

Abstract
An attractive feature of targeted radionuclide therapy is the ability to select radionuclides and targeting vehicles with characteristics that are best suited for a particular clinical application. One combination that has been receiving increasing attention is the use of monoclonal antibodies specifically reactive to receptors and antigens that are expressed on tumor cells to selectively deliver the α-particle emitting radiohalogen 211At to malignant cell populations. Promising results have been obtained in preclinical models with multiple 211At-labeled mAbs; however, translation of concept to the clinic has been slow. Impediments to this process include limited radionuclide availability, the need for suitable radiochemistry methods operant at high activity levels, and the lack of data concerning toxicity of α-particle emitters in humans. Nonetheless, two clinical trials have been initiated to date with 211At-labeled monoclonal antibodies and others are planned for the near future.
Keywords: Astatine-211, radioimmunotherapy, targeted radiotherapy, monoclonal antibodies
1. Introduction
Targeted radionuclide therapy (aka targeted radiotherapy, molecular radiotherapy) is an exciting strategy for tumor therapy that has some similarities to the most commonly used approaches for cancer treatment. It is similar to external beam therapy in that cancer cell destruction is achieved by means of radiation induced damage to cellular DNA. Moreover, targeted radiotherapy is like chemotherapy; it is a systemic treatment that uses a molecule, in this case bearing a radiolabel, to deliver a cytotoxic substance to disease sites. However, unlike conventional drugs and toxins, which kill only the directly targeted cells, a unique feature of radionuclides is that they can exert a “bystander” or “crossfire” effect, potentially destroying adjacent tumor cells even if they lack the specific tumor-associated receptor, enzyme or antigen. Furthermore, a systemically administered targeted radiotherapeutic, which combines the exquisite specificity of molecular specific cellular recognition with the anti-neoplastic effects of ionizing radiation has the potential to simultaneously eliminate both a primary tumor site and cancer that has spread throughout the body, including malignant cell populations undetectable by diagnostic imaging.
One of the attractive features of molecular radiotherapy is its modular nature. One can in principle select a targeting vehicle and radionuclide with properties that not only are well matched to each other, but also are compatible with the constraints, both clinical and geometrical, of the intended therapeutic application. There are three types of particulate radiation of consequence for targeted radionuclide therapy – β-particles, α-particles and Auger electrons – which can irradiate tissue volumes with multi-cellular, cellular and sub-cellular dimensions, respectively. Moreover, there are multiple radionuclides decaying by each of these three modes with a variety of tissue ranges, half lives and chemistries, offering the attractive possibility of tailor-making the properties of a targeted radionuclide therapeutic to a particular type of cancer and perhaps ultimately, the needs of an individual patient.
Targeting α-particle emitting radionuclides to cancer cells has emerged as a particularly promising approach to molecular radiotherapy based on accumulating evidence from preclinical studies, as summarized in several recent reviews [1-3]. Because the range of α-particles in tissue is only about 50-80 μm, equivalent to only a few cell diameters, at least on a conceptual level, they are well matched to the cellular specificity of molecularly specific targeting vehicles, and minimize irradiation of tumor-adjacent normal tissues. On the other hand, this property could be detrimental in compensating for heterogeneities in the delivery of the labeled therapeutic and/or the expression of the tumor associated molecular target [4]. A potential compensating factor, however, is the recently identified radiation induced biological bystander effect, in which cells not directly exposed to particulate-emitting radiopharmaceuticals can be killed through as yet unidentified mechanisms [5].
Compared with β-emitters such as 90Y and 131I, the radionuclides utilized in current FDA-approved radiotherapeutics, radionuclides decaying by the emission of α-particles offer several significant advantages from a radiobiological perspective [6]. These are derived from their short tissue range, noted above, and their relatively high decay energy (generally 4-9 MeV), properties which combine to make them radiation of high linear energy transfer (LET). For example, the LET of α-particles emitted by 211At is 97 keV/μm, a value more than 400 times higher than that for the high energy β-particles emitted by 90Y. A fortuitous consequence of the LET of α-particles is that the distance between ionizing events is almost the same as that between the two strands of DNA, yielding a high probability of creating non-repairable DNA double stranded breaks. Numerous studies performed in cell culture with human cancer cell lines have confirmed the exquisite cytotoxicity of targeted α-particles, reduction in cell survival to 37% (D0, the classical parameter for evaluating cell kill) achieved after only about 1-10 α-particle hits per cell [2,4]. Furthermore, the ability of high LET radiation to kill cancer cells is not compromised by hypoxia, dose rate effects or cell cycle position, enhancing their attractiveness for targeted radiotherapy.
