

The signaling pathways that mediate intercellular communication during development are inextricably tied to the cellular organization of multiprotein signaling complexes. These complexes include activated transmembrane receptors associated with adaptors and signaling effectors (Hoeller et al., 2005), the β-catenin destruction complex (Kimelman and Xu, 2006), and complexes associated with endosomes (Fischer et al., 2006). In each of these cases, clustering the signal transduction machinery in a small, defined subcellular locale promotes efficient and rapid interaction and modification of signaling proteins in response to signal.
Hedgehog signaling is essential for many different aspects of the development of both invertebrates and vertebrates (McMahon et al., 2003) and dysregulated Hh signaling contributes to a variety of human tumors (Rubin and de Sauvage, 2006). The Drosophila Hedgehog (Hh) signaling pathway depends on a protein complex between the transmembrane protein Smoothened (Smo), the transcription factor Ci that implements Hh signals, and several associated proteins, analogous to receptor-associated complexes in other pathways. Recent results on mammalian Hedgehog signaling have revealed a unique variation on this theme. In mammals, it appears that Smo, Glis (the homologues of Ci) and associated proteins are localized to a specific organelle, the cilium. Here we provide an overview of cilia, the evidence that cilia are essential for mammalian Hedgehog signaling, the available data on the steps in Hh signal transduction that take place in cilia, and the observations that suggest cilia are important in other developmental signaling pathways.
The nature of cilia
Cilia and flagella are slender projections from cells with a stereotyped microtubule-based structure that includes a ring of nine double microtubules (“the outer doublets”); each cilium grows from a basal body, a modified centrosome. Mammalian cilia can be motile or immotile and can have a variety of cell-type specific structures. Among cells with motile cilia, some, like those of the embryonic node, have a single cilium, while ependymal cells of the adult brain can have multiple cilia and cells in the bronchial epithelium can have hundreds of cilia (Afzelius, 2004).
The physiological importance of motile cilia has been clear since the mid 1970s. Manes Kartagener (1933) defined an inherited human syndrome that showed the unusual combination of bronchiectasis (a type of chronic pulmonary obstruction), chronic sinus infections, and reversal of left-right asymmetry (situs inversus). The identification of an additional aspect of the syndrome, sperm with immotile flagella, led to the recognition that immotile cilia were responsible for the lung and sinus defects and suggested that normal left-right asymmetry was likely to depend on cilia as well (Camner et al., 1975; Afzelius, 1976). Subsequent studies in the mouse embryo have defined many aspects of how the motile cilia of the mouse node generate left-right asymmetry (for a recent review, see Shiratori and Hamada, 2006).
Non-motile cilia are found as a single cilium per cell. Non-motile cilia can be specialized to form structures as elaborate as the outer segments of photoreceptors and the sensory apparatus of olfactory neurons in which signal transduction components are enriched in the cilium. In addition, nearly all interphase and non-dividing cells in vertebrates have a single, non-motile apparently unspecialized cilium (called a primary cilium). The role of primary cilia came into prominence beginning in 2000, when connections became apparent between primary cilia and polycystic kidney disease (PKD), and it is currently believed that renal cilia act as mechanosensors for flow within kidney tubules, although it is not yet clear how disrupted mechanosensation leads to polycystic kidneys (Nauli and Zhou, 2004). Here we focus on the ubiquitous, yet still mysterious, non-motile primary cilia found on other cell types in the adult and developing embryo.
Building cilia: Intraflagellar transport
Our current understanding of the structure, construction and function of cilia is largely grounded in genetic and biochemical studies in the unicellular alga Chlamydomonas. Chlamydomonas has two long flagella (which have the same structure as cilia) that it uses to swim; the flagella are also essential for recognition and fusion of the two mating types that precedes meiosis. The isolation and characterization of mutants defective in swimming defined many of the components necessary for construction or motility of the flagella (Dutcher, 1995).
Kozminski et al. (1993) observed particles that moved from the base to the tip of the Chlamydomonas flagellum and back again by DIC microscopy; they named this process intraflagellar transport (IFT; summarized in Figure 1). They went on to correlate the IFT particles with large raft-like structures visible in transmission EM between the outer doublet microtubules and the ciliary plasma membrane. The FLA10 gene was required for presence of flagella and analysis of the process of intraflagellar transport in temperature-sensitive fla10 alleles showed that the gene was required for the movement of IFT particles (Kozminski et al., 1995). FLA10 turned out to encode a subunit of kinesin-II, a plus end-directed, microtubule-based motor (Walther et al., 1994) that was associated with outer doublets (Kozminski et al., 1995).
Figure 1. Cilia and IFT.
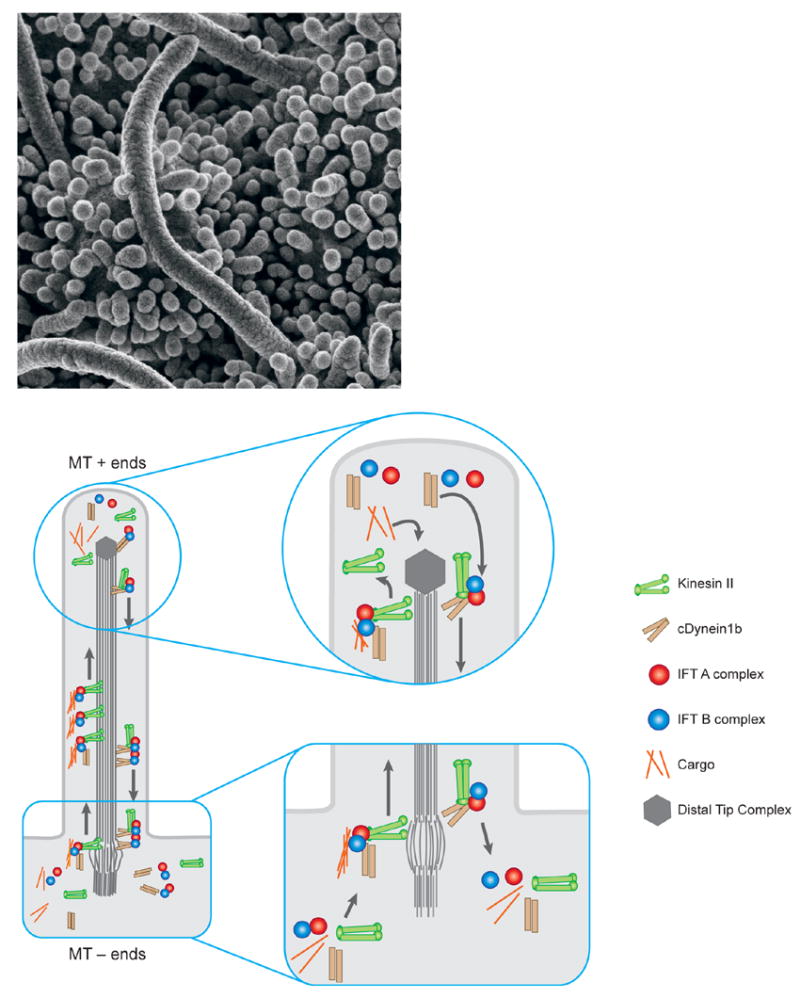
a. Cilia on the node of the e8.0 mouse embryo. These cilia are roughly 3–4 microns long, relatively longer than typical primary cilia.
b. A schematic of the process of Intraflagellar Transport (IFT). Construction and maintenance of cilia and flagella depends on a steady source of new components, which are assembled at the distal tip complex (DTC, at the plus ends of the microtubules). Transport to the distal tip involves assembly of the anterograde IFT platform on transition fibers at the base of the growing cilium and anterograde movement powered by the kinesin II motor. Though still mechanistically unclear, events at the DTC are thought to involve disassembly of the anterograde transport machinery, assembly of structural components into the growing axoneme, and assembly/engagement of the retrograde transport apparatus. IFT machinery and residual structural cargo are recycled by retrograde transport from the tip to the base of the cilium powered by the cytoplasmic dynein motor. At the base, the retrograde apparatus is disassembled, potentially allowing IFT components to be reused.
Subsequent studies showed that IFT depends on a trimeric kinesin-II motor (one subunit of which is encoded by FLA10) that carries proteins to the distal tip of the cilium where they can be assembled into the axoneme of the cilium (or flagellum) and a dimeric cytoplasmic dynein that powers retrograde transport of ciliary proteins back to the cell body. At least seventeen proteins in two large complexes (A and B) create the raft-like structures seen in the EM. In the absence of the kinesin subunits or of IFT complex B proteins analyzed, IFT fails and no flagellum can be constructed.
Movement of IFT particles in cilia has been visualized in the cilia of chemosensory neurons in living C. elegans using transgenic worms that express GFP-tagged components of the IFT machinery (Orozco et al., 1999). Similarly, the movement mammalian GFP-tagged IFT20 along the cilium has been visualized in mammalian cells (Follit et al., 2006). Genetic studies have confirmed that IFT proteins are essential for the construction of cilia in C. elegans (Cole et al., 1998), zebrafish (Sun et al., 2004; Tsujikawa and Malicki, 2004) and mice (Pazour et al., 2000; Huangfu et al., 2003).
Mouse IFT mutants demonstrate that Hh signaling is coupled to cilia
The first indications that intraflagellar transport proteins play roles in developmental processes in addition to the establishment of left-right asymmetry came from analysis of mutations in the mouse Ift88 gene (also called Tg737, orpk or polaris). The transgene insertion allele Tg737 is a partial loss-of-function mutation that causes both PKD and a clear defect in limb patterning, preaxial polydactyly, in homozygotes. Although animals homozygous for the Tg737 mutation survive until after birth, a targeted null allele of mouse Ift88/polaris caused lethality at midgestation (Murcia et al., 2000). The mutant embryos showed randomized left-right asymmetry associated with loss of cilia in the node. These embryos also showed defects in other midline structures that could not be readily explained by defects in nodal cilia. Similarly, targeted mutations in mouse Kif3a and Kif3b subunits of the anterograde kinesin-II IFT motor, which cause the absence of cilia and defects in left-right patterning, also caused midgestation lethality that could not be attributed to defects in left-right asymmetry (Nonaka et al., 1998; Takeda et al., 1999; Marszalek et al., 1999).
