

Abstract
The E1 and E2 proteins from bovine papillomavirus bind cooperatively to binding sites in the viral origin of DNA replication. The DNA-binding domains (DBDs) of the two proteins interact with each other, and the E2 transactivation domain interacts with the helicase domain of E1. Mutations that disrupt the interaction between the two DBDs also disrupt the interaction between the E2 activation domain and the E1 helicase domain, demonstrating interdependence of the two interactions. Cooperative binding of the two DBDs generates a sharp bend in the DNA that is required for interaction between the E2 activation domain and E1. This indicates that interaction between the two DBDs plays an architectural role, ‘triggering’ a productive interaction between the E2 transactivation domain and E1 through introduction of a sharp bend in the DNA. This two-step mechanism may be a required feature for cooperative DNA binding to proximal binding sites.
Keywords: cooperative binding/DNA bending/papillomavirus
Introduction
Cooperative DNA binding is an important component of many regulatory systems and forms a cornerstone for combinatorial regulation. Cooperativity allows occupancy of low affinity sites that would be poorly bound by the individual proteins and therefore can serve as a conditional switch. Cooperative binding may also serve other functions. Protein–protein interactions that result in cooperative DNA binding frequently also result in bending or looping of the intervening DNA, contributing to the formation of higher order structured DNA–protein complexes. Recent characterizations of a number of DNA-bound complexes from both pro- and eukaryotes indicate that the three-dimensional arrangement of factors and DNA contributes to the formation of an active complex (Grosschedl et al., 1994; Grosschedl, 1995; Thanos and Maniatis, 1995; Perez-Martin and de Lorenzo, 1997). The individual interactions that generate these structures are likely to involve a combination of architectural components that affect the DNA structure as well as long- and short-range protein–protein interactions. Short-range interactions between DNA-binding domains (DBDs, e.g. homeodomains) have been studied extensively, and the consequences and mechanisms of interaction are well established in several cases (Li et al., 1995; Wilson and Desplan, 1995; Wolberger, 1996; Chen et al., 1998; Tan and Richmond, 1998; Piper et al., 1999). Similarly, several factors that appear to serve exclusively architectural functions, such as HMG proteins, have been identified and their interactions with DNA characterized (Schumacher et al., 1994; Love et al., 1995; Werner et al., 1995, 1996). The role played by long-range inter actions, as well as how the combination of protein–protein interactions and architectural effects cooperate to form higher order complexes, are less well understood.
Cooperative DNA binding between the initiator E1 and the transcription factor E2 provides an example where short- and long-range interactions and architectural effects participate in the formation of a simple complex. This complex performs a specific and essential function in initiation of papillomavirus DNA replication (Sedman and Stenlund, 1995). The initiator E1 binds with low specificity to the replication origin (ori), and specific and efficient recognition is accomplished by cooperative binding of E1 and E2 to immediately adjacent sites (Mohr et al., 1990; Lusky et al., 1993; Seo et al., 1993; Sedman and Stenlund, 1995). Once the complex is formed, in an ATP-dependent step E2 can be displaced and additional E1 molecules added to the complex (Sanders and Stenlund, 1998).
The interaction between the E1 and E2 proteins has two components. The DBD of E2 interacts with the DBD of E1 and the transcriptional activation domain of E2 interacts with the C-terminal helicase domain of E1 (Mohr et al., 1990; Benson and Howley, 1995; Abroi et al., 1996; Ferguson and Botchan, 1996; Berg and Stenlund, 1997; Masterson et al., 1998; Chen and Stenlund, 1998). The interaction between the DBDs is weak in the absence of DNA, and in the presence of DNA shows strong dependence on precise distance and positioning of the two binding sites, indicating that it corresponds to a short-range interaction (Berg and Stenlund, 1997; Chen and Stenlund, 1998). In contrast, the interaction between the E2 activation domain and E1 can be detected readily both in the absence and presence of DNA and shows little dependence on the relative position of the respective binding sites (Mohr et al., 1990; Lusky and Fontane, 1991; Benson and Howley, 1995; Ferguson and Botchan, 1996; Berg and Stenlund, 1997; Masterson et al., 1998). Indeed, in vivo replication assays indicate that this interaction can occur over distances of several kilobase pairs (Ustav et al., 1993). Furthermore, whereas the interaction between the E2 activation domain and E1 by itself is sufficient for DNA replication, the interaction between the E2 and E1 DBDs by itself does not allow DNA replication: indeed, an E2 lacking the activation domain has no activity for DNA replication in vivo (Ustav and Stenlund, 1991; Berg and Stenlund, 1997; Lim et al., 1998).
Previous experiments have indicated that although these two interactions can be detected in isolation, in some contexts they are not independent. In experiments with chimeric proteins, replacement of the bovine papillomavirus (BPV) E2 DBD with the highly homologous human papillomavirus 11 (HPV-11) E2 DBD, which is unable to interact with BPV E1 DBD, also abolished the interaction between the BPV E2 activation domain and E1. However, when the distance between the binding sites was increased to two turns of the helix, no dependence on the identity of the DBD was observed, and both the interaction between the E2 activation domain and E1 as well as DNA replication could be detected (Berg and Stenlund, 1997; Chen and Stenlund, 1998). Thus, the interaction between the two DBDs only plays a role when the binding sites for the two proteins are proximal, as in the wild-type ori.
Here we have investigated the nature of the interaction between the E1 and E2 DBDs. We find that the interaction between the two DBDs generates a sharp bend in the DNA and that this bend is a prerequisite for cooperative binding of the two DBDs. Furthermore, the induction of the bend is also required for the interaction between the E2 activation domain and E1 both in vitro and in vivo. Thus, cooperative binding of E1 and E2 to the ori is a two-step process where the bend generated by the interaction between the two DBDs is required for the productive interaction between the E2 activation domain and E1. The interaction between the DBDs can be viewed as being analogous to the action of separate and distinct factors such as HMG proteins, which can modify the architecture of a complex by introducing bends in DNA, allowing interactions between other DNA-bound factors. Thus, the role played by interaction between the E1 and E2 DBDs appears to be architechtural, ‘triggering’ the productive interaction between E1 and the E2 activation domain.
Results
Disruption of the interaction between the E1 and E2 DBDs results in loss of interaction between the full-length E1 and E2 proteins
Previous studies have indicated that the interaction between the E1 and E2 DBDs is important for cooperative binding of the two proteins in the context of the BPV-1 ori (Berg and Stenlund, 1997). Specifically, when the DBD of the BPV-1 E2 protein was replaced with the E2 DBD from HPV-11, the interaction between E1 and the chimeric E2 protein was abolished. We have recently isolated specific mutations in the E2 DBD that affect the interaction between the E1 and E2 DBDs. These single point alanine substitutions at positions 385, 388, 390 and 401, which map to two patches on the E2 DBD, as well as the double mutants 390/385 and 390/388 severely reduce the interaction with the E1 DBD. The wild-type E2 DBD stimulates binding of the E1 DBD 70-fold. The point mutations 390 and 401 show significant reduction of cooperativity and stimulate binding of E1 DBD 2- and 5-fold, respectively, while the double mutant 390/388 fails to stimulate binding of E1 DBD (Chen and Stenlund, 2000). In addition, these E2 DBD mutations strongly affect DNA replication and transformation in the context of the viral genome (Chen and Stenlund, 2000).
