


Abstract
Correlation of motif occurrences with gene expression intensity is an effective strategy for elucidating transcriptional cis-regulatory logic. Here we demonstrate that this approach can also identify cis-regulatory elements for alternative pre-mRNA splicing. Using data from a human exon microarray, we identified 56 cassette exons that exhibited higher transcript-normalized expression in muscle than in other normal adult tissues. Intron sequences flanking these exons were then analyzed to identify candidate regulatory motifs for muscle-specific alternative splicing. Correlation of motif parameters with gene-normalized exon expression levels was examined using linear regression and linear splines on RNA words and degenerate weight matrices, respectively. Our unbiased analysis uncovered multiple candidate regulatory motifs for muscle-specific splicing, many of which are phylogenetically conserved among vertebrate genomes. The most prominent downstream motifs were binding sites for Fox1- and CELF-related splicing factors, and a branchpoint-like element acuaac; pyrimidine-rich elements resembling PTB-binding sites were most significant in upstream introns. Intriguingly, our systematic study indicates a paucity of novel muscle-specific elements that are dominant in short proximal intronic regions. We propose that Fox and CELF proteins play major roles in enforcing the muscle-specific alternative splicing program, facilitating expression of unique isoforms of cytoskeletal proteins critical to muscle cell function.
INTRODUCTION
Alternative pre-mRNA splicing is a critical mechanism for regulating gene expression in metazoan organisms, and leads to tremendous protein diversity from a relatively small number of genes. A majority of human genes exhibit some form of alternative splicing. In particular, the human genome encodes a complex alternative splicing program that switches alternative exons on and off according to the needs of individual differentiated cell types. Despite intensive study in recent years, the mechanisms regulating the human alternative splicing program are not yet well understood. The complex decision process, involving which subset of exons on the primary RNA transcript (henceforth, pre-mRNA) will get spliced into the mature mRNA isoform, is mediated by a combination of cis-regulatory elements organized across exons and introns (1), quite analogous to the cis-regulation of transcription. Global identification of splicing regulatory elements has been difficult and has been primarily restricted to exonic elements (2–6), while limited computational information is available on intronic elements (7–13). However, availability of splicing microarrays (14–16), which can interrogate expression levels of exons genome-wide under any particular biological condition, has opened up new possibilities. In this work, we demonstrate that one can now apply analogous computational approaches used for dissecting transcriptional regulation (17) to decipher the splicing regulatory elements, with genes replaced by exons and promoters by pre-mRNA regions proximal to the splice sites.
A new set of approaches based on correlation with expression has been particularly successful in identifying cis-regulatory elements governing transcription (18–21). Here, the premise is that gene expression results from integration of multiple signals within the promoter region, as mediated by binding of trans-factors to the cis-elements. This implies that for an active cis-regulatory motif, its parameters [occurrence frequencies and position weight matrix (PWM) scores] must be significantly correlated with the expression levels across genes under any specific biological condition. Multiple studies have eseished that, using this strategy, one can identify the motifs that are functional under the tested condition. Furthermore, expression data from a single test condition and a reference condition are often sufficient for the analysis. In addition, unlike clustering-based approaches, interacting combinations of motifs can be inferred with high confidence (19,22). Finally, a recent study based on linear splines, which model the sigmoidal nature of transcriptional response, shows that such approaches can accurately identify direct targets of trans-factors binding to the active motifs, even when the motifs are very degenerate (22). Target identification in such situations has been quite challenging. Thus, one can delineate the key elements of transcriptional regulatory networks using correlation with expression. This has proven effective in both lower eukaryotes, e.g. yeast (18,19,23), and in mammals (22).
Here we report the first application, to our knowledge, of the correlation with expression approach for identification of cis-elements that regulate alternative splicing by integrating pre-mRNA sequence information with the exon microarray data. Specifically, we focused on tissue-specific splicing, as tissue-specific pre-mRNA regions are largely conserved across species (8,24,25), and thus, phylogenetic conservation can be used to evaluate the predictions. We employed an Affymetrix exon microarray (26) to identify 56 muscle-enriched alternative cassette exons, a number of which are predicted to alter the expression of cytoskeletal related genes. We used both linear regression (18) and linear splines (22) to examine whether cis-elements in introns adjoining these exons correlate with gene-normalized exon expression in muscle. Multiple motifs that demonstrated statistically significant correlation were also found to be conserved in mouse, chicken and frog. In addition, several of these elements have been previously characterized experimentally as regulators of muscle-specific splicing via binding to members of the Fox (27–33), CELF (34) and PTB (35) families of splicing factors. Taken together, our study shows that correlation with expression is indeed effective in deciphering splicing regulatory elements, and provides the most comprehensive picture yet available of muscle-specific alternative splicing program in humans.