By an independent approach, two mutations were identified in a forward genetic screen for ENU-induced mutations that prevented specification of set of ventral cell types in the developing neural tube (Huangfu et al., 2003). Sonic hedgehog (Shh) is required to specify the cell types that were missing in these mutants, but the mutations mapped to regions of the genome that did not include the evolutionarily conserved components of the Hedgehog signaling pathway. Positional cloning showed that the mutations affected two IFT complex B proteins, IFT172 and IFT88. Subsequent studies showed that mutants that lack IFT52, Kif3a or the heavy chain of the retrograde dynein (Dynch2; formerly called Dnchc2) show a similar loss of Shh-dependent ventral neural cell types (Huangfu et al., 2003; Liu et al., 2005; Huangfu and Anderson, 2005). Although less well characterized, mutations in several other mouse IFT genes appear to have similar effects on neural patterning, including mutations in the second subunit of the retrograde dynein motor, D2lic, and another complex B protein, IFT57/hippi. Thus seven mouse genes that encode IFT proteins and motors appear to be required for specification of Shh-dependent cell types in the ventral neural tube (Table 1).
Table 1.
Mutations that affect cilia structure and their effects on embryonic patterninga
| Mutation | Effect on ciliab | Effect on neural patterningc | Effect on limb patterningd | Reference(s) |
|---|---|---|---|---|
| Ift172 | Absent | Loss of ventral cell types | * | Huangfu et al. (2003) |
| Ift88 | Absent | Loss of ventral cell types | Preaxial polydactyly | Liu et al. (2005), Haycraft et al. (2007) |
| Ift52 | n.d. | Loss of ventral cell types | Preaxial polydactyly | Liu et al. (2005) |
| Ift57/bippi | Absent? | Loss of ventral cell types | * | Houde et al. (2006) |
| Kif3a | Absent | Loss of ventral cell types | * | Huangfu et al. (2003) |
| Kif3b | Absent | Loss of ventral cell types (inferred) | * | Nonaka et al. (1998) |
| Dyncb2 | Abnormal bulges | Loss of ventral cell types | * | Huangfu & Anderson (2005), May et al. (2005) |
| D2lic | Absent | Loss of ventral cell types | * | Rana et al. (2004) |
| Ofd1 | Absent | Loss of ventral cell types | Polysyndactyly | Ferrante et al. (2006) |
| Ift122 | Abnormal bulges | Expansion of ventral cell types | Preaxial polydactyly | J.Qin & J.T.Eggenschwiler, Manuscript in preparation |
| talpid3 (chick) | n.d. | Loss of ventral cell types | Polydactyly | Davey et al. (2006) |
| Hnn (Arl13b) | Short, abnormal | Expansion of lateral cell types | Axial polydactyly | Caspary et al. (2007) |
| Rfx3 | Short | None (inferred) | None | Bonnafe et al. (2004) |
| Lrd(Dnabc11) | Immotile | None (inferred) | None | Supp et al. (1999) |
All known mutations that prevent formation of cilia disrupt patterning of the neural tube and the limb. Patterning is not affected in mutants that have short or immotile cilia.
n.d. denotes not determined
It is inferred that mutations that are homozygous viable do not affect neural patterning.
indicates that an effect on limb patterning could not be determined owing to early lethality of mutants
Analysis of neural tube patterning shows that IFT proteins are required at the core of the cytoplasmic Hh signal transduction machinery
Hedgehog signaling is evolutionarily conserved and extensive genetic studies in Drosophila had not revealed any role for IFT proteins in the process, so the correlation between loss of Shh signaling and loss of IFT proteins in mice was a surprise. Signaling by the three mammalian Hedgehog ligands (Sonic, Indian and Desert) works through two membrane proteins, Patched and Smoothened, to regulate the activities of three transcription factors of the Gli family, Gli1, Gli2 and Gli3 (reviewed in Ingham and McMahon, 2001). Analysis of neural patterning in double mutant embryos demonstrated that the IFT mutations block Hh signal transduction downstream of Patched1 and Smoothened. For example, Patched1, which binds Hh ligands, is an unusual receptor, because its activity is required to keep the pathway off in the absence of ligand. As a result, the downstream pathway is activated in Patched1 mutant cells. For example, while ventral neural cell types are not specified in Shh mutants, all neural cells adopt a ventral fate in Patched1 mutant embryos. In mutants that lack IFT complex B proteins Ift88 or Ift172, ventral neural cell types are absent, as in Shh mutants. The neural patterning phenotype of Patched1 Ift88 or Patched1 Ift172 double mutants is identical to that of the Ift single mutants: ventral neural cell types are not specified (Huangfu et al., 2003). Thus while removal of Patched1 activates downstream steps in the signaling pathway, this cannot occur when IFT proteins are absent, which indicates that the IFT proteins affect a step in the Shh signaling pathway that is downstream of Patched1.
The Gli proteins that implement Hh signals can act as both transcriptional repressors and transcriptional activators, and experiments have demonstrated that IFT proteins are required for both repression and activation by Gli proteins. Gli1 and Gli2 act primarily as transcriptional activators, and Gli2 is the primary activator of Hh target genes in neural tube patterning. Gli3 can act as either a transcriptional activator, or, if proteolytically processed, can act as a transcriptional repressor that keeps target genes inactive in the absence of ligand. Hh activity promotes transcriptional activation of target genes in two ways: it promotes formation of Gli activators and it blocks the proteolytic processing event that creates the Gli3 repressor.
In the absence of IFT proteins, there appears to be no Gli activator: the targets of Gli2 are not activated in the ventral neural tube (Huangfu et al., 2003) and a Gli-reporter cannot be activated in response to Shh in cultured IFT mutant fibroblasts (Haycraft et al., 2005; P.J. Ocbina and KVA, unpublished). IFT proteins are also required for normal proteolytic processing of Gli3 to its repressor form (Huangfu and Anderson, 2005; Liu et al., 2005; May et al., 2005). Thus in the absence of IFT proteins, all modulation of target genes in response to ligand is blocked: Hh ligands can neither activate nor repress target gene expression and the basal level of target gene expression is elevated.
Because IFT proteins are required for both Gli activator and Gli repressor, the phenotypes caused by loss of the IFT proteins are not identical to those of Shh mutants and instead resemble that of double mutants that lack both Gli2 and Gli3 or double mutants that lack both Shh and Gli3. For example, lateral neural cell types that depend on Shh (e.g. V2 interneurons) can be specified in the absence of IFT proteins (Huangfu et al., 2003), presumably because these cell types require the low levels of Shh target gene expression caused by the lack of Gli3 repressor.
IFT proteins and Shh signaling in limb patterning
Shh is required for normal patterning of the limb, where it regulates growth, digit number, and anterior-posterior polarity (reviewed in Tickle, 2006). In the mouse, loss of Shh leads to severe hypoplasia and the formation of a single digit of anterior identity, whereas loss of Gli3 leads to supernumerary digits in the anterior limb bud (preaxial polydactyly). The Shh Gli3 double mutant phenotype is very similar to that of Gli3 single mutants, which demonstrated that Gli3 mediates the effects of Shh on digit patterning (te Welscher et al., 2002; Litingtung et al., 2002). Thus in contrast to the neural tube, where Gli-activators play a central role in patterning, most of the effects of Shh in the limb arise through the regulation of Gli3-repressor.
As in the neural tube, IFT proteins are required for transduction the Shh signal in the developing mouse limb. Although a number of IFT mutants die at approximately e10.5, before limbs are patterned, loss of function alleles of Ift88, Dynch2, Ift52, and Ift122 (Haycraft et al., 2005; May et al., 2005; Liu et al., 2005, J. Qin and JTE, in preparation) cause preaxial polydactyly.
Although all these IFT mutants show preaxial polydactyly similar to that of Gli3 mutants, the analysis of molecular reporters of Hh signaling in Ift88 and Dynch2 mutants revealed that they affect both Gli-repressor and Gli-activator function in the limb. For example, the mutant limbs exhibit ectopic expression of genes normally repressed by Gli3-repressor, such as HoxD genes and Gremlin, in the anterior regions, indicating that Gli3-repressor is dependent on IFT, as predicted by their polydactylous phenotype (Liu et al., 2005; May et al., 2005). IFT88 and Dynch2 are required for proteolytic processing of full-length Gli3, and transfection of a construct that expressed a truncated form of Gli3 was functional in Ift88 mutant limb bud cells, which confirmed that Ift88 is required for processing and not activity of Gli3 repressor (Haycraft et al., 2005). Ift88 and Dynch2 mutants also show defects in the positive arm of the Shh pathway, including diminished expression of Gli1 and Patched1 in the posterior limb bud (Liu et al., 2005; May et al., 2005). Thus, as in the neural tube, the patterning phenotype seen in mutant limbs defective in IFT represents a combined disruption of both Gli activator and Gli repressor functions. These findings indicate that the effect of loss of IFT proteins in a specific tissue will depend on the relative importance of Gli-activator and Gli-repressor in the tissue; thus the phenotypes of IFT mutants may resemble that of Shh, that of Gli3 or a combination of the two in any particular tissue (Figure 2).
Figure 2. Tissue-specific effects of IFT mutants depend on the importance of Gli activator and Gli repressor in the tissue.
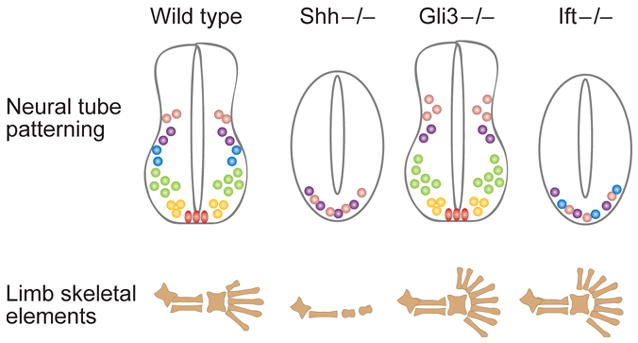
All Hedgehog signaling in the mouse depends on IFT proteins. Mammalian Hedgehog signaling has two outputs: production of Gli activator and prevention of processing of Gli3 repressor. In the neural tube, Gli activators play a central role in neural patterning, so loss of IFT in that tissue has a phenotype similar to that of Shh mutants. The IFT mutant neural tube phenotype is not identical to that of Shh mutants because Gli3 repressor (which is present and active in Shh mutants) is absent in IFT mutants; thus there is a low level of pathway activation. In the limb, Gli3 repressor plays a central role in digit patterning, and the phenotype caused by loss of IFT is similar to that caused by loss of Gli3. Nevertheless, analysis of molecular targets shows that Gli activators also fail to function in the IFT mutant limb.
