

Abstract
Diverse guanine nucleotide exchange factors (GEFs) regulate the activity of GTP binding proteins. One of the most complicated pairs is eukaryotic initiation factor 2B (eIF2B) and eIF2, which function during protein synthesis initiation in eukaryotes. We have mutated conserved surface residues within the eIF2B GEF domain, located at the eIF2Bɛ C terminus. Extensive genetic and biochemical characterization established how these residues contribute to GEF activity. We find that the universally conserved residue E569 is critical for activity and that even a conservative E569D substitution is lethal in vivo. Several mutations within residues close to E569 have no discernible effect on growth or GCN4 expression, but an alanine substitution at the adjacent L568 is cold sensitive and deregulates GCN4 activity at 15°C. The mutation of W699, found on a separate surface approximately 40 Å from E569, is also lethal. Binding studies show that W699 is critical for interaction with eIF2β, while L568 and E569 are not. In contrast, all three residues are critical for interaction with eIF2γ. These data show that multiple contacts between eIF2γ and eIF2Bɛ mediate nucleotide exchange.
The cap-dependent pathway for the initiation of translation requires the assembly of eukaryotic initiation factor (eIF) ribosomal subunits and a selected mRNA. Central to the initiation process is the GTP binding protein eIF2, which delivers aminoacylated initiator methionyl tRNA (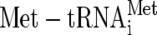 ) to the 40S ribosome as part of a multifactor complex containing eIFs 1, 3, and 5 (5). This 43S preinitiation complex associates with the 5′ end of an mRNA and migrates along it to locate an AUG initiator codon with the aid of other factors. AUG codon recognition, eIF5-promoted hydrolysis of GTP bound to eIF2, and phosphate release stimulate eIF2-GDP and eIF5 dissociation from the initiation complex, probably as an eIF2-GDP/eIF5 complex (33).
) to the 40S ribosome as part of a multifactor complex containing eIFs 1, 3, and 5 (5). This 43S preinitiation complex associates with the 5′ end of an mRNA and migrates along it to locate an AUG initiator codon with the aid of other factors. AUG codon recognition, eIF5-promoted hydrolysis of GTP bound to eIF2, and phosphate release stimulate eIF2-GDP and eIF5 dissociation from the initiation complex, probably as an eIF2-GDP/eIF5 complex (33). 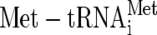 remains bound to the 40S ribosomal subunit at the AUG and is probably stabilized by eIF1A/eIF5B-GTP. 60S ribosomal subunit joining is accelerated by GTP hydrolysis and the release of eIF5B-GDP. At this point, translation elongation can commence (20).
remains bound to the 40S ribosomal subunit at the AUG and is probably stabilized by eIF1A/eIF5B-GTP. 60S ribosomal subunit joining is accelerated by GTP hydrolysis and the release of eIF5B-GDP. At this point, translation elongation can commence (20).
The regeneration of eIF2-GTP from the inactive GDP-bound complex released from the initiation complex is carried out by the guanine nucleotide exchange factor (GEF) eIF2B. eIF2B accelerates the otherwise slow dissociation of GDP from eIF2, allowing its replacement with GTP. This process is regulated by the phosphorylation of eIF2α on the conserved ser51 residue in response to a diverse set of cellular stresses (20). For example, in yeast cells (Saccharomyces cerevisiae), translational control of GCN4 expression proceeds by the following pathway: amino acid starvation activates Gcn2p, which phosphorylates eIF2α and inhibits eIF2B activity. This lowers the rate of nucleotide exchange and slows recruitment of the eIF2-GTP/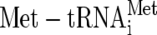 ternary complex (TC) to mRNAs. GCN4 translation is activated by this response, as it contains short upstream open reading frames that normally limit the flow of scanning ribosomes to the GCN4 initiator codon and repress its translation. Starvation for one or more amino acids affects reinitiation within the GCN4 5′ leader such that inhibitory upstream open reading frames are bypassed and GCN4 translation is enhanced by up to 10-fold (23).
ternary complex (TC) to mRNAs. GCN4 translation is activated by this response, as it contains short upstream open reading frames that normally limit the flow of scanning ribosomes to the GCN4 initiator codon and repress its translation. Starvation for one or more amino acids affects reinitiation within the GCN4 5′ leader such that inhibitory upstream open reading frames are bypassed and GCN4 translation is enhanced by up to 10-fold (23).
Inherited eIF2B mutations are responsible for a fatal human disorder known as childhood ataxia with central nervous system hypomyelination or leukoencephalopathy with vanishing white matter (18). This chronic progressive disease results in the aberrant development or destruction of glial cells within the brain. Cellular disease models show that eIF2B mutations lower the nucleotide exchange activity of eIF2B and can elevate cellular stress responses in both nonglial cells (19, 25, 31) and glia (39) and impair the development of astrocytes (14). As would be expected for a general protein synthesis factor defect, mutations that are associated with more severe disease pathologies affect other tissues and result in early mortality (38). Disease-causing mutations have been found in all eIF2B subunits.
eIF2B is a heteropentameric complex, consisting of subunits eIF2Bα to eIF2Bɛ, encoded by five different genes. The eIF2Bα, -β, and -δ subunits form the “regulatory” subcomplex that down-regulates eIF2B activity in response to the phosphorylation of ser51 on eIF2α. The phosphorylated form of eIF2 is more tightly bound by the regulatory subcomplex but prevents the exchange of guanine nucleotide, resulting in a reduction in the availability of eIF2B to regenerate active eIF2 (28). The eIF2Bγ and eIF2Bɛ subunits form the “catalytic” subcomplex that is required for accelerating the rate of guanine nucleotide exchange. eIF2Bγ and eIF2Bɛ share significant sequence homology with each other and possess motifs also present in other protein families, including a large family of nucleotidyl transferases (pfam00483) and a hexapeptide repeat common in acyltransferases (pfam00132) (3). The functional significance of these sequence similarities is not clear. In addition, the C-terminal domain (CTD) of eIF2Bɛ is homologous both to the eIF5-CTD and to eIF4G (pfam02020) (8, 9, 40). It is this last domain that contains residues sufficient for GEF activity of eIF2B, albeit at a reduced rate compared with that of the full five-subunit complex (21). The domain consists of stacked pairs of α-helices and has been called a modified HEAT repeat.
To characterize residues within this catalytic domain (ɛcat) that are essential for nucleotide exchange, we have undertaken a site-directed mutagenesis (SDM) approach, targeting conserved surface residues, followed by extensive genetic and biochemical studies. We find that at least two residues on opposing sides of the domain are critical for the nucleotide exchange function by eIF2B, E569 and W699, as mutations of these residues are lethal in vivo. Nonlethal mutations within residues on the same surface as E569 are cold sensitive for growth and GCN4 activity and salt sensitive for eIF2 interaction. By examining interactions between individual eIF2 subunits, we show that W699 is critical for interaction with eIF2β, while L568 and E569 mutations do not significantly alter this interaction. Instead, mutations to all three residues are impaired for eIF2γ binding. The combined genetic and biochemical data support the ideas that interactions between ɛcat and eIF2γ are extensive and that contacts between ɛcat and both eIF2β and eIF2γ contribute to eIF2B nucleotide exchange function.
