

Abstract
Opioid compounds with mixed μ agonist / δ antagonist properties could be used as analgesics with low propensity to induce tolerance and dependence. Here we report the synthesis of a new designed multiple ligand deriving from the μ selective agonist endomorphin-2 and the δ selective antagonist pharmacophore Dmt-Tic. As predicted, the resulting bivalent ligand showed a μ agonist / δ antagonist profile deriving from the corresponding activities of each pharmacophore.
Keywords: Designed multiple ligand, Endomorphin-2, Dmt-Tic pharmacophore, Analgesia, Physical dependence
1. Introduction
The development of tolerance and physical dependence induced by chronic morphine administration limits its prolonged use in the treatment of pain. Analgesia and tolerance to morphine are abolished in μ opioid receptor knock-out mice, implicating the μ opioid receptor as the primary receptor type mediating both of these effects.1–3 However, several lines of evidence suggest the additional involvement of the δ opioid receptor in morphine tolerance. Initial studies using δ opioid receptor antagonists4 and more recent studies using δ opioid receptor knock-out mice5 were shown to disrupt the development of tolerance. In pharmacological studies, the selective δ receptor antagonist naltrindole has been shown to interact with alternative receptors because naltrindole binding was still detected in the μ/δ/κ triple knock-out mice.6
Furthermore, at high concentrations, naltrindole has been shown to lose its δ selectivity and act as an agonist in some cell types.7 These observations suggest that the development of opioid ligands possessing mixed μ agonist / δ antagonist activity may provide a novel approach for the development of analgesic agents with low propensity to produce tolerance, physical dependence and other side effects. In this context, interesting compounds were reported. As an example, the pseudotetrapeptide DIPP[Ψ] displayed mixed μ agonist / δ antagonist properties in vitro, and analgesia with reduced physical dependence and tolerance when administered icv in rats.8 In our previous SAR studies, we demonstrated that the C-terminal elongation of the Dmt-Tic (Dmt, 2′,6′-dimethyl-L-tyrosine; Tic, 1.2.3.4-tetrahydroisoquinoline3-carboxylic acid) δ selective dipeptide antagonist pharmacophore, yielded the mixed μ agonist / δ antagonist compound, H-Dmt-Tic-Gly-NH-Bzl [pEC50 (Guinea Pig Ileum, GPI) = 8.57 and pA2 (Mouse Vas Deferens, MVD) = 9.25].9
A new interesting strategy in the synthesis of such compounds is actually developed through the Designed Multiple Ligands (DML) obtained by the linkage of two different selective pharmacophores.10 For example, a mixed μ agonist / δ antagonist pseudopeptide was obtained by linking tail to tail through an ethylene spacer, a selective δ antagonist (H-Tyr-TicΨ[CH2-NH]Cha-Phe-OH) with a selective μ agonist (H-Dmt-D-Arg-Phe-Lys-NH2).11 Recently, Neumeyer et al. reported the synthesis and pharmacological evaluation of homo and heterodimeric designed multiple ligands