

Abstract
Mitochondrial morphology depends on balanced fusion and fission events. A central component of the mitochondrial fusion apparatus is the conserved GTPase Fzo1 in the outer membrane of mitochondria. Mdm30, an F-box protein required for mitochondrial fusion in vegetatively growing cells, affects the cellular Fzo1 concentration in an unknown manner. We demonstrate that mitochondrial fusion requires a tight control of Fzo1 levels, which is ensured by Fzo1 turnover. Mdm30 binds to Fzo1 and, dependent on its F-box, mediates proteolysis of Fzo1. Unexpectedly, degradation occurs along a novel proteolytic pathway not involving ubiquitylation, Skp1–Cdc53–F-box (SCF) E3 ubiquitin ligase complexes, or 26S proteasomes, indicating a novel function of an F-box protein. This contrasts to the ubiquitin- and proteasome-dependent turnover of Fzo1 in α-factor–arrested yeast cells. Our results therefore reveal not only a critical role of Fzo1 degradation for mitochondrial fusion in vegetatively growing cells but also the existence of two distinct proteolytic pathways for the turnover of mitochondrial outer membrane proteins.
Introduction
Mitochondria are essential organelles whose structure and function adapt to different cellular conditions through continuous fusion and fission events (Okamoto and Shaw, 2005). Mitochondrial dynamics exert essential physiological and developmental roles, regulate apoptotic processes, and affect energy production within mitochondria (Chen and Chan, 2005). Two neuropathies, Charcot-Marie-Tooth type 2A and autosomal dominant optic atrophy, are caused by mutations in essential fusion components, namely, mitofusin 2 or OPA1 (Chen and Chan, 2005). Although several components involved in fusion have been identified, many of them in yeast, molecular mechanisms underlying mitochondrial fusion events are poorly understood. A central role has been assigned to yeast Fzo1 in the outer membrane of mitochondria (or to its mammalian homologues mitofusin 1 and 2) as part of a fusion complex that also contains Ugo1 and Mgm1 in yeast (Meeusen and Nunnari, 2005).
Less clear is the role of the F-box protein Mdm30, whose loss leads to the accumulation of aggregated and fragmented mitochondria (Fritz et al., 2003). Among the 21 annotated proteins of Saccharomyces cerevisiae with an F-box motif (Willems et al., 2004), some assemble in Skp1–Cdc53–F-box (SCF) E3 ubiquitin ligase complexes, which mediate proteasomal proteolysis of specific substrates (Petroski and Deshaies, 2005). Indeed, Mdm30 has been linked to the turnover of the transcription factor Gal4 in the nucleus (Muratani et al., 2005). Mdm30 was not only localized to the cytosolic fraction but also found in association with mitochondria (Fritz et al., 2003). Therefore, in analogy to the degradation of resident ER proteins, Mdm30 may affect mitochondrial dynamics by coupling the mitochondrial fusion machinery to the ubiquitin–proteasome system (UPS) in the cytosol. Consistently, accumulation of Fzo1 has been observed in cells lacking Mdm30 (Fritz et al., 2003). However, it remained unclear whether this indeed reflects Mdm30-dependent proteolysis of Fzo1 and whether the UPS is involved. Notably, 26S proteasomes have been linked to the degradation of Fzo1 in α-factor–arrested yeast cells (Neutzner and Youle, 2005). However, degradation does not depend on Mdm30 under these conditions (Neutzner and Youle, 2005). Moreover, the involvement of 26S proteasomes remained controversial, as only proteasome inhibitors have been used, which are known to be effective only in yeast cells with increased membrane permeability or lacking drug-efflux pumps (Lee and Goldberg, 1996).
We have analyzed the role of Mdm30 for the regulation of mitochondrial dynamics and demonstrate for the first time Mdm30-dependent proteolysis of Fzo1 in vegetatively growing yeast cells. Mdm30 is part of a novel proteolytic pathway that does not involve SCF complexes and 26S proteasomes and thus is strikingly different to the proteasomal degradation of Fzo1 in α-factor–arrested yeast cells.
Results and discussion
Mdm30-dependent and -independent degradation of Fzo1
The accumulation of Fzo1 in Δmdm30 cells suggests that the F-box protein Mdm30 is involved in the degradation of Fzo1 (Fig. 1 A; Fritz et al., 2003). We therefore assessed the stability of Fzo1 in wild-type and Δmdm30 cells after inhibition of protein synthesis with cycloheximide. This analysis revealed that Fzo1 is constitutively degraded in wild-type cells, whereas it remained stable in the absence of Mdm30 (Fig. 1 A). Similar experiments were performed in yeast cells in the exponential or post–diauxic-shift phase, but a significant dependence of Fzo1 stability on the growth phase was not observed (Fig. 1 A). To distinguish between complete Fzo1 turnover and processing, we followed Fzo1 degradation using antibodies directed against either the NH2-terminal GTPase domain or a peptide located at the COOH-terminal segment of Fzo1. Moreover, the stability of an Fzo1 variant carrying an NH2-terminal HA tag was examined. As no proteolytic fragments of Fzo1 were detected, we conclude that Fzo1 is completely degraded in an Mdm30-dependent manner.