To determine the importance of the interaction between E2 DBD and E1 in the context of full-length proteins, the alanine substitutions 390, 390/385 and 390/388 were introduced into full-length E2. These full-length E2 mutant proteins were then assayed for their ability to stimulate E1 binding to the wild-type minimal BPV ori by a McKay assay in which three different probes were used, as shown in Figure 1A. The shortest probe (probe III) contains an E1-binding site, but lacks the E2-binding site. Probe II contains the BPV minimal ori with the wild-type E2-binding site adjacent to the E1-binding site. Probe I contains a high affinity E2-binding site located 22 bp, or two DNA helical turns, away from the E1-binding site (Berg and Stenlund, 1997). Using these probes, we can measure simultaneously binding of E1 alone (probe III), or binding of E1 stimulated by E2 from either the proximal (probe II) or distal site (probe I). Since we have shown previously that the activation domain of E2 is the only determinant required for cooperative binding when E2 is bound at a distal binding site, probe I serves as an internal control for E2 activity (Berg and Stenlund, 1997).


Fig. 1. (A) Mutations that disrupt the ability of the E2 DBD to interact with E1 abolish cooperative binding of full-length E2 to a proximal E2-binding site, but not a distal E2-binding site. Three different probes containing a high affinity E2-binding site distal to the E1-binding site (I), the wild-type E2-binding site proximal to the E1-binding site (II) and an E1-binding site in the absence of an E2-binding site (III) were incubated with 6 ng of GST–E1 alone (lane 1), or together with three 2-fold titrations of partially purified wild-type full-length E2 (lanes 2–4), or full-length E2 containing the mutations 390 (lanes 5–7), 390/388 (lanes 8–10) or 390/385 (lanes 11–13). Binding reactions were in a final volume of 10 µl and were incubated at room temperature for 30 min. Probes bound by GST–E1 were recovered using glutathione–agarose beads and analyzed on a 6% urea gel. Recovery of the probes was quantified using a Fuji BAS imaging system. The recovery of probe I compared with probe III is a measure of the stimulation of E1 binding by E2 bound to the distal E2-binding site and also serves as a measure of the total E2 activity present in each reaction. The recovery of probe II compared with probe III is a measure of the stimulation of E1 binding by E2 bound to the proximal E2-binding site. This ratio is shown graphically in the diagram at the bottom of the figure. (B) Mutations in the E2 DBD that affect cooperativity with E1 significantly reduce DNA replication in vivo. Full-length E2 with the mutations 390, 401 or 390/388 were tested for their ability to support replication in a transient replication assay. Expression vectors for wild-type E2, or E2 mutants, and E1 were co-transfected with the wild-type BPV minimal ori with a proximal E2-binding site (ori B) and an ori containing a distal high affinity E2-binding site (ori A) into CHO cells. Low molecular weight DNA was recovered at 48 and 72 h post-transfection, digested with DpnI, XbaI and EcoO109, and analyzed by Southern blotting. Ori A lacks the EcoO109 restriction site and gives rise to a 2.9 kb band, while the wild-type BPV minimal ori (ori B) gives rise to a 2.5 kb band. Each set of two lanes contains replicated ori A and B at 72 and 48 h post-transfection, respectively, in the presence of either wild-type E2 (lanes 1 and 2), or E2 containing mutations 401 (lanes 3 and 4), 390 (lanes 5 and 6) or 390/388 (lanes 7 and 8).
In the absence of E2, glutathione S-transferase (GST)–E1 bound to all three probes equally well (Figure 1A, lane 1). In the presence of wild-type E2, binding of GST–E1 to both probes I and II was stimulated strongly (e.g. ratios I/III and II/III are 16.2 and 11.0, respectively; Figure 1A, lane 3). In contrast, for mutant 390, despite equivalent levels of E2 activity as indicated by the levels of E1 binding to probe III relative to probe I (ratio I/III), the degree of stimulation of binding to probe II (ratio II/III) is reduced 3-fold compared with wild type (see lane 6, ratios I/III and II/III are 16.5 and 3.5, respectively, and compare with lane 3 for wild type). This demonstrates that mutation 390 greatly reduces the ability of the full-length E2 to interact with E1 from the proximal E2-binding site, although the activity from the distal E2-binding site is maintained at the wild-type level. The double mutation 390/388 had an even more severe effect. No cooperativity between E1 and E2 could be detected from the proximal site, with a ratio of probe II/III close to 1 (Figure 1A, lanes 8–10). The lack of cooperativity was not due to reduced E2 activity, since the levels of E2 stimulation to the internal control probe I were virtually identical to those observed for wild-type E2 (e.g. compare lane 9 where ratios I/III and II/III are 18.1 and 1.5, respectively, with lane 3). The effect of 390/385 was less severe, showing a weak stimulation of E1 binding despite virtually identical levels of E2 activity (lanes 11–13, ratios II/III are 2 and 4). Thus, mutations that disrupt the interaction between E1 and the E2 DBD, most notably 390/388, could abolish the ability of full-length E2 to interact with E1 on the ori despite the presence of the intact E2 activation domain. The complete loss of cooperative binding from the proximal site by the mutant 390/388 strongly suggests that the interaction between the E2 activation domain and E1 critically depends on the interaction between the E1 and E2 DBDs when the binding sites are proximal.
The interaction between E1 and E2 DBDs is required for DNA replication in vivo
DNA replication in vivo is the ultimate test of the significance of the interaction between the E1 and E2 DBDs. We therefore inserted E2 with the mutations 390, 401 and 390/388 into the mammalian expression vector pCG E2 and tested the mutant E2s for their ability to support DNA replication in a transient replication assay (Figure 1B). To obtain an accurate quantitation of the efficiency of replication, transient replication assays were performed by co-transfecting two different ori constructs. One (ori A) contains a high affinity E2-binding site distal to the E1-binding site. This construct, like probe III in the above McKay assay, served as an internal control. The second (ori B) was the wild-type minimal BPV ori, which contains the wild-type E2-binding site 12 directly adjacent to the E1-binding site. To distinguish between the two plasmids, an EcoO109 restriction site that is present in the pUC19 backbone was destroyed in ori A but left intact in ori B. Expression vectors for E1 and either wild-type E2 or the mutant E2s were co-transfected together with ori A and B into CHO cells. Cells were harvested at two different time points after transfection, and low molecular weight DNA was recovered by alkaline lysis. After digestion with DpnI, which digests only unmethylated, unreplicated input DNA, the DNA samples were digested with XbaI and EcoO109. Ori A gives rise to a 2.9 kb band, while ori B, which contains the EcoO109 site, gives rise to a smaller band of 2.5 kb.
As shown in Figure 1B, in the presence of wild-type E1 and E2, both plasmids replicated at equivalent levels, demonstrating that wild-type E2 functions equally well for replication of both oris (lanes 1 and 2). Co-transfection with the E2 mutant 401 resulted in a decrease in the amount of ori B replication compared with ori A (lanes 3 and 4), and similar effects were observed with mutant 390. Thus, the single mutations in either 390 or 401, which affect the E2 DBD interaction with E1, cause a severe reduction in replication of ori B. The double mutant 390/388, which in the context of the E2 DBD and the full-length protein abolished the E1–E2 interaction, was inactive for replication of ori B. Thus, the levels of replication of ori B for the three mutants correlated with the decreased ability of the E2 DBD to bind cooperatively to E1 in vitro. These results demonstrate that the mutant E2s are incapable of functioning efficiently from the proximal E2-binding site in ori B, strongly suggesting that the E1–E2 DBD interaction is required in order for the E2 activation domain to interact with E1 and for replication to occur in vivo. In addition, because none of these mutations have an effect on replication of ori A, the mutations in the E2 DBD appear to be completely selective for the ori with a proximal E2-binding site.