deriving from morphinans,12 and from the μ/κ agonist morphinan derivative butorphan, and the δ antagonist dipeptide Dmt-Tic.13 With the aim to extend our studies in this field, we now report the synthesis of the first DML obtained through the tail to tail condensation of the δ selective Dmt-Tic pharmacophore with the endogenous opioid μ selective agonist endomorphin-2, connected by an ethylendiamine spacer. Although opioid bivalent ligands were developed by tail to tail condensation, such as the μ-opioid selective dermorphin analogues14 and the opioid mimetics containing the bis-[H-Dmt-NH(CH2)n]-2(1H)pyrazinone,15 or the δ-opioid selective enkephalin16 and the 1,6-bis-(N,N-dimethyl-Dmt-Tic-NH)hexyl;17 this is the first report of a C-terminally extended endomorphin-2 with the Dmt-Tic pharmacophore in which both moieties of the new DML retained their inherent opioid receptor preference and biological activities. Bifunctional ligands were also formed between disparate receptor systems, for example combining the weakly μ-opioid selective casomorphine and a substance P antagonist.18
2. Chemistry
The DML pseudopeptide was prepared by standard solution peptide synthesis reactions as outlined in Scheme 1. Boc-N-protected endomorphin-2 tetrapeptide (Boc-Tyr-Pro-Phe-Phe-OH)15e was obtained by a 2 + 2 condensation via WSC/HOBt (WSC, N-(3-dimethyaminopropyl)-N′-ethylcarbodiimide; HOBt, 1-hydroxybenzotriazole) starting from the corresponding dipeptides Boc-Tyr-Pro-OH19 and H-Phe-Phe-OBzl.20 Z-N-monoprotected ethylendiamine was condensed with Boc-Tic-OH via WSC/HOBt to give Z-NH-CH2-CH2-NH←Tic←Boc. After Boc deprotection (TFA, trifluoroacetic acid), it was condensed with Boc-Dmt-OH (WSC/HOBt) to obtain Z-NH-CH2-CH2-NH←Tic←Dmt←Boc. This compound was Z (benzyloxycarbonyl) deprotected by catalytic hydrogenation and then condensed (WSC/HOBt) with the Boc-N-protected endomorphin-2 tetrapeptide previously deprotected at the C-terminus by catalytic hydrogenation. Removal of Boc (tert-butyloxycarbonyl) protecting groups (TFA) gave the final pseudopeptides DML that was purified by preparative reverse phase HPLC. The final compounds (1, 4) were identified by HPLC, elemental analysis, mass spectrometry and NMR spectroscopy.
Scheme 1.
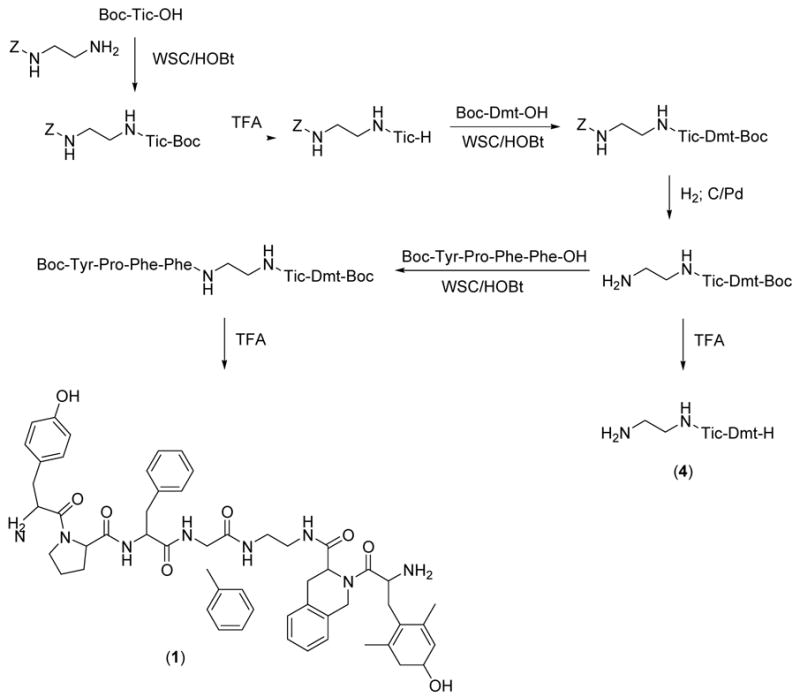
Synthesis of H-Tyr-Pro-Phe-Phe-NH-CH2-CH2-NH←Tic←Dmt-H
3. Results and discussion
3.1. Receptor affinity analysis
Receptor binding and functional bioactivities of H-Tyr-Pro-Phe-Phe-NH-CH2-CH2-NH←Tic←Dmt-H (1) and the reference compounds (2–4) are reported in Table 1. Opioid receptor binding studies were performed using a rat brain synaptosome (P2) fraction.9, 21 In this assay, (1) displayed high μ-receptor binding affinity (Kiμ = 1.03 nM), which is similar to the μ selective reference compound endomorphin-2 (3) (Kiμ = 0.69 nM),22 and high δ receptor binding affinity (Kiδ =1.45 nM) comparable to the reference δ selective antagonist H-Dmt-Tic-NH2 (Kiμ = 1.22 nM).21 Compound (1) did not show significant κ receptor binding affinity (Kiκ > 1 μM).