IFT proteins are required for signaling by both Sonic and Indian hedgehog
Indian hedgehog (Ihh) is required for normal development of the long bones of the limb, through its effects on proliferation and differentiaion of the chondrocyte lineage (St. Jacques et al., 1999). Conditional deletion of Ift88 in limb mesenchyme causes shortening of the proximodistal axis of the limb, similar to the phenotype of Ihh mutants. Gli1 and Ptch1 (Hh target genes) fail to be expressed in developing bones of the Ift88 conditional mutant (Haycraft et al., 2007). Similarly, the initial specification of the lung depends on both Ihh and Shh, and no lung primordium is established in IFT mutants (Huangfu and Anderson, 2006). No experiments have addressed the question of whether Desert Hh (required for spermatogenesis) also depends in IFT proteins, but it seems likely that all Hh signaling depends on IFT proteins.
Hedgehog pathway components are enriched in cilia
The phenotypes of the IFT mutants described above demonstrate that there is a tight correlation between the presence of cilia and the ability of cells to transduce Hh signals. These results raised the interesting possibility that cilia might provide a milieu that is critical for Hh signaling and that some components of the Hh signal transduction machinery might localize to cilia.
Smoothened, a multipass membrane protein similar in structure to G-protein coupled receptors, is required for Hh pathway activity. Smo acts a step downstream of Patched, the Hh receptor, and activity of Smo both promotes Gli activator function and blocks the processing event that generates Gli3 repressor. Smo is not detectable in cilia in resting cells, but becomes enriched in cilia when cultured cells are exposed to Hh, and is found in cilia in the embryonic node (cells that are actively signaling in response to Hh; Corbit et al., 2005). A sequence similar to that which targets C. elegans olfactory receptors to cilia is present in Smo, and that sequence is necessary for both localization of Smo to cilia and Smo activity. Conversely, a constitutively activated form of Smo is localized to cilia in the absence of Hh. Thus, activity of Smo is correlated with its presence in cilia.
More remarkably, the transcription factors that mediate Hh signaling also appear to be enriched in cilia. Tagged, overexpressed Gli1, Gli2 and Gli3 are all enriched at the distal tip of the cilium in primary cultures of limb bud mesenchyme cells (Haycraft et al., 2005). Endogenous full-length Gli3 protein has also been detected at the tip of cilia in the same cells (Haycraft et al., 2005), and endogenous Gli1, which is only produced when the Hedgehog pathway has been activated, is also found at cilia tips (JTE, unpublished). However, it has not been tested whether the endogenous Gli proteins move out of cilia in response to ligand.
Suppressor of fused (Sufu), an essential negative regulator of the pathway, is enriched at cilia tips in the absence of Hh ligands (Haycraft et al., 2005). In Drosophila, Sufu helps tether Ci (the Gli homologue) in the cytoplasm in the absence of signaling, so it might serve a similar function in the cilia of mammalian cells (Kogerman et al., 1999). However, mammalian Sufu may have additional functions in the nucleus (Paces-Fessy et al., 2004; Barnfield et al., 2005; Svard et al., 2006), so it would be interesting to determine how Sufu localization is affected by Hh ligand.
A model that accounts for the observations is that Sufu and Gli proteins are present in cilia in the absence of ligand. When cells are not exposed to ligand, Gli3 is processed to generate a transcriptional repressor, and processing can only occur if Gli3 has resided within the cilium. In response to ligand, Smo moves into cilia where it can associates with Sufu and Gli proteins. Once Smo is in the cilium, Smo can interact with signaling components present there to form a Smo-Sufu-Gli complex (or complexes) which modify Gli2 (and probably Gli3) to make it a transcriptional activator and prevent the proteolytic processing of Gli3 to the repressor form, thus fully activating the pathway. Thus, in this model, both ligand-dependent and ligand-independent aspects of Gli regulation depend on cilia (Figure 3).
Figure 3.
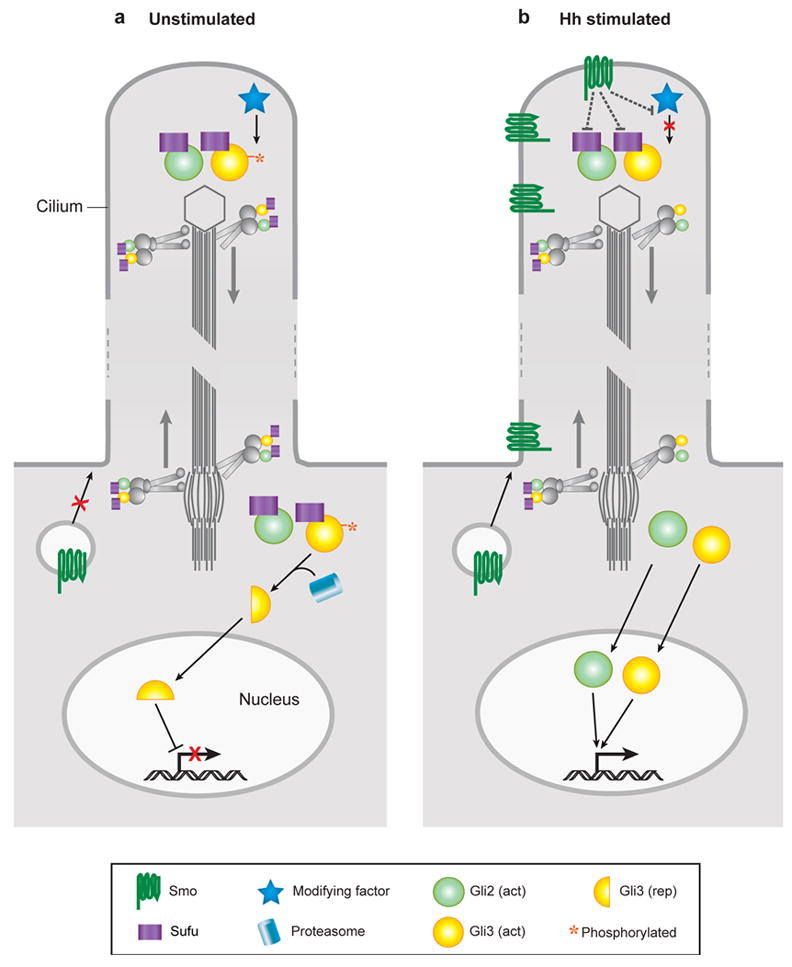
A model for Hedgehog signal transduction within the cilium. In the absence of stimulation, Gli proteins and Su(fu) are transported to and from the tip of the cilium. Within the cilium, Gli3 protein is phosphorylated, targeting it for cleavage into the transcriptional repressor form once it encounters the proteasome at the cilium base. Gli proteins are maintained in an inactive state by Su(fu) and other components. Upon stimulation by Hh, activated Smoothened in vesicles is targeted to the cilium membrane where it can spread to the cilium tip. Here, Smo antagonizes downstream inhibitors such as Su(fu) and blocks phosphorylation of Gli3. Released from inhibition, activated forms of Gli proteins are transported to the cell body and ultimately enter the nucleus where they stimulate the expression of Hh target genes.
This model does not, however, define the biochemical events that take place in the cilium. Gli3 processing is thought to be mediated the proteasome (Wang and Li, 2006; Pan et al., 2006), and proteasomes have not seen in cilia by EM. However proteasomes are enriched in the basal bodies of cilia (Wigley et al., 1999). Thus it is likely that some other event required for proteolytic processing takes place in cilia; perhaps phosphorylation by one of the kinases necessary for processing (PKA, and GSK3β; Wang et al., 2000; Tempe et al., 2006) and that limited proteolysis occurs once Gli proteins are transported back to the basal bodies. In contrast, the nature of the events that regulate the formation of Gli activators remains mysterious, even for Drosophila Ci (Méthot and Basler, 2000).
Are cilia more than a site for enrichment of the Hh signal transduction machinery?
The results described thus far demonstrate that IFT proteins are required for Hh signaling and that some Hh signaling proteins are enriched in cilia. These findings do not exclude the possibility that IFT proteins traffic Hh pathway components outside of cilia and that the enrichment of Smo, Glis and Sufu in cilia is not essential for their activity. However, mutations in several mouse cilia-associated proteins also show altered Hh signaling. Analysis of these mutants argues that cilia, not just the intraflagellar transport machinery, are required for Hh signaling. In addition, some of these mutants suggest that normal Hh signal transduction depends on active participation of ciliary proteins.
Loss of function of mouse Ofd1, which was first identified as the gene affected in human Oral-Facial-Digital Sydrome 1, causes phenotypes similar to those of IFT mutants. The OFD1 protein has been seen localized to the basal body and centrosome rather than in the cilium (Romio et al., 2004), but it is required for cilia formation (Ferrante et al., 2006). Ofd1 is X-linked, although the human Ofd1 gene is not subject to X-inactivation; females heterozygous for Ofd1 mutations have a syndrome that includes craniofacial and digit abnormalities and, occasionally, polycystic kidneys, presumably due to haploinsufficiency of the gene. Ofd1 mutations are apparently lethal to hemizygous human males. In mice, the Ofd1 gene is subject to X-inactivation and, as a result, Ofd1 mutant female mice have a more severe phenotype than the human females, presumably because of mosaic loss of function due to random inactivation of the X-chromosome (Ferrante et al., 2006). Ofd1 null mutant mice fail to form cilia in the embryonic node, and show the associated randomization of left-right patterning. Ventral neural cell fates are not specified in Ofd1 mutant male embryos and the mutants express the direct Hh-target gene Patched1 at low levels, indicating that Hh signaling is defective in these embryos. Thus loss of this centrosome/basal body protein blocks the formation of cilia and that disrupts Hh signaling, which supports the hypothesis that cilia are required for Hh signaling, beyond the requirement for IFT proteins.
Ofd1 mutant limb buds exhibit ectopic expression of Gli3R target genes such as HoxD genes in the anterior limb buds, suggesting that, as in IFT mutants, formation of Gli3R is impaired, which is consistent with the polydactylous phenotype observed in Ofd1/+ female embryos. However, in contrast to Ift88 and Dynch2 mutants, Ofd1-deficient male limb buds exhibit normal levels of Patched1 and Gli1 expression in the posterior limb bud. These data suggest that OFD1 is not required for Gli activator function in the limb bud, although it does appear to be required for Gli activator function in the neural tube. Because it is not known whether cilia are present on Ofd1 mutant limb bud cells, it is possible that these mutant cells retain some aspects of basal body function and/or ciliogenesis that permits normal Gli activator function but blocks formation of Gli3R. Further studies may pinpoint whether the Ofd1 mutation separates the functions of the cilium and the basal body in the regulation of Gli protein activity in the limb.