Figure 1.

Fzo1 stability is controlled by two independent proteolytic pathways. (A, top) The stability of Fzo1 in exponentially growing (exp) or post–diauxic-shift cultures (PDS) after adding cycloheximide (CHX) was monitored by SDS-PAGE and immunoblotting. (bottom) A quantification including standard deviation of three independent experiments. (B) Cellular subfractionation. Exponentially growing wild-type (wt) and Δmdm30 cells were split by differential centrifugation into a mitochondrial (pellet) and a cytosolic (sup) fraction as described previously (Rapaport et al., 1998). The fractions were analyzed by SDS-PAGE and immunoblotting for the presence of Fzo1. The mitochondrial outer membrane protein Tom40 and the cytosolic protein Tpi1 were used as marker proteins to validate the fractionation. (C) Complex formation of Fzo1 in the mitochondrial outer membrane. Mitochondrial membranes of wild-type and Δmdm30 cells were solubilized in Triton X-100 and fractionated by Superose 6 gel filtration chromatography. Eluate fractions were analyzed by immunoblotting using Fzo1-specific antibodies. (D) Fzo1 degradation upon α-factor– induced cell cycle arrest. Fzo1 stability in cell cycle–arrested wild-type and Δmdm30 cells was analyzed as in A. The position of standard molecular mass markers during electrophoresis corresponding to 116, 43, and 27 kD are indicated by the small bars in panels showing Fzo1, Tom40, or Tpi1, respectively.
Mdm30 might affect the localization and/or complex assembly of Fzo1 and thereby affect Fzo1 stability and mitochondrial fusion. Therefore, mitochondrial and postmitochondrial supernatant fractions of wild-type and Δmdm30 cells were analyzed for the presence of Fzo1 (Fig. 1 B). Regardless of the presence of Mdm30, Fzo1 was recovered from the mitochondrial fraction, which also contained the mitochondrial outer membrane protein Tom40 but was devoid of the soluble cytosolic protein Tpi1 (Fig. 1 B). Fzo1 assembles into an 850-kD complex in wild-type cells (Rapaport et al., 1998) and was found to be present in a complex of the same size in Δmdm30 cells (Fig. 1 C). Thus, Mdm30 does not affect the assembly of Fzo1 in the outer membrane of mitochondria.
Fzo1 was recently reported to be degraded upon mating factor–induced cell cycle arrest (Neutzner and Youle, 2005; Fig. 1 D). Intriguingly, under these conditions, proteolysis occurs independent of Mdm30 (Neutzner and Youle, 2005; Fig. 1 D) or any other nonessential yeast gene encoding an F-box protein (unpublished data). We therefore conclude that Fzo1 is degraded along two physiologically and mechanistically distinct pathways, one prevalent in vegetatively growing cells and the other in α-factor–arrested cells.
Mdm30 binds to Fzo1 and mediates proteolysis in an F-box–dependent manner
To assess the requirement of the Mdm30 F-box motif for Fzo1 degradation, four conserved amino acid residues within the motif were replaced, namely, L19P, P20A, E22Q, and I23A (Fig. 2 A). In a second construct, the entire F-box was deleted (amino acids 1–58; Fig. 2 A, ΔF-box). Wild-type or mutant forms of Mdm30 were found in equal association with mitochondria upon subcellular fractionation (unpublished data). After expression in wild-type and Δmdm30 strains, the steady-state level of Fzo1 was monitored by immunoblotting. In wild-type cells, the steady-state concentration of Fzo1 was unaffected by the presence of the mutant Mdm30 forms (Fig. 2 B). In Δmdm30 cells, however, only wild-type Mdm30 but not Mdm30 variants with a defective F-box were able to restore degradation and low Fzo1 steady-state concentrations (Fig. 2 B). These results demonstrate that proteolysis of Fzo1 depends on the integrity of the Mdm30 F-box.
Figure 2.