MATERIALS AND METHODS
S. cerevisiae strains and genetic methods.
Previously described yeast strains GP3750 (MATa ura3-52::HIS4-lacZ leu2-3-112 ino1 gcn2Δ::hisG gcd6Δ [GCD6 URA3]), GP3889 (MATa ura3-52 leu2-3-112 trp1-Δ63 gcn2Δ pep4::LEU2), GP4115 (MATa ura3-52::HIS4-lacZ leu2-3-112 ino1 gcn2Δ::hisG gcd6Δ [high-copy-number {hc} eIF2 LEU2]) (21), and their derivatives were transformed with plasmids by the lithium acetate method (1). Plasmid shuffling employed 5-fluoroorotic acid (5-FOA) (1). 3-Aminotriazole (3-AT) was added where indicated. An analysis of growth on selective media was achieved by growing liquid cell cultures to an A600 of 0.3, from which fivefold dilutions were made. Three microliters of each dilution was then spotted onto the appropriate medium and incubated at 30°C unless indicated.
Plasmids.
Saccharomyces cerevisiae eIF2Bɛ is encoded by the GCD6 gene. eIF2α, -β, and -γ are encoded by the SUI2, SUI3, and GCD11 genes, respectively. For consistency in the Results and Discussion sections, all proteins/mutants are referred to as eIF2 or eIF2Bɛ derivatives, but here yeast genetic nomenclature is used to specify DNA constructs. pAV1767 [GCD6 CEN LEU2] and several site-directed mutant derivatives (pAV1762 [gcd6-E548A CEN LEU2], pAV1763 [gcd6-R555A], pAV1764 [gcd6-D564A], pAV1768 [gcd6-L570A], pAV1782 [gcd6-E569A], and pAV1783 [gcd6-L568A]) were described previously (9). Other gcd6 plasmids used here include pAV1427 [pGALFH6-GCD6 URA3 2μm] (where FH6 represents tandem FLAG (DYKDDDDK) and polyhistidine (HHHHHH) epitope tags and pGAL represents the galactose-inducible GAL1 promoter), pAV1458 [pGALFH6-gcd6-S576N URA3; 2μm], pAV1513 [GCD6 CEN URA3], pAV1524 [gcd6-F250L CEN URA3], pAV1582 [gcd6-T552I CEN URA3], and pAV1586 [gcd6-S576N CEN URA3] (22). Plasmids overexpressing different combinations of yeast eIF2 subunits from YEp24-derived plasmids (eIF2β, p927 [SUI3 URA3; 2μm]; eIF2γ, p1781 [GCD11 URA3; 2μm]; and eIF2αβγ, p1780 [SUI2 SUI3 GCD11 URA3; 2μm]) have been described previously (13), as were plasmids expressing GCN2, (p722 [GCN2 CEN URA3]) (42) and gcn2-507 (p561 [gcn2-507 CEN URA3]). gcn2-507 is a weakened mutant allele caused by a two-codon (E-L) insertion at residue 1246 within the HisRS domain of GCN2 (41).
Mutant plasmids made for this study by the QuikChange method (Stratagene) were pAV1800 [gcd6-E569D CEN LEU2], pAV1813 [gcd6-E569R], pAV1814 [gcd6-W699A], and pAV1815 [gcd6-E569K], using mutagenesis oligonucleotides (data not shown). The DNA sequence of GCD6 was confirmed after each reaction. Mutants with phenotypes were subcloned into pRS316 (URA3 CEN) to create pAV1781 [gcd6-E569A], pAV1816 [gcd6-E569D], pAV1817 [gcd6-E569R], pAV1818 [gcd6-W699A], and pAV1819 [gcd6-E569K]. To overexpress the minimal “catalytic” domain (ɛcat), residues 518 to 712 were subcloned as PCR fragments using MluI and SpeI cut fragments ligated into a derivative of the pGAL1 expression plasmid pEMBLyex4 containing the FH6 epitope tags, pAV1411 [pGAL1GCN2 2μm URA3 leu2-d], replacing the GCN2 DNA with the indicated ɛcat allele (21). The plasmids generated were pAV1689 [pGAL1FH6-ɛcat 2μm URA3 leu2-d], pAV1833 [pGAL1FH6-ɛcat-L568A 2μm URA3 leu2-d], pAV1834 [pGAL1FH6-ɛcat-E569A 2μm URA3 leu2-d], pAV1835 [pGAL1FH6- ɛcat-E569D 2μm URA3 leu2-d], pAV1836 [pGAL1FH6-ɛcat-E569K 2μm URA3 leu2-d], pAV1837 [pGAL1FH6-ɛcat-E569R 2μm URA3 leu2-d], and pAV1838 [pGAL1FH6-ɛcat-W699A 2μm URA3 leu2-d].
β-Galactosidase assay.
Activity from a HIS4-lacZ fusion integrated at ura3-52 was assessed as described previously (12).
Purification of proteins.
Yeast eIF2 was purified as described previously (7). FLAG affinity purification of eIF2Bɛ proteins and minimal fragments from yeast cells was described in detail recently (27).
Guanine nucleotide exchange assay.
This assay was performed as described previously (7). Reactions were carried out at 0°C.
FH6-ɛcat and eIF2 interaction assays. (i) interaction between prepurified proteins.
Two hundred nanomolar of FH6-ɛcat, FH6-ɛcat-E569A, or FLAG peptide was immobilized on preequilibrated anti-Flag M2 resin (20 μl; Sigma) in binding buffer (100 mM KCl, 20 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 1 μg bovine serum albumin [BSA], 0.1% Triton X-100, 5 mM β-mercaptoethanol, and 1× complete EDTA-free protease inhibitors [Roche]) to a final volume of 100 μl at 4°C for 2 h. After being washed with binding buffer (3× 100 μl), purified eIF2 was added to each reaction mixture at a concentration of 40, 20, 10, or 5 nM and incubated for 2 h at 4°C and washed as before. Finally, the resin was resuspended in Laemmli sample loading buffer and denatured for 5 min at 100°C before 3 μl was loaded onto a 10% polyacrylamide-sodium dodecyl sulfate (SDS) gel. Bound proteins were detected by immunoblotting (using anti-eIF2α rabbit polyclonal or anti-M2-FLAG mouse monoclonal primary antisera [Sigma], followed by horseradish peroxidase-linked secondary antibody and enhanced chemiluminescence [GE Healthcare]).
(ii) Salt dependence of eIF2Bɛ-eIF2 interaction.
Reactions were performed as described above except that the binding and wash buffers contained 10 μg BSA and the indicated concentrations of NaCl in place of KCl. Ten microliters of EZview anti-FLAG resin (Sigma) and 20 nM of purified eIF2 were used.
(iii) ɛcat-eIF2 subunit interaction assays.
A total of 500 μg extract from GP3889 yeast cells overexpressing wild-type FH6-ɛcat or the indicated missense mutant was immobilized on 10 μl anti-M2-FLAG EZview resin (Sigma) in binding buffer BB-0.5 [20 mM HEPES, pH 7.4, 0.5 M CH3COOK, 3 mM (CH3COO)2Mg, 5 mM β-mercaptoethanol, 0.1% Triton X-100, 1× complete EDTA-free protease inhibitor cocktail (Roche), 1 mM phenylmethylsulfonyl fluoride, 0.7 μg/ml pepstatin, 1 μg/ml leupeptin, 5 μg/ml aprotinin] for 2 h at 4°C. Control reaction mixtures included 500 μg BSA and 6 μg of 3× FLAG peptide in place of FH6-ɛcat extracts. Collected resin was washed once in BB-0.5 and twice in BB-0.1 (as in BB-0.5 but with 0.1 M CH3COOK). Each resin pellet containing immobilized FH6-ɛcat was incubated with 250 μg extract from strain GP3889 overexpressing either eIF2β or eIF2γ alone, eIF2αβγ, or a vector control in 100 μl BB-0.1 at 4°C for 2 h. After three washes in BB-0.1 resin, bound complexes were eluted into Laemmli sample loading buffer at 95°C and resolved by electrophoresis on 10% polyacrylamide-SDS gels.