As noted above, one of the underlying design principles for developing targeted radiotherapeutics is to match the characteristics of the radionuclide with those of the molecular carrier being used to affect its delivery to malignant cell populations. Given the short tissue range of α-particles, it would seem counterintuitive to combine them with monoclonal antibodies (mAbs) which diffuse through tissue very slowly, drastically limiting delivery of the radionuclide to the majority of tumor cells. Nonetheless, mAbs have been the most widely evaluated molecular carrier for targeted α-particle therapy and the results to date have been encouraging [7-9]. This apparent disconnect is due to the fact that in general, α-emitter radioimmunotherapy is generally pursued in settings in which rapid exposure of the tumor to the labeled mAb can be achieved, for example, by targeting tumors of the blood such as leukemia or those amenable to intracavitary delivery such as ovarian carcinoma. Moreover, by focusing on malignancies with limited tumor depth such as residual margins following tumor debulking, or neoplastic cells spread on the surface of and within body cavities, the short range of α-particles should actually be an advantage.
2. Astatine-211 Labeled mAbs: Promise and Problems
Of the more than 100 known α-particle emitting radionuclides, three have received the most attention for radioimmunotherapy – 213Bi, 211At and 225Ac with half lives of 46 min, 7.2 h and 10 days, respectively [1,8,9]. The half life of 213Bi is so short that it is problematic from a logistical perspective, both in terms of labeling chemistry and patient management. At the other extreme, the 10 day half life of 225Ac, while most advantageous from a convenience perspective, presents a major hurdle to the radiochemist who must devise strategies to avoid escape of 225Ac from the tumor over a multi-week time period. This problem is further complicated by the need to retain the four α-particle emitting daughters in tumor and minimize their deleterious effects on normal tissues in the face of differences in daughter radionuclide chemistry and recoil nuclei induced bond rupture. Although the development of clinically useful 211At-labeled radioimmunotherapeutics certainly is also a daunting task, it holds great promise because of the properties of this radionuclide.
The 7.2-h half life of 211At is long enough to permit multi-step mAb labeling procedures and while not ideal for many intravenous applications, is a reasonable match with the pharmacokinetics of intact mAbs and fragments administered in non-intravenous settings where targeted α-particle radiotherapy has the greatest chance of having meaningful clinical impact. More than 99% of the radiation energy from 211At comes from α-particles [10]. As shown in Figure 1, the emission of an α-particle is associated with 100% of 211At decays, either directly by α-decay to 207Bi (42%), followed by electron capture decay to stable 207Pb, or by electron capture decay to 211Po (58%) followed by α-emission to stable 207Pb. This second decay branch could lead to escape of radioactivity from the 211At uptake site. However, the problem is much less than noted above for 225Ac because the half life of the 211Po intermediate is only 520 msec, limiting its diffusion distance prior to emission of its α-particle. In addition, the potential destabilizing effect of α-particle recoil nuclei on chemical integrity is not a factor because the 211Po is not a daughter created by α-decay. The second decay branch, via electron capture, actually provides an important advantage for 211At -- the emission of 77-92 keV polonium K x-rays, which provide a valuable means for imaging and quantifying 211At activity levels by planar and SPECT imaging [11].
Figure 1.
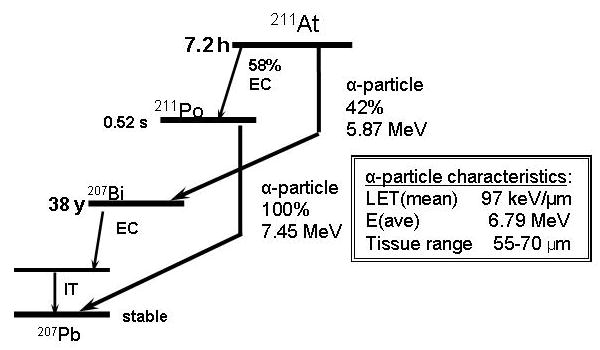
Simplified decay scheme for 211At and properties of 211At α-particles.
One of the most significant impediments to the clinical application of 211At-labeled radioimmunotherapeutic agents is the limited supply of this radionuclide. This does not reflect complexities in production and purification technologies but the limited availability of accelerators with appropriate beam characteristics. Astatine-211 is generally produced by the bombardment of natural bismuth metal targets utilizing the 209Bi(α, 2n)211At reaction. With the availability of internal target systems [12] and efficient dry distillation techniques [13], clinically relevant levels of 211At can be readily obtained by utilizing incident α beams of 28-29 MeV. Unfortunately, however, there are only a few cyclotrons in the United States currently equipped with beams of this type, with the result that during the past 5 years, 211At production has been limited to only three cyclotron facilities, those at University of Washington, the National Institutes of Health, and Duke University. The number of centers capable of producing 211At in Europe is about the same. When the 70-MeV cyclotron in Nantes is operational at the end of 2008, the situation should improve because IBA has announced that it will use this machine to make 211At commercially available.