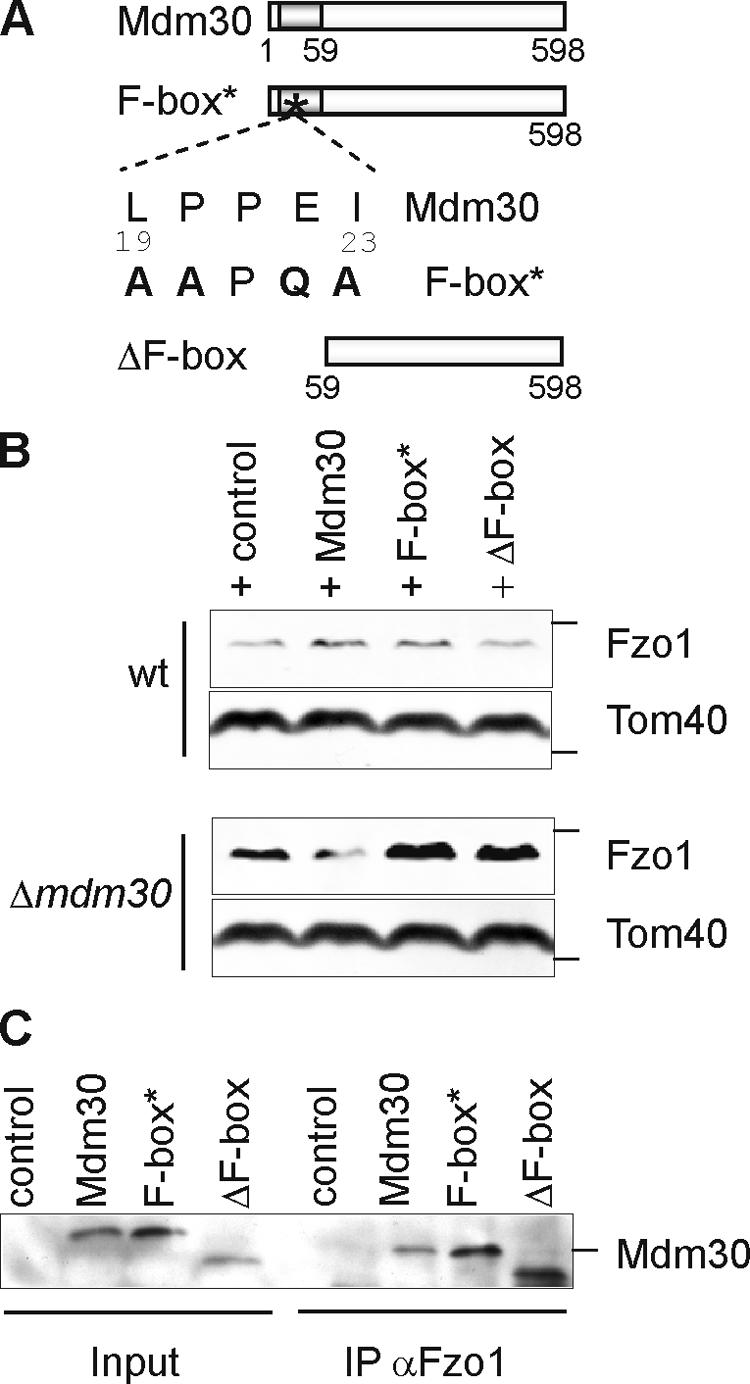
The constitutive Fzo1 turnover depends on the F-box motif of its interacting partner, Mdm30. (A) Schematic representation of Mdm30 variants. The Mdm30 derivative F-box* was obtained, replacing amino acid residues 19, 20, 22, and 23 within consensus residues of the F-box motif of Mdm30 as shown. The variant ΔF-box was generated by deleting amino acid residues 1–58 of Mdm30. (B) Mitochondrial extracts of wild-type (wt) and Δmdm30 strains expressing Mdm30 variants or transformed with the vector control were analyzed by immunoblotting using Fzo1-specific and, as a loading control, Tom40-specific antibodies. (C) Mitochondria derived from wild-type cells expressing several Mdm30 variants harboring an NH2-terminal Flag epitope or cells transformed with the vector control were solubilized in digitonin and subjected to immunoprecipitation using Fzo1-specific antibodies. Total extracts (input; 5%) and immunoprecipitates (IP; 100%) were analyzed by immunoblotting with antibodies directed against the Flag epitope of the Mdm30 proteins. The position of standard molecular mass markers during electrophoresis corresponding to 116, 43, and 66 kD are indicated by the small bars in panels showing Fzo1, Tom40, or Mdm30, respectively.