RESULTS
eIF2Bɛ E569A mutant is catalytically inactive in vitro.
Previously we presented genetic evidence that within the eIF2Bɛ coding region, a single amino acid substitution at glutamic acid 569 to alanine (E569A) was lethal in an otherwise wild-type cell when present as the only source of eIF2Bɛ. In contrast, similar alanine substitutions at other highly conserved residues within this region had no obvious deleterious effects (highlighted in the sequence alignment in Fig. 1A). As GCD6, the gene encoding eIF2Bɛ in yeast, is essential, we used a genetic trick to overcome this lethality by cooverexpressing all three subunits of eIF2 and a 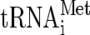 gene (termed hcTC). This strain (GP4115) bypasses the otherwise essential function of eIF2B but remains extremely slow growing. When mutated eIF2BɛE569A was expressed in this strain, we were able to determine that the mutant protein was expressed normally, but as the growth rate of the resulting cells remained unchanged, we concluded that the mutant retained no function in vivo (9).
gene (termed hcTC). This strain (GP4115) bypasses the otherwise essential function of eIF2B but remains extremely slow growing. When mutated eIF2BɛE569A was expressed in this strain, we were able to determine that the mutant protein was expressed normally, but as the growth rate of the resulting cells remained unchanged, we concluded that the mutant retained no function in vivo (9).
FIG. 1.

The catalytic domain of eIF2Bɛ. (A) Multiple sequence alignment of the catalytic domains of eIF2Bɛ performed using Clustal X (37) for the organisms listed. Residues identical in all six sequences are in reverse type, while those identical in four or five sequences are shaded. *, residues targeted for mutagenesis; □, residues found essential; •, T552 and S576 mutated previously (22). Secondary structure helices are indicated as barrels numbered I to VIII, and the extents of the conserved AA box motifs are also indicted. (B) Representations of the ɛcat helical structure (top), indicating the locations of residues studied here with space-filled side chains and a partially transparent surface to show surface exposure (bottom). Images were created with MacPyMOL using the 1PAQ coordinates from the Protein Data Bank.
We hypothesized that the E569A mutant was not functional in vivo, either because it could not interact with eIF2 or because the protein had lost a charged surface residue (Fig. 1B) that was critically important for directly mediating the guanine-nucleotide exchange reaction. As eIF2B is a large, multisubunit, multidomain protein with many eIF2 interaction sites (28), we assumed that any defect might be more readily apparent in a minimal system. We therefore decided to focus our attention on the previously defined minimal eIF2Bɛ domain responsible for stimulating nucleotide exchange in vitro, which extends from residues 518 to 712, here called ɛcat (21). An in vitro guanine nucleotide exchange assay was carried out to assess the rate of release of 3H-GDP from an eIF2-3H-GDP binary complex. As previously reported, purified wild-type, FLAG-tagged ɛcat promoted the release of 3H-GDP, while the mutant protein ɛcat-E569A could not enhance nucleotide release over the intrinsic dissociation rate (Fig. 2A). The ɛcat structure is composed of four pairs of α-helices stacked in HEAT repeats (Fig. 1B) (9). We used circular dichroism (CD) spectroscopy to assess whether the introduced mutation altered the overall secondary structure of the purified protein. The results (see Fig. S1 in the supplemental material) show that the wild-type and mutated CD spectra are indistinguishable and confirm that the α-helical nature has not been altered by the mutation.
FIG. 2.

E569A mutation eliminates catalytic activity but not eIF2 binding. (A) Four micrograms of purified ɛcat, ɛcatE568A, or buffer only were added, and the dissociation of 3H-GDP from preformed eIF2-3H-GDP binary complexes was monitored by liquid scintillation counting. Mean results (n = 5) ± standard deviations are shown. (B) ɛcatE568A binds eIF2 in vitro. Immunoblots of FLAG affinity interaction between 200 mM tagged wild type (lanes 3 to 6) or E569A mutant (lanes 7 to 10) ɛcat or a FLAG peptide control (lanes 2) and the indicated concentration of purified eIF2. Ten percent of the reaction mixture was loaded in each lane. Lanes 1, 11, and 12 contain the indicated input loadings. +, presence of.
To assess whether catalytic inactivity was caused by reduced substrate (eIF2) binding activity, we used protein affinity chromatography to assess the binding of various eIF2 concentrations to excess FLAG-immobilized ɛcat and ɛcat-E569A. After extensive washing, the extent of immobilized ɛcat/eIF2 complexes was assessed by immunoblotting. We found that ɛcat and ɛcat-E569A bound similar substrate amounts over the range of eIF2 concentrations tested (Fig. 2B). Together with previously published experiments, these results are consistent with the idea that the E569A mutation reduces guanine-nucleotide exchange activity to undetectable levels without disrupting the normal secondary structure or the ability to interact with eIF2. We conclude that the mutation has altered a residue that is critical for directly stimulating the guanine nucleotide exchange.
Alanine mutations to residues surrounding E569 do not impair GCN4 activation.
There was a dramatic contrast between the effects of the E569A mutation, which was lethal in vivo and inactive in vitro, and those of the other alanine mutations we introduced into residues surrounding E569 that exhibited no growth phenotype at 30°C in our initial studies (9). This was especially unanticipated because we included the adjacent residues L568 and L570 in this analysis, and the mutation S576N impairs eIF2B function dramatically (22). We decided to examine the nonlethal mutants in more detail in an attempt to reveal any minor phenotypes. As in previous studies, we exploited the fact that down-regulation of eIF2B activity is necessary for the activation of GCN4 translation to assess any minor defect in eIF2B function. As described in the introduction, GCN4 is a sensitive indicator of eIF2B function. Under conditions of histidine starvation induced by 3-AT, gcn2Δ cells expressing wild-type eIF2B are unable to induce GCN4 expression via the activation of Gcn2p and fail to grow (see Fig. S2A in the supplemental material). Mutations in eIF2B that reduce eIF2B activity enhance growth of cells on 3-AT medium as the eIF2B defect lowers TC (for an example, see reference 31). In this assay, none of the mutants tested conferred enhanced growth on 3-AT. Transforming these cells with a plasmid encoding wild-type GCN2 or a mutant kinase with reduced activity (called gcn2-507) produced the expected growth on 3-AT medium for wild-type eIF2B cells and an identical growth pattern for each mutant tested. As the cells contain a β-galactosidase fusion to the Gcn4p-target gene HIS4, the expression levels of HIS4-lacZ were assessed in each strain as a quantitative measure of GCN4 activity (see Fig. S2B in the supplemental material). Consistent with the plating results, no significant differences were observed between strains in these assays; all strains show a GCN2-dependent and 3-AT-dependent increase in HIS4 activity. These results show that none of the mutant strains tested exhibit a phenotype different from that of the wild-type strain at the optimal growth temperature for yeast (30°C). These observations make the results obtained with the E569A mutant even more striking.
Genetic analysis reveals that eIF2Bɛ-L568A has a cold-sensitive phenotype.