Table 1.
Receptor binding and functional bioactivity
| Receptor affinity (nM)c | Selectivity | Functional bioactivity | |||||
|---|---|---|---|---|---|---|---|
| Compounds | Kiδ | Kiμ | δ/μ | μ/δ | MVD (IC50) (nM) | MVD pA2d | GPI (IC50) (nM)e |
| 1 H-Tyr-Pro-Phe-Phe-NH-CH2-CH2-NH←Tic←Dmt-H | 1.45 ± 0.20 (3) | 1.03 ± 0.17 (4) | 1.41 | - | 8.9 | 25.1 ± 3 | |
| 2 H-Dmt-Tic-NH2a | 1.22 ± 0.09 | 277 ± 26 | - | 227 | - | 7.2 | > 10000 |
| 3 H-Tyr-Pro-Phe-Phe-NH2b | 9230 ± 200 | 0.69 ± 0.16 | 13400 | - | 344 ± 93 | - | 15 ± 2 |
| 4 H-Dmt-Tic-NH-CH2-CH2-NH2 | 1.81±0.12 (4) | 2.72±0.18 (3) | 3.36 | - | 7.6 | 285.6±21.4 | |
Ref.21.
Ref. 22.
The Ki values (nM) were determined according to Chang and Prusoff.23 The mean ± SE with n repetitions in parentheses is based on independent duplicate binding assays with five to eight peptide doses using several different synaptosomal preparations.
pA2 is the negative logarithm to base 10 of the molar concentration of an antagonist that is necessary to double the concentration of agonist needed to elicit the original submaximal response; the antagonist properties of these compounds were tested using deltorphin II as a δ selective opioid agonist.
Agonist activity was expressed as IC50 obtained from dose-response curves using guinea-pig ileum (GPI). These values represent the mean ± SE for at least five fresh tissue samples. Deltorphin II and dermorphin were the internal standards for mouse vas deferens (MVD, δ-opioid receptor bioactivity) and GPI (μ-opioid receptor bioactivity) tissue preparations, respectively.
3.2 Functional bioactivity
The GPI and MVD bioassays were carried out as reported in the experimental section9, 21 The functional bioactivity of compound (1) showed a μ agonist activity (IC50, GPI = 25.1 nM) in the same order of magnitude of the reference endomorphin-2 (IC50, GPI = 15 nM).22 At the same time, its δ antagonist activity (pA2, MVD = 8.9) was 50 times higher than the reference δ antagonist H-Dmt-Tic-NH2 (pA2, MVD = 7.2).21 This new DML compound endowed of μ agonist / δ antagonist activity showed a functional bioactivity that is in quite good accord with our best μ agonist / δ antagonist compound derived from the Dmt-Tic pharmacophore (H-Dmt-Tic-Gly-NH-Bzl; IC50, GPI = 8.57 nM; pA2, MVD = 9.25).9 With the aim to demonstrate that μ activity is independent from the presence of the ethylendiamine spacer we synthesized compound (4) which maintained the δ antagonism but is endowed with only poor μ agonism.