Despite the limited availability of 211At, a variety of 211At-labeled mAbs have been evaluated in preclinical models and two have reached the stage of initial clinical trials. Table 1 summaries the properties of some of the most extensively investigated 211At-labeled immunoconjugates investigated to date and further details can be found in References 14-27. The most widely utilized procedure for labeling mAbs and mAb fragments is a two step procedure that involves the synthesis of N-succinimidyl 3- or 4-[211At]astatobenzoate (SAB) from the corresponding N-succinimidyl trialkylstannybenzoate precursor followed by coupling of SAB to ε-amino groups of mAb lysine residues [14]. Paired-label comparisons to correspondingly radioiodinated mAbs have shown that at least for intact mAbs, the SAB method provides an 211At-labeled immunoconjugate with sufficient stability to permit clinical studies [16]. Attempts have been made to develop better reagents for linking 211At to mAbs by varying the nature of the benzoate substituent ortho to the 211At as well as the spacer between the aryl ring and the active ester [28,29]. Results to date from in vivo experiments have not demonstrated any conclusive advantage for these labeling methods compared with the original SAB reagent. Astatine-211 labeling of mAbs that rapidly internalize into cancer cells after binding to their molecular target present an additional challenge for the radiopharmaceutical chemist. One approach for accomplishing this task relies on the generation of positively charged catabolites that become trapped inside the cell after lysosomal degradation of the labeled mAb [27].
Table 1.
Some 211At-labeled monoclonal antibodies and fragments under investigation for use as targeted radiotherapeutics
| Antibody | Molecular Target | Labeling Method | Clinical Clinical Application | Reference |
|---|---|---|---|---|
| Me1-14 F(ab’)2 | Proteoglycan chondroitin sulfate | SABa | Melanoma | Zalutsky et al. [14] |
| A6H F(ab’)2 | 120-140 kDa antigen | SAB | Renal cell carcinoma | Wilbur et al. [15] |
| 81C6 | Tenascin | SAB | Glioma | Zalutsky et al.[16] |
| rituximab | CD20 | SAB | B-cell lymphoma | Aurlien et al. [17] |
| L19 scFv | Fibronectin ED-B domain | SAB | Tumor vasculature | Demartis et al. [18] |
| C215 | Not reported | SAB | Colorectal carcinoma | Palm et al. [19] |
| TP-1, TP-3 | Folate receptor | SAB | Various | Henriksen et al. [20] |
| A33 | A33 antigen (43 kDa glycoprotein) | SAB | Colorectal carcinoma | Almqvist et al [21] |
| U36 | CD44v6 | SAB | Head and neck squamous cell carcinoma | Nestor et al. [22] |
| trastuzumab | HER2 | SAB | Breast carcinoma | Akabani et al. [4] |
| 7G7/B6 | CD25 | SAPSb | T-cell leukemia | Zhang et al. [23] |
| MOV18 | 38 kDa folate binding glycoprotein | SAB | Ovarian carcinoma | Andersson et al. [24] |
| MX35 F(ab’)2 | 95 kDa glycoprotein | SAB | Ovarian carcinoma | Elgqvist et al. [25] |
| Anti-TAC | IL-2 receptor | SAPS | T-cell leukemia | Wesley et al. [26] |
| L8A4 | EGFRvIII | SGMABc | glioma | Vaidyanathan et al. [27] |
SAB, N-succinimidyl 3- or 4-[211At]astatobenzoate;
SAPS, N-succinimidyl N-(4-[211At]astatophenethyl) succinamate;
SGMAB, N-succinimidyl 3-[211At]astato-4-guanidinomethylbenzoate
In common with other short half life radionuclides that deposit large amounts of energy in a highly focal manner, an additional hurdle that must be overcome in developing 211At-labeled mAbs as therapeutic radiopharmaceuticals is the potentially deleterious effects of these high radiation fields on labeling chemistry [30]. The SAB method has been shown to be quite reliable for preparing 211At-labeled mAbs at the activity levels needed for preclinical experimentation; however, our experience in labeling a mAb with this reagent for clinical studies demonstrated that this was not the case when doses of labeled mAb of 370 MBq were required [31]. Three problems were encountered: decline in SAB yield, decrease in immunoreactivity and nonspecific binding of the labeled mAb to the walls of the reaction vessel.
Subsequent investigations demonstrated that radiolytic factors can have a major impact on the synthesis of SAB at the activity levels needed for patient treatment. These include generation of byproducts from solvents such as chloroform that can compete with 211At for the tin precursor, reaction with the solvent (benzene), and generation of uneractive 211At species [32,33]. At certain radiation dose levels, SAB could be synthesized in reasonable yields in the absence of acetic acid and oxidant, two components previously thought to be essential for astatodestannylation. Based on these results, we have developed a revised procedure in which the SAB reaction is performed in methanol instead of chloroform, and parallel reaction vials are used to keep the dose in each vial below 1500 Gy. For example, under these conditions, 900 MBq of SAB could be produced from 1375 MBq of 211At divided among four reaction vessels [33].
