.1073/pnas.95.18.10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK, et al. Effect of vascular normalization by antiangiogenic therapy on inerstitial hypertension, peritumor edema, and lymphatic metastasis: Insighs from a mathematical model. Cancer Research. 2007;67:2729–2735. doi: 10.1158/0008-5472.CAN-06-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi S, et al. NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. J Clin Invest. 2005;115:1816–1827. doi: 10.1172/JCI24015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbel RS, Kamen BA. THE ANTI-ANGIOGENIC BASIS OF METRONOMIC CHEMOTHERAPY. Nature Reviews Cancer. 2004;4:423–436. doi: 10.1038/nrc1369. [DOI] [PubMed] [Google Scholar]
- Kozin SV, et al. Vascular endothelial growth factor receptor-2-blocking antibody potentiates radiation-induced long-term control of human tumor xenografts. Cancer Research. 2001;61:39–44. [PubMed] [Google Scholar]
- Krogh A. The Anantomy and Physiology of Capillaries. Yale University Press; New York: 1922. [Google Scholar]
- Lee CG, et al. Anti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Research. 2000;60:5565–5570. [PubMed] [Google Scholar]
- Leu AJ, et al. Absence of functional lymphatics within a murine sarcoma: A molecular and functional evaluation. Cancer Research. 2000;60:4324–4327. [PubMed] [Google Scholar]
- Li G, et al. Function and regulation of melanoma–stromal fibroblast interactions: when seeds meet soil. Oncogene. 2003;22:3162–3171. doi: 10.1038/sj.onc.1206455. [DOI] [PubMed] [Google Scholar]
- Liotta LA, Kohn EC. The microenvironment of the tumour–host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- Martin GR, Jain RK. Fluorescence ratio imaging measurement of pH gradients: calibration and application in normal and tumor tissues. Microvasc Res. 1993;46:216–30. doi: 10.1006/mvre.1993.1048. [DOI] [PubMed] [Google Scholar]
- McDonald DM, Choyke PL. Imaging of angiogenesis: from microscope to clinic. Nat Med. 2003;9:713–725. doi: 10.1038/nm0603-713. [DOI] [PubMed] [Google Scholar]
- Monsky WL, et al. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad vs. cranial tumors. Clinical Cancer Research. 2002;8:1008–1013. [PubMed] [Google Scholar]
- Monsky WL, et al. Augmentation of transvascular transport of macromolecules and nanoparticles in tumors using vascular endothelial growth factor. Cancer Research. 1999;59:4129–4135. [PubMed] [Google Scholar]
- Netti PA, et al. Time-Dependent Behavior of Interstitial Fluid Pressure in Solid Tumors: Implications for Drug-Delivery. Cancer Research. 1995;55:5451–5458. [PubMed] [Google Scholar]
- Noel A, et al. Enhancement of tumorigenicity of human breast adenocarcinoma cells in nude mice by matrigel and fibroblasts. British Journal of Cancer. 1993;68:909–15. doi: 10.1038/bjc.1993.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, et al. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Padera TP, et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. 2002;296:1883–1886. doi: 10.1126/science.1071420. [DOI] [PubMed] [Google Scholar]
- Padera TP, et al. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427:695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- Pennacchietti S, et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Pouyssegur J, et al. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- Rofstad EK, et al. Acidic Extracellular pH Promotes Experimental Metastasis of Human Melanoma Cells in Athymic Nude Mice. Cancer Res. 2006;66:6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- Roose T, et al. Solid stress generated by spheroid growth estimated using a linear poroelasticity model. Microvascular Research. 2003;66:204–212. doi: 10.1016/s0026-2862(03)00057-8. [DOI] [PubMed] [Google Scholar]
- Ruiter DJ, et al. Tumour metastasis: is tissue an issue? Lancet Oncology. 2001;2:109–12. doi: 10.1016/S1470-2045(00)00229-1. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Singh RK, et al. Organ site-dependent expression of basic fibroblast growth factor in human renal cell carcinoma cells. American Journal of Pathology. 1994;145:365–374. [PMC free article] [PubMed] [Google Scholar]