Cooperative binding of the E1 and E2 DBDs to the BPV ori generates a sharp bend in the DNA
We had observed that the complexes formed between E1 DBD and mutant E2 DBDs migrated faster than the complex formed between the wild-type E1 and E2 DBDs. One explanation for this difference in mobility could be that cooperative binding of the two DBDs results in changes in template structure. The ability of proteins to induce bends in DNA upon binding, and techniques for analyzing these bends, have been well established (Crothers et al., 1991). To determine the effects of binding of E1 and E2 DBDs individually and in combination, we performed circular permutation assays using purified E1 and E2 DBDs. The DNA probes used to examine bending were made in such a way that the position of the binding site for the proteins in the probe can be changed while maintaining the same overall length of the probe, as shown in Figure 2A. If binding of the protein induces a bend in the DNA, a complex formed on a probe with the binding site near the end of the probe will migrate faster in the gel shift than a complex formed on a probe with the binding site in the center of the probe. The probes are labeled 1–7, with the binding site for the protein closest to the ends in probes 1 and 7. Binding of a protein that does not bend the DNA will have little or no differential effect on the migration of the various protein–probe complexes.


Fig. 2. Circular permutation assays. (A) Schematic representation of the probes used for circular permutation assays. The positions of the E1- and E2-binding sites are indicated. Digestion of an internally labeled PCR product with seven different restriction enzymes yields seven probes with the same overall length, but with the E1- and E2-binding sites in different positions relative to the end of the probe. (B) Circular permutation assays were performed with the E1 DBD alone (left panel) or with E1 DBD in the presence of the E2 DBD (right panel) using the seven probes shown in (A). Lane 1 is a mixture of all seven probes in the absence of added protein, indicating the migration of the free probe. E1 DBD was incubated with the seven probes alone (lanes 2–7) or in the presence of E2 DBD (lanes 9–15). The arrows labeled E11 and E12 indicate the mobility of the E1 monomer and dimer complexes, respectively. The migration of the E2 dimer complex is indicated by the E22 arrow. The arrow labeled E12E22 indicates the migration of the combined E1–E2 complex.
Figure 2 shows the results when gel shifts were generated with these seven probes with either E1 DBD alone or E1 DBD and E2 DBD together (Figure 2B). The lower band corresponds to free probe (lane 1). The upper complex in lanes 2–8 corresponds to a dimer of the E1 DBD bound to the probes shown in Figure 2A, while the lower complex corresponds to a monomer of bound E1 DBD. Binding of the dimer of E1 DBD produces a slight bend indicated by the slight change in the mobility of the complex. Interestingly, the monomer complex of E1 (E11) does not induce a bend, as indicated by the absence of a change in the mobility of this complex. Binding by E2 DBD alone similarly caused only minor differences in mobility of the seven probes, indicating a modest bend (lanes 9–15). This is consistent with the E2 DBD–DNA co-crystal structure, which shows that E2 DBD bends the DNA upon binding (Hegde et al., 1992). The upper complex corresponds to the complex formed by the cooperative binding of the E1 and E2 DBDs and contains a dimer of E1 and a dimer of E2. The difference in mobility between this complex formed on the seven different probes is much greater than for either E1 DBD or E2 DBD alone, indicating that the bend is greater for the combined complex than for the individual complexes. Circular permutation assays may be influenced by position-dependent effects on probe migration that are not due to bending (Gartenberg et al., 1990). To verify that the differences in migration were due to bending, we also performed phasing analysis (Zinkel and Crothers, 1987; see Materials and methods). An intrinsically bent DNA sequence is introduced at different positions relative to the binding site for the protein of interest. Depending on the direction and phase of an induced bend relative to the intrinsinc bend, the mobility of the complex will vary for the different probes. In the absence of an induced bend, no change in complex mobility is expected for the different probes. The results of these assays were consistent with bending of the probe upon binding of E1 and E2 DBDs (data not shown).
Cooperative DNA binding correlates with DNA bending
The extent of bending by the combined E1–E2 complex seemed much greater than the sum of the bends induced by E1 and E2 alone. Empirical estimation of the bend angle from the changes in complex mobility indicated that the bend for E12 was 40–50°, for E22 ∼40° and for the combined E12–E22 complex 120–130° (Thompson and Landy, 1988; see Materials and methods). We therefore considered the possibility that interaction between E1 and E2 could generate an additional bend in the sequences between the E1- and E2-binding sites. To determine if this was the case, we wanted to test the E2 DBD mutants that fail to bind cooperatively to E1 for their ability to bend DNA in conjunction with the E1 DBD. The bend generated by cooperative binding of E1 and E2 can be thought of as composed of three components. Both the E1 dimer and the E2 dimer individually induce slight bends, and any contribution from the cooperative interaction between the two proteins would constitute the third component. To compare the degree of bending directly, we wished to eliminate the influence of at least one of these factors. We had observed previously that the monomer E1–DNA complex did not appear to bend the DNA appreciably (Figure 2B). We also knew that a monomer of E1 can bind cooperatively to E2 if the binding sites are adjacent to each other (Chen and Stenlund, 1998). Therefore, we generated probes containing only half of the E1-binding site palindrome adjacent to the E2-binding site. In such a complex, the degree of bending would consist of two components, the bend produced by the E2 dimer and the putative bend produced by the interaction between the E1 and E2 DBDs. Thus, the degree of bending by the E2 dimer and the E11–E22 complex can be compared directly. We first performed cooperative binding assays using the half-site probe (Figure 3A). Cooperative binding was readily observed with the wild-type E2 DBD (compare lanes 2 and 3 with 5 and 6; ∼5-fold stimulation), although the degree of stimulation of E1 binding was reduced compared with when the full-length probe was used. The mutants 390, 401 and 390/388 all failed to show detectable stimulation of E1 binding, presumably due to the general reduction in cooperative binding to the half-site probe (Figure 3A, lanes 8 and 9, 11 and 12, and 14 and 15). Indeed, these mutants were as inactive as the HPV-11 E2 DBD (lanes 17 and 18).


Fig. 3. Mutant E2 DBDs fail to bend DNA in combination with E1 DBD. (A) Binding assays were performed using the half-site probe. Two concentrations of E1 DBD (20 and 10 ng) were bound to the probe either alone (lanes 2 and 3) or in the presence of wild-type E2 DBD or mutant 390 (lanes 8 and 9), mutant 401 (lanes 11 and 12) or the double mutant 390/388 (lanes 14 and 15), or the HPV-11 E2 DBD (lanes 17 and 18). Lane 1 contains probe alone. In lanes 4, 7, 10, 13 and 16, the wild-type and mutant E2 DBDs were added in the absence of E1 DBD. Migration of free probe is indicated by the arrow labeled FP. The complex formed by the binding of a monomer of the E1 DBD is indicated by the arrow labeled E11. The migration of the E2 DBD dimer complex is indicated by the arrow labeled E22. The complex formed by the binding of a dimer of the E2 DBD together with a monomer of the E1 DBD is indicated by the arrow labeled E11E22. (B) Three circularly permuted probes, labeled 1, 4 and 7 in Figure 2A, were used. Binding by E1 DBD alone to these three probes is shown in lanes 2–4. Binding, in the absence and presence of E1 DBD, is shown for wild-type E2 DBD (lanes 5–10), mutant 390 (lanes 11–16), mutant 401 (lanes 17–22) and mutant 390/388 (lanes 23–28).