4. Conclusion
The C-terminal joining of endomorphin-2 and the H-Dmt-Tic pharmacophore via an ethylendiamine linker yielded a new DML pseudopeptide, that maintained the high receptor affinity and in vitro biological potency of the parent peptide ligands. In summary, here we confirmed once more the usefulness of the δ selective Dmt-Tic pharmacophore in the synthesis of DML compounds;13, 15e, 24 but, more important, here we reported for the first time the possibility to use endomorphin-2 (an endogenous ligand for μ opioid receptors) in the synthesis of a new DML. As an example, endomorphin-2 could be used in the synthesis of compounds useful for Magnetic Resonance Imaging or PET imaging. H-Tyr-Pro-Phe-Phe-NH-CH2-CH2-NH-CO-C6H4-pF, could represent a potential pharmacological tool for PET imaging of μ opioid receptors).25
5. Experimental
5.1. Chemistry
5.1.1. General methods
Crude peptides were purified by preparative reversed-phase HPLC [Waters Delta Prep 4000 system with Waters Prep LC 40 mm Assembly column C18 (30 × 4 cm, 15 μm particle size)] and eluted at a flow rate of 25 mL/min with mobile phase solvent A (10% acetonitrile + 0.1% TFA in H2O, v/v), and a linear gradient from 25% to 75% B (60%, acetonitrile + 0.1% TFA in H2O, v/v) in 25 min. Analytical HPLC analyses were performed with a Beckman System Gold (Beckman ultrasphere ODS column, 250 × 4.6 mm, 5 μm particle size). Analytical determinations and capacity factor (K′) of the products used HPLC in solvents A and B programmed at flow rate of 1 mL/min with linear gradient from 0% to 100% B in 25 min. Analogues had less than 1% impurities at 220 and 254 nm. TLC was performed on precoated plates of silica gel F254 (Merck, Darmstadt, Germany): (A) 1-butanol/AcOH/H2O (3:1:1, v/v/v); (B) CH2Cl2/toluene/methanol (17:1:2). Ninhydrin (1% ethanol, Merck), fluorescamine (Hoffman-La Roche) and chlorine spray reagents. Melting points were determined on a Kofler apparatus and are uncorrected. Optical rotations were assessed at 10 mg/mL in methanol with a Perkin-Elmer 241 polarimeter in a 10 cm water-jacketed cell. Molecular weights of the compounds were determined by a MALDI-TOF analysis (Hewlett Packard G2025A LD-TOF system mass spectrometer) and α-cyano-4-hydroxycinnamic acid as a matrix. 1H NMR (δ) spectra were measured, when not specified, in DMSO-d6 solution using a Bruker AC-200 spectrometer, and peak positions are given in parts per million downfield from tetramethylsilane as internal standard.
5.2. Peptide synthesis
5.2.1. Boc-Tyr-Pro-Phe-Phe-OBzl
To a solution of Boc-Tyr-Pro-OH 19 (0.37 g, 0.97 mmol) and TFA · H-Phe-Phe-OBzl 20 (0.5 g, 0.97 mmol) in DMF (10 mL) at 0 °C, NMM (4-methylmorpholine) (0.11 mL, 0.97 mmol), HOBt (0.16 g, 1.07 mmol), and WSC (0.2 g, 1.07 mmol) were added. The reaction mixture was stirred for 3 h at 0 °C and 24 h at room temperature. After DMF (N,N-dimethylformamide) was evaporated, the residue was dissolved in EtOAc (ethyl acetate) and washed with citric acid (10% in H2O), NaHCO3 (5% in H2O), and brine. The organic phase was dried (Na2SO4) and evaporated to dryness. The residue was precipitated from Et2O/Pe (diethyl ether/petroleum ether) (1:9, v/v): yield 0.66 g (89%); Rf (B) 0.91; HPLC K′ 9.14; mp 133–135 °C; [α]20D -22.6; m/z 764 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 1.92-2.34 (m, 4H), 2.92-3.29 (m, 6H), 3.41-3.51 (m, 2H), 4.40-4.92 (m, 4H), 5.34 (s, 2H), 6.68-7.21 (m, 19H).
5.2.2. Boc-Tyr-Pro-Phe-Phe-OH
15e To a solution of Boc-Tyr-Pro-Phe-Phe-OBzl (0.66 g, 0.86 mmol) in methanol (30 mL) was added Pd/C (10%, 0.1 g), and H2 was bubbled for 1 h at room temperature. After filtration, the solution was evaporated to dryness. The residue was crystallized from Et2O/Pe (1:9, v/v): yield 0.56 g (96%); Rf (B) 0.82; HPLC K′ 7.39; mp 142–144 °C; [α]20D -25.1; m/z 674 (M+H)+.