Taken together, these observations demonstrate the importance of understanding the effects of radiolysis on the synthesis of 211At-labeled mAbs needed for clinical radioimmunotherapy and presumably, other α-particle emitting radiopharmaceuticals. If 211At-labeled mAbs are to have meaningful impact on patient care, future research will be needed to identify radiochemical strategies that circumvent the deleterious effects of radiolysis on the labeling reaction. The fact that 211At is only available at a few centers intensifies the importance of the problem because of the requirement that the 211At be shipped from the cyclotron production site to distant medical centers. Whether this can be best accomplished with the activity embedded in the target, in distilled form, as SAB or as the labeled mAb remains to be ascertained.
3. Translation of 211At-labeled mAbs from Bench to Bedside: Regulatory Hurdles
The conceptual appeal of utilizing 211At-labeled mAbs as cancer therapeutics has been recognized for at least twenty years, and as noted above, more than 15 mAbs and mAb fragments have been labeled with 211At and evaluated in preclinical models. Two impediments to the clinical evaluation of 211At-labled mAbs noted in the previous section were the need for production and purification methods for high activity levels of 211At as well as methodologies for labeling mAbs with good in vivo stability and preservation of immunoreactivity. Another barrier that needed to be overcome was obtaining the data necessary to obtain an Investigational New Drug Permit to allow initiation of clinical studies.
Because no 211At-labled compounds had been investigated previously in humans, to facilitate this process, our strategy was to select a disease setting where improved patient treatment was critically needed and the prospect for minimizing excessive irradiation of normal organs was high. On this basis, we sought FDA approval for a clinical trial to evaluate 211At-labeled chimeric 81C6 anti-tenacin mAb administered into surgically created (tumor) resection cavities (SCRC) in patients with recurrent malignant gliomas. From a clinical significance perspective, conventional methods for treating brain tumors are not effective due to dose limiting toxicity to normal brain. The median survival for patients with glioblastoma multiforme (GBM), the most aggressive malignant brain tumor, is less than 1 year, and the tumors in nearly all patients recur adjacent to the original tumor site, after which median survival, even after surgical debulking, is generally only 16-24 weeks [34]. From a safety perspective, more than 100 newly diagnosed and recurrent brain tumor patients had already been treated with 131I-labeled murine 81C6 mAb, labeled using the Iodogen method, and these studies demonstrated excellent retention of radionuclide within the SCRC and little systemic uptake over the first 24 h after mAb administration. Moreover, laboratory studies had demonstrated that the mouse/human IgG2 construct was considerably more stable than its murine parent both in vivo and in the presence of SCRC cyst fluid [35]. For this reason, chimeric 81C6 was selected for use instead of murine 81C6 as the targeting vehicle for 211At labeling in the clinical trial.
Although administration of a 211At-labeled mAb via the SCRC presents advantages in terms of maximizing efficacy and safety, the lack of a rodent SCRC model complicates acquisition of preclinical data documenting in vivo stability, dosimetry and toxicity profile after administering the labeled mAb via this route. For this reason, it was necessary to evaluate these parameters after intravenous administration, assuming that this represents the worst case scenario, i.e. immediate and complete leakage of the mAb from the SCRC into the systemic circulation. The studies described below provided the critical information upon which an IND from the FDA was obtained for evaluating 211At-labeled chimeric 81C6 in brain tumor patients.
A potential concern with the use of 211At-labeled molecules in humans is that the relatively low bond strength of the carbon-astatine bond could result in loss of label in vivo. To address this issue, a paired-label tissue distribution study was performed in athymic mice with subcutaneous D-54 MG human glioma xenografts to compare the tissue distribution of 211At- and 131I-labeled chimeric 81C6 [16]. Tumor accumulation of 211At-labeled chimeric 81C6 peaked at 16 h and remained constant through the end of the 48-h study. Importantly, the cumulative tumor activity concentration (after correcting for differences in radionuclide half life) was essentially the same for 211At and 131I. The activity levels of the two radiohalogens were similar in most normal tissues; however at some time points, spleen and stomach levels were higher for 211At compared with 131I; presumably reflecting the generation of [211At]astatide in vivo. These results demonstrated that 211At- labeled chimeric 81C6 was reasonably stable in vivo.
Dosimetry calculations were performed based on these intravenous biodistribution data using a quality factor of 5 to reflect the higher radiobiological effectiveness of α-particles; however, these were not used in the IND submission because the intended clinical application involved administration of the labeled mAb into the SCRC. Particularly given the similar retention of radioiodinated and 211At-labeled chimeric 81C6 in tumor noted above, patient SCRC clearance and blood data obtained with 131I-labeled 81C6 mAb was utilized for these calculations. Bone marrow radiation dose was estimated to be 0.4 cGy/mCi and using a relative biological effectiveness of 5 for α-particles, this yielded an estimated dose of 2 cSv/mCi.