- Tatum JL, et al. Hypoxia: Importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. International Journal of Radiation Biology. 2006;82:699–757. doi: 10.1080/09553000601002324. [DOI] [PubMed] [Google Scholar]
- Tlsty TD. Stromal cells can contribute oncogenic signals. Cancer Biology. 2001;11:97–104. doi: 10.1006/scbi.2000.0361. [DOI] [PubMed] [Google Scholar]
- Tong RT, et al. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–6. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- Torres-Filho IP, et al. Noninvasive Measurement of Microvascular and Interstitial Oxygen Profiles in a Human Tumor in SCID Mice. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:2081–2085. doi: 10.1073/pnas.91.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki Y, et al. Pancreas microenvironment promotes VEGF expression and tumor growth: Novel window models for pancreatic tumor angiogenesis and microcirculation. Laboratory Investigation. 2001;81:1439–1452. doi: 10.1038/labinvest.3780357. [DOI] [PubMed] [Google Scholar]
- Tsuzuki Y, et al. Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia inducible factor-1a Æ Hypoxia response element Æ VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Research. 2000;60:6248–6252. [PubMed] [Google Scholar]
- Vosseler S, et al. Angiogenesis inhibition by vascular endothelial growth factor receptor-2 blockade reduces stromal matrix metalloproteinase expression, normalizes stromal tissue, and reverts epithelial tumor phenotype in surface heterotransplants. Cancer Res. 2005;65:1294–305. doi: 10.1158/0008-5472.CAN-03-3986. [DOI] [PubMed] [Google Scholar]
- Vukovic V, Tannock IF. Influence of low pH on cytotoxicity of paclitaxel, mitoxantrone and topotecan. British Journal of Cancer. 1997;75:1167–1172. doi: 10.1038/bjc.1997.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The Biology of Cancer. Garland Science Publishing; New York: 2006. [Google Scholar]
- Wildiers H, et al. Effect of antivascular endothelial growth factor treatment on the intratumoral uptake of CPT-11. Br J Cancer. 2003;88:1979–86. doi: 10.1038/sj.bjc.6601005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett CG, et al. Direct evidence that the anti-VEGF antibody Bevacizumab has anti-vascular effects in human rectal cancer. Nature Medicine. 2004;10:145–147. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett CG, et al. Surrogate Markers for Antiangiogenic Therapy and Dose-Limiting Toxicities for Bevacizumab With Radiation and Chemotherapy: Continued Experience of a Phase I Trial in Rectal Cancer Patients. J Clin Oncol. 2005;23:8136–8139. doi: 10.1200/JCO.2005.02.5635. [DOI] [PubMed] [Google Scholar]
- Winkler F, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1 and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Xu L, et al. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway - Mechanism of low pH-induced VEGF. Journal of Biological Chemistry. 2002;277:11368–11374. doi: 10.1074/jbc.M108347200. [DOI] [PubMed] [Google Scholar]
- Xu L, et al. Hypoxia-induced activation of p38 mitogen-activated protein kinase and phosphatidylinositol 3′-kinase signaling pathways contributes to expression of interleukin-8 in human ovarian carcinoma cells. Clinical Cancer Research. 2004;10:701–707. doi: 10.1158/1078-0432.ccr-0953-03. [DOI] [PubMed] [Google Scholar]
- Yuan F, et al. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor vascular permeability factor antibody. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:14765–14770. doi: 10.1073/pnas.93.25.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F, et al. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994a;54:3352–3356. [PubMed] [Google Scholar]
- Yuan F, et al. Vascular Permeability and Microcirculation of Gliomas and Mammary Carcinomas Transplanted in Rat and Mouse Cranial Windows. Cancer Research. 1994b;54:4564–4568. [PubMed] [Google Scholar]