In the course of our studies, we transformed yeast strains bearing either the wild type or the eIF2Bɛ-S576N slow-growing mutant with compatible plasmids, each expressing an alanine-substituted eIF2Bɛ mutant. Thus, in these strains there were two sources of eIF2Bɛ, each encoded on a centromeric plasmid. In the presence of wild-type eIF2B, all eIF2Bɛ-alanine substitutions were recessive, but when combined with eIF2Bɛ-S576N, both E569A and L568A conferred a dominant reduction in growth rate (Fig. 3A). We reasoned that in cells expressing both the S576N and the E569A alleles of eIF2Bɛ, each was competing for inclusion within the eIF2B complex and that the inclusion of the nonfunctional ɛE569A reduced the concentration of active eIF2B complexes containing ɛS576N. Because in this strain, eIF2B activity is limiting for growth, any further reduction in eIF2B activity resulted in an enhancement of the growth defect. Surprisingly, we found that ɛL568A also caused a dominant growth phenotype similar to that of ɛE569A (Fig. 3A). This was the first genetic evidence that ɛL568A is a reduced-function allele.
FIG. 3.

E568A and other catalytic center residues are cold sensitive. (A) Growth of serially diluted yeast strains (derived from GP3750 [gcd6Δ gcn2Δ HIS4-lacZ]), bearing two different eIF2Bɛ alleles on separate plasmids on minimal synthetic dextrose medium. Plates were grown for 3 days at 30°C. (B) β-Galactosidase (β-Gal) reporter assay of HIS4-lacZ activity after growth at 15°C. Error bars show standard deviations (n = 6; P < 0.01). (C) Growth of serially diluted yeast strains (derived from GP3750) bearing a single eIF2Bɛ plasmid at the indicated incubation temperatures. The growth score represents the rate of growth at each temperature, where “5” represents wild-type (WT) growth and “0” is inviable. (D) Immunoblotting of eIF2Bɛ protein levels for the strains used in panel C after growth at 15°C.
In our hunt for further phenotypes, we screened strains with single eIF2B mutations at low (15°C) and high (36°C) growth temperatures and found that ɛL568A conferred a reduced growth rate at 15°C, while all other alleles tested behaved like the wild type (data not shown). β-Galactosidase assays of the HIS4-lacZ reporter using extracts from cells grown at 15°C revealed a twofold increase in lacZ activity in L568A mutant cells (Fig. 3B), consistent with the growth defect. These data show that L568 is an important residue involved in the nucleotide exchange reaction, especially during low-temperature growth.
Cold sensitivity is specific to mutations within the catalytic domain.
In light of the cold sensitivity of the L568A allele, the growth at low temperature of other previously described, reduced-function eIF2Bɛ missense mutants was assessed. These were F250L, T552I, and S576N, of which F250L is outside the catalytic domain. Serial dilutions taken from liquid cell cultures growing in exponential phase were spotted onto yeast extract-peptone-dextrose (YPD) medium and grown at either 30°C or 15°C (Fig. 3C). At 30°C, T552I has a moderate slow-growth phenotype, while F250L and S576N are more severe. All strains grew slower at 15°C, but we noted that relative to the wild-type strain, those bearing mutations within the catalytic domain grew slower at 15°C, with the strain carrying S576N having the most extreme phenotype observed. However, the F250L mutant was relatively unaffected by low temperature. These similar phenotypes suggested that these mutations in ɛcat helices I and II all affected eIF2B activity via a similar mechanism. Immunoblotting extracts from cells grown at 15°C confirmed that the phenotypes were not caused by a reduction in protein expression or stability of the cold-sensitive alleles (Fig. 3D).
The interaction between ɛS576N and eIF2 is salt sensitive.
It has been suggested by several authors that cold-sensitive phenotypes are caused by protein-protein interaction defects (29). This would indicate that either an eIF2B subunit interaction defect or an eIF2B/eIF2 interaction defect is likely. In a previous work, neither was found (22). To assess the eIF2/eIF2B interaction more rigorously, we examined the salt sensitivity of binding. eIF2 and eIF2B copurify from cells in 100 mM salt-containing buffers but are dissociated in buffers containing 500 mM salt (11). To assess whether the mutants may have a defect in eIF2/eIF2Bɛ complex formation, we examined the interaction of ɛS576N and eIF2 and compared it with that of wild-type ɛ and eIF2 in buffers containing increasing NaCl concentrations. We chose this mutant as it appeared to be the most cold sensitive in vivo. Differences in the extent of eIF2/eIF2Bɛ complex formation were detected (Fig. 4). A sharp reduction of eIF2 binding to ɛS576N was observed at 150 mM NaCl (a loss of ∼66% compared with the ∼25% loss from wild-type ɛ at this concentration). This result provided evidence that the cold-sensitive eIF2Bɛ alleles may be partially defective in eIF2 binding.
FIG. 4.

The interaction between eIF2Bɛ and eIF2 is salt sensitive. (A) Immunoblots from interaction assays similar to that shown in Fig. 2, except that a single eIF2 concentration (20 mM) was used and incubation and wash buffers contained NaCl concentrations starting at 100 mM (lanes 1 and 6) and increasing by 50 mM per lane to 300 mM (lanes 5 and 10). Lanes 11 and 12 contain 20 nM of each purified input protein indicated (10% of that used in the reaction mixture). Ten and 25% of the total reaction mixture were loaded on blots for the top and bottom panels, respectively. (B) Plot of densitometry readings of eIF2 that were detected, calculated using NIH Image gel plotting densitometry software, and presented as percentages of eIF2 bound by wild-type eIF2Bɛ in 100 mM NaCl buffer. Open bars show results for the wild type; closed bars show results for the S576N mutant. Error bars show standard deviations (n = 3). Asterisks indicate pairs that are significantly different (P ≤ 0.01).
Substitution of other amino acids at position E569.
As described above, E569 was the only residue that when mutated caused the most severe phenotype in that E569A eliminated eIF2B nucleotide exchange function, a result observed previously with only alleles lacking the catalytic domain entirely (21). To examine the role of the charged side chain of E569 more closely, we used SDM to introduce either a conservative Asp or a charge reversal Arg or Lys residue in place of the Glu residue within the full eIF2Bɛ coding region. When introduced into eIF2BɛΔ yeast (strain GP3750) on a centromeric plasmid and subjected to plasmid shuffling on medium containing 5-fluoroorotic acid (5-FOA), all mutations, including the conservative E569D, were found to be lethal (Fig. 5A). When introduced into the slow-growing eIF2BɛΔ hcTC strain (GP4115) as the sole source of eIF2Bɛ, only the conservative E569D allele indicated any residual function, as it partially rescued the doubling time of the strain from 6 to 5 h (Fig. 5B). This made E569D identical in growth behavior to the previously described lethal allele N249K (Fig. 5B). Although lethal, purified five-subunit eIF2B containing N249K did retain some nucleotide exchange activity in vitro that was consistent with these results (22). Immunoblotting confirmed that none of the mutations introduced affected the expression of the resulting protein (Fig. 5C). As E569D is a very modest change to the protein, shortening the side chain of this residue by a single -CH2- group, we were surprised by this dramatic reduction in function.
FIG. 5.