Most interesting is that some cilia-associated proteins have different roles in Hh signaling than seen in the kinesin, dynein and IFT complex B proteins described above. For example, in direct contrast to loss of complex B IFT proteins where Gli activator function is disrupted, a null mutation in mouse Ift122, which encodes a complex A protein, leads to constitutive activation of the Hedgehog pathway and a ventralization of the neural tube (J. Qin and JTE, in preparation). Genetic studies show that mouse IFT122 functions at a step in the pathway downstream of Shh and Smoothened, but upstream of the transcription factor Gli2. In contrast to complex B Ift mutant cells, Ift122 mutant cells have primary cilia, but these cilia are morphologically abnormal and exhibit pronounced accumulation of IFT proteins within the ciliary axoneme and at its tip, a phenotype similar to that of mutants that lack the retrograde IFT dynein motor (Huangfu and Anderson, 2005), suggesting that retrograde transport is severely impaired in Ift122 mutants.
Studies in C. elegans and Chlamydomonas have also distinguished between the functions of complex A and complex B proteins. While loss of complex B proteins leads to absence of flagella, disruption of complex A proteins causes less severe effects on cilia length (Brazelton et al., 2001; Piperno et al., 1998; Perkins et al., 1986; Blacque et al., 2006). In mutants defective for complex A, flagella show swellings or bulges, accompanied by accumulation of IFT proteins within or at the tips of axonemes (Perkins et al., 1986, Blacque et al., 2006). A temperature sensitive allele of Chlamydamonas fla15, which exhibits significant depletion of complex A IFT proteins yet normal levels of complex B IFT proteins, shows normal rates of anterograde but reduced rates of retrograde transport at the permissive temperature (Piperno et al., 1998). These data have lead to the hypothesis that complex B IFT proteins are important for anterograde transport, whereas complex A IFT proteins may be dedicated to retrograde IFT (Piperno et al, 1998, Blacque et al, 2006).
Nevertheless, the neural patterning phenotype of the Ift122 mutants is surprising because mutant embryos deficient for Dynch2, a subunit of the retrograde IFT motor, fail to activate Hh target genes, similar to complex B mutants, which is the opposite phenotype to that of Ift122 mutants. Recent data suggest that complex A and B could each participate in several aspects of the transport process. For example, complex A IFT proteins can associate with Kinesin II in a complex B-independent manner (Pederson et al., 2006; Ou et al., 2005). Thus it is possible that complex B and complex A both have roles in anterograde transport and have different cargo specificities. Complex B may be generally required for anterograde transport of cargo used to assemble the cilium (e.g. tubulin dimers, radial spoke proteins, and other structural components of the cilium), whereas complex A IFT proteins may be important for the transport of machinery used for turnaround at the distal tip or retrograde transport. As a result, loss of IFT122 could lead to constitutive activation of the pathway because of a defect in anterograde transport of a specific factor that antagonizes Gli function within the cilium.
Arl13b (formerly called Arl2l1, the homologue of the zebrafish scorpion gene; Sun et al., 2004) is a small GTPase of the Arl/Arf subgroup of the Ras superfamily and plays a unique role of formation of the cilium and in Hh signaling. The Arl13b protein is localized to the shaft of the cilium, and mouse mutants that lack Arl13b have short cilia of abnormal structure, in which the B-tubule of the outer doublet is open (T. Caspary and KVA, submitted). Coupled to this defect in cilia structure, the mutant embryos show a novel defect in neural patterning, in which both the most dorsal and the most ventral cell types are not specified and the domain of motor neurons, which arise at an intermediate position, is greatly expanded. Analysis of double mutants with the core components of the Hh pathway indicate that Gli3 repressor activity is normal in the Arl13b mutant embryos, but Gli activators are constitutively active at low levels. Thus while normal cilia are required for both activator and repressor activities of Gli proteins, those events are separated in the Arl13b cilia, which can regulate Gli repressor but not activator.
The phenotypes of the Ift122 and Arl13b mutants suggest that the cilium is more than a sensory antenna where Hh pathway components are concentrated. Instead, the structure and dynamics of the cilium appear to have a more integral role in Hh signaling. Additional studies will be needed to address how proteins in the cilium interact with and regulate Hh signaling components, and how the spatially regulated expression of genes encoding IFT proteins and other components of the cilium (e.g. Huangfu and Anderson, 2005) modulates Hh signaling. A number of other vertebrate-specific genes have been identified that affect Hh signaling at the same step as IFT proteins, downstream of Smo and upstream of Gli proteins. These include both positive and negative regulators of the pathway (mouse Rab23, Fkbp8, and Sil, zebrafish Iguana and chicken Talpid3; Eggenschwiler et al., 2001; Bulgakov et al., 2004; Izraeli et al., 2001; Sekimizu et al., 2004; Wolff et al., 2004; Davey et al., 2006), and could also affect cilia. Studies on the structure of cilia in these mutants and of the subcellular localization of these proteins will be necessary to see if these proteins also affect Hh signaling through cilia.
Are cilia required for Hh signaling in all vertebrates?
The studies described above have established that cilia are required for all responses to Hedgehog ligands in the mouse embryo. In Drosophila, mutations that block the formation of cilia have no effect on Hh signaling and cilia are restricted to sensory neurons (Avidor-Reiss et al., 2004; Han et al., 2003). Thus this evolutionarily conserved signaling pathway either became coupled to cilia at some stage in the vertebrate lineage or, alternatively, the coupling to cilia was lost in the lineage that led to dipterans.
Indeed, it is not yet clear whether all vertebrates require cilia for Hh signaling. Xenopus embryos with disrupted cilia show a loss of Hedgehog signaling (Park et al., 2006). However, the best genetic data in another vertebrate comes from zebrafish, where no connection has been reported between IFT mutants and Hh signaling. Mutations in Ift88, Ift172, Ift81 and Ift57 as well as morpholino knockdown of Ift52, all lead to cystic kidneys, as well as defects in structure of cilia in the kidney and other tissues (Sun et al., 2004; Tsujikawa and Malicki. 2004). However, the mutants do not show the defects in somite patterning characteristic of zebrafish Hh pathway mutants, although they do show a ventrally curved tail, a relatively nonspecific phenotype seen in some Hh pathway mutants. The lack of overt Hh phenotype could be due to the perdurance of maternal gene products, as cilia are still detectable even in the Ift mutants. The sequence motif in Smo that is required for its localization to cilia in MDCK cells is also required for activity of Smo in zebrafish (Corbit et al., 2005), which suggests that cilia could be important for Hh signaling in zebrafish as well. Transplantation of zebrafish Ift mutant germ cells into hosts that lack PGCs (Ciruna et al., 2002) would make it possible to generate animals that lack both maternal and zygotic sources of IFT proteins and then to test whether IFT is required for Hh signaling in zebrafish.
Are there other developmental signals that depend on cilia?
A striking aspect of the mouse IFT mutant embryos is that all the obvious visible phenotypes can be explained as lack the result of lack of Hh signaling: the open neural tube, the changes in neural patterning, the polydactyly and the failure to initiate lung development can all be traced directly to disruptions of Hh signaling. This give the impression that, during development, primary cilia are organelles that are dedicated to Hh signaling. Indeed, at this time, there is no definitive evidence that any other developmental signaling pathway depends on cilia. However, the lethality of IFT mutants at midgestation and the obvious defects caused by disruption of Hh signaling might mask other roles for cilia, and there are some links between cilia and other developmental signaling pathways that we explore below.
PDGFRα signaling
The Platelet-Derived Growth Factor (PDGF) signaling pathway is used for a variety of cellular processes during development, including proliferation, survival, and migration of many cell types (reviewed in Heldin and Westermark, 1999). Like the Hedgehog signaling pathway, inappropriate activation of the PDGF pathway is intimately linked to a variety of human malignancies (Pietras et al., 2004). There are two major forms of both the PDGF ligands and their receptors, and both can act as either homo- or hetero-dimers. Of relevance here, the PDGF-AA ligand can only bind and signal through PDGFRαα homodimers. Two important responses downstream of activated PDGF receptors are activation of the PI3K-AKT and MEK1/2-ERK1/2 pathways, which mediate several cellular functions of the pathway.
Recently, elegant experiments in NIH3T3 fibroblasts have documented a relationship between cilia and signaling by the PDGF receptor α (PDGFRα) (Schneider et al., 2005). These experiments showed that PDGFRα is upregulated and localized to cilia in growth-arrested cells, that its presence in cilia correlates with the ability of cells to respond to its ligand (PDGF-AA), that the PDGFRα in cilia becomes phosphorylated in response to ligand and that the downstream target MEK1/2 is also phosphorylated within cilia in response to ligand stimulation. In contrast to PDGFRα, PDGFRβ is localized primarily to clusters at the cell surface although a fraction appears to be localized to cilia. Consistent with the cilium representing the primary site of PDGFRα but not PDGFRβ function, mouse embryonic fibroblasts from Tg737orpk mice (which are homozygous for a partial loss of function allele of Polaris/Ift88), have short cilia and fail to show normal responses to PDGF-AA without significant decrease in PDGF-BB responsiveness.
The upregulation and ciliary localization of PDGFRα in G0 cells is unlikely to be coincidental. Cilia are present on cultured fibroblasts when they are the G0 phase (Tucker and Pardee, 1979a), and a primary role for PDGF signaling is to promote entry into the cell cycle from the G0 arrested state by activation of intermediate-early genes such as c-fos (Stiles et al., 1979; Pledger et al., 1981; Greenberg and Ziff, 1984). Targeting of PDGFRα to cilia could thus restrict the ability of cells to respond to PDGF-AA to the G0 cell cycle phase. Consistent with this possibility, in response to PDGF, NIH3T3 fibroblasts begin entry into the cell cycle and their centrioles become rapidly deciliated (Tucker et al., 1979b). This loss of cilia could, in turn, suppress further responses to PDGF stimulation.
Another potential relationship between the primary cilium and the PDGF signaling pathway lies in their association with cell migration. NIH3T3 fibroblasts exhibit directed migration (chemotaxis) towards a source of PDGF-AA (Uren et al., 1994) and migration of oligodendrocyte progenitors (02A) is dependent on PDGFRα in vivo and in vitro (Klinghoffer et al., 2002; Forsberg-Nilsson et al., 1998). Do cilia also play a role in cellular migration? In cultured migrating NIH3T3 fibroblasts, nonmotile cilia were found to orient parallel to the direction of migration (Albrecht-Buehler, 1977), suggesting that they play a cell autonomous, instructive role in cellular migration. This, in turn, raises the possibility that if the apparatus used to sense and respond to PDGF-AA is specifically localized to the primary cilium, orientation of this structure along a gradient of PDGF-AA may aid in sensing or responding to that gradient.