The half-site sequence was inserted into the pBend-vector, probes were generated and the mutant and wild-type E2 DBDs were assayed for their ability to bend alone and together with the E1 DBD (Figure 3B). The number of bending probes used in this and subsequent bending experiments was reduced from seven to three probes for simplicity. An estimate of the degree of bending can be obtained from the difference in migration of the complexes formed on the first two probes. As observed previously, the E1 DBD binds predominantly as a monomer to the half-site probe and does not induce a bend. The ability of the wild-type and mutant BPV E2 DBDs to bind and bend DNA is not affected by the point mutations in the E2 DBD (complexes labeled E22) (Figure 3B, lanes 11–13, 17–19 and 23–25). As expected, E1 DBD binds cooperatively to the wild-type E2 DBD and simultaneously induces a substantial bend as measured by the mobility difference between the first two probes (E11E22) (lanes 8–10). E1 DBD in combination with the mutant E2 DBDs shows a reduced degree of cooperativity. Furthermore, the bend generated by E1 DBD in combination with the mutant E2 DBDs is significantly reduced or absent. The mutants 390, 401 and 390/388 (lanes 14–16, 20–22 and 26–28), which have little or no activity for cooperative binding to E1 DBD, show no increase in bending beyond the bend induced by the E2 DBDs alone. These results demonstrate that the loss of cooperative DNA binding observed for the E2 DBD mutants is accompanied by a reduction in DNA bending, indicating that the sharp bend in the DNA may be produced by the interaction between the E1 and E2 DBDs.
Decreasing the bendability of the ori sequence between the E1- and E2-binding sites affects cooperative binding
A prediction based on these results where DNA bending accompanies cooperative binding and vice versa is that a change of the bendability of the DNA sequence between the E1- and E2-binding sites might affect cooperative binding by the E1 and E2 DBDs. The sequence between the two binding sites contains 3 bp that are not required for binding of E1 or E2 (Sedman et al., 1997). This sequence, AAT, is involved in binding of additional E1 molecules for the formation of larger E1 complexes. We changed the two As in this sequence to Ts individually and together to generate a T4 stretch (Figure 4A). A–T stretches of this kind are known to have increased rigidity (Rhodes, 1979; Nelson et al., 1987; Yoon et al., 1988). Direct measurement of the flexibility of a DNA sequence is difficult but a general consequence of increased stiffness of a DNA sequence appears to be reduced susceptibility to DNase digestion (Drew and Travers, 1985; Hogan et al., 1989; Lahm and Suck, 1991). DNase I generates a bend toward the major groove when binding to DNA. Cleavability by DNase I can thus serve as an indicator of bendability of a particular DNA sequence. We utilized this property and analyzed the DNase cleavage of the wild-type and mutant templates (Figure 4B). The T7 and T8 mutations individually showed some changes in cleavage pattern compared with the wild-type sequence but no general reduction in DNase cleavage. Interestingly, the T7T8 mutations decreased the rate of DNase cleavage consistent with increased rigidity of the T4 sequence. The mutants T7, T8 and T7T8 were subsequently tested for binding of E1 and E2 DBDs alone and in combination. These mutations had no detectable effect on binding of E1 alone (Figure 4A, lanes 5–8) or E2 alone (Figure 4A, lanes 9–12). The individual mutations also had small effects on the combined binding of E1 and E2 (Figure 4A, lanes 15 and 16). E2 stimulated binding of E1 2.2-fold on the wild-type and the T8 template, while stimulation of E1 binding for the T7 mutant was 1.5-fold. Interestingly, the T7T8 mutation resulted in complete loss of cooperativity; no stimulation of E1 binding by E2 was observed (Figure 4A, lane 14), as indicated by the 1:1 ratio of the E11 and E11–E22 complexes in lane 14.

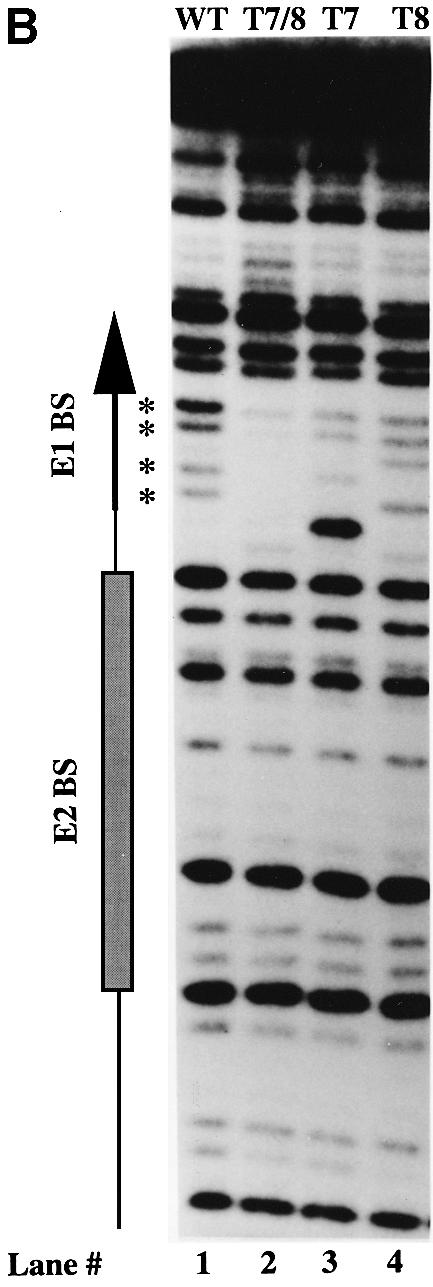
Fig. 4. (A) Cooperative binding of E1 and E2 DBDs is disrupted by a double point mutation between the two sites. Gel shift assays were performed using wild-type or three mutant probes (T7, T8 and T7T8). Binding of E1 DBD alone, E2 DBD alone, and E1 and E2 DBDs together was compared. With E1 DBD alone (lanes 5–8), 11–12% of the probe was shifted for the different probes. With E2 DBD alone (lanes 9–12), 37–38% of the probe was shifted for the different probes. Stimulation of E1 binding by E2 was measured by comparing the level of E1 DBD binding in lanes 5–8 and lanes 13–16. The stimulation observed with the wild-type, T7 and T8 probes was 2.2-, 1.5- and 2.1-fold, respectively. No stimulation by E2 was observed with the T7T8 probe. The migration of the different complexes is indicated by arrows. Below is shown a schematic representation of the half-site probe and the mutations that were tested. (B) DNase digestion indicates increased rigidity of the the T7T8 mutant. The wild-type and the T7, T8 and T7T8 mutant probes were subjected to DNase digestion, and the extent of cleavage was determined by analysis on a denaturing polyacrylamide gel. Positions of reduced cleavage in the T7T8 probe are indicated by asterisks.
Figure 5 compares the ability of the wild-type E1 and E2 DBDs to bend the mutant and wild-type half-site probes. The E1 monomer complex (E11) formed on all the probes with equal efficiency (Figure 5, compare lanes 3–5 and 6–8). Similarly, binding and bending by the E2 DBD alone to the wild-type (lanes 9–11) and mutant (lanes 12–14) probes was indistinguishable. However, when formation of the combined complex was measured, a significant difference was observed between the two probes. E1 and E2 DBDs failed to induce a bend of the T7T8 probe (Figure 5, lanes 18–20) while a significant bend could be observed with the wild-type probe (lanes 15–17). Thus, in addition to the strong correlation between cooperative binding and bending, we conclude from these results that the ability to bend the DNA is a prerequisite for the interaction between the two DBDs.

Fig. 5. The T7T8 mutation inhibits bending. Circular permutation assays were performed using the probes 1, 4 and 7 in Figure 2A containing either the wild-type sequence (lanes 3–5, 9–11 and 15–17) or the T7T8 mutations (lanes 6–8, 12–14 and 18–20). Binding of E1 DBD alone to the two sets of probes is shown in lanes 3–8. Binding of E2 DBD alone to the two sets of probes is shown in lanes 9–14. E1 DBD and E2 DBD were combined in lanes 15–20. The migration of the different complexes is indicated by arrows.