Because of the lack of human studies with 211At-labeled compounds in humans, extensive toxicology studies were required. The first set of studies involved evaluation of the acute and chronic toxicity of intravenously administered [211At]astatide measured over a 1 yr period [36], which was requested based on the fact that this represented a worst possible case scenario, i.e. the toxicity that would result if the labeled mAb underwent total and immediate dehalogenation in vivo. In the second, we investigated the toxicity and established the LD10 for intravenously administered 211At-labeled chimeric 81C6, again, over a 1 yr period [37]. The LD10 was found to be 45.7 kBq/g body weight in females and 101.5 kBq/g in males, equivalent to intravenous patient doses of 2.66 GBq in females and 7.10 GBq in males. The toxicity of intravenous 211At-labeled chimeric 81C6 was about half observed for [211At]astatide even though the residence time of the labeled mAb in most organs and blood was higher . Although the reason for this apparently contradictory behavior is not know at this time, it could reflect differences in the heterogeneity of labeled molecule distribution that are likely to occur in dimensions corresponding to the range of α-particles in tissues.
4. Clinical Evaluation of 211At-labeled Chimeric 81C6 mAb
On the basis of the studies outlined above, we received FDA approval to perform a Phase 1 evaluation of the pharmacokinetics and toxicity of 211At-labeled chimeric 81C6 mAb in patients with recurrent malignant brain tumors [38]. The trial has been completed and a manuscript describing details of this study will be submitted in the near future. Briefly, 18 patients received single doses of 10 mg of 211At-labeled chimeric 81C6 mAb labeled with escalating doses of 211At (74 to 370 MBq) via the SCRC. The patient population consisted of 14 GBM, 3 anaplastic astrocytoma and 1 patient with anaplastic oligodendroglioma. Serial gamma camera imaging and blood sampling performed over the first 24 h after injection demonstrated that leakage of 211At from the cavity was slow; an average of 96.7 ± 3.6% of the 211At decays occurred within the cavity and the maximum activity found in the blood pool was 0.26 ± 0.43% of the injected dose. The average [range] radiation absorbed dose delivered to the tumor SCRC margin was 2764 [155-35,000] Gy, reflecting in large part the fact that cavity volumes for these patients varied from 0.2 cm3 to 37.2 cm.3 As shown in Figure 2, median survival for GBM patients (n=14) and all patients (n=18) was 52 and 56.5 weeks, respectively. Particularly encouraging is the fact that 2 of these recurrent GBM patients survived for 150 and 151 weeks after 211At-labeled mAb treatment, which is considerably better than that obtained with conventional treatments (25-30 weeks). In addition, the patient with anaplastic oligodendroglioma survived for nearly 5 years after treatment.
Figure 2.

Kaplan-Meier survival of all recurrent patients and those with GBM after treatment with 211At-labeled chimeric 81C6 anti-tenascin mAb.
5. Potential Impact on Clinical Outcome
A discussion of the potential impact of 211At-labeled mAbs on clinical cancer treatment and patient outcome is exceedingly premature. Until such time as this radionuclide becomes more widely available, routine utilization of these promising α-particle therapeutics cannot be realistically considered. Increased supply of 211At would be most conveniently accomplished from the commercial sector and there are preliminary plans to do this, at least on a trial basis, utilizing the 70-MeV cyclotron currently being constructed in Nantes. Unfortunately, the best motivation for establishing 211At production facilities would come from the demonstration of efficacy in clinical trials which, of course, is difficult to accomplish without a reliable supply of 211At. A further conundrum is that because of α-particle induced, radiolytic effects, astatine becomes less chemically tractable with the passage of time, complicating its use at centers remote from the original production site.
Despite these challenges, two clinical trials have been initiated to evaluate the toxicity, pharmacokinetics and efficacy of 211At-labeled mAbs in cancer patients. In addition to the study performed at Duke University Medical Center and summarized above, the group in Göteborg, Sweden, have recently initiated a Phase 1 study of intraperitoneally administered 211At-lableled MX-35 F(ab’)2 in ovarian cancer patients (R. Hultborn and L. Jacobsson, personal communication). Both trials are similar in that they are directed at the treatment of residual disease present on the surface of body cavities, settings which benefit from the short range of 211At α-particles. They also involve administration of the labeled mAb directly into the body compartment in which the tumor cells are located, minimizing irradiation of normal tissues and expediting exposure of malignant cells before a significant fraction of the 211At atoms have decayed. Perhaps the most important implication of these studies is that they demonstrate that it is feasible to perform clinical trials with 211At-labeled mAbs.
Operating under the optimistic assumption that all the radionuclide supply and radiochemistry issues have been addressed, we can speculate about the potential impact of 211At-labeled chimeric 81C6 on the clinical outcome of patients with glioma. As with other targeted therapeutics, it would be preferable to administer 211At-labeled mAbs to newly diagnosed brain tumor patients instead of waiting for recurrence. In this way, infiltration of glioma cells into neighboring normal brain might be reduced and cumulative dose limiting toxicity from largely ineffective conventional treatments could be avoided. Recent trials in newly diagnosed GBM patients have shown that addition of temozolomide concomitant with radiotherapy increased median survival to 14.6 months [39], the first meaningful increase in survival in this patient population in about 25 years. Even with this advance, there is an obvious need for further advances in brain tumor treatment, particularly because most classes of patients do not receive a significant increase in survival with the addition of temozolomide [40]. Furthermore, if the effect of temozolomide in combination with a targeted radionuclide agent is the same as observed with external beam radiation, then investigation of 211At-labeled chimeric 81C6 plus temozolomide should be considered.