MATERIALS AND METHODS
Identification of muscle-enriched alternative exon and control exon datasets
Total RNA from three biological replicates (three separate individuals) of 16 normal adult human tissues was purchased from BioChain (Hayward, CA, USA). Labeled target was generated from ∼200 ng of total RNA and hybridized to a prototype version of the Affymetrix Human Exon Array as described (26). The set of microarrays contain ∼1.4 million probesets designed to interrogate, as comprehensively as possible, more than 1 million exon clusters derived from a variety of input sources including annotated genes, cDNA sequences and exon prediction algorithms. Design information and microarray data is available at the GEO database (http://www.ncbi.nlm.nih.gov/geo/; accession number: GSE5791).
Candidate muscle-enriched probesets were identified using the splicing index approach (26,36,37). Exon-level expression was normalized to the expression level of the parent gene by dividing probeset intensities by the median intensity of probesets from exons supported by RefSeq or Ensembl annotations. Exons that exhibited statistically significant differences in inclusion rate were identified using a student's t-test on the gene-level normalized probeset intensities (NI). NI values from the three biological replicates of heart and skeletal muscle tissues were compared as a group to the replicates of 14 other non-muscle tissues as a second group. The magnitude of inclusion rate change (splicing index) was estimated by calculating a log ratio (base 2) of the median muscle NI and the median non-muscle NI (26,37). After filtering out non-expressed probesets and genes with low expression, probesets with t-test P-values <0.001 and splicing index magnitudes of >0.5 were considered candidates for muscle-enriched exons.
Manual filtering of the initial list was performed to select further for high confidence internal cassette exons, by mapping candidate muscle-enriched probeset to their genomic context using the BLAT tool (38) at the UCSC genome browser (http://genome.ucsc.edu). Probesets that overlapped annotated alternative transcriptional starts, alternative polyadenylation sites, or regions with alternative 5′ or 3′ splice sites, were removed from consideration in this study. Exon-level probeset intensities were additionally observed using BLIS (Biotique Systems, Inc. Reno, NV), an integrated genome browser that enables exon expression data from the microarray to be viewed in genomic context. Only probesets that showed clear patterns of muscle enrichment were kept for further analysis. Probesets had to demonstrate higher intensity levels in the muscle tissues and have exon-level data for surrounding probesets consistent with exon skipping in a majority of non-muscle tissues. Probesets were subsequently mapped to the May 2004 human genome (NCBI Build 35) using BLAT (38). Exact exon boundaries were determined by comparison to EST and mRNA sequences requiring consensus splice sites.
For phylogenetic analysis, the orthologous exons were identified in another mammalian genome (mouse; Mus musculus), in an avian genome (chicken; Gallus gallus) and in an amphibian genome (frog; Xenopus tropicalis) using VISTA alignment tools. Automatic alignment was successful at finding most of the longer alternative exons directly, but in a few cases the alignments were adjusted manually. The upstream 200 nt (U200) intronic region was selected as the base 1 to base 200 adjoining the exon in the upstream direction, while downstream 200 nt (D200) intronic region was selected as the base 1 to base 200 downstream of the exon. Alignments of orthologus introns and exons sequences were generated by LAGAN using default parameters (39).
The ‘tissue-non-specific alternative’ exon dataset was derived as described previously (8) from the European Bioinformatics Institute database of human alternative exons (http://www.ebi.ac.uk/asd/altextron/index.html). ‘Control exon datasets’ were generated from randomly selected chromosomal regions by extraction from RefSeq annotation databases to get exon coordinates. Control groups for the mammalian and chicken genomes were described previously (8). The muscle-enriched datasets and the control datasets is available at: http://vision.lbl.gov/People/ddas/NAR_SPLICE1/
Validation of muscle-enriched expression
A random subset of candidate muscle-enriched exons was selected for validation by RT–PCR, focusing (for ease of amplification) on those ⩽155 nt in length. RNAs from different human tissues, including heart, skeletal muscle and six non-muscle sources, were purchased from Clontech. One microgram of each RNA source was transcribed into cDNA using random hexamer primers in a total volume of 10 μl. Then, 2 μl cDNA was amplified in a volume of 25 μl, using primers located in the flanking constitutive exons (Supplementary Table 2), for 35 cycles under the following conditions: 30 s at 94°C; 30s at 55°C; 45 s at 72°C. The identity of PCR products was confirmed by DNA sequence analysis.