Cooperative binding of full-length E1 and E2 proteins is reduced by the T7T8 mutation
The loss of cooperative binding between full-length E1 and E2 as a consequence of the mutations in the E2 DBD (Figure 1A) indicated that bending of the DNA may be the required trigger for interaction between the E2 activation domain and E1. We reasoned that if we could inhibit bending by the use of the T7T8 mutant, the importance of the bend on binding of the two full-length proteins could be determined (Figure 6). Full-length E1 and E2 proteins were incubated in the presence of either the wild-type (lanes 1, 3, 5 and 7) or T7T8 (lanes 2, 4, 6 and 8) half-site probes. Full-length E1 alone bound to both probes with equal efficiency, forming the complex E11 (lanes 3 and 4). Full-length E2 alone also bound to both probes equally well, forming the E2 dimer complex (lanes 5 and 6). Cooperative binding of full-length E1 and E2 on the wild-type probe resulted in a strong stimulation of E1 binding, although the exact level of stimulation is hard to determine since the low level of E1 (0.6 ng) used for binding in the presence of E2 does not by itself give rise to a detectable complex. Nevertheless, stimulation of E1 binding was reduced significantly using the T7T8 probe compared with the wild-type probe (3- to 4-fold, compare lanes 7 and 8), indicating that bending of the DNA and interaction between the two DBDs contributes significantly to interaction between the two full-length proteins. Thus, interaction between full-length E1 and E2 proteins can be disrupted either by mutations in the E2 DBD or by mutations that increase the stiffness of the DNA between the E1- and E2-binding sites. Both of these results are consistent with a requirement for an interaction between the E1 and E2 DBDs, which can only occur through bending of the DNA sequence between the two sites.
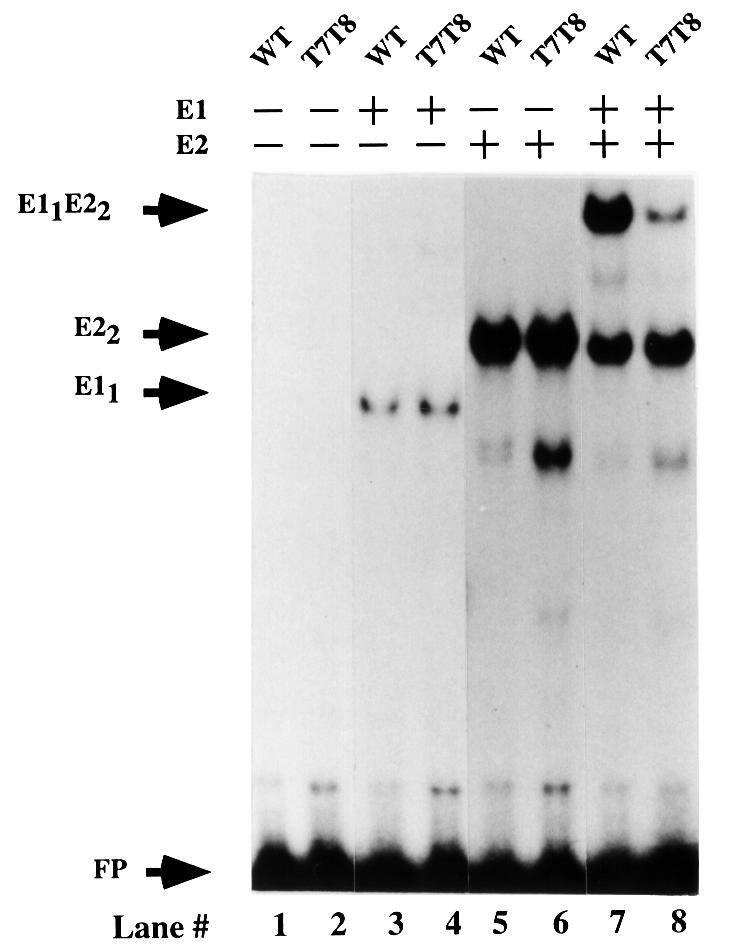
Fig. 6. Decreased bendability of the probe results in reduced cooperative binding between full-length E1 and E2. Gel shift assays were performed with full-length E1 and E2 proteins with a wild-type half-site probe (lanes 1, 3, 5 and 7) and with the T7T8 mutant probe (lanes 2, 4, 6 and 8). Probe in the absence of added protein is shown in lanes 1 and 2. In lanes 3 and 4, 30 ng of full-length E1 were added. In lanes 5 and 6, 20 pg of E2 were added. In lanes 7 and 8, 0.6 ng of E1 and 20 pg of E2 were incubated with the two probes. With E1 alone (lanes 3 and 4), 1.2 and 1.8% of the probes were shifted. With E2 alone (lanes 5 and 6), 20 and 22.5% of the probes were shifted. In the presence of both E1 and E2 (lanes 7 and 8), the E11–E22 complex constituted 16.2 and 4.9% of the total probe, respectively. The migration of the complex formed by the binding of a monomer of E1 and a dimer of E2 is indicated by the arrow E11E22.
Discussion
The BPV genome contains a large number of E2-binding sites other than the ori-proximal site. The majority of these sites have much higher affinities, and previous studies have indicated that when high affinity E2-binding sites are multimerized they can function for replication from great distances, consistent with the ability of E2 to interact from distant sites and loop the intervening DNA (Knight et al., 1991; Ustav et al., 1993). An interesting question is how the proximal E2 site and its specialized mode of cooperative binding relates to the function of other E2-binding sites present in the viral genome. A distinction between proximal and distal positions is that in the proximal position very low affinity E2-binding sites can function for interaction with E1 and replication, but, as the distance is increased even slightly, the low affinity E2-binding site becomes inactive (Ustav et al., 1993). Activity can be restored by increasing the affinity of the E2-binding site. One interpretation of these results is that in the absence of cooperating E1, the low affinity ori-proximal E2-binding site may not be occupied by E2. Thus, whether or not E2 is bound at the ori may be determined by the levels of E1. If, for example, E2 bound to the ori-proximal site can control E1 expression negatively, or control expression of an antagonist of E1 expression positively, an autoregulatory loop would be established that would link the control of E1 expression to levels of E1, as well as to the frequency of initiation of DNA replication.
The interaction between the E1 and E2 DBDs is a prerequisite for the interaction between the E2 activation domain and E1
The E1 and E2 proteins bind cooperatively to adjacent binding sites, resulting in a substantial increase in specificity and affinity compared with binding of the individual proteins. The interaction between the two proteins has two components. Two distinct domains in E2 interact with two separate regions in E1, resulting in a complex in the pathway to a replication initiation complex (Sanders and Stenlund, 1998). The interaction between the activation domain of E2 and E1 is in itself sufficient for both cooperative binding in vitro and DNA replication in vivo when the binding sites for the two proteins are in a distal position. The interaction between the E1 and E2 DBDs is required only in a specific context, i.e. when the E1- and E2-binding sites are positioned immediately adjacent to each other. In that context, mutations in the E2 DBD that disrupt the interaction with E1 DBD result in complete loss of interaction between E1 and E2, as well as loss of replication activity in vivo (Figure 1A and B).
Formally, these results have two possible interpretations. (i) The E2 activation domain could interact with E1 when the binding sites are distal, and the E2 DBD could perform the same function when the binding sites are proximal. (ii) Alternatively, the interaction between the E2 transactivation domain and E1 is required when the binding sites are both proximal and distal; however, the contact between the E1 and E2 DBDs is required to facilitate this interaction only when the binding sites are proximal. The evidence indicates that the second alternative is correct. Comparison of cooperative binding demonstrates that interaction of full-length E1 with full-length E2 is at least 10-fold stronger than the interaction with E2 DBD, consistent with a significant contribution of the E2 activation domain when the proteins are bound in the proximal position (Chen and Stenlund, 1998). Thus, cooperative binding of E1 and E2 to the ori involves cooperation of two elements in each protein, where the interaction between the E1 and E2 DBDs facilitates the interaction between the E2 activation domain and E1.