To examine whether Mdm30 interacts directly with Fzo1, we performed coimmunoprecipitation experiments using Fzo1-specific antibodies in cells expressing Mdm30 or F-box variants thereof. Mdm30 was precipitated with Fzo1-specific antibodies, demonstrating complex formation of both proteins in wild-type cells (Fig. 2 C). Thus, in agreement with studies on other F-box proteins (Skowyra et al., 1997), Fzo1 recognition by Mdm30 does not depend on its F-box (Fig. 2 C). It should be noted that after solubization of mitochondrial membranes with digitonin, Mdm30 or mutant variants did not coelute with Fzo1 upon sizing chromatography, indicating a rather dynamic interaction (unpublished data).
Mdm30-mediated Fzo1 turnover is independent of the SCF complex or of ubiquitylation
Mdm30 was found associated with both Skp1 and Cdc53 in high throughput protein–protein interaction studies (Uetz et al., 2000) and in vitro (Kus et al., 2004). Its requirement for Fzo1 proteolysis therefore suggests that degradation occurs through ubiquitylation by SCF complexes (Petroski and Deshaies, 2005). Protein shut-off experiments were performed in cells deficient for core components of SCF complexes (skp1-11, skp1-12, and cdc53-1) or for the E2 enzyme Cdc34 (cdc34-2). Fzo1 was degraded in these SCF-deficient cells with kinetics similar to those of wild-type cells (Fig. 3 A). This was in contrast to Grr1, a known substrate of the SCF complex (Galan and Peter, 1999), which stably accumulated in these mutants (Fig. 3 B). We also analyzed the stability of Fzo1 in cells that allow tetracycline-sensitive expression of the essential genes CDC53 and CDC34 (Mnaimneh et al., 2004) but did not observe a stabilization of Fzo1 upon repression of CDC53 or CDC34 (unpublished data). Thus, Mdm30-dependent proteolysis of Fzo1 does not require SCF ubiquitin ligase complexes. These findings are reminiscent of the F-box proteins Ctf13 and Rcy1, which do not assemble in SCF complexes (Russell et al., 1999; Galan et al., 2001), and substantiate that F-box proteins may exert cellular functions other than ubiquitin-dependent protein degradation. It should be emphasized, however, that Mdm30 could mediate the degradation of other substrate proteins by interaction with SCF complexes. This hypothesis is supported by the recently observed Mdm30-dependent ubiquitylation of Gal4 in the nucleus (Muratani et al., 2005).
Figure 3.

Differential requirement of ubiquitylation for constitutive and induced Fzo1 degradation. (A–C) The stability of Fzo1 and Grr1-myc was analyzed after inhibition of cytosolic protein synthesis with cycloheximide (CHX) in vegetatively growing wild-type (wt), Δmdm30, cdc34-2, cdc53-1, skp1-11, skp1-12, and uba1ts cells at 37°C, as described in Fig. 1 A. (D) Proteolysis of Fzo1 in the presence of α-factor was monitored at 37°C in wild-type and uba1ts cells. The position of standard molecular mass markers during electrophoresis corresponding to 158 and 116 kD are indicated by the small bars in panels showing Grr1 or Fzo1, respectively.
To directly examine a possible ubiquitylation of Fzo1 by other E3 ubiquitin ligases, cellular ubiquitin pools were limited either by using a mutant E1 enzyme (uba1ts) or through the overexpression of mutant ubiquitin forms, unable to form ubiquitin chains (K29R, K48R, K63R, or RRR). Neither approach caused a stabilization of Fzo1 (Fig. 3 C). We conclude that Mdm30-dependent constitutive Fzo1 degradation is independent of SCF and ubiquitin.
We also examined the role of ubiquitylation for Fzo1 proteolysis upon mating factor–induced cell cycle arrest in uba1ts cells and found it to be impaired (Fig. 3 D). Thus, in striking contrast to its constitutive and Mdm30-dependent degradation, the proteolytic breakdown of Fzo1 in arrested cells is ubiquitin dependent (Fig. 3 D).
Proteasome independence and ATP dependence of Fzo1 degradation
We next examined the stability of Fzo1 in yeast cells with deficient proteasomal activity. Proteolysis of Fzo1 was not impaired in pre1-1 or cim5-1 cells carrying mutations in a proteolytic subunit of the 20S core particle or in an ATPase subunit of the 19S regulatory complex, respectively (Fig. 4 A). Similarly, deletion of the proteasomal assembly factor Ump1 did not interfere with the proteolytic breakdown of Fzo1 in vegetatively growing cells (Fig. 4 A). This is in contrast to Fzo1 proteolysis in cells arrested in the presence of mating factor (Fig. 4 B). In agreement with previous inhibitor studies (Neutzner and Youle, 2005), Fzo1 remained stable after treating pre1-1 or cim5-1 cells with α-factor (Fig. 4 B). We conclude that the constitutive Mdm30-dependent turnover of Fzo1 occurs independently of the 26S proteasome, whereas degradation of Fzo1 upon α-factor arrest involves ubiquitylation and 26S proteasomes.