5.2.3. Boc-Tic-NH-CH2-CH2-NH-Z
To a solution of Boc-Tic-OH (0.2 g, 0.72 mmol) and HCl H2N-CH2-CH2-NH-Z (0.17 g, 0.72 mmol) in DMF (10 mL) at 0 °C, NMM (0.08 mL, 0.72 mmol), HOBt (0.12 g, 0.79 mmol), and WSC (0.15 g, 0.79 mmol) were added. The reaction mixture was stirred for 3 h at 0 °C and 24 h at room temperature. After DMF was evaporated, the residue was dissolved in EtOAc and washed with citric acid (10% in H2O), NaHCO3 (5% in H2O), and brine. The organic phase was dried (Na2SO4) and evaporated to dryness. The residue was precipitated from Et2O/Pe (1:9, v/v): yield 0.29 g (89%); Rf (B) 0.81; HPLC K′ 5.18; mp 116–118 °C; [α]20D +7.3; m/z 455 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 2.92-3.46 (m, 6H), 4.17-4.27 (m, 2H), 4.92-5.34 (m, 3H), 6.96-7.19 (m, 9H).
5.2.4. TFA · H-Tic-NH-CH2-CH2-NH-Z
Boc-Tic-NH-CH2-CH2-NH-Z (0.25 g, 0.55 mmol) was treated with TFA (2 mL) for 0.5 h at room temperature. Et2O/Pe (1:1, v/v) were added to the solution until the product precipitated: yield 0.24 g (92%); Rf (A) 0.45; HPLC K′ 3.44; mp 127–129 °C; [α]20D +6.9; m/z 354 (M+H)+.
5.2.5. Boc-Dmt-Tic-NH-CH2-CH2-NH-Z
To asolution of Boc-Dmt-OH (0.18 g, 0.59 mmol) and TFA · H-Tic-NH-CH2-CH2-NH-Z (0.28 g, 0.59 mmol) in DMF (10 mL) at 0 °C, NMM (0.06 mL, 0.59 mmol), HOBt (0.1 g, 0.65 mmol), and WSC (0.12 g, 0.65 mmol) were added. The reaction mixture was stirred for 3 h at 0 °C and 24 h at room temperature. After DMF was evaporated, the residue was dissolved in EtOAc and washed with citric acid (10% in H2O), NaHCO3 (5% in H2O), and brine. The organic phase was dried (Na2SO4) and evaporated to dryness. The residue was precipitated from Et2O/Pe (1:9, v/v): yield 0.33 g (87%); Rf (B) 0.78; HPLC K′ 5.14; mp 134–136 °C; [α]20D +13.7; m/z 646 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 2.35 (s, 6H), 2.92-3.46 (m, 8H), 4.41-4.51 (m, 2H), 4.92-5.34 (m, 4H), 6.29 (s, 2H), 6.96-7.19 (m, 9H).
5.2.6. Boc-Dmt-Tic-NH-CH2-CH2-NH2
To a solution of Boc-Dmt-Tic-NH-CH2-CH2-NH-Z (0.30 g, 0.47 mmol) in methanol (30 mL) was added Pd/C (10%, 0.1 g), and H2 was bubbled for 1 h at room temperature. After filtration, the solution was evaporated to dryness. The residue was crystallized from Et2O/Pe (1:9, v/v): yield 0.20 g (86%); Rf (B) 0.61; HPLC K′ 4.03; mp 132–134 °C; [α]20D +15.9; m/z 512 (M+H)+.
5.2.7. Boc-Tyr-Pro-Phe-Phe-NH-CH2-CH2-NH←Tic←Dmt←Boc
To a solution of Boc-Tyr-Pro-Phe-Phe-OH (0.13 g, 0.2 mmol) and Boc-Dmt-Tic-NH-CH2-CH2-NH2 (0.1 g, 0.2 mmol) in DMF (10 mL) at 0 °C, HOBt (0.03 g, 0.22 mmol), and WSC (0.04 g, 0.22 mmol) were added. The reaction mixture was stirred for 3 h at 0 °C and 24 h at room temperature. After DMF was evaporated, the residue was dissolved in EtOAc and washed with citric acid (10% in H2O), NaHCO3 (5% in H2O), and brine. The organic phase was dried (Na2SO4) and evaporated to dryness. The residue was precipitated from Et2O/Pe (1:9, v/v): yield 0.2 g (84%); Rf (B) 0.92; HPLC K′ 5.30; mp 147–149 °C; [α]20D -17.2; m/z 1166 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40-1.44 (m, 18H), 1.92-2.34 (m, 4H), 2.35 (s, 6H), 2.92-3.51 (m, 16H), 4.41-4.51 (m, 3H), 4.92-4.97 (m, 5H), 6.29 (s, 2H), 6.68-7.21 (m, 18H).