The bend generated by interaction between the DBDs facilitates interaction between the E2 activation domain and E1
The interaction between the E1 and E2 DBDs causes a sharp bend in the DNA at the binding sites for the two proteins. Since bending of the DNA is a prerequisite for interaction between the two DBDs, our experiments cannot determine directly whether the physical interaction, or the resulting bend, constitutes the required trigger for interaction between the E2 activation domain and E1. However, since the interaction between the DBDs is dispensable when E2 is bound at a distance, it seems likely that the bend in the DNA constitutes the critical factor. The bend can account for the importance of the DBD contact for the interaction between the E2 activation domain and E1 as illustrated in Figure 7. The bend caused by the interaction between the two DBDs may place the E2 activation domain physically closer to its interaction partner in E1, thereby facilitating the interaction between these two domains. In the absence of the bend, the E2 activation domain and E1 fail to make physical contact.

Fig. 7. Model for the two-step process required for the cooperative binding of E1 and E2 to the BPV minimal ori. (A) The E1 and E2 DBDs bind to adjacent sites on the DNA. Interaction between the two DBDs results in the generation of a sharp bend of the DNA. Mutations in the E2 DBD that abolish interaction between the two DBDs (indicated by a filled circle) result in co-occupancy of the two DBDs, but loss of cooperativity and bending. (B) In the context of the full-length E1 and E2 proteins, the mutations in the E2 DBD that interfere with the interaction between the DBDs also result in the loss of interaction between the E2 activation domain (E2 AD) and the E1 helicase domain (E1 H). The bend induced by the interaction between the E1 and E2 DBDs, however, places the E2 activation domain and the E1 helicase domain physically closer to each other, allowing interaction.
An interesting question is why cooperative binding of E1 and E2 requires a ‘facilitating’ interaction. The answer is clearly related to the positioning of the binding sites since the DBD interaction is dispensable when the E2-binding site is in the distal position. It is well established that in long DNA fragments, small changes in bend angle at many individual base steps result in substantial bends with very low cost in free energy. However, short DNA sequences are inherently stiff because generation of a sharp bend requires greater changes in individual bend angles, since only a small number of base steps are involved (Wang and Giaever, 1988). The very close juxtaposition of the proximal E1- and E2-binding sites will impose constraints on bending, possibly requiring substantial structural alterations in the DNA as has been observed for binding of proteins such as TBP, LEF and IHF (Werner and Burley, 1997). It therefore seems likely that the ‘facilitation’, i.e. bending of the DNA, requires an activity unique to the DBDs. The two DBDs interact only over short distances and the interaction is highly dependent on DNA binding and positioning of the binding sites, consistent with such a role. The interaction between the E2 activation domain and E1, in contrast, is readily detectable in the absence of DNA and shows great flexibility in terms of distance and positioning of the binding sites, consistent with the ability to loop the DNA when the binding sites are distal.
The combination of one interaction that contributes architecturally and facilitates a second, productive, interaction resembles the role that HMG proteins such as LEF-1 and SRY play in the assembly of promoter and enhancer complexes (Grosschedl, 1995). These are DNA-binding proteins that can bend DNA to bring together interaction partners that would otherwise interact poorly because of stiffness of the DNA or the distance between the two sites. A unique feature of the interaction between E1 and E2 is that these distinct, but complementary, activities reside in different domains of the same polypeptide.
The bipartite, interdependent, interaction that we observe between the E1 and E2 proteins to our knowledge represents a novel arrangement to accomplish cooperative DNA binding. Although many examples of interactions between DBDs exist, and transactivation domains of transcription factors are generally believed to interact with other proteins, a coupling of these interactions as observed here has not been described. A question that is raised by these results is whether interactions between other DBDs that have been characterized represent ‘productive’ interactions or are architectural, facilitating other as yet undetected interactions between other domains of the same proteins. The complete loss of cooperative binding that we observe as a consequence of single mutations in the E2 DBD generally would be taken as evidence that only a single interaction exists and a bipartite interaction of this kind could easily go undetected even for well characterized proteins.
A related question is whether close juxtaposition of interacting factors in general necessitates alterations of the DNA structure. Interestingly, in several of the ternary protein–DNA complexes whose X-ray crystal structures have been determined, sharp bends in the DNA caused by interacting DBDs are observed (Li et al., 1995; Chen et al., 1998; Tan and Richmond, 1998). Many of the interactions appear to be of the type observed for the E1 and E2 DBDs, i.e. distance- and position-dependent short-range interactions. A particularly interesting and well studied example of this kind is the Mat a1–α2 complex, which shows similaritites to E1 and E2 at several levels. a1 and α2 individually bind poorly to DNA but bind cooperatively with high affinity and specificity to the hsg operator. Cooperative binding of a1 and α2 DBDs generates a sharp bend in the DNA (Li et al., 1995; Wolberger, 1996). Interestingly, the binding affinity using the two isolated homeodomains is 10-fold lower than for the full-length proteins, indicating that other interactions exist outside the DBDs for the a1–α2 pair just as for the E1–E2 pair and that those interactions may contribute to the cooperative interaction of the two proteins (Goutte and Johnson, 1993). It would be interesting to determine whether the sharp DNA bend that is generated by cooperative binding of the a1 and α2 homeodomains is required for additional interactions between other domains of a1 and α2.
Materials and methods
Ori constructs
All full-length ori constructs have been described previously (Sedman and Stenlund, 1995; Berg and Stenlund, 1997). The half-site ori constructs used for cooperative binding assays were constructed by PCR amplification using an ori template containing an XhoI linker insertion in the center of the E1 palindrome (Ustav et al., 1991). Primers used for amplification were the universal primer RSP, and a primer with the sequence GGTCTAGACCTCGAGGAACAATAATCACACC for the wild-type half-site probe. The resulting PCR product was digested with XbaI and HindIII and inserted into the polylinker of pUC19. The resulting construct contains one half of the E1 palindrome and the downstream, proximal E2-binding site (see also Figure 3).
Constructs for circular permutation assays
The HindIII restriction site in pBEND 2 (Kim et al., 1989) was destroyed and the unique SalI cloning site was converted to a HindIII site by insertion of a HindIII linker. We now refer to this vector as pBEND 2H. The fragments to be tested for bending were generated by PCR and inserted between the XbaI and HindIII sites, and the sequences of the inserts were verified by DNA sequencing. All constructs used for circular permutation assays contain the high affinity E2-binding site 9 in place of E2-binding site 12.
Probes
Probes for cooperative binding studies were generated by PCR amplification of ori constructs using the universal primers USP and RSP. The labeled PCR products were gel purified and eluted before use. The probes used for circular permuation assays were generated by internal labeling of PCR fragments with [α-32P]dATP using primers Bend 1 and 2 that anneal on either side of the permuted sequence in the pBEND 2H vector. The sequences of the primers Bend 1 and Bend 2 are TAGGCGTATCACGAGGCCCT and CGTTAGCAATTTAACTGT GAT, respectively. After PCR amplification, the PCR product was digested using the enzymes MluI, NheI, XhoI, EcoRV, StuI, NruI and BamHI to generate the seven circularly permuted probes. After digestion, the probes were gel purified, eluted and precipitated.