Figure 4.

Fzo1 degradation is mediated by a novel proteolytic system. (A) The stability of Fzo1 was monitored in wild-type (wt), Δmdm30, pre1-1, cim5-1, Δump1, Δyme1, and Δpep4 cells after addition of cycloheximide (CHX) as in Fig. 1 A. (B) Fzo1 stability upon α-factor–induced cell cycle arrest was examined in wild-type, pre1-1, and cim5-1 cells, which also lacked BAR1. (C) Exponentially growing wild-type cells were treated with cycloheximide (CHX) and additionally with sodium azide (NaN3) or CCCP as indicated. The stability of Fzo1 was determined as in Fig. 1 A. The position of the standard molecular mass marker during electrophoresis corresponding to 116 kD is indicated by the small bars.
To further characterize the Mdm30-mediated Fzo1 proteolysis, we examined a potential ATP dependence of this proteolytic pathway. Two chemical inhibitors of ATP production in mitochondria were used: NaN3, an inhibitor of the cytochrome c oxidase, and carbonylcyanide m-chlorophenylhydrazone (CCCP), an uncoupler dissipating the membrane potential across the mitochondrial inner membrane. Both treatments completely blocked degradation of Fzo1, which appears to be ATP dependent (Fig. 4 C).
We therefore analyzed the stability of Fzo1 in mitochondria lacking the ATP-dependent i-AAA protease subunit Yme1, which is active in the intermembrane space of mitochondria (Leonhard et al., 1996), but did not observe any effect on Fzo1 degradation (Fig. 4 A). Moreover, we examined the involvement of vacuolar degradation, which is responsible for the general removal of mitochondrial proteins under rapamycin-induced autophagic conditions (Kissova et al., 2004). However, Fzo1 was not stabilized in pep4 cells lacking active vacuolar peptidases (Fig. 4 A). We conclude that Fzo1 is constitutively degraded by a yet unknown proteolytic system whose activity is apparently ATP dependent.
Mdm30 controls mitochondrial fusion exclusively through Fzo1 proteolysis
Deletion of MDM30 impairs mitochondrial fusion and results in the formation of fragmented mitochondria accompanied by the accumulation of Fzo1 (Fig. 5 A; Fritz et al., 2003). To examine whether Fzo1 accumulation in Δmdm30 cells is a general consequence of impaired mitochondrial fusion, the steady-state concentrations of Fzo1 were determined in Δugo1, Δmgm1, and Δpcp1 mutant cells. Fzo1 did not accumulate in either of these mutant cells, demonstrating that there is no direct link between the cellular level of Fzo1 and ongoing mitochondrial fusion (Fig. 5 B). To further substantiate the specific effect of Mdm30 on Fzo1 stability and mitochondrial fusion, we analyzed the effect on the steady-state level of Fzo1 of the 131 nonessential deletion strains and in the 16 tet-off essential genes known to have fragmented mitochondria (Dimmer et al., 2002; Altmann and Westermann, 2005). However, apart from Δmdm30 cells, we did not observe altered Fzo1 levels (unpublished data). It therefore seems likely that Mdm30 affects mitochondrial dynamics by specifically regulating the stability of Fzo1.
Figure 5.

Control of mitochondrial fusion by Mdm30-dependent proteolysis of Fzo1. (A) Cellular (Nomarski) and mitochondrial (GFP) morphology was visualized by fluorescence microscopy after expressing mtGFP in wild-type (wt), Δfzo1, and Δmdm30 cells or in wild-type cells expressing Fzo1 from a multicopy plasmid under the control of the CUP1 promoter in the presence of added copper (50 μM; wt + Fzo12μ). (B) Steady-state concentrations of Fzo1 in extracts of the indicated strains were analyzed by immunoblotting using Fzo1-specific and, as a loading control, Tom40-specific antibodies. (C) Stability of Fzo1 present at different levels in wild-type and Δmdm30 cells. Protein synthesis shut-off experiments were performed as in Fig. 1 A using exponentially growing wild-type or Δmdm30 cells expressing HA-Fzo1 from either a centromeric (cen) or a multicopy (2μ) plasmid under the control of the endogenous FZO1 promoter. (D) Wild-type and Δmdm30 cells expressing mtGFP and harboring a chromosomal integration of the GAL1 promoter upstream of the FZO1 coding region were grown on glucose-, raffinose-, or galactose-containing medium. Cellular (Nomarski) and mitochondrial (GFP) morphology was visualized after expression of mtGFP by fluorescence microscopy. (E) Wild-type cells expressing mtGFP and harboring a chromosomal integration of the GAL1 promoter upstream of the FZO1 coding region were grown on glucose- or galactose-containing medium. Cells were expressing MDM30 from a multicopy plasmid under the control of the CUP1 promoter when indicated. Cellular (Nomarski) and mitochondrial (GFP) morphology was visualized by fluorescence microscopy. The position of standard molecular mass markers during electrophoresis corresponding to 116 and 43 kD are indicated by the small bars in panels showing Fzo1 or Tom40, respectively. Bars, 5 μm.