Further refinements in treatment strategy beyond application in newly diagnosed patients and concomitant administration of temozolomide could lead to enhanced clinical efficacy of 211At-labeled chimeric 81C6 in patients with malignant gliomas. Particularly given the fact that this is a short half life, short effective range radiotherapeutic, administration of the labeled mAb in multi-dose regimens might be of benefit. Perhaps the most important tactic is to develop treatment protocols that are tailored to the characteristics of individual patients. For example, instead of administering 211At-labeled chimeric 81C6 at a fixed radioactivity dose, a desired SCRC margin radiation dose could be achieved by taking into account SCRC volume and cavity clearance determined by imaging as has been done with 131I-labeled 81C6 [34]. Finally, it is envisioned that the ideal treatment scenario might be to administer the 211At-labled mAb in tandem with, or perhaps subsequent to, the 131I-labeled mAb to maximize dose deposition in residual tumor margins. Clearly, the selection of optimal radionuclide for this purpose would benefit greatly from knowledge of the extent of tumor remaining after surgical debulking; unfortunately, such information is currently not available.
6. Conclusions
Despite the long recognized conceptual appeal of eliminating malignant cell populations by means of the selective delivery of α-particle emitting 211At via tumor-directed mAbs, translation of concept to clinic has only just begun. Although it would be premature to speculate about the practical impact of this promising class of targeted radiotherapeutics on patient outcome, recent work has achieved an important first step -- demonstration that clinical investigation of 211At-labeled mAbs is feasible. It is envisioned that by focusing on minimal residual disease settings, the chances for clinical success will be greatest. However, progress in this exciting field is critically dependent upon developing strategies for improving the availability of 211At to basic and clinical researchers.
Acknowledgments
The authors’ work is supported by grants CA42324 and NS20023 from the National Institutes of Health and a grant from the Pediatric Brain Tumor Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mulford DA, Scheinberg DA, Jurcic JG. The promise of targeted α-particle therapy. J Nucl Med. 2005;46:199S–204S. [PubMed] [Google Scholar]
- 2.Zalutsky MR, Vaidyanathan G. Astaine-211-labeled radiotherapeutics: an emerging approach to targeted alpha particle therapy. Curr Pharm Design. 2000;6:1433–55. doi: 10.2174/1381612003399275. [DOI] [PubMed] [Google Scholar]
- 3.Allen BJ, Raja C, Rizvi S, Li Y, Tsui W, Zhang D, et al. Targeted alpha therapy for cancer. Phys Med Biol. 2004;49:3703–3712. doi: 10.1088/0031-9155/49/16/016. [DOI] [PubMed] [Google Scholar]
- 4.Akabani G, Carlin S, Welsh P, Zalutsky MR. In vitro cytotoxicity of 211At-labeled trastuzumab in human breast cancer cell lines: Effect of specific activity and HER2 receptor heterogeneity on survival fraction. Nucl Med Biol. 2006;33:333–47. doi: 10.1016/j.nucmedbio.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Boyd M, Ross SC, Dorrens J, Fullerton NE, Tan KW, Zalutsky MR, et al. Radiation induced biological bystander effect elicited in vitro by targeted radiopharmaceuticals labeled with α–, β– and Auger Electron emitting radionuclides. J Nucl Med. 2006;47:1007–15. [PubMed] [Google Scholar]
- 6.Zalutsky MR. Radionuclide therapy. In: Roesch F, editor. Handbook of nuclear chemistry Volume 4: radiochemistry and radiopharmaceutical chemistry in life sciences. Kluwer Academic; Dordrecht, Netherlands: 2003. pp. 315–48. [Google Scholar]
- 7.Zalutsky MR, Pozzi OR. Radioimmunotherapy with α-particle emitting radionuclides. Q J Nucl Med Mol Imaging. 2004;48:289–96. [PubMed] [Google Scholar]
- 8.Kotzerke J, Bunjes D, Scheinberg DA. Radioimmunoconjugates in acute leukemia treatment: the future is radiant. Bone Marrow Transpantation. 2005;36:1021–26. doi: 10.1038/sj.bmt.1705182. [DOI] [PubMed] [Google Scholar]
- 9.Couturier O, Supiot S, Degraef-Mougin M, Faivre-Chauvet A, Carlier T, Chatal J-F, et al. Cancer radioimmunotherapy with alpha-emitting nuclides. Eur J Nucl Med Mol Imaging. 2005;32:601–14. doi: 10.1007/s00259-005-1803-2. [DOI] [PubMed] [Google Scholar]