5.2.8. TFA · H-Tyr-Pro-Phe-Phe-NH-CH2-CH2-NH←Tic←Dmt-H · TFA (1)
Boc-Tyr-Pro-Phe-Phe-NH-CH2-CH2-NH←Tic←Dmt←Boc (0.17 g, 0.15 mmol) was treated with TFA (1.5 mL) for 0.5 h at room temperature. Et2O/Pe (1:1, v/v) were added to the solution until the product precipitated: yield 0.17 g (95%); Rf (A) 0.46; HPLC K′ 5.16; mp 153–155 °C; [α]20D -18.6; m/z 966 (M+H)+, 1H-NMR (DMSO-d6) δ 1.92-2.34 (m, 4H), 2.35 (s, 6H), 2.92-3.51 (m, 16H), 3.93-3.97 (m, 2H), 4.40-4.51 (m, 3H), 4.92-4.97 (m, 3H), 6.29 (s, 2H), 6.68-7.21 (m, 18H), Anal Calcd for C59H66F6N8O12: C, 59.39; H, 5.58; N, 9.39. Found: C, 58.82; H, 5.91; N, 9.52.
5.2.9. TFA.H-Dmt-Tic-NH-CH2-CH2-NH2 · TFA (4)
Boc-Dmt-Tic-NH-CH2-CH2-NH2 (0.07 g, 0.14 mmol) was treated with TFA (1 mL) for 0.5 h at room temperature. Et2O/Pe (1:1, v/v) were added to the solution until the product precipitated: yield 0.08 g (93%); Rf (A) 0.36; HPLC K′ 3.27; mp 142–146 °C; [α]20D +14.2; m/z 412 (M+H)+, 1H-NMR (DMSO-d6) δ 2.35 (s, 6H), 2.91-3.17 (m, 6H), 3.46-3.95 (m, 3H), 4.41-4.92 (m, 3H), 6.29 (s, 2H), 6.96-7.02 (m, 4H), Anal Calcd for C27H32F6N4O7: C, 50.78; H, 5.05; N, 8.77. Found: C, 50.62; H, 5.18; N, 8.95.
5.3. Pharmacology
5.3.1.Radioreceptor binding assays
Opioid receptor affinity was determined under equilibrium conditions [2.5 h at room temperature (23 °C)] in a competition assay using brain P2 synaptosomal membranes prepared from Sprague–Dawley rats.26, 27 Synaptosomes were preincubated to remove endogenous opioid peptides and stored at −80 °C in buffered 20% glycerol.26, 28 Each analogue was analyzed in duplicate assays using 5–8 dosages and 3–5 independent repetitions with different synaptosomal preparations (n values are listed in Table 1 in parentheses and results are means ± SE). Unlabelled peptide (2 μM) was used to determine non-specific binding in the presence of 1.9 nM [3H]deltorphin II (45.0 Ci/mmol, Perkin-Elmer, Boston, MA; KD = 1.4 nM) for δ-opioid receptors and 3.5 nM [3H]DAMGO (50.0 Ci/mmol, Amersham Bioscience, Buckinghamshire, UK; KD = 1.5 nM) for μ-opioid receptors. Glass fibre filters (Whatman GFC) were soaked in 0.1% polyethylenimine in order to enhance the signal-to-noise ratio of the bound radiolabelled-synaptosome complex, and the filters were washed thrice in ice-cold buffered BSA (bovine serum albumin).26 The affinity constants (Ki) were calculated according to Cheng and Prusoff.23
5.3.2. Biological activity in isolated tissue preparations
The myenteric plexus longitudinal muscle preparations (2–3 cm segments) from the small intestine of male Hartley strain guinea pigs (GPI) measured μ-opioid receptor agonism, and a single mouse vas deferens (MVD) was used to determine δ-opioid receptor agonism as described previously.6, 29 The isolated tissues were suspended in organ baths containing balanced salt solutions in a physiological buffer, pH 7.5. Agonists were tested for the inhibition of electrically evoked contraction and expressed as IC50 (nM) obtained from the dose–response curves. The IC50 values represent means ± SE of five or six separate assays. δ-antagonist potencies in the MVD assay were determined against the δ-agonist deltorphin II and are expressed as pA2 determined using the Schild Plot.30