To substantiate the specific effect of Mdm30 on Fzo1, we assessed mitochondrial morphology upon overexpression of Fzo1 in wild-type cells. Strikingly, mitochondria in cells overexpressing Fzo1 in the presence of an MDM30 wild-type allele resembled the fragmented mitochondria seen in Δmdm30 cells (Fig. 5 A). In agreement with this observation, proteolysis of Fzo1 was only observed when Fzo1 was expressed from a centromeric low-copy plasmid but not when expressed from a multicopy plasmid (Fig. 5 C). These findings were further supported by modulating Fzo1 expression in wild-type and Δmdm30 cells using the inducible GAL1 promoter. Growing yeast cells in the presence of glucose, raffinose, or galactose results in repression, nonrepression, or induction of Fzo1 synthesis. Importantly, tubular mitochondria were observed in Δmdm30 cells grown under nonrepressed conditions in the presence of raffinose, i.e., at intermediate expression levels of Fzo1 (Fig. 5 D and Table I). This confirms that mitochondrial morphology can be modulated by changing Fzo1 expression levels. Thus, mitochondrial fusion requires a tight control of the steady-state concentration of Fzo1. We conclude that lowering Fzo1 protein levels allows bypassing of Mdm30. Therefore, Mdm30 promotes mitochondrial fusion exclusively by controlling the steady-state level of Fzo1.
Table I.
Mitochondrial phenotypes upon different Fzo1 expression levels
|
|
Mitochondrial morphology (percentage of cells)
|
||
|---|---|---|---|
| Strain/media | Fragmented | Aggregated | Tubular-like |
| wt/GAL1-FZO1 | |||
| Glucose | 92 | 7 | 1 |
| Raffinose | 90 | 8 | 2 |
| Galactose | 16 | 79 | 5 |
| Δmdm30/GAL1-FZO1 | |||
| Glucose | 89 | 5 | 6 |
| Raffinose | 8 | 6 | 86 |
| Galactose | 10 | 86 | 4 |
Wild-type (wt) and Δmdm30 strains expressing mtGFP and harboring a chromosomal integration of the GAL1 promoter upstream of the FZO1 coding region were grown on glucose, raffinose, or galactose, and mitochondrial morphology of ∼100 cells was classified as fragmented, aggregated, or tubular. Bold numbers indicate the most represented class on each condition tested. Notably, mitochondrial tubules observed in Δmdm30 cells were usually shorter and clearly distinct from the tubular morphology of wild-type cells.
Overexpressing Fzo1 in wild-type cells prevents its degradation, suggesting that Mdm30 could be the limiting factor. Therefore, Mdm30 was overexpressed in cells containing a galactose-inducible FZO1 allele. Surprisingly, we found that the simultaneous overexpression of Fzo1 and Mdm30 is deleterious for cellular growth and leads to abnormal cellular and mitochondrial morphologies (Fig. 5 E). It is therefore conceivable that the Fzo1–Mdm30 complex sequesters an as-yet-unknown factor that is essential for growth.
Conclusions
Our experiments demonstrate a crucial role of Fzo1 proteolysis for the maintenance of mitochondrial morphology. The stability of Fzo1 is controlled by a novel, apparently ATP-dependent proteolytic system. This system involves the F-box protein Mdm30 but not ubiquitylation and SCF complexes, opening new perspectives for cellular activities of F-box proteins. Our results also demonstrate that proteins in the outer membrane of mitochondria can be degraded in at least two ways, only one of which is dependent on the UPS. At the same time, they suggest a mechanism that regulates organellar morphology by the use of two different proteolytic systems during different physiological states of a eukaryotic cell.
Materials and methods
Yeast strains and growth media
Yeast strains were grown according to standard procedures on complete or synthetic media supplemented with 2% (wt/vol) glucose or, when indicated, 2% (wt/vol) galactose or raffinose and 0.02% (wt/vol) glucose. 100 μg/ml cycloheximide, 10 μM α-factor (or 0.1 μM in the case of Δbar1 cells), 2 mM NaN3, or 1 mM CCCP was added when indicated. The cell cycle arrest in the presence of α-factor was verified microscopically.