- 10.Larsen RH, Murud KM, Hoff P, Bruland ØS, Zalutsky MR. 211At- and 131I-labeled bisphosphonates with high in vivo stability and bone accumulation. J Nucl Med. 1999;40:1197–1203. [PubMed] [Google Scholar]
- 11.Johnson EL, Turkington TG, Jaszczak RJ, Vaidyanathan G, Green KL, Coleman RE, et al. Quantitation of 211At in small volumes for evaluation of targeted radiotherapy in animal models. Nucl Med Biol. 1995;22:45–54. doi: 10.1016/0969-8051(94)00077-w. [DOI] [PubMed] [Google Scholar]
- 12.Larsen RH, Wieland BW, Zalutsky MR. Evaluation of an internal cyclotron target for the production of astatine-211 via the 209Bi(,2n)211At reaction. Appl Radiat Isotop. 1996;47:135–143. doi: 10.1016/0969-8043(95)00285-5. [DOI] [PubMed] [Google Scholar]
- 13.Lindegren S, Bäck T, Jensen HJ. Dry-distillation of astatine-211 from irradiated bismuth targets: A time-saving procedure with high recovery yields. Appl Radiat Isotopes. 2001;55:157–160. doi: 10.1016/s0969-8043(01)00044-6. [DOI] [PubMed] [Google Scholar]
- 14.Zalutsky MR, Garg PK, Friedman HS, Bigner DD. Labeling monoclonal antibodies and F(ab’)2 fragments with the alpha particle emitting nuclide astatine-211: preservation of immunoreactivity and in vivo localizing capacity. Proc Natl Acad Sci USA. 1989;86:7149–7153. doi: 10.1073/pnas.86.18.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilbur DS, Vessella RL, Stray JE, Goffe DK, Blouke KA, Atcher RW. Preparation and evaluation of para-[211At]astatobenzoyl labeled anti-renal cell carcinoma antibody A6H F(ab’)2. In vivo distribution comparison with para-[125I]iodobenzoyl labeled A6H F(ab’)2. Nucl Med Biol. 1993;20:917–27. doi: 10.1016/0969-8051(93)90092-9. [DOI] [PubMed] [Google Scholar]
- 16.Zalutsky MR, Stabin M, Larsen RH, Bigner DD. Tissue distribution and radiation dosimetry of astatine-211-labeled chimeric 81C6, an α-particle emitting immunoconjugate. Nucl Med Biol. 1997;24:255–262. doi: 10.1016/s0969-8051(97)00060-7. [DOI] [PubMed] [Google Scholar]
- 17.Aurlien E, Larsen RH, Kvalheim G, Bruland ØS. Demonstration of highly specific toxicity of the alpha-emitting radioimmunoconjugate 211At-rituximab against non-Hodgkin’s lymphoma cells. Br J Cancer. 2000;83:1375–9. doi: 10.1054/bjoc.2000.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demartis S, Tarli L, Borsi L, Zardi L, Neri D. Selective targeting of tumour neovasculature by a radiohalogenated human antibody fragment specific for the ED-B domain of fibronectin. Eur J Nucl Med. 2001;28:534–9. doi: 10.1007/s002590100480. [DOI] [PubMed] [Google Scholar]
- 19.Palm S, Back T, Claesson I, Delle U, Hultborn R, Lindegren S, et al. Single-cell irradiation from [211At]astatine-labeled C215 monoclonal antibody: improved estimates of radiosensitivity from measurements on cellular uptake and retention. Anticancer Res. 2003;23:1219–21. [PubMed] [Google Scholar]
- 20.Henriksen G, Bruland ØS, Larsen RH. Preparation and preclinical assessment of folate-conjugated, radiolabelled antibodies. Anticancer Res. 2005;25:9–15. [PubMed] [Google Scholar]
- 21.Almqvist Y, Orlova A, Sjostrom A, Jensen HJ, Lundqvist H, Sundin A, et al. In vitro characterization of 211At-labeled antibody A33--a potential therapeutic agent against metastatic colorectal carcinoma. Cancer Biother Radiopharm. 2005;20:514–23. doi: 10.1089/cbr.2005.20.514. [DOI] [PubMed] [Google Scholar]
- 22.Nestor M, Persson M, van Dongen GA, Jensen HJ, Lundqvist H, Anniko M, et al. In vitro evaluation of the astatinated chimeric monoclonal antibody U36, a potential candidate for treatment of head and neck squamous cell carcinoma. Eur J Nucl Med Mol Imaging. 2005;32:1296–304. doi: 10.1007/s00259-005-1848-2. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Zhang M, Garmestani K, Talanov VS, Plascjak PS, Beck B, et al. Effective treatment of a murine model of adult T-cell leukemia using 211At-7G7/B6 and its combination with unmodified anti-Tac (daclizumab) directed toward CD25. Blood. 2006;108:1007–12. doi: 10.1182/blood-2005-11-4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersson H, Palm S, Lindegren S, Back T, Jacobsson L, Leser G, et al. Comparison of the therapeutic efficacy of 211At- and 131I-labelled monoclonal antibody MOv18 in nude mice with intraperitoneal growth of human ovarian cancer. Anticancer Res. 2001;21:409–12. [PubMed] [Google Scholar]