An important open question is whether cilia are required for normal responses to PDGF-AA in vivo. If this were true, some aspects of the phenotypes seen in mice deficient for IFT function could be due to disruption of PDGF-A signaling. Mice lacking PDGF-A and PDGFRα have a variety of embryonic phenotypes including severe growth retardation, apoptosis within neural crest derivatives, defective oligodendrocyte proliferation, abnormal patterning of the somites, neural tube closure defects, and skeletal abnormalities (Bostrom et al, 1996; Soriano, 1997). IFT mutant embryos do show defects in the tissues that require PDGF signaling; however these phenotypes could be caused by defective Hedgehog signaling since proper regulation of Hh signaling is also important for embryonic growth, neural crest survival, neural tube closure, somite development and oligodendrocyte specification (Chiang et al, 1996; Brito et al., 2006; Hui and Joyner, 1993; Buttitta et al., 2003; Orentas et al., 1999). Thus, confirmation of a requirement for cilia in PDGF signaling in vivo will ultimately depend on conditional disruption of IFT in developmental contexts in which defects in Hedgehog signaling would have little effect.
Canonical Wnt signaling
The Hh and the canonical Wnt signaling pathways have a number of striking similarities (Nusse, 2003; Lum and Beachy, 2004). Both rely on serpentine receptors for pathway activation (Smo; Frizzleds). Both depend on regulation of proteosome-dependent events (processing of Gli3; regulated degradation of β-catenin). It is therefore reasonable to wonder if Wnt signaling might also depend on cilia, and some observations have raised considerable interest about the possibility (Simons et al., 2005; Ross et al., 2005). If cilia do play a role in Wnt signaling, the cilium could provide a site for integration of the Hh and Wnt pathways in cells that are simultaneously exposed to both signals, as in the ventral neural tube (Lei et al., 2006).
Results from several experiments have suggested that loss of cilia may lead to elevated canonical Wnt signaling. Although the origins of the cystic kidneys caused by loss of IFT proteins are controversial, it has been suggested that increased calcium caused by loss of polycystins may activate Wnt signaling and hence proliferation. Expression of an activated form of β-catenin in the kidney causes overproliferation and renal cysts (Saadi-Kheddouci et al., 2001), and elevated levels of cytoplasmic and nuclear β-catenin were reported in the postnatal cystic kidneys caused by conditional deletion of Kif3a in the kidney (Lin et al., 2003). Similar increases in β-catenin were seen in the postnatal pancreas of animals with defective IFT (Cano et al., 2004). However, the β-catenin levels in the conditional Kif3a mutants were assayed after birth, while cystic organs are detectable as soon as e14.5–15.5. It will therefore be necessary to analyze signaling pathways at that early stage in development or to use conditional inactivation in the adult (as in Piontek et al., 2004) to see if elevated nuclear β-catenin is a cause or consequence of the cystic kidneys caused by loss of IFT.
Despite the potential connections between IFT and Wnt signaling in the kidney and pancreas, the phenotypes of the IFT mutants demonstrate that cilia cannot be essential for canonical Wnt signaling. Mouse mutants that lack canonical Wnt signaling should arrest very early in development and show phenotypes characteristic of loss of canonical Wnt signaling. For example, Wnt3 mutants die at gastrulation and Lef1 Tcf1 double mutants (which lack two of the transcription factors that mediate much of Wnt signaling) die at e10.5 with a characteristic accumulation of cells in the tailbud (Liu et al., 1999; Galceran et al., 1999); neither of these phenotypes is seen in either null or partial loss-of-function IFT mutants. Elevated Wnt signaling also causes severe disruption in early development, as seen in mutants that lack the negative regulators Axin or APC (Zeng et al., 1997; Chazaud and Rossant, 2006), which die very early in development with characteristic phenotypes that, again, do not overlap the defects seen in IFT mutants embryos.
Expression of transgenic reporters of canonical Wnt signaling is normal in Ift172 mutant embryos, which provides a direct demonstration that canonical Wnt signaling in the midgestation embryo is neither decreased nor elevated in the absence of cilia (Figure 4). Therefore because of the complex relationships between signaling pathways during development, it is possible that loss of IFT in the developing kidney disrupts Hh signaling and that, in turn, leads to inappropriate activation of Wnt signaling. Loss of either Shh leads to failure of development of one or both kidneys, but those mutant kidneys that do develop show dilated tubules (Hu et al., 2006). It therefore remains possible that the cystic kidney phenotype could be due to defects in Hh signaling at a particular stage in kidney development. To test this possibility it will be necessary to keep in mind that loss of IFT can have cell type-specific effects on the activity of Hh target genes, depending on the relative importance of Gli-activator and Gli-repressor in the relevant cells (Figure 2), so the effect of loss of IFT on Hh signaling in the developing kidney may be different than that of either Shh or Gli3 mutants.
Figure 4.
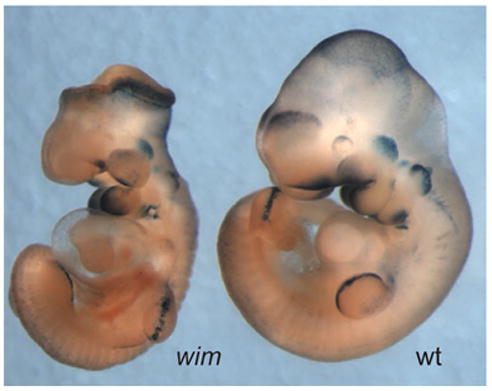
Canonical Wnt signaling in the mouse embryo is normal in Ift172 mutant embryos. The Topgal transgene contains multimerized Lef/Tcf binding sites driving the expression of β-galactosidase and acts as a faithful reporter for sites of endogenous Wnt signaling (DasGupta et al, 1999). a. A wild type e10.5 embryo that carries a copy of the Topgal reporter. β-galactosidase is expressed in many distinct sites where canonical Wnt signaling is active at this stage of development. B. Homozygous Ift172 (wim) embryos that carry a copy of the Topgal reporter show the characteristic morphology of Ift mutants; the most obvious phenotype is an open neural tube that lacks the normal ventral groove of the neural tissue. The Topgal reporter is expressed in the same set of cell types in the mutant as in the wild type, and at indistinguishable levels.
When considering the role of cilia in the kidney and in the pancreas, it is important to remember that there are likely to be different proteins associated with cilia in specialized cell types. There are obvious morphological differences between motile and non-motile cilia, between specialized motile cilia on cells in the node, brain ventricles and bronchial cells, and between non-motile cilia in photoreceptor and olfactory cells (reviewed in Rosenbaum and Witman, 2002). In addition, there are molecular differences among similar classes of cilia. For example, the left-right dynein (lrd) is required for motility of node cells, but is apparently not essential for motility of bronchial or ependymal cells (Supp et al., 1999) and Polycystin-1 is expressed in the mechanosensory cilia of the kidney but not in the embryonic node (Karcher et al., 2005). Thus there is cell-type specific regulation of which proteins are trafficked to cilia. It will therefore be important to test whether Wnt signaling pathway components are localized to cilia in specific developing tissues. For example, Inversin protein, which is required for left-right asymmetry and normal kidney development, is associated with cilia and centrosomes (Watanabe et al., 2003). Inversin can bind Disheveled1 (Dvl1) (Simons et al., 2005), a component of both the canonical and non-canonical Wnt pathways, and it would be interesting to test whether Dvl1 is associated with cilia in specific cell types, such as the developing kidney.
Non-canonical Wnt signaling
It has also been proposed that cilia are coupled to non-canonical Wnt (planar cell polarity/PCP) signaling. PCP signaling in Drosophila provides proximo-distal orientation for cells within an epithelium (Adler, 2002) and has been shown to have similar functions in the orientation of hairs on mouse skin (Guo et al., 2004) and in cells of the inner ear (Montcouquiol et al., 2006). In addition, the most striking effect of loss of PCP in the mouse embryo is the complete failure to close the spinal cord, which is believed to represent a failure in a particular type of cell rearrangement (convergent extension) that depends on cell polarization (for review, see Wang and Nathans, 2007).
Two different types of connection between cilia and PCP signaling have been proposed. Several studies have suggested that cilia are required for PCP signaling or may be involved in a switch between canonical and non-canonical Wnt signaling (e.g. Ross et al., 2005; Simons et al., 2005). In contrast, another study suggested the PCP effector proteins may be required to construct normal cilia (Park et al., 2006).
Some studies have argued that the core components of the non-canonical Wnt pathway might depend on cilia for their function. Vangl2 (the protein affected by the Loop-tail mutation), a core component of the non-canonical Wnt pathway is enriched in basal bodies and cilia of IMCD3 kidney and respiratory epithelium cells and weak genetic interactions have been observed between mutations in Bbs proteins, which are localized to basal bodies, and Loop-tail/Vangl2 (Ross et al., 2005). However, in the inner ear, where Vangl2 is required for polarity of the stereocilia (actin-based structures), Vangl2 protein colocalizes with β-catenin at the membrane. Vangl2 is asymmetrically localized in these cells, but opposite the kinocilium, the conventional microtubule-based cilium found in these cells (Montcouquiol et al., 2006).
The phenotypes of the IFT mutants show that cilia cannot be essential for non-canonical Wnt signaling. Mouse mutants that lack non-canoncial Wnt signaling have a shortened body axis and completely fail to close the neural tube in the trunk region; these phenotypes are not seen in IFT mutants that lack cilia altogether. Because there are no molecular readouts of non-canonical Wnt signaling in the early embryo, it was possible that the loss Hh signaling in IFT mutants might mask the effects of loss of PCP signaling. However double mutants that lack both the core component of the noncanonical pathway Vangl2 and IFT proteins show a simple additive phenotype (K. Liem and KVA, unpublished), which indicates that loss of Hh signaling does not mask a loss of non-canonical Wnt signaling.