Yeast strains used were derivatives of W303 and BY4741. The strains Δmdm30, Δfzo1, Δugo1, Δpcp1, or Δmgm1 and the isogenic wild-type strain BY4741 were obtained from Euroscarf. For expression of Fzo1 in wild-type and Δmdm30 cells under the control of the GAL1 promoter, previously described strains were used (Fritz et al., 2003). MDM30 and PEP4 were deleted by PCR-based homologous recombination in W303. To increase the efficiency of the α-factor arrest, BAR1 was deleted by PCR-based homologous recombination in wild-type, cim5-1, and pre1-1 cells.
Plasmids
FZO1, MDM30, and mdm30 mutant variants (F-box*, with the L19P, P20A, E22Q, and I23A mutations; ΔF-box, without the residues 1–58) encoding an NH2-terminal FLAG tag were cloned into the multicopy vector pJDCEX2 (2μ; LEU2 and CUP1 promoter). Fzo1 harboring an NH2-terminal HA tag was expressed under the control of the endogenous promoter using the centromeric plasmid pRS315 or the multicopy plasmid YEplac181. For visualizing mitochondria, the plasmid pVT100U-mtGFP (mitochondria-targeted GFP) was used.
Protein synthesis shut off and α-factor cell cycle arrest
To monitor constitutive proteolysis of Fzo1, cycloheximide was added to logarithmically growing yeast cultures at a final concentration of 100 μg/ml. Thermosensitive strains were incubated for 1 h at 37°C before adding cycloheximide. To examine Fzo1 degradation upon α-factor–induced cell cycle arrest, early log phase cells grown on YPD were treated with 20 mM sodium citrate and 10 μM α-factor (or 0.1 μM in Δbar1 strains). At the indicated time points, cells corresponding to 3 OD units were collected and lysed at alkaline pH (Tatsuta and Langer, 2006), and protein extracts were analyzed by SDS-PAGE and immunoblotting. Mean values of at least three independent experiments are shown.
Coimmunoprecipitation of Fzo1 and Mdm30
Wild-type cells expressing Mdm30 and variants thereof from a copper-responsive promoter were grown on synthetic media without an additional copper supplement. Crude mitochondria obtained from 60 OD units of these cells were solubilized for 15 min in 1% (wt/vol) digitonin in immunoprecipitation buffer (150 mM KAc, 4 mM MgAc, 30 mM Tris/HCl, pH 7.4, 1 mM PMSF, and 1 mM ATP). After a clarifying spin for 15 min at 45,000 g, supernatants were incubated for 1 h at 4°C under constant mixing with protein A–Sepharose and polyclonal antibodies against the GTPase domain of Fzo1. After two washing steps in immunoprecipitation buffer and one washing step in 10 mM Tris/HCl, pH 7.4, bound material was eluted with SDS sample buffer and used for Western blot analysis.
Antibodies
Antisera against the COOH terminus of Fzo1 (Rapaport et al., 1998), the GTPase domain of Fzo1 (provided by J. Nunnari, University of California, Davis, Davis, CA), the HA epitope (3F10; Roche), the FLAG epitope (M2-F3165; Sigma-Aldrich), or the myc epitope (9B11; Cell Signaling Technology) were used.
Microscopy
Overnight cultures were analyzed by epifluorescence microscopy on a microscope (Axioplan; Carl Zeiss MicroImaging, Inc.) using a 100× oil-immersion objective. GFP fluorescence was visualized in living cells. Images were acquired with a camera (Quantix; Photometrix) and processed with MetaMorph 4.5 software (Universal Imaging Corp.).
Acknowledgments
We are grateful to J. Dohmen for the ump1ts and uba1ts strains, the plasmid pJDCEX2, the Tpi1 antibodies, and his helpful advice; R. Deshaies for the skp1-11 and skp1-12 strains; J.M. Galan for yeast strains expressing Grr1-Myc; C. Mann for the cim5-1 strain; K. Nasmyth for the cdc34-2 strain; J. Nunnari for the GTPase Fzo1 antibody; M. Tyers for cdc53-1; and D. Wolf for the strains pre1-1 and cim3-1. The expert technical assistance of D. Tils and G. Zimmer and helpful advice of M. Collart and B. Daignan-Fornier are gratefully acknowledged.
This work was supported by a European Molecular Biology Organization fellowship to M. Escobar-Henriques (ALTF 586-2002) and funds of the Deutsche Forschungsgemeinschaft to T. Langer (SFB635, P18) and B. Westermann (WE 2174/2-4).
Abbreviations used in this paper: CCCP, carbonylcyanide m-chlorophenylhydrazone; mtGFP, mitochondria-targeted GFP; SCF, Skp1–Cdc53–F-box; UPS, ubiquitin–proteasome system.