Genetics of E569 and W699 mutants. (A) Growth of transformants of strain GP3750 on minimal (synthetic dextrose [SD]) and 5-FOA-containing media. (B) Growth of transformants of strain GP4115 (gcd6Δ gcn2Δ hcTC) grown on synthetic complete medium (SC) and doubling times (Td) in liquid SC medium. Error bars show standard deviations (n = 3). Asterisks indicate growth rates statistically different from that of the wild type. *, P < 0.05; **, P < 0.01. (C) Immunoblotting of eIF2Bɛ protein levels for strains used in panel B.
W699 is a third critical residue in vivo.
When examining interactions between eIF2Bɛ and its substrate, we have used intact eIF2 heterotrimer as a source of eIF2. Hinnebusch and colleagues found that multiple alanine substitutions of conserved residues within the AA boxes at the C terminus of eIF2Bɛ disrupted its interaction with repeated lysine clusters within the eIF2β subunit (6). The subsequent ɛcat crystal structure indicated that many of the AA box residues mutated in that study are important for the structural integrity of the domain but that the conserved tryptophan residue at 699 is exposed on the surface (Fig. 1B) and may therefore be important in mediating protein-protein interactions (9). We therefore decided to investigate the role of W699 and used SDM to change this to alanine. In vivo W699A behaved identically to E569A in that it was unable to support the growth of the eIF2BɛΔ strain (Fig. 5A), could not rescue growth of the eIF2BɛΔ/hcTC strain (Fig. 5B), and did not destabilize the protein (Fig. 5C). This analysis therefore identified a second residue essential for eIF2B function in vivo, situated on an opposite surface of the ɛcat structure ∼40 Å from E569 (Fig. 1B).
Effects of L568, E569, and W699 mutations on interactions between ɛcat and eIF2 subunits.
As stated above, previous work has indicated a critical interaction between eIF2β and eIF2Bɛ. More recently, Alone and Dever showed that both eIF5 and eIF2Bɛ could interact directly with the nucleotide binding G-domain of eIF2γ (2). They further showed that the ɛcat domain was sufficient for the interaction. This provided the first evidence that eIF2B might function by direct interaction with eIF2γ rather than via a more indirect mechanism of action.
To examine the interaction of ɛcat and mutated versions with eIF2 subunits, we developed an assay to measure the interaction between wild-type and mutant forms of ɛcat and eIF2β and eIF2γ. Because eIF2α interacts with the eIF2Bαβδ subcomplex (as opposed to ɛcat), direct binding between ɛcat and eIF2α was not assessed. FLAG-tagged versions of ɛcat were purified to homogeneity. As these mutants lack catalytic function, we assessed the CD spectrum of each and found that all mutants retained an α-helical structure identical to that of the wild-type protein (see Fig. S1 in the supplemental material), indicating that each mutation does not adversely affect the protein's overall helical structure. We developed an interaction assay whereby ɛcat was immobilized on anti-FLAG affinity resin and then incubated with a yeast extract (GP3889) overexpressing different eIF2 subunits (eIF2αβγ together, eIF2β alone, or eIF2γ alone) or bearing an empty plasmid vector control (Fig. 6A). After extensive washing, eIF2 subunits remaining bound to the resin were assessed by SDS-polyacrylamide gel electrophoresis and immunoblotting. Control reactions where FLAG peptide was immobilized showed that the eIF2 subunit interactions were specific (Fig. 6C to F, lanes 1). In the absence of artificial overexpression, both wild-type ɛcat and E569A alleles bound significant endogenous eIF2 (Fig. 6C, lanes 2 and 3), while the other mutations introduced at E569, L568A, and W699A all significantly reduced eIF2 binding. The overexpression of all eIF2 subunits (∼10-fold) (Fig. 6B and D) enhanced binding between the eIF2 subunits and wild-type ɛcat or all ɛ mutants except W699A. Similarly, excess eIF2β alone enhanced binding between eIF2β and all forms of ɛcat assessed. But in this experiment, W699A was the only mutant tested with significantly reduced eIF2β binding, suggesting that W699 is important for the eIF2β/ɛcat interaction (Fig. 6B and E). In contrast, excess eIF2γ enhanced binding to the wild type and E569A but had only a minor influence on interactions between eIF2γ and the other E569 alleles, W699A and L568A (Fig. 6B and F). Taken together, these data show that the impaired binding of the L568A and E569 alleles to eIF2 (Fig. 6C, lanes 5 to 8) is primarily due to the disruption of the eIF2γ interface, while the W699A allele disrupts both eIF2β and eIF2γ interactions (Fig. 6E and F, lanes 4). These results reveal that both eIF2β and eIF2γ interfaces with eIF2Bɛcat are critical for nucleotide exchange.
FIG. 6.

Interaction of ɛcat alleles with eIF2αβγ, eIF2β, and eIF2γ. (A) Immunoblots of cell extracts overexpressing the indicated eIF2 subunits probed with indicated sera; 20 μg cell protein was loaded per lane. (B) Quantification table for binding results shown in panels C to F, determined by using Quantity One software (Bio-Rad). Relative ratios of eIF2 subunit/ɛcat immunoblot signals are shown, with values for wild-type combinations normalized to 1 for each panel. (C to F) Blots of pellet fractions for interaction of indicated FLAG-immobilized ɛcat protein (lanes 2 to 8) or FLAG peptide control (lane 1) with eIF2β and eIF2γ. Fifty percent of each reaction mixture was probed with αFLAG, and 50% was probed with both αeIF2β and αeIF2γ rabbit polyclonal antisera. Cell extracts without overexpression of proteins (C) and extracts overexpressing all three eIF2 subunits (D), eIF2β alone (E), or eIF2γ alone (F) were used.
DISCUSSION
To uncover how eIF2B functions as a GEF, we used a yeast genetics approach to identify and characterize the importance of conserved residues within the eIF2Bɛ catalytic domain and followed this analysis with biochemical studies to uncover eIF2 interaction defects. We identified three evolutionarily conserved surface residues that are critical for the nucleotide exchange function, L568, E569, and W699 (Fig. 1). W699 is located at the extreme C terminus within helix VIII, while L568 and E569 are within helix II at the N terminus of the domain ∼40 Å away. Residues were targeted for analysis based on both sequence conservation and structural information. We focused on charged and hydrophobic residues, as these are known to be important in other known structurally diverse GEFs (10, 30, 35) Importantly, alanine substitutions at five conserved residues within helices I and II failed to exhibit any growth or GCN4 phenotype (see Fig. S2 in the supplemental material). These negative results underscore the importance of L568, E569, and W699 in the nucleotide exchange reaction.
Evidence that E569 is the principal catalytic residue.
Our initial genetic analysis suggested that E569 was critical for nucleotide exchange in vivo. An alanine substitution at this residue within the full-length protein failed to support the growth of yeast (9). Here we extended our studies and found that the purified ɛcat domain (residues 518 to 712) containing the E569A substitution was inactive in our exchange assay but did not significantly alter the interaction with the eIF2 heterotrimer (Fig. 2 and 6). Acidic residues have been found important in other GEFs, including EF-Ts (asp80) (35), Sec7 (glu156) (30), and RopGEF8 (glu338) (36). Interestingly, a charge reversal mutation in ARNO (E156K) resulted in a stabilized inactive Arf-GDP/Sec7 complex (30). Although all GEFs are structurally diverse and ɛcat shares no homology with ARNO, we hypothesized that an equivalent charge reversal in eIF2B might also stabilize an eIF2/eIF2B complex. So we introduced changes at E569, including the conservative Asp and the charge reversal Lys or Arg residues. All three additional substitutions at E569 were also lethal. In our most sensitive in vivo assay for eIF2B function, cells lacking eIF2B activity can be kept alive by cooverexpressing eIF2 and TC (Fig. 5B) (17, 21). These cells are extremely slow growing, but reintroducing functional eIF2B alleles on plasmids rescues growth. In this assay, only the conservative E569D allele modestly improved growth, consistent with the notion that it retained some function. We were surprised by the severity of this phenotype, which provides additional strong evidence for the importance of E569.