Recent studies of the PCP pathway proteins Fuzzy and Inturned in Xenopus embryos suggest that they may participate indirectly in Hh signaling. Drosophila Fuzzy and Inturned act downstream of the core PCP proteins and are thought to link the core PCP pathway to the polarized organization of the cytoskeleton (Adler, 2002). Injection of morpholinos to inactivate Xenopus Fuzzy or Inturned prevented neural tube closure in the head, but not the spinal cord, a pattern different than that seen in PCP mutants (Park et al., 2006). Further analysis showed that the defect in neural tube closure was correlated with a loss of both Hedgehog signaling and a loss of cilia in the neural tube. These findings suggest that Fuzzy and Inturned are required for proper formation of cilia and that disruption of cilia causes disruption of Hh signaling. The morphant embryos also showed mild defects in convergent extension, suggesting that Fuzzy and Inturned contribute to both PCP and to cilia formation in Xenopus. Other studies have also indicated that organization of the apical cytoskeletal domain of the cell is important for the formation of cilia (Omori and Malicki, 2006; Fan et al., 2004).
The possibility that PCP could be important for construction or positioning of cilia is an attractive hypothesis to explain how cilia are placed asymmetrically on some cells. For example, the motile cilia of the mouse node must be asymmetrically positioned and tilted toward the posterior of the cell in order to generate the leftward nodal flow that appears to be required for the initial establishment of left-right asymmetry (Nonaka et al., 2005). That type of asymmetric positioning of cellular specializations is exactly the type of process controlled by PCP in Drosophila.
Do human genetic diseases associated with cilia reflect disruption of signaling pathways?
Over the past ten years, it has become clear that defects in cilia and basal body function are intimately involved in human disease. In addition to the disorders resulting from primary cilium dyskinesia discussed above (e.g. Kartagener’s syndrome), several dozen human congenital disorders have been associated with defects in cilia and/or basal bodies. The connections between cilia, basal bodies and human disease have been facilitated by the identification of a set of evolutionarily conserved genes that are specifically associated with cilia and basal bodies. Whole genome sequences comparisons led to the recognition that cilia were present in the eukaryotic ancestor of both plants and animals, but have been lost independently in the lineages of current organisms. For example, Chlamydomonas has flagella (the structural equivalent of cilia) but Arabidopsis (another plant) does not. Based on these whole genome sequence comparisons, expression studies and proteomic analysis of purified flagella carried out in ten different labs a ciliary proteome of ~1200 different proteins that are found in cilia and/or basal bodies has been defined; http://www.ciliaproteome.org/browse.shtml). This parts-list provides a valuable resource to identify genes responsible for cilia-related phenotypes. Syndromes that include polycystic kidney disease, retinal degeneration, anosmia, polydactyly or situs reversal are candidates to affect cilia. These include Bardet-Biedl syndrome (BBS), Alstrom syndrome (ALMS), Meckel-Gruber syndrome (MKS), and Oral-facial-digital (OFD) syndrome. Several of these syndromes include features not obviously related to cilia, including obesity and mental retardation. Rather than providing a review of the literature on these “ciliopathies” (for recent review, see Badano et al, 2006), we consider here whether these disorders could stem from disruption of developmental signaling processes.
As described above, human females heterozygous for mutations in OFD1 (the gene responsible for Oral-Facial-Digital syndrome type I) can have polycystic kidney disease, in combination with abnormalities in the digits and the face and the OFD1 protein localizes to the basal body and is required for assembly of the cilium. The features exhibited by OFD type I patients can generally be ascribed to defective cilia function and some of these features seen in OFD1 females are likely to reflect defective control of the Hedgehog pathway. For example, limb abnormalities such as syndactyly and polydactyly may result from failure to generate the Gli3 repressor during development, given that mice and humans harboring mutations in Gli3 (Greig cephalopolysyndactyly and Pallister-Hall syndromes) show similar features. In addition, features such as cleft palate, cleft lip, and agensis of the corpus collosum, all features associated with holoprosencephaly, are associated with both OFD1 and Gli2 loss-of-function (Mo et al.1997, Vieira et al., 2005).
BBS is a genetically heterogeneous syndrome: mutations in 11 different genes have been shown to cause BBS. The BBS4, BBS5, BBS6, and BBS8 proteins (as well as the Alstrom syndome and Meckel-Gruber syndrome proteins ALMS1 and MKS1) localize to basal bodies and centrosomes (Ansley et al., 2003; Li et al., 2004; Kim et al., 2005; Kim et al., 2004; Hearn et al., 2005; Dawe et al, 2007). Mouse mutations in Bbs1, Bbs2, Bbs4 and Bbs6 (Mkks) mimic some of the phenotypes of human Bardet-Biedl syndrome, including obesity, anosmia, rod-cone dystrophy, hearing loss, polycystic kidney disease, situs inversus, and polydactyly. Most of these features can be explained by defects in the specialized cilia of the visual and olfactory systems and defects in renal cilia. Situs inversus in BBS patients could result from a defect in motile or mechanosensory cilia in the embryonic node (McGrath et al., 2003). It may be possible to investigate nodal cilia in the mouse mutants, although defects in laterality have not been described in the mouse mutants at this time. The polydactyly associated with human BBS could be related to dysregulated Hedgehog signaling, although polydactyly is not seen in the mouse BBS mutants, which makes it difficult to test this hypothesis.
The origin of other features of these syndromes, such as obesity, remains obscure. It is also interesting that mice mutant for the tubby gene share features in common with BBS patients, such as obesity, retinal degeneration, and hearing loss (Ohlemiller et al., 1995). Other members of Tubby protein family also have cilia-related activities. For example, in Bbs2 and Tubby-like protein 1 (Tulp1) mouse mutants, rhodopsin is not effectively transported via the connecting cilium to the outer segment of retinal photoreceptor cells, apparently leading to death of photoreceptors (Nishimura et al., 2004; Hagstrom et al., 1998; Hagstrom et al., 2001). Mouse Tubby-like protein 3 (Tulp3) localizes to the basal bodies and the tips of cilia and functions as an antagonist of Shh signaling in the nervous system and the limb (R. Norman. A. Ikeda, and JTE, in preparation). Like BBS patients, Tulp3 mouse mutants are polydactylous. C. elegans Tub1 and BBS1 are both expressed in ciliated neurons and are involved in controlling fat storage. The C. elegans bbs1;tub1 double mutants exhibit the identical phenotype observed in bbs-1 and tub-1 single mutants, suggesting the two factors function in the same pathway (Mak et al., 2006). Tubby has been implicated in several signaling processes and the protein has been shown to translocate from the membrane, where it associated with PIP2, to the nucleus, where it is capable of DNA binding, in response to activation of specific G protein-coupled receptors, such as 5-HT (serotonin) receptors (Santagata et al., 2001). Given that 5-HT signaling is intimately involved in controlling weight, feeding behavior, and fat deposition (reviewed in Meguid et al., 2000), and that the 5-HT(6) receptor has been shown to localize to cilia (Brailov et al., 2000), it is tempting to speculate that BBS and Tubby proteins could control obesity by permitting proper serotonergic signaling occurring within cilia.
Mechanistic studies in C. elegans have shown that BBS proteins have a specific function in specialized cilia that have singlet microtubules extended from the distal tip of the standard outer doublets. C. elegans BBS7 and BBS8 are required for assembly of the distal segment of the cilium and are believed to coordinate the activities of two anterograde motors, kinesin-II and Osm-3 (Ou et al., 2005). This raises the possibility that mammalian BBS proteins may not have a role in all cilia but, rather, could play specialized functions in particular types of cilia, such in construction or maintenance of the distal segment in photoreceptor cells.
Meckel-Gruber syndrome type 3 and Joubert syndrome type 6 also show renal cystic dysplasia and malformations of the CNS, and both are associated with mutations in the MKS3 gene (Smith et al., 2006; Baala et al., 2007). MKS3 encodes Meckelin, a novel seven-pass transmembrane protein related to Frizzleds, components of the Wnt receptors. Like several other proteins of similar structure such as Smoothened and 5-HT receptors, Meckelin localizes to primary cilia and the plasma membrane and Meckelin co-immunoprecipitates with MKS1, the basal body-associated factor mutated in Meckel-Gruber syndrome type 1 (Dawe et al, 2007). These findings suggest that Meckelin and MKS1 may act in specialized cilia to participate in an unidentified signal transduction pathway that is important for development of the kidney and the CNS.
Are primary cilia signal transduction machines?
It has been suggested that cilia, which protrude into the extracellular space, act as antennae to receive extracellular signals (Singla and Reiter, 2006). If Hedgehog ligands bind the Patched receptor at the cilium, the cilium may indeed act as such an antenna. Nevertheless, it is also useful to think of cilia as signaling machines. An organelle like the ribosome contains dozens of proteins and acts as a processive machine that creates an ordered series of chemical reactions. A cilium is at least as complex an organelle as the ribosome, with hundreds of proteins dedicated to cilia organization. The complexity of the events that take place in cilia suggests it may be useful to think of the cilium as another organelle that acts as a processive cellular machine. Mammalian Hedgehog signaling depends on an ordered series of events that apparently take place in the cilium. In the absence of ligand, the cilium provides baseline regulation of Gli transcription factors, which must be localized to cilia to be processed. Activation of the pathway by ligand promotes the relocalization of the transmembrane protein Smo to the base of the cilium, allowing it to be transported to the tip of the cilium, where it may undergo an ordered set of interactions with Sufu, Glis and other proteins. When these reactions are complete, retrograde IFT selects that appropriately modified Glis for transport back to the base of the cilium, and ultimately to cellular nuclei. If additional signaling pathways depend on cilia in vivo, the complex organization of the cilium may direct specific interactions between pathways. Cell-type specific modulation of cilia structure would add flexibility to the processes of signal transduction and cross-talk. Future studies will show whether the cilium can act as a signal integrator as well as a signal assembly line.
Acknowledgments
We thank Miquel Tuson and Tim Bestor for comments on the manuscript. We also thank Miquel Tuson, Karel Liem, Tamara Caspary, Jian Qin, and Ryan Norman for sharing their unpublished data. Work on Hedgehog signaling in the Eggenschwiler and Anderson laboratories is supported by grants from the National Institutes of Health.