Acknowledgments
This research was supported in part by the University of Cagliari, University of Ferrara, and the intramural Research Program of NIH and NIEHS. The authors appreciate the professional expertise and assistance of the library staff and the Comparative Medicine Branch at NIEHS,
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fang FG, Fields HL, Lee NM. J Pharmacol Exp Ther. 1986;238:1039. [PubMed] [Google Scholar]
- 2.Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Nature. 1996;383:819. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 3.Sora I, Funada M, Uhl GR. Eur J Pharmacol. 1997;324:R1. doi: 10.1016/s0014-2999(97)10016-4. [DOI] [PubMed] [Google Scholar]
- 4.Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. J Pharmacol Exp Ther. 1991;258:299. [PubMed] [Google Scholar]
- 5.Nitsche F, Schuller AG, King MA, Zengh M, Pasternak GW, Pintar JE. J Neurosci. 2002;22:10906. doi: 10.1523/JNEUROSCI.22-24-10906.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaveriaux-Ruff C, Filliol D, Simonin F, Matthes HW, Kieffer BL. J Pharmacol Exp Ther. 2001;298:1193. [PubMed] [Google Scholar]
- 7.Chen YL, Law PY, Loh HH. Cancer Res. 2004;64:8723. doi: 10.1158/0008-5472.CAN-03-3091. [DOI] [PubMed] [Google Scholar]
- 8.Schiller PW, Fundytus ME, Merovitz L, Weltrowska G, Nguyen TM-D, Lemieux C, Chung NN, Coderre TJ. J Med Chem. 1999;42:3520. doi: 10.1021/jm980724+. [DOI] [PubMed] [Google Scholar]
- 9.Balboni G, Guerrini R, Salvadori S, Bianchi C, Rizzi D, Bryant SD, Lazarus LH. J Med Chem. 2002;45:713. doi: 10.1021/jm010449i. [DOI] [PubMed] [Google Scholar]
- 10.Morphy R, Rankovic Z. J Med Chem. 2005;48:6523. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 11.Weltrowska G, Lemieux C, Chung NN, Schiller PW. J Pept Res. 2004;63:63. doi: 10.1111/j.1399-3011.2003.00108.x. [DOI] [PubMed] [Google Scholar]
- 12.Peng X, Knapp BI, Bidlack JM, Neumeyer JL. J Med Chem. 2006;49:256. doi: 10.1021/jm050577x. [DOI] [PubMed] [Google Scholar]
- 13.Neumeyer JL, Peng X, Knapp BI, Bidlack JM, Lazarus LH, Salvadori S, Trapella C, Balboni G. J Med Chem. 2006;49:5640. doi: 10.1021/jm0605785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lazarus LH, Guglietta A, Wilson WE, Irons BJ, De Castiglione R. J Biol Chem. 1989;264:354. [PubMed] [Google Scholar]
- 15.a) Okada Y, Tsuda Y, Yokoi T, Sasaki Y, Ambo A, Nagata M, Jinsmaa Y, Bryant SD, Lazarus LH. J Med Chem. 2003;46:3201. doi: 10.1021/jm020459z. [DOI] [PubMed] [Google Scholar]; b) Jinsmaa Y, Miyazaki A, Fujita Y, Fujisawa Y, Shiotani K, Li T, Tsuda Y, Yokoi T, Ambo A, Sasaki Y, Bryant SD, Lazarus LH, Okada Y. J Med Chem. 2004;47:2599. doi: 10.1021/jm0304616. [DOI] [PubMed] [Google Scholar]; c) Jinsmaa Y, Okada Y, Tsuda Y, Shiotani K, Sasaki Y, Ambo A, Bryant SD, Lazarus LH. J Pharmacol Exp Ther. 2004;309:432. doi: 10.1124/jpet.103.060061. [DOI] [PubMed] [Google