To examine any defects in eIF2 binding, we developed an assay that allowed us to monitor interactions with both eIF2β and eIF2γ. Binding between eIF2β and eIF2Bɛ is mediated by three runs of lysine residues (K boxes) in the eIF2β N terminus and the AA box regions of eIF2Bɛ (6). This interface is also critical for interaction between eIF2 and eIF5. In addition, it was recently shown that the ɛcat domain interacted directly with a glutathione S-transferase-eIF2γ G domain fusion in vitro, while glutathione S-transferase alone did not (2). In contrast to the effects of charge reversals on the Arf-GDP/Sec7 complex (30), we found that E569D/K/R substitutions all destabilized the interaction with eIF2γ (Fig. 6F) but retained the ability to interact with eIF2β (Fig. 6E). It is likely that the eIF2γ-interaction defect was important for the overall reduced affinity for the eIF2 heterotrimer observed with these mutants (Fig. 6C and D). In contrast, the neutral E569A mutation retained the ability to interact with eIF2 (Fig. 2 and 6). Thus, the E569A mutant retains interaction but lacks any activity, while the E569D mutant interaction with eIF2 is weakened but retains some minimal GEF activity in vivo. In silico modeling of the effects of the E569D mutation on the ɛcat structure suggests that the center of the negative charge at residue 569 moves so that it is ∼3 to 4 Å further from the adjacent charged surface residues E548 and R555 and instead ∼2 to 4 Å closer to H561, T565, L568, and L573 side chains. This may therefore form altered interactions that destabilize any contributions to overall eIF2γ binding made by these surface residues. Overall, these data are consistent with the idea that the E569/eIF2γ interaction is essential for the nucleotide exchange reaction.
Mutations altering residues surrounding E569 are cold sensitive.
We also found that the eIF2Bɛ mutation L568A, T552I, or S576N conferred cold sensitive growth at 15°C (Fig. 3C). The L568A allele was defective at only a low temperature and increased GCN4-dependent HIS3 expression under non-amino acid starvation conditions at a low temperature (Fig. 3B) but not at the normal growth temperature (data not shown). This result suggested either that yeast cells require higher eIF2B activity at this low growth temperature or that the low temperature exacerbates the mutant protein activity in some manner. As growth of the F250L allele was not exacerbated relative to that of the wild type, the latter idea is more likely. It has been noted that cold-sensitive mutations are indicative of protein-protein interaction defects (29). We examined this possibility for both S576N and L568A alleles. The interaction of S576N with eIF2 was more sensitive to elevated salt conditions (Fig. 4), suggesting that ionic interactions are disrupted more easily in this mutant. The eIF2-ɛcat-L568A interaction was also weakened. The interaction defects observed with this mutant were similar to that with eIF2-ɛcat-E569D described above, indicating that the eIF2γ/ɛcat interaction was most disrupted. We speculate that because our protein-protein interaction assays were performed at 4°C and the mutations are cold sensitive, the interaction defect observed may have been enhanced by the assay conditions. These observations further support the idea that ɛcat helices I and II make contacts with eIF2γ that are critical for nucleotide exchange.
W699A disrupts multiple interactions between eIF2 and ɛcat.
Our data show that W699 is critical for interaction with both eIF2β and eIF2γ. W699 is highly conserved among both eIF2Bɛ and eIF5 sequences and part of the AA box 2 motif. The mutation of seven conserved residues within this AA box (termed the gcd6-7A mutation, which included W699A) was shown to weaken the interaction between eIF2β and eIF2B. Surprisingly perhaps, the multiple changes in the 7A allele did not result in a lethal phenotype but did lower eIF2B activity in vivo as indicated by the Gcn2p-independent activation of GCN4 (6). The eIF2Bɛ/eIF2β interaction was shown to be dependent upon conserved repeated lysine residues within the eIF2β N terminus (termed K boxes 1 to 3). The equivalent eIF5-7A mutation had similar effects on function and eIF5/eIF2β interaction, leading to the idea that GEF and GTPase-activating protein share a common interaction site on eIF2β (6). The crystal structures of both ɛcat and eIF5-CTD confirm that they share a highly similar helical HEAT repeat structure and that the ɛW699 (W391 in yeast eIF5) side chain is exposed on the surface at one end of the protein, predicting that it would be important for eIF2 interaction (8, 9, 40) (Fig. 1B). Our finding that a single W699A mutation completely inactivated eIF2B in vivo (Fig. 5) confirmed its importance but made extensive genetic analysis challenging. As W699A failed to enhance growth in the strain kept alive by overexpression of TC (GP4115) (Fig. 5B), this result provides strong genetic evidence that eIF2B function is eliminated by this mutation. Our binding studies show that the mutation within the context of the ɛcat domain dramatically impairs binding to eIF2αβγ complexes as well as to either eIF2β or eIF2γ alone (Fig. 6). W699A was the only mutation tested that reduced eIF2β binding (Fig. 6E), demonstrating that this surface region is critical for eIF2β interaction, while the helix I/II surface is not.
W699A also dramatically impaired interaction with eIF2γ (Fig. 6F). This unexpected result indicates that this mutation disrupts ɛcat interaction with both eIF2β and eIF2γ interfaces. As W699 is located approximately 40 Å from E569 at the opposite end of the structure (Fig. 7), this result suggests that ɛcat makes extensive interactions with eIF2γ. One explanation for W699 being critical for both eIF2β and eIF2γ binding is an analogy with eIF5-CTD, where adjacent surface regions interact with eIF2β and eIF3 (43). As W699 is located at one end of the domain, perhaps this residue is important for the initial contact with both subunits.
FIG. 7.

Model for direct interaction between ɛcat and eIF2 for nucleotide exchange. The eIF2 heterotrimer is shown as a series of ovals. eIF2γ has three domains with γIbinding GDP (white lines) and Mg2+ (sphere) and containing the key regions P-loop, Sw1, and Sw2 (black lines), which are remodeled in other G proteins to effect nucleotide and Mg2+ binding/release. The relative positions of these elements and that of the eIF2β subunit are based upon the aIF2βγ-GDP-Mg2+ structure from the archaebacterium Pyrococcus furiosus (34). The crosshatched “KKK” region of eIF2β represents the conserved lysine boxes found only in eukaryotic eIF2β, whose relative positions and structures are unknown. The position of eIF2α is inferred from mutational analysis (32) and the aIF2αγ heterodimer structure from Sulfolobus solfataricus (44). A ribbon image of ɛcat is shown as in Fig. 1, with solid arrows to indicate contacts that mediate GDP and Mg2+ release.
Implications for exchange mechanism.
Together, our data show the importance of two regions of ɛcat for interaction with eIF2β and γ and catalysis of nucleotide exchange. Mutations within ɛcat helix II disrupt interaction with eIF2γ, thus clearly establishing, for the first time, a direct interaction between the GDP binding subunit of eIF2 and residues of eIF2B that are critical for nucleotide exchange. The E569A mutant is effectively catalytically inactive in vitro and in vivo but did not disrupt eIF2β or -γ or -αβγ binding (Fig. 2 and 6), providing strong evidence that E569 is critically important for nucleotide exchange.