Literature Cited
- Adler PN. Planar signaling and morphogenesis in Drosophila. Dev Cell. 2002;2(5):525–35. doi: 10.1016/s1534-5807(02)00176-4. [DOI] [PubMed] [Google Scholar]
- Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193(4250):317–9. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- Afzelius BA. Cilia-related diseases. J Pathol. 2004;204(4):470–7. doi: 10.1002/path.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht-Buehler G. Phagokinetic tracks of 3T3 cells: parallels between the orientation of track segments and of cellular structures which contain actin or tubulin. Cell. 1977;12(2):333–9. doi: 10.1016/0092-8674(77)90109-x. [DOI] [PubMed] [Google Scholar]
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425(6958):628–33. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, et al. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117(4):527–39. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu Rev Genomics Hum Genet. 2006;7:125–48. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- Barnfield PC, Zhang X, Thanabalasingham V, Yoshida M, Hui CC. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation. 2005;73(8):397–405. doi: 10.1111/j.1432-0436.2005.00042.x. [DOI] [PubMed] [Google Scholar]
- Blacque OE, Li C, Inglis PN, Esmail MA, Ou G, et al. The WD repeat-containing protein IFTA-1 is required for retrograde intraflagellar transport. Mol Biol Cell. 2006;17(12):5053–62. doi: 10.1091/mbc.E06-06-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacque OE, Leroux MR. Bardet-Biedl syndrome: an emerging pathomechanism of intracellular transport. Cell Mol Life Sci. 2006;63(18):2145–61. doi: 10.1007/s00018-006-6180-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom H, Willetts K, Pekny M, Leveen P, Lindahl P, et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell. 1996;85(6):863–73. doi: 10.1016/s0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, Verge D. Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res. 2000;872(1–2):271–5. doi: 10.1016/s0006-8993(00)02519-1. [DOI] [PubMed] [Google Scholar]
- Brazelton WJ, Amundsen CD, Silflow CD, Lefebvre PA. bld1 mutation identifies the Chlamydomonas osm-6 homolog as a gene required for flagellar assembly. Curr Biol. 2001;11(20):1591–4. doi: 10.1016/s0960-9822(01)00485-7. [DOI] [PubMed] [Google Scholar]
- Brito JM, Teillet MA, Le Douarin NM. An early role for sonic hedgehog from foregut endoderm in jaw development: ensuring neural crest cell survival. Proc Natl Acad Sci USA. 2006;103(31):11607–12. doi: 10.1073/pnas.0604751103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulgakov OV, Eggenschwiler JT, Hong DH, Anderson KV, Li T. FKBP8 is a negative regulator of mouse sonic hedgehog signaling in neural tissues. Development. 2004;131(9):2149–59. doi: 10.1242/dev.01122. [DOI] [PubMed] [Google Scholar]
- Buttitta L, Mo R, Hui CC, Fan CM. Interplays of Gli2 and Gli3 and their requirement in mediating Shh-dependent sclerotome induction. Development. 2003;130(25):6233–43. doi: 10.1242/dev.00851. [DOI] [PubMed] [Google Scholar]
- Camner P, Mossberg B, Afzelius BA. Evidence of congenitally nonfunctioning cilia in the tracheobronchial tract in two subjects. Am Rev Respir Dis. 1975;112(6):807–9. doi: 10.1164/arrd.1975.112.6.807. [DOI] [PubMed] [Google Scholar]
- Cano DA, Murcia NS, Pazour GJ, Hebrok M. Orpk mouse model of polycystic kidney disease reveals essential role of primary cilia in pancreatic tissue organization. Development. 2004;131(14):3457–67. doi: 10.1242/dev.01189. [DOI] [PubMed] [Google Scholar]
- Chazaud C, Rossant J. Disruption of early proximodistal patterning and AVE formation in Apc mutants. Development. 2006;133(17):3379–87. doi: 10.1242/dev.02523. [DOI] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383(6599):407–13. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- Ciruna B, Weidinger G, Knaut H, Thisse B, Thisse C, et al. Production of maternal-zygotic mutant zebrafish by germ-line replacement. Proc Natl Acad Sci USA. 2002;99(23):14919–24. doi: 10.1073/pnas.222459999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol. 1998;141(4):993–1008. doi: 10.1083/jcb.141.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–21. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126(20):4557–68. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, et al. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16(2):173–86. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- Dutcher SK. Flagellar assembly in two hundred and fifty easy-to-follow steps. Trends Genet. 1995;11(10):398–404. doi: 10.1016/s0168-9525(00)89123-4. [DOI] [PubMed] [Google Scholar]
- Eggenschwiler JT, Espinoza E, Anderson KV. Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature. 2001;412(6843):194–8. doi: 10.1038/35084089. [DOI] [PubMed] [Google Scholar]
- Fan S, Hurd TW, Liu CJ, Straight SW, Weimbs T, et al. Polarity proteins control ciliogenesis via kinesin motor interactions. Curr Biol. 2004;14(16):1451–61. doi: 10.1016/j.cub.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38(1):112–7. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- Fischer JA, Eun SH, Doolan BT. Endocytosis, endosome trafficking, and the regulation of Drosophila development. Annu Rev Cell Dev Biol. 2006;22:181–206. doi: 10.1146/annurev.cellbio.22.010605.093205. [DOI] [PubMed] [Google Scholar]
- Follit JA, Tuft RA, Fogarty KE, Pazour GJ. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol Biol Cell. 2006;17(9):3781–92. doi: 10.1091/mbc.E06-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg-Nilsson K, Behar TN, Afrakhte M, Barker JL, McKay RD. Platelet-derived growth factor induces chemotaxis of neuroepithelial stem cells. J Neurosci Res. 1998;53(5):521–30. doi: 10.1002/(SICI)1097-4547(19980901)53:5<521::AID-JNR2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Franco B, Ballabio A. X-inactivation and human disease: X-linked dominant male-lethal disorders. Curr Opin Genet Dev. 2006;16(3):254–9. doi: 10.1016/j.gde.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Galceran J, Farinas I, Depew MJ, Clevers H, Grosschedl R. Wnt3a−/−-like phenotype and limb deficiency in Lef1(−/−)Tcf1(−/−) mice. Genes Dev. 1999;13(6):709–17. doi: 10.1101/gad.13.6.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Ziff EB. Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature. 1984;311(5985):433–8. doi: 10.1038/311433a0. [DOI] [PubMed] [Google Scholar]
- Guo N, Hawkins C, Nathans J. Frizzled6 controls hair patterning in mice. Proc Natl Acad Sci USA. 2004;101(25):9277–81. doi: 10.1073/pnas.0402802101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagstrom SA, Adamian M, Scimeca M, Pawlyk BS, Yue G, Li T. A role for the Tubby-like protein 1 in rhodopsin transport. Invest Ophthalmol Vis Sci. 2001;42(9):1955–62. [PubMed] [Google Scholar]
- Han YG, Kwok BH, Kernan MJ. Intraflagellar transport is required in Drosophila to differentiate sensory cilia but not sperm. Curr Biol. 2003;13(19):1679–86. doi: 10.1016/j.cub.2003.08.034. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1(4):e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, et al. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134(2):307–16. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- Hearn T, Spalluto C, Phillips VJ, Renforth GL, Copin N, et al. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes. 2005;54(5):1581–7. doi: 10.2337/diabetes.54.5.1581. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79(4):1283–316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Volarevic S, Dikic I. Compartmentalization of growth factor receptor signalling. Curr Opin Cell Biol. 2005;17(2):107–11. doi: 10.1016/j.ceb.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Hu MC, Mo R, Bhella S, Wilson CW, Chuang PT, et al. GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes disrupts renal morphogenesis. Development. 2006;133(3):569–78. doi: 10.1242/dev.02220. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426(6962):83–7. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA. 2005;102(32):11325–30. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development. 2006;133(1):3–14. doi: 10.1242/dev.02169. [DOI] [PubMed] [Google Scholar]
- Hui CC, Joyner AL. A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet. 1993;3(3):241–6. doi: 10.1038/ng0393-241. [DOI] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059–87. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Izraeli S, Lowe LA, Bertness VL, Campaner S, Hahn H, et al. Genetic evidence that Sil is required for the Sonic Hedgehog response pathway. Genesis. 2001;31(2):72–7. doi: 10.1002/gene.10004. [DOI] [PubMed] [Google Scholar]
- Karcher C, Fischer A, Schweickert A, Bitzer E, Horie S, et al. Lack of a laterality phenotype in Pkd1 knock-out embryos correlates with absence of polycystin-1 in nodal cilia. Differentiation. 2005;73(8):425–32. doi: 10.1111/j.1432-0436.2005.00048.x. [DOI] [PubMed] [Google Scholar]
- Kartagener M. Zur Pathogenese der Bronchiektasien: Bronchiektasien bei Situs viscerum inversus. Beiträge zum Klinik der Tuberkulose. 1933;83:489–501. [Google Scholar]
- Kim JC, Badano JL, Sibold S, Esmail MA, Hill J, et al. The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet. 2004;36(5):462–70. doi: 10.1038/ng1352. [DOI] [PubMed] [Google Scholar]
- Kim JC, Ou YY, Badano JL, Esmail MA, Leitch CC, et al. MKKS/BBS6, a divergent chaperonin-like protein linked to the obesity disorder Bardet-Biedl syndrome, is a novel centrosomal component required for cytokinesis. J Cell Sci. 2005;118(Pt 5):1007–20. doi: 10.1242/jcs.01676. [DOI] [PubMed] [Google Scholar]
- Kimelman D, Xu W. β-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25(57):7482–91. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA, Hamilton TG, Hoch R, Soriano P. An allelic series at the PDGFalphaR locus indicates unequal contributions of distinct signaling pathways during development. Dev Cell. 2002;2(1):103–13. doi: 10.1016/s1534-5807(01)00103-4. [DOI] [PubMed] [Google Scholar]
- Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, et al. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1(5):312–9. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- Kozminski KG, Johnson KA, Forscher P, Rosenbaum JL. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci USA. 1993;90(12):5519–23. doi: 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski KG, Beech PL, Rosenbaum JL. The Chlamydomonas kinesin-like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol. 1995;131(6 Pt 1):1517–27. doi: 10.1083/jcb.131.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Q, Jeong Y, Misra K, Li S, Zelman AK, et al. Wnt signaling inhibitors regulate the transcriptional response to morphogenetic Shh-Gli signaling in the neural tube. Dev Cell. 2006;11(3):325–37. doi: 10.1016/j.devcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117(4):541–52. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA. 2003;100(9):5286–91. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litingtung Y, Dahn RD, Li Y, Fallon JF, Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418(6901):979–83. doi: 10.1038/nature01033. [DOI] [PubMed] [Google Scholar]
- Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22(4):361–5. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132(13):3103–11. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304(5678):1755–9. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- Mak HY, Nelson LS, Basson M, Johnson CD, Ruvkun G. Polygenic control of Caenorhabditis elegans fat storage. Nat Genet. 2006;38(3):363–8. doi: 10.1038/ng1739. [DOI] [PubMed] [Google Scholar]