References
- Altmann, K., and B. Westermann. 2005. Role of essential genes in mitochondrial morphogenesis in Saccharomyces cerevisiae. Mol. Biol. Cell. 16:5410–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H., and D.C. Chan. 2005. Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet. 14:R283–R289. [DOI] [PubMed] [Google Scholar]
- Dimmer, K.S., S. Fritz, F. Fuchs, M. Messerschmitt, N. Weinbach, W. Neupert, and B. Westermann. 2002. Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol. Biol. Cell. 13:847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz, S., N. Weinbach, and B. Westermann. 2003. Mdm30 is an F-box protein required for maintenance of fusion-competent mitochondria in yeast. Mol. Biol. Cell. 14:2303–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan, J.M., and M. Peter. 1999. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc. Natl. Acad. Sci. USA. 96:9124–9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan, J.M., A. Wiederkehr, J.H. Seol, R. Haguenauer-Tsapis, R.J. Deshaies, H. Riezman, and M. Peter. 2001. Skp1p and the F-box protein Rcy1p form a non-SCF complex involved in recycling of the SNARE Snc1p in yeast. Mol. Cell. Biol. 21:3105–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissova, I., M. Deffieu, S. Manon, and N. Camougrand. 2004. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 279:39068–39074. [DOI] [PubMed] [Google Scholar]
- Kus, B.M., C.E. Caldon, R. Andorn-Broza, and A.M. Edwards. 2004. Functional interaction of 13 yeast SCF complexes with a set of yeast E2 enzymes in vitro. Proteins. 54:455–467. [DOI] [PubMed] [Google Scholar]
- Lee, D.H., and A.L. Goldberg. 1996. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J. Biol. Chem. 271:27280–27284. [DOI] [PubMed] [Google Scholar]
- Leonhard, K., J.M. Herrmann, R.A. Stuart, G. Mannhaupt, W. Neupert, and T. Langer. 1996. AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondria. EMBO J. 15:4218–4229. [PMC free article] [PubMed] [Google Scholar]
- Meeusen, S.L., and J. Nunnari. 2005. How mitochondria fuse. Curr. Opin. Cell Biol. 17:389–394. [DOI] [PubMed] [Google Scholar]
- Mnaimneh, S., A.P. Davierwala, J. Haynes, J. Moffat, W.T. Peng, W. Zhang, X. Yang, J. Pootoolal, G. Chua, A. Lopez, et al. 2004. Exploration of essential gene functions via titratable promoter alleles. Cell. 118:31–44. [DOI] [PubMed] [Google Scholar]
- Muratani, M., C. Kung, K.M. Shokat, and W.P. Tansey. 2005. The F box protein Dsg1/Mdm30 is a transcriptional coactivator that stimulates Gal4 turnover and cotranscriptional mRNA processing. Cell. 120:887–899. [DOI] [PubMed] [Google Scholar]
- Neutzner, A., and R.J. Youle. 2005. Instability of the mitofusin Fzo1 regulates mitochondrial morphology during the mating response of the yeast Saccharomyces cerevisiae. J. Biol. Chem. 280:18598–18603. [DOI] [PubMed] [Google Scholar]
- Okamoto, K., and J.M. Shaw. 2005. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 39:503–536. [DOI] [PubMed] [Google Scholar]
- Petroski, M.D., and R.J. Deshaies. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6:9–20. [DOI] [PubMed] [Google Scholar]
- Rapaport, D., M. Brunner, W. Neupert, and B. Westermann. 1998. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J. Biol. Chem. 273:20150–20155. [DOI] [PubMed] [Google Scholar]
- Russell, I.D., A.S. Grancell, and P.K. Sorger. 1999. The unstable F-box protein p58-Ctf13 forms the structural core of the CBF3 kinetochore complex. J. Cell Biol. 145:933–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra, D., K.L. Craig, M. Tyers, S.J. Elledge, and J.W. Harper. 1997. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 91:209–219. [DOI] [PubMed] [Google Scholar]
- Tatsuta, T., and T. Langer. 2006. Studying proteolysis within mitochondria. Curr. Topics Gen. In press. [DOI] [PubMed]
- Uetz, P., L. Giot, G. Cagney, T.A. Mansfield, R.S. Judson, J.R. Knight, D. Lockshon, V. Narayan, M. Srinivasan, P. Pochart, et al. 2000. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 403:623–627. [DOI] [PubMed] [Google Scholar]
- Willems, A.R., M. Schwab, and M. Tyers. 2004. A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim. Biophys. Acta. 1695:133–170. [DOI] [PubMed] [Google Scholar]