Costructures between G proteins and their GEFs exist for small G proteins and translation elongation factors, and the structural rearrangements in the G protein have been reviewed previously by others (10, 35). For these GEFs, binding promotes rearrangement of the phosphate binding P-loop (GXXXXGKS/T) and the switch regions (Sw1 and Sw2) that are important for magnesium ion and phosphate binding. The rearrangements in the P-loop region particularly reduce the Mg2+-GDP affinity that facilitates nucleotide release. A glutamate residue has been identified as critical for nucleotide exchange in several GEFs, including the Sec7 domain of ARNO. ARNO Glu156 interacts with and distorts the P-loop of the G protein ARF by binding Lys30 within the P-loop. ARNO also makes contacts with and stabilizes the conformation of Sw2 (30).
In contrast to the small G proteins, the translation elongation factor G proteins EF-Tu and eEF1A possess three domains, a G domain and two additional domains (domains 2 and 3) that play roles in GEF binding. EF-Ts makes extensive contacts with both the G domain and domain 3 of EF-Tu (26), while eEF1Bα interacts with both the G domain and domain 2 of eEF1A (4). eIF2γ is highly homologous to both elongation factors, similarly possessing domains 2 and 3, but is further complicated by additional eIF2 subunits (eIF2α and eIF2β). A recently determined structure for an archaeal translation initiation factor 2βγ (aIF2βγ) complex reveals that aIF2β sits astride the aIF2γ G domain over the zinc binding region and extends to the Sw1 region (34). Similarly X-ray structure and mutagenesis studies show that eIF2α interacts with eIF2γ domain 2 (32) (44). Using these data and that for other GEF/G protein interactions, we propose a model to explain our observations on the importance of E569 and W699 in ɛcat (Fig. 7). We suggest that ɛcat binds with W699 across an eIF2β/γ interface and that E569, in conjunction with other residues, remodels the P-loop and the Sw1/Sw2 regions to facilitate nucleotide exchange. However, it is possible that eIF2B instead reduces the affinity of eIF2γ for the GDP guanine base. Prior genetic and biochemical analyses of eIF2γ alleles suggest this because mutations altering single Sw1 or P-loop residues were found to impair  interactions rather than nucleotide binding. Instead, alteration of the guanine ring binding residue K250 within the conserved NKXD loop enhanced GDP/GTP release and mimicked eIF2B function (15, 16, 24). The genetic tools generated here should facilitate further molecular studies to address these alternative ideas for the mechanism of eIF2B-mediated nucleotide exchange.
interactions rather than nucleotide binding. Instead, alteration of the guanine ring binding residue K250 within the conserved NKXD loop enhanced GDP/GTP release and mimicked eIF2B function (15, 16, 24). The genetic tools generated here should facilitate further molecular studies to address these alternative ideas for the mechanism of eIF2B-mediated nucleotide exchange.
Supplementary Material
Acknowledgments
We thank Thomas Jowitt at the University of Manchester FLS Biomolecular analysis facility for assistance with CD spectra; Thomas Boesen, Gregers Rom Andersen (University of Aarhus, Aarhus, Denmark), Mark Ashe (Manchester, United Kingdom), and other members of the Pavitt team for helpful discussions; and the anonymous reviewers for their suggestions.
This work was supported by grants to G.D.P. from The Wellcome Trust.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
Published ahead of print on 25 May 2007.
REFERENCES
- 1.Adams, A., D. E. Gottschling, C. A. Kaiser, and T. Stearns. 1998. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 2.Alone, P. V., and T. E. Dever. 2006. Direct binding of translation initiation factor eIF2γ-G domain to its GTPase-activating and GDP-GTP exchange factors eIF5 and eIF2B epsilon. J. Biol. Chem. 281:12636-12644. [DOI] [PubMed] [Google Scholar]
- 3.Anantharaman, V., and L. Aravind. 2006. Diversification of catalytic activities and ligand interactions in the protein fold shared by the sugar isomerases, eIF2B, DeoR transcription factors, acyl-CoA transferases and methenyltetrahydrofolate synthetase. J. Mol. Biol. 356:823-842. [DOI] [PubMed] [Google Scholar]
- 4.Andersen, G. R., L. Pedersen, L. Valente, I. Chatterjee, T. G. Kinzy, M. Kjeldgaard, and J. Nyborg. 2000. Structural basis for nucleotide exchange and competition with tRNA in the yeast elongation factor complex eEF1A:eEF1Bα. Mol. Cell 6:1261-1266. [DOI] [PubMed] [Google Scholar]
- 5.Asano, K., J. Clayton, A. Shalev, and A. G. Hinnebusch. 2000. A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev. 14:2534-2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asano, K., T. Krishnamoorthy, L. Phan, G. D. Pavitt, and A. G. Hinnebusch. 1999. Conserved bipartite motifs in yeast eIF5 and eIF2Bɛ, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J. 18:1673-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asano, K., L. Phan, T. Krishnamoorthy, G. D. Pavitt, E. Gomez, E. M. Hannig, J. Nika, T. F. Donahue, H. K. Huang, and A. G. Hinnebusch. 2002. Analysis and reconstitution of translation initiation in vitro. Methods Enzymol. 351:221-247. [DOI] [PubMed] [Google Scholar]
- 8.Bieniossek, C., P. Schutz, M. Bumann, A. Limacher, I. Uson, and U. Baumann. 2006. The crystal structure of the carboxy-terminal domain of human translation initiation factor eIF5. J. Mol. Biol. 360:457-465. [DOI] [PubMed] [Google Scholar]
- 9.Boesen, T., S. S. Mohammad, G. D. Pavitt, and G. R. Andersen. 2004. Structure of the catalytic fragment of translation initiation factor 2B and identification of a critically important catalytic residue. J. Biol. Chem. 279:10584-10592. [DOI] [PubMed] [Google Scholar]
- 10.Cherfils, J., and P. Chardin. 1999. GEFs: structural basis for their activation of small GTP-binding proteins. Trends Biochem. Sci. 24:306-311. [DOI] [PubMed] [Google Scholar]
- 11.Cigan, A. M., M. Foiani, E. M. Hannig, and A. G. Hinnebusch. 1991. Complex formation by positive and negative translational regulators of GCN4. Mol. Cell. Biol. 11:3217-3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dever, T. E. 1997. Using GCN4 as a reporter of eIF2α phosphorylation and translational regulation in yeast. Methods 11:403-417. [DOI] [PubMed] [Google Scholar]
-
13.Dever, T. E., W. Yang, S. Astrom, A. S. Bystrom, and A. G. Hinnebusch. 1995. Modulation of
 , eIF-2, and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2·GTP·Met-tRNAiMet ternary complexes. Mol. Cell. Biol. 15:6351-6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
, eIF-2, and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2·GTP·Met-tRNAiMet ternary complexes. Mol. Cell. Biol. 15:6351-6363. [DOI] [PMC free article] [PubMed] [Google Scholar] - 14.Dietrich, J., M. Lacagnina, D. Gass, E. Richfield, M. Mayer-Proschel, M. Noble, C. Torres, and C. Proschel. 2005. EIF2B5 mutations compromise GFAP+ astrocyte generation in vanishing white matter leukodystrophy. Nat. Med. 11:277-283. [DOI] [PubMed] [Google Scholar]