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci USA. 1999;96(9):5043–8. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, et al. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287(2):378–89. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114(1):61–73. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- Meguid MM, Fetissov SO, Varma M, Sato T, Zhang L, et al. Hypothalamic dopamine and serotonin in the regulation of food intake. Nutrition. 2000;16(10):843–57. doi: 10.1016/s0899-9007(00)00449-4. [DOI] [PubMed] [Google Scholar]
- Methot N, Basler K. Suppressor of fused opposes hedgehog signal transduction by impeding nuclear accumulation of the activator form of Cubitus interruptus. Development. 2000;127(18):4001–10. doi: 10.1242/dev.127.18.4001. [DOI] [PubMed] [Google Scholar]
- Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124(1):113–23. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- Montcouquiol M, Sans N, Huss D, Kach J, Dickman JD, et al. Asymmetric localization of Vangl2 and Fz3 indicate novel mechanisms for planar cell polarity in mammals. J Neurosci. 2006;26(19):5265–75. doi: 10.1523/JNEUROSCI.4680-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development. 2000;127(11):2347–55. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. 2004;26(8):844–56. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci USA. 2004;101(47):16588–93. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95(6):829–37. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Yoshiba S, Watanabe D, Ikeuchi S, Goto T, et al. De novo formation of left-right asymmetry by posterior tilt of nodal cilia. PLoS Biol. 2005;3(8):e268. doi: 10.1371/journal.pbio.0030268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R. Wnts and Hedgehogs: lipid-modified proteins and similarities in signaling mechanisms at the cell surface. Development. 2003;130(22):5297–305. doi: 10.1242/dev.00821. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK, Hughes RM, Mosinger-Ogilvie J, Speck JD, Grosof DH, Silverman MS. Cochlear and retinal degeneration in the tubby mouse. Neuroreport. 1995;6(6):845–9. doi: 10.1097/00001756-199504190-00005. [DOI] [PubMed] [Google Scholar]
- Omori Y, Malicki J. oko meduzy and related crumbs genes are determinants of apical cell features in the vertebrate embryo. Curr Biol. 2006;16(10):945–57. doi: 10.1016/j.cub.2006.03.058. [DOI] [PubMed] [Google Scholar]
- Orentas DM, Hayes JE, Dyer KL, Miller RH. Sonic hedgehog signaling is required during the appearance of spinal cord oligodendrocyte precursors. Development. 1999;126(11):2419–29. doi: 10.1242/dev.126.11.2419. [DOI] [PubMed] [Google Scholar]
- Orozco JT, Wedaman KP, Signor D, Brown H, Rose L, Scholey JM. Movement of motor and cargo along cilia. Nature. 1999;398(6729):674. doi: 10.1038/19448. [DOI] [PubMed] [Google Scholar]
- Ou G, Blacque OE, Snow JJ, Leroux MR, Scholey JM. Functional coordination of intraflagellar transport motors. Nature. 2005;436(7050):583–7. doi: 10.1038/nature03818. [DOI] [PubMed] [Google Scholar]
- Paces-Fessy M, Boucher D, Petit E, Paute-Briand S, Blanchet-Tournier MF. The negative regulator of Gli, Suppressor of fused (Sufu), interacts with SAP18, Galectin3 and other nuclear proteins. Biochem J. 2004;378(Pt 2):353–62. doi: 10.1042/BJ20030786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B. Hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26(9):3365–77. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Ou G, Civelekoglu-Scholey G, Blacque OE, Endres NF, et al. Mechanism of transport of IFT particles in C. elegans cilia by the concerted action of kinesin-II and OSM-3 motors. J Cell Biol. 2006;174:1035–45. doi: 10.1083/jcb.200606003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TJ, Haigo SL, Wallingford JB. Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nat Genet. 2006;38(3):303–11. doi: 10.1038/ng1753. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151(3):709–18. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LB, Geimer S, Rosenbaum JL. Dissecting the molecular mechanisms of intraflagellar transport in chlamydomonas. Curr Biol. 2006;16(5):450–9. doi: 10.1016/j.cub.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Perkins LA, Hedgecock EM, Thomson JN, Culotti JG. Mutant sensory cilia in the nematode Caenorhabditis elegans. Dev Biol. 1986;117(2):456–87. doi: 10.1016/0012-1606(86)90314-3. [DOI] [PubMed] [Google Scholar]
- Pietras K, Sjoblom T, Rubin K, Heldin CH, Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3(5):439–43. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
- Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, et al. Functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol. 2004;15(12):3035–43. doi: 10.1097/01.ASN.0000144204.01352.86. [DOI] [PubMed] [Google Scholar]
- Piperno G, Siuda E, Henderson S, Segil M, Vaananen H, Sassaroli M. Distinct mutants of retrograde intraflagellar transport (IFT) share similar morphological and molecular defects. J Cell Biol. 1998;143(6):1591–601. doi: 10.1083/jcb.143.6.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pledger WJ, Hart CA, Locatell KL, Scher CD. Platelet-derived growth factor-modulated proteins: constitutive synthesis by a transformed cell line. Proc Natl Acad Sci USA. 1981;78(7):4358–62. doi: 10.1073/pnas.78.7.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA. OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. J Am Soc Nephrol. 2004;15(10):2556–68. doi: 10.1097/01.ASN.0000140220.46477.5C. [DOI] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002 Nov;3(11):813–25. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet. 2005;37(10):1135–40. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5(12):1026–33. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- Saadi-Kheddouci S, Berrebi D, Romagnolo B, Cluzeaud F, Peuchmaur M, et al. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene. 2001;20(42):5972–81. doi: 10.1038/sj.onc.1204825. [DOI] [PubMed] [Google Scholar]
- Santagata S, Boggon TJ, Baird CL, Gomez CA, Zhao J, et al. G-protein signaling through tubby proteins. Science. 2001;292(5524):2041–50. doi: 10.1126/science.1061233. [DOI] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, et al. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15(20):1861–6. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Sekimizu K, Nishioka N, Sasaki H, Takeda H, Karlstrom RO, Kawakami A. The zebrafish iguana locus encodes Dzip1, a novel zinc-finger protein required for proper regulation of Hedgehog signaling. Development. 2004;131(11):2521–32. doi: 10.1242/dev.01059. [DOI] [PubMed] [Google Scholar]
- Shiratori H, Hamada H. The left-right axis in the mouse: from origin to morphology. Development. 2006;133(11):2095–104. doi: 10.1242/dev.02384. [DOI] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37(5):537–43. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. 2006;313(5787):629–33. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- Soriano P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124(14):2691–700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- Stiles CD, Capone GT, Scher CD, Antoniades HN, Van Wyk JJ, Pledger WJ. Dual control of cell growth by somatomedins and platelet-derived growth factor. Proc Natl Acad Sci USA. 1979;76(3):1279–83. doi: 10.1073/pnas.76.3.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13(16):2072–86. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131(16):4085–93. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- Supp DM, Brueckner M, Kuehn MR, Witte DP, Lowe LA, et al. Targeted deletion of the ATP binding domain of left-right dynein confirms its role in specifying development of left-right asymmetries. Development. 1999;126(23):5495–504. doi: 10.1242/dev.126.23.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10(2):187–97. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N. Left-right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A−/− mice analysis. J Cell Biol. 1999;145(4):825–36. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Welscher P, Zuniga A, Kuijper S, Drenth T, Goedemans HJ, Meijlink F, Zeller R. Progression of vertebrate limb development through SHH-mediated counteraction of GLI3. Science. 2002 Oct 25;298(5594):827–30. doi: 10.1126/science.1075620. [DOI] [PubMed] [Google Scholar]
- Tempe D, Casas M, Karaz S, Blanchet-Tournier MF, Concordet JP. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol Cell Biol. 2006;26(11):4316–26. doi: 10.1128/MCB.02183-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickle C. Making digit patterns in the vertebrate limb. Nat Rev Mol Cell Biol. 2006 Jan;7(1):45–53. doi: 10.1038/nrm1830. [DOI] [PubMed] [Google Scholar]
- Tsujikawa M, Malicki J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron. 2004;42(5):703–16. doi: 10.1016/s0896-6273(04)00268-5. [DOI] [PubMed] [Google Scholar]
- Tucker RW, Pardee AB, Fujiwara K. Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell. 1979a;17(3):527–35. doi: 10.1016/0092-8674(79)90261-7. [DOI] [PubMed] [Google Scholar]
- Tucker RW, Scher CD, Stiles CD. Centriole deciliation associated with the early response of 3T3 cells to growth factors but not to SV40. Cell. 1979b;18(4):1065–72. doi: 10.1016/0092-8674(79)90219-8. [DOI] [PubMed] [Google Scholar]
- Uren A, Yu JC, Gholami NS, Pierce JH, Heidaran MA. The alpha PDGFR tyrosine kinase mediates locomotion of two different cell types through chemotaxis and chemokinesis. Biochem Biophys Res Commun. 1994;204(2):628–34. doi: 10.1006/bbrc.1994.2505. [DOI] [PubMed] [Google Scholar]
- Vieira AR, Avila JR, Daack-Hirsch S, Dragan E, Felix TM, et al. Medical sequencing of candidate genes for nonsyndromic cleft lip and palate. PLoS Genet. 2005;1(6):e64. doi: 10.1371/journal.pgen.0010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther Z, Vashishtha M, Hall JL. The Chlamydomonas FLA10 gene encodes a novel kinesin-homologous protein. J Cell Biol. 1994;126(1):175–88. doi: 10.1083/jcb.126.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100(4):423–34. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- Wang B, Li Y. Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc Natl Acad Sci USA. 2006;103(1):33–8. doi: 10.1073/pnas.0509927103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nathans J. Tissue/planar cell polarity in vertebrates: new insights and new questions. Development. 2007;134(4):647–58. doi: 10.1242/dev.02772. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Saijoh Y, Nonaka S, Sasaki G, Ikawa Y, et al. The left-right determinant Inversin is a component of node monocilia and other 9+0 cilia. Development. 2003;130(9):1725–34. doi: 10.1242/dev.00407. [DOI] [PubMed] [Google Scholar]
- Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, et al. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145(3):481–90. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff C, Roy S, Lewis KE, Schauerte H, Joerg-Rauch G, et al. Iguana encodes a novel zinc-finger protein with coiled-coil domains essential for Hedgehog signal transduction in the zebrafish embryo. Genes Dev. 2004;18(13):1565–76. doi: 10.1101/gad.296004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, et al. The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell. 1997;90(1):181–92. doi: 10.1016/s0092-8674(00)80324-4. [DOI] [PubMed] [Google Scholar]