Gel mobility shift assays
Probe (5000 c.p.m./reaction) was incubated with protein and 20 ng of non-specific competitor (pUC119) in 10 µl of binding buffer [20 mM potassium phosphate pH 7.4, 100 mM NaCl, 1 mM EDTA, 10% glycerol, 0.1% NP-40, 5 mM dithiothreitol (DTT), 3 mg/ml bovine serum albumin (BSA)]. After a 30 min incubation at room temperature, the samples were loaded onto 5, 6 or 10% 80:1 (acrylamide/bis-acrylamide) pre-run polyacrylamide gels and electrophoresed in 0.5× TBE at 225 V. After electrophoresis, the gels were dried and subjected to autoradiography. Quantitation was performed using a FUJI BAS 1000 using Image-Gauge software.
Phasing analysis
Determination of the phasing of the putative bends was perfomed using the procedure of Zinkel and Crothers (1987). The vector used for phasing analysis is based on the pBEND 2 plasmid (Kim et al., 1989) as described in Natesan and Gilman (1993). These vectors contain an intrinsically bent DNA sequence inserted adjacent to a SalI site used for cloning. PCR fragments containing the E1- and E2-binding sites were inserted such that 0, 2, 5, 7 and 10 bp separated the inserted fragment and the intrinsic bend. Proteins that induce bends in the DNA upon binding will cause alterations of the mobility of the DNA–protein complex relative to the free probe. When the induced and intrinsic bends are in-phase, the mobility of the complex will reach a maximum. When the induced and intrinsic bends are out-of-phase, the mobility will reach a minimum. In the absence of an induced bend, no change in complex mobility relative to free probe is expected for the different probes. This assay was performed analyzing the E1 dimer complex, the E2 dimer complex and the combined E1–E2 complex. The E1 dimer complex and the E1–E2 complex showed minimal mobility with the +5 probe while the E2 dimer complex showed minimal mobility with the +3 probe.
Estimation of bend angles
Estimation of bend angles from circular permutation assays was performed using the empirical method developed by Thompson and Landy (1988). By measuring the mobilities of the shifted complexes, the ratio of slowest to fastest migrating complexes can be calculated (µm/µE). These values were applied to the formula µm/µE = cos(α/2) where α represents the bend angle. The estimated bend angles were ∼40° for the E2 dimer complex, 40–50° for the E1 dimer complex and 120–130° for the E12–E22 complex. The bend angle for the E2 DBD in the X-ray crystal structure is 42.7–44° (Hegde et al., 1998).
Protein expression and purification
The expression and purification of full-length E1 and E2 have been described previously (Sedman et al., 1997). Expression and purification of the E1 DBD were performed as described (Chen and Stenlund, 1998). Wild-type and mutant E2 DBDs were expressed using the Escherichia coli expression vector pET 11C E2 DBD (Berg and Stenlund, 1997) in the strain BL21 (DE3). Liquid cultures were inoculated and grown at 18°C until an optical density at 600 nm of 0.6–0.8 was reached. Cultures were then induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and grown for an additional 6 h at 18°C. Bacterial pellets were resuspended in lysis buffer [50 mM Tris pH 7.5, 0.1 M NaCl, 5 mM EDTA, 5 mM DTT, 20% sucrose and 1 mM phenylmethylsulfonyl fluoride (PMSF)] and treated with lysozyme (100 µg/ml) for 10 min on ice. The lysate was cleared by centrifugation at 20 000 g for 30 min at 4°C. The cleared cell lysate was then applied to a 1 ml S-Sepharose column. The column was washed with 10 column volumes of buffer A (20 mM MES pH 6.2, 200 mM NaCl, 10 mM DTT, 1 mM PMSF) and the bound protein eluted with a 0.1–1 M NaCl gradient in the same buffer. Peak fractions were pooled, diluted 5-fold with buffer B (20 mM MES pH 6.5, 100 mM NaCl, 1 mM DTT, 1 mM PMSF) and loaded onto a 1 ml Mono-S column. The column was washed with buffer B and eluted with a 10 ml gradient of 0.1–1 M NaCl in buffer B. Glycerol was added to 10% final concentration in the peak fractions, the fractions diluted, aliquoted, frozen in liquid nitrogen and stored at –70°C for use. The protein was deemed to be >95% pure.
McKay assay
Purified full-length E1 with an N-terminal GST fusion was incubated together with partially purified wild-type or mutant E2 proteins in binding buffer and 100 ng of non-specific competitor DNA [poly(dA/dT)] together with the three ori probes. After incubation for 30 min, 2.5 µl of glutathione–agarose beads were added to the 10 µl binding reactions, and the total volume was brought up to 50 µl by the addition of binding buffer. After 20 min of rotational mixing at room temperature, the beads were washed three times with 200 µl of binding buffer. A 100 µl aliquot of stop buffer (1% SDS, 50 mM EDTA, 0.1 M NaCl, 25 µg/ml tRNA) together with 5 µg of mussel glycogen carrier was added, and the samples were extracted with phenol/chloroform, and ethanol precipitated. The DNA pellets were resuspended in formamide loading buffer and loaded onto a 6% denaturing polyacrylamide gel.
Transient replication assays
Transient replication assays were performed in CHO cells as previously described (Ustav and Stenlund, 1991; Sedman et al., 1997).
Acknowledgments
Acknowledgements
We thank Leemor Joshua-Tor for critical reading of the manuscript. This work was supported by a grant NIH CA 13106 to A.S.