- 15.Dorris, D. R., F. L. Erickson, and E. M. Hannig. 1995. Mutations in GCD11, the structural gene for eIF-2 gamma in yeast, alter translational regulation of GCN4 and the selection of the start site for protein synthesis. EMBO J. 14:2239-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erickson, F. L., and E. M. Hannig. 1996. Ligand interactions with eukaryotic translation initiation factor 2: role of the γ-subunit. EMBO J. 15:6311-6320. [PMC free article] [PubMed] [Google Scholar]
- 17.Erickson, F. L., J. Nika, S. Rippel, and E. M. Hannig. 2001. Minimum requirements for the function of eukaryotic translation initiation factor 2. Genetics 158:123-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fogli, A., and O. Boespflug-Tanguy. 2006. The large spectrum of eIF2B-related diseases. Biochem. Soc. Trans. 34:22-29. [DOI] [PubMed] [Google Scholar]
- 19.Fogli, A., R. Schiffmann, L. Hugendubler, P. Combes, E. Bertini, D. Rodriguez, S. R. Kimball, and O. Boespflug-Tanguy. 2004. Decreased guanine nucleotide exchange factor activity in eIF2B-mutated patients. Eur. J. Hum. Genet. 12:561-566. [DOI] [PubMed] [Google Scholar]
- 20.Gebauer, F., and M. W. Hentze. 2004. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 5:827-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomez, E., S. S. Mohammad, and G. D. Pavitt. 2002. Characterization of the minimal catalytic domain within eIF2B: the guanine-nucleotide exchange factor for translation initiation. EMBO J. 21:5292-5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomez, E., and G. D. Pavitt. 2000. Identification of domains and residues within the ɛ subunit of eukaryotic translation initiation factor 2B (eIF2Bɛ) required for guanine nucleotide exchange reveals a novel activation function promoted by eIF2B complex formation. Mol. Cell. Biol. 20:3965-3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinnebusch, A. G. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 59:407-450. [DOI] [PubMed] [Google Scholar]
- 24.Huang, H. K., H. Yoon, E. M. Hannig, and T. F. Donahue. 1997. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 11:2396-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kantor, L., H. P. Harding, D. Ron, R. Schiffmann, C. R. Kaneski, S. R. Kimball, and O. Elroy-Stein. 2005. Heightened stress response in primary fibroblasts expressing mutant eIF2B genes from CACH/VWM leukodystrophy patients. Hum. Genet. 118:99-106. [DOI] [PubMed] [Google Scholar]
- 26.Kawashima, T., C. Berthet-Colominas, M. Wulff, S. Cusack, and R. Leberman. 1996. The structure of the Escherichia coli EF-Tu EF-Ts complex at 2.5A resolution. Nature 379:511-518. [DOI] [PubMed] [Google Scholar]
- 27.Mohammad-Qureshi, S. S., R. Haddad, K. S. Palmer, J. P. Richardson, E. Gomez, and G. D. Pavitt. Purification of FLAG-tagged eukaryotic initiation factor 2B complexes, sub-complexes and fragments from Saccharomyces cerevisiae. Methods Enzymol., in press. [DOI] [PubMed]
- 28.Pavitt, G. D. 2005. eIF2B, a mediator of general and gene-specific translational control. Biochem. Soc. Trans. 33:1487-1492. [DOI] [PubMed] [Google Scholar]
- 29.Phizicky, E. M., and S. Fields. 1995. Protein-protein interactions: methods for detection and analysis. Microbiol. Rev. 59:94-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Renault, L., B. Guibert, and J. Cherfils. 2003. Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature 426:525-530. [DOI] [PubMed] [Google Scholar]
- 31.Richardson, J. P., S. S. Mohammad, and G. D. Pavitt. 2004. Mutations causing childhood ataxia with central nervous system hypomyelination reduce eukaryotic initiation factor 2B complex formation and activity. Mol. Cell. Biol. 24:2352-2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roll-Mecak, A., P. Alone, C. Cao, T. E. Dever, and S. K. Burley. 2004. X-ray structure of translation initiation factor eIF2γ: implications for tRNA and eIF2α binding. J. Biol. Chem. 279:10634-10642. [DOI] [PubMed] [Google Scholar]
- 33.Singh, C. R., B. Lee, T. Udagawa, S. S. Mohammad-Qureshi, Y. Yamamoto, G. D. Pavitt, and K. Asano. 2006. An eIF5/eIF2 complex antagonizes guanine nucleotide exchange by eIF2B during translation initiation. EMBO J. 25:4537-4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sokabe, M., M. Yao, N. Sakai, S. Toya, and I. Tanaka. 2006. Structure of archaeal translational initiation factor 2βγ-GDP reveals significant conformational change of the β-subunit and switch 1 region. Proc. Natl. Acad. Sci. USA 103:13016-13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sprang, S. R., and D. E. Coleman. 1998. Invasion of the nucleotide snatchers: structural insights into the mechanism of G protein GEFs. Cell 95:155-158. [DOI] [PubMed] [Google Scholar]
- 36.Thomas, C., I. Fricke, A. Scrima, A. Berken, and A. Wittinghofer. 2007. Structural evidence for a common intermediate in small G protein-GEF reactions. Mol. Cell 25:141-149. [DOI] [PubMed] [Google Scholar]
- 37.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Knaap, M. S., C. G. van Berkel, J. Herms, R. van Coster, M. Baethmann, S. Naidu, E. Boltshauser, M. A. Willemsen, B. Plecko, G. F. Hoffmann, C. G. Proud, G. C. Scheper, and J. C. Pronk. 2003. eIF2B-related disorders: antenatal onset and involvement of multiple organs. Am. J. Hum. Genet. 73:1199-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Kollenburg, B., J. van Dijk, J. Garbern, A. A. Thomas, G. C. Scheper, J. M. Powers, and M. S. van der Knaap. 2006. Glia-specific activation of all pathways of the unfolded protein response in vanishing white matter disease. J. Neuropathol. Exp. Neurol. 65:707-715. [DOI] [PubMed] [Google Scholar]
- 40.Wei, Z., Y. Xue, H. Xu, and W. Gong. 2006. Crystal structure of the C-terminal domain of S. cerevisiae eIF5. J. Mol. Biol. 359:1-9. [DOI] [PubMed] [Google Scholar]
- 41.Wek, R. C., B. M. Jackson, and A. G. Hinnebusch. 1989. Juxtaposition of domains homologous to protein kinases and histidyl-tRNA synthetases in GCN2 protein suggests a mechanism for coupling GCN4 expression to amino acid availability. Proc. Natl. Acad. Sci. USA 86:4579-4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wek, R. C., M. Ramirez, B. M. Jackson, and A. G. Hinnebusch. 1990. Identification of positive-acting domains in GCN2 protein kinase required for translational activation of GCN4 expression. Mol. Cell. Biol. 10:2820-2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamamoto, Y., C. R. Singh, A. Marintchev, N. S. Hall, E. M. Hannig, G. Wagner, and K. Asano. 2005. The eukaryotic initiation factor (eIF) 5 HEAT domain mediates multifactor assembly and scanning with distinct interfaces to eIF1, eIF2, eIF3, and eIF4G. Proc. Natl. Acad. Sci. USA 102:16164-16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yatime, L., Y. Mechulam, S. Blanquet, and E. Schmitt. 2006. Structural switch of the γ subunit in an archaeal aIF2 αγ heterodimer. Structure 14:119-128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.