Scholar]; d) Gao Y, Liu X, Wei J, Zhu B, Chen Q, Wang R. Bioorg Med Chem Lett. 2005;15:1847. doi: 10.1016/j.bmcl.2005.02.021. [DOI] [PubMed] [Google Scholar]; e) Vázquez ME, Blanco JB, Salvadori S, Trapella C, Argazzi R, Bryant SD, Jinsmaa Y, Lazarus LH, Negri L, Giannini E, Lattanzi R, Colucci M, Balboni G. J Med Chem. 2006;49:3653. doi: 10.1021/jm060343t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Shimohigashi Y, Costa T, Chen H-C, Rodbard D. Nature. 1982;297:333. doi: 10.1038/297333a0. [DOI] [PubMed] [Google Scholar]; b) Lipkowski AW, Knonecka AM, Sroczynska I. Peptides. 1982;3:697. doi: 10.1016/0196-9781(82)90173-5. [DOI] [PubMed] [Google Scholar]
- 17.Li T, Fujita Y, Shiotani K, Miyazaki A, Tsuda Y, Ambo A, Sasaki Y, Jinsmaa Y, Marczak E, Bryant SD, Salvadori S, Lazarus LH, Okada Y. J Med Chem. 2005;48:8035. doi: 10.1021/jm050377l. [DOI] [PubMed] [Google Scholar]
- 18.Lipkowski AW, Misterek K. Pol J Pharmacol Pharm. 1992;44:25. [PubMed] [Google Scholar]
- 19.Salvadori S, Marastoni M, Balboni G, Borea PA, Tomatis R. Eur J Med Chem. 1990;25:171. [Google Scholar]
- 20.Oya M, Takahashi T. Bull Chem Soc Jpn. 1981;54:2075. [Google Scholar]
- 21.Salvadori S, Attila M, Balboni G, Bianchi C, Bryant SD, Crescenzi O, Guerrini R, Picone D, Tancredi T, Temessi PA, Lazarus LH. Mol Med. 1995;1:678. [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Zadina JE, Hacker L, Ge LJ, Kastin AB. Nature. 1997;386:499. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]; (b) Fujita Y, Motoyama T, Takahashi M, Shimizu Y, Tsuda Y, Yokoi T, Li T, Sasaki Y, Ambo A, Kita A, Jinsmaa Y, Bryant SD, Lazarus LH, Okada Y. J Med Chem. 2004;47:3591. doi: 10.1021/jm030649p. [DOI] [PubMed] [Google Scholar]
- 23.Cheng Y-C, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 24.Balboni G, Onnis V, Congiu C, Zotti M, Sasaki Y, Ambo A, Bryant SD, Jinsmaa Y, Lazarus LH, Lazzari I, Trapella C, Salvadori S. Bioorg Med Chem. 2007;15:3143–3151. doi: 10.1016/j.bmc.2007.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Zhang X, Cai W, Cao F, Schreibmann E, Wu Y, Wu JC, Xing L, Chen X. J Nucl Med. 2006;47:492. [PubMed] [Google Scholar]; (b) Tyake RJ, Robinson ESJ, Schnabel R, Lewis JW, Husbands SM, Nutt DJ, Hudson AL. Nucl Med Biol. 2002;29:455. doi: 10.1016/s0969-8051(02)00300-1. [DOI] [PubMed] [Google Scholar]
- 26.Lazarus LH, Salvadori S, Santagada V, Tomatis R, Wilson WE. J Med Chem. 1991;34:1350. doi: 10.1021/jm00108a017. [DOI] [PubMed] [Google Scholar]
- 27.Lazarus LH, Salvadori S, Attila M, Grieco P, Bundy DM, Wilson WE, Tomatis R. Peptides. 1993;14:21. doi: 10.1016/0196-9781(93)90006-3. [DOI] [PubMed] [Google Scholar]
- 28.Lazarus LH, Wilson WE, De Castiglione R, Guglietta A. J Biol Chem. 1989;264:3047. [PubMed] [Google Scholar]
- 29.Sasaki Y, Sasaki A, Niizuma H, Goto H, Ambo A. Bioorg Med Chem. 2003;11:675. doi: 10.1016/s0968-0896(02)00601-6. [DOI] [PubMed] [Google Scholar]
- 30.Arunlakshana Q, Schild HO. Br J Pharmacol. 1959;14:48. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]