- 25.Elgqvist J, Andersson H, Back T, Claesson I, Hultborn R, Jensen H, et al. Alpha-radioimmunotherapy of intraperitoneally growing OVCAR-3 tumors of variable dimensions: Outcome related to measured tumor size and mean absorbed dose. J Nucl Med. 2006;47:1342–50. [PubMed] [Google Scholar]
- 26.Wesley JN, McGee EC, Garmestani K, Brechbiel MW, Yordanov AT, Wu C, et al. Systemic radioimmunotherapy using a monoclonal antibody, anti-Tac directed toward the alpha subunit of the IL-2 receptor armed with the α-emitting radionuclides 212Bi or 211At. Nucl Med Biol. 2004;31:357–64. doi: 10.1016/j.nucmedbio.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Vaidyanathan G, Affleck DJ, Bigner DD, Zalutsky MR. N-succinimidyl 3-[211At]astato-4-guanidinomethylbenzoate: an acylation agent for labeling internalizing antibodies with α-particle emitting 211At. Nucl Med Biol. 2003;30:351–359. doi: 10.1016/s0969-8051(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 28.Talanov VS, Yordanov AT, Garmestani K, Milenic DE, Arora HC, Plascjak PS, et al. Preparation and in vivo evaluation of novel linkers for 211At labeling of proteins. Nucl Med Biol. 2004;31:1061–71. doi: 10.1016/j.nucmedbio.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Talanov VS, Garmestani K, Regino CA, Milenic DE, Plascjak PS, Waldmann TA, et al. Preparation and in vivo evaluation of a novel stabilized linker for 211At labeling of protein. Nucl Med Biol. 2006;33:469–80. doi: 10.1016/j.nucmedbio.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Welch MJ. Potentials and pitfalls with α-particles. J Nucl Med. 2005;46:1254–1255. [PubMed] [Google Scholar]
- 31.Zalutsky MR, Zhao X-G, Alston KL, Bigner DD. High-level production of α-particle-emitting 211At and preparation of 211At-labeled antibodies for clinical use. J Nucl Med. 2001;42:1508–1515. [PubMed] [Google Scholar]
- 32.Pozzi OR, Zalutsky MR. Radiopharmaceutical chemistry of targeted radiotherapeutics I: Effects of solvent on the degradation of radiohalogenation precursors by astatine-211 α-particles. J Nucl Med. 2005;46:700–706. [PubMed] [Google Scholar]
- 33.Pozzi OR, Zalutsky MR. Radiopharmaceutical chemistry of targeted radiotherapeutics II: Radiolytic effects of astatine-211 α-particles influence N-succinimidyl 3-[211At]astatobenzoate synthesis. J Nucl Med. 2005;46:1393–1400. [PubMed] [Google Scholar]
- 34.Zalutsky MR. Current status of treatment of solid tumors: brain tumor therapy. J Nucl Med. 2005;46:151S–156S. [PubMed] [Google Scholar]
- 35.Reist CJ, Bigner DD, Zalutsky MR. Human IgG2 constant region enhances in vivo stability of anti-tenascin antibody 81C6 compared with its murine parent. Clinical Cancer Res. 1998;4:2495–2502. [PubMed] [Google Scholar]
- 36.McLendon RE, Archer GE, Garg PK, Bigner DD, Zalutsky MR. Radiotoxicity of systemically administered [211At]astatide in B6C3F1 and BALB/c (nu/nu) mice: a long term survival study with histologic analysis. Int J Radiat Oncol Biol Phys. 1996;35:69–80. doi: 10.1016/s0360-3016(96)85013-9. [DOI] [PubMed] [Google Scholar]
- 37.McLendon RE, Archer GE, Larsen RH, Akabani G, Bigner DD, Zalutsky MR. Radiotoxicity of systemically administered 211At-labeled human/mouse chimeric monoclonal antibody: a long-term survival study with histological analysis. Int J Radiat Oncol Biol Phys. 1999;45:491–499. doi: 10.1016/s0360-3016(99)00206-0. [DOI] [PubMed] [Google Scholar]
- 38.Zalutsky M, Reardon D, Akabani G, Friedman A, Friedman H, Herndon J, et al. Astatine-211 labeled human/mouse chimeric anti-tenascin monoclonal antibody via surgically created resection cavities for patients with recurrent glioma: Phase I study. Neuro-Oncol. 2002;4:S103. [Google Scholar]
- 39.Stupp R, Mason W, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 40.Mirimanoff R-O, Gorlia T, Mason W, van den Bent MJ, Kortmann R-D, Fisher B, et al. Radiotherapy and temozolomide for newly diagnosed glioblastoma: recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3 phase III randomized trial. J Clin Oncol. 2006;24:2563–69. doi: 10.1200/JCO.2005.04.5963. [DOI] [PubMed] [Google Scholar]