References
- Abroi A., Kurt,R. and Ustav,M. (1996) Transcriptional and replicational activation functions in the bovine papillomavirus type 1 E2 protein are encoded by different structural determinants. J. Virol., 70, 6169–6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson J.D. and Howley,P.M. (1995) Amino-terminal domains of the bovine papillomavirus type 1 E1 and E2 proteins participate in complex formation. J. Virol., 69, 4364–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg M. and Stenlund,A. (1997) Functional interactions between papillomavirus E1 and E2 proteins. J. Virol., 71, 3853–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. and Stenlund,A. (1998) Characterization of the DNA-binding domain of the bovine papillomavirus replication initiator E1. J. Virol., 72, 2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. and Stenlund,A. (2000) Two patches of amino acids on the E2 DNA binding domain define the surface for interaction with E1. J. Virol., 74, 1506–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Glover,J.N.M., Hogan,P.G., Rao,A. and Harrison,S.C. (1998) Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature, 392, 42–48. [DOI] [PubMed] [Google Scholar]
- Crothers D.M., Gartenberg,M.R. and Shrader,T.E. (1991) DNA bending in protein–DNA complexes. Methods Enzymol., 208, 118–145. [DOI] [PubMed] [Google Scholar]
- Drew H.R. and Travers,A.A. (1985) DNA bending and its relation to nucleosome positioning. Nucleic Acids Res., 13, 4445–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson M.K. and Botchan,M.R. (1996) Genetic analysis of the activation domain of bovine papillomavirus protein E2: its role in transcription and replication. J. Virol., 70, 4193–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartenberg M.R., Ampe,C. Steitz,T.A. and Crothers,D.M. (1990) Molecular characterization of the GCN4–DNA complex. Proc. Natl Acad. Sci. USA, 87, 6034–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutte C. and Johnson,A.D. (1993) Yeast a1 and α2 homeodomain proteins form a DNA-binding activity with properties distinct from those of either protein. J. Mol. Biol., 233, 359–371. [DOI] [PubMed] [Google Scholar]
- Grosschedl R. (1995) Higher-order nucleoprotein complexes in transcription: analogies with site-specific recombination. Curr. Opin. Cell Biol., 7, 362–370. [DOI] [PubMed] [Google Scholar]
- Grosschedl R., Giese,K. and Pagel,J. (1994) HMG domain proteins: architechtural elements in the assembly of nucleoprotein structures. Trends Genet., 10, 94–100. [DOI] [PubMed] [Google Scholar]
- Hegde R.S., Grossman,S.R., Laimins,L.A. and Sigler,P.B. (1992) Crystal structure at 1.7 Å of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature, 359, 505–512. [DOI] [PubMed] [Google Scholar]
- Hegde R.S., Wang,A.F., Kim,S.S. and Schapira,M. (1998) Subunit rearrangement accompanies sequence-specific DNA binding by the bovine papillomavirus-1 E2 protein. J. Mol. Biol., 276, 797–808. [DOI] [PubMed] [Google Scholar]
- Hogan M.E., Roberson,M.W. and Austin,R.H. (1989) DNA flexibility variation may dominate DNase cleavage. Proc. Natl Acad. Sci. USA, 86, 9273–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Zweib,C. and Adhya,S. (1989) Bending of DNA by gene-regulatory proteins: construction and use of a DNA bending vector. Gene, 85, 15–23. [DOI] [PubMed] [Google Scholar]
- Knight J.D., Li,R. and Botchan,M. (1991) The activation domain of the bovine papillomavirus E2 protein mediates association of DNA bound dimers to form DNA loops. Proc. Natl Acad. Sci. USA, 88, 3204–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahm A. and Suck,D. (1991) DNase I-induced DNA conformation: 2 Å structure of a DNase I–octamer complex. J. Mol. Biol., 222, 645–667. [DOI] [PubMed] [Google Scholar]
- Li T., Stark,M.R., Johnson,A.D. and Wolberger,C. (1995) Crystal structure of the MATa1/MATα2 homeodomain heterodimer bound to DNA. Science, 270, 262–269. [DOI] [PubMed] [Google Scholar]
- Lim D.A., Gossen,M., Lehman,C.W. and Botchan,M.R. (1998) Competition for DNA binding sites between the short and long forms of E2 dimers underlies repression in bovine papillomavirus type 1 DNA replication control. J. Virol., 72, 1931–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love J.J., Li,X., Case,D.A., Giese,K., Grosschedl,R. and Wright,P.E. (1995) Structural basis for DNA bending by the architetural transcription factor LEF-1. Nature, 376, 791–795. [DOI] [PubMed] [Google Scholar]
- Lusky M. and Fontane,E. (1991) Formation of the complex of bovine papillomavirus E1 and E2 proteins is modulated by E2 phosphorylation and depends upon sequences within the carboxyl terminus of E1. Proc. Natl Acad. Sci. USA, 88, 6363–6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusky M., Hurwitz,J. and Seo,Y.S. (1993) Cooperative assembly of the bovine papilloma virus E1 and E2 proteins on the replication origin requires an intact E2 binding site. J. Biol. Chem., 268, 15795–15803. [PubMed] [Google Scholar]
- Masterson P.J., Stanley,M.A., Lewis,A.P. and Romanos,M.A. (1998) A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase α-primase p68 subunit. J. Virol., 72, 7407–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr I.J., Clark,R., Sun,S., Androphy,E.J., MacPherson,P. and Botchan,M.R. (1990) Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science, 250, 1694–1699. [DOI] [PubMed] [Google Scholar]
- Natesan S. and Gilman,M.Z. (1993) DNA bending and orientation-dependent function of YY1 in the c-fos promoter. Genes Dev., 7, 2497–2509. [DOI] [PubMed] [Google Scholar]
- Nelson H.C.M., Finch,J.T., Bonaventura,F.L. and Klug,A. (1987) The structure of an oligo(dT) tract and its biological implications. Nature, 330, 221–226. [DOI] [PubMed] [Google Scholar]
- Perez-Martin J. and de Lorenzo,V. (1997) Clues and consequences of DNA bending in transcription. Annu. Rev. Microbiol., 51, 593–628. [DOI] [PubMed] [Google Scholar]
- Piper D.E., Batchelor,A.H., Chang,C.-P., Cleary,M.L. and Wolberger,C. (1999) Structure of a HoxB1–Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell, 96, 587–597. [DOI] [PubMed] [Google Scholar]
- Rhodes D. (1979) Nucleosome cores reconstituted from poly(dA–dT) and the octamer of histones. Nucleic Acids Res., 6, 1805–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C.M. and Stenlund,A. (1998) Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalysed by a transcription factor. EMBO J., 17, 7044–7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M.A., Choi,K.Y., Zalkian,H. and Brennan,R.G. (1994) Crystal structure of a Lac I member, Pur R, bound to DNA: minor groove binding by α helices. Science, 266, 763–770. [DOI] [PubMed] [Google Scholar]
- Sedman J. and Stenlund,A. (1995) Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J., 14, 6218–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedman T., Sedman,J. and Stenlund,A. (1997) Binding of the E1 and E2 proteins to the origin of replication of bovine papillomavirus. J. Virol., 71, 2887–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y.S., Muller,F., Lusky,M. and Hurwitz,J. (1993) Bovine papilloma virus (BPV)-encoded E1 protein contains multiple activities required for BPV DNA replication. Proc. Natl Acad. Sci. USA, 90, 702–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S. and Richmond,T.J. (1998) Crystal structure of the yeast MATα2/MCM1/DNA ternary complex. Nature, 391, 660–666. [DOI] [PubMed] [Google Scholar]
- Thanos D. and Maniatis,T. (1995) Virus induction of human IFN β gene expression requires the assembly of an enhanceosome. Cell, 83, 1091–1100. [DOI] [PubMed] [Google Scholar]
- Thompson J.F. and Landy,A. (1988) Empirical estimation of protein-induced DNA bending angles: applications to λ site-specific recombination complexes. Nucleic Acids Res., 16, 9687–9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustav E., Ustav,M., Szymanski,P. and Stenlund,A. (1993) The bovine papillomavirus origin of replication requires a binding site for the E2 transcriptional activator. Proc. Natl Acad. Sci. USA, 90, 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustav M. and Stenlund,A. (1991) Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J., 10, 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.C. and Giaever,G.N. (1988) Action at a distance along a DNA. Science, 240, 300–304. [DOI] [PubMed] [Google Scholar]
- Werner M.H., Clore,M., Fisher,C.L., Fisher,R.J., Trinh,L., Shiloach,J. and Gronenborn,A.M. (1995) The solution structure of the human ETS1–DNA complex reveals a novel mode of binding and true side chain intercalation. Cell, 83, 761–771. [DOI] [PubMed] [Google Scholar]
- Werner M.H. and Burley,S.K. (1997) Architectural transcription factors: proteins that remodel DNA. Cell, 88, 733–736. [DOI] [PubMed] [Google Scholar]
- Werner M.H., Gronenborn,A.M. and Clore,G.M. (1996) Intercalation, DNA kinking and the control of transcription. Science, 271, 778–784. [DOI] [PubMed] [Google Scholar]
- Wilson D.S. and Desplan,C. (1995) Homeodomain proteins. Cooperating to be different. Curr. Biol., 5, 32–34. [DOI] [PubMed] [Google Scholar]
- Wolberger C. (1996) Homeodomain interactions. Curr. Opin. Struct. Biol., 6, 62–68. [DOI] [PubMed] [Google Scholar]
- Yoon C., Privé,G.G., Goodsell,D.S. and Dickerson,R.E. (1988) Structure of an alternating B-DNA helix and its relationhip to A-tract DNA. Proc. Natl Acad. Sci. USA, 85, 6332–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkel S.S. and Crothers,D.M. (1987) DNA bend direction by phase sensitive detection. Nature, 328, 178–181. [DOI] [PubMed] [Google Scholar]