Correlation with expression
Linear correlation
Counts of hexamers were obtained in a specific pre-mRNA sequence region (upstream or downstream proximal intron). For each region, a linear model was fitted between the logarithm of ratios of gene-normalized exon expression levels and count of each 6-mer word w across a set of exons, 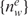 :
:
NIe is the gene-normalized expression level of exon e in muscle, and C refers to a reference sample. The reference data was taken as the average NI across all tissues. The coefficients aw and bw were obtained by a least squares fit. P-values were calculated using an F-test, as described previously (40).
The best fit was obtained for a set of sequences that included the muscle-specific exons (foreground set) and a background set of m sequences (m = 300), drawn randomly from a set of manually curated 957 cassette exons across the human genome (11). Since we started with a prioritized set of tissue-enriched sequences, a background set was necessary to model the correct dependence of log ratios on word count. n such random draws were performed (n = 25), and a linear fit was obtained for each such draw. A geometric mean of the P-values from all iterations reflects the overall significance of the word.
Linear splines
Linear splines differ from lines by introducing a threshold, called knot, below which the function is constant and linear above it (19,22). A significant difficulty in modeling binding sites via PWMs is that they give rise to a continuous distribution of scores across all possible binding sites. Consequently, a cutoff score needs to be determined to discriminate the true sites from false sites. Such cutoff scores are often based on predetermined background sequences and thresholds, and as a result, are complicated by subjective choices (41). In a linear spline model, the cutoff score corresponds to the knot, and thus, is learnt directly from the input data (22). For each PWM μ of width L, each L-mer in the input sequence was assigned a probability score M:
where pi(bi) is the probability of observing the base bi at the position i. Thus, the score M always assumes a value between 0 and 1. It is related to binding affinity (42). PWM scores across exons 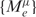 for a given motif μ were fitted to the splicing ratios {log (NIe / NIeC)} using the following model:
for a given motif μ were fitted to the splicing ratios {log (NIe / NIeC)} using the following model:
where θ(x,0) is a linear spline: it is x, when x⩾0, and zero, otherwise. ξμ, termed knot, corresponds to the cutoff score. The coefficients aμ and bμ and the location of the knot ξμ were determined by a least squares fit. This leads to an unbiased and adaptive determination of the knot ξμ for any given PWM. Importantly, in contrast to previous approaches (22), where contribution from only the maximum scoring site was considered, we systematically accounted for contribution from active sites with weaker scores as well. The number of such active sites is adaptively learnt, as displayed in the equation above. Thus, both binding affinity and occurrences of active motifs are accounted for in our approach. The significance of the fit was assessed using an F-test (40). The overall significance of each PWM was enumerated using the same iterative procedure as for the linear regression discussed above.
Over-representation analysis
RNA words
We examined over-representation of candidate oligonucleotide sequences (RNA words) in each tissue-specific dataset, relative to the control datasets, using a hypergeometric distribution. The results were corrected for multiple testing using the false discovery rate (FDR) method (43). The results of this test were generally consistent with the non-parametric approach that we have described previously (7). Furthermore, for each word a contrast score was also calculated as the difference in frequency in the tissue-specific dataset versus the control dataset. Similar results were obtained using two control sets, one composed of predominantly constitutive exons, and the other containing alternative but nontissue-specific exons (8). Like standard motif analyses, repeat elements were not explicitly excluded from this analysis. They are automatically filtered by the correlational analyses. Moreover, only non-overlapping motifs were counted in the word frequency calculations. Manual examination of the sequences revealed no cases of long repeating elements that would influence frequency calculations of the candidate regulatory motifs.
PWMs
Over-represented PWMs were obtained using the DME (Discriminating Matrix Enumerator) algorithm (44,45). DME is an enumerative search algorithm that finds the PWMs over-represented in a foreground set relative to a background set. Both intronic regions (upstream and downstream) were searched for over-represented matrices of width 6 nt, using the background sets as above. Default parameter settings were used, except that we varied the average information content of the PWM from 1.0 to 2.0 in steps of 0.1. Fifteen PWMs were obtained for each such setting. Correlation analysis was performed on non-redundant sets of matrices. Matrix similarity was assessed using MatCompare (46).
RESULTS
Identification and characterization of muscle-enriched alternative exons
The human muscle-enriched exon dataset analyzed in this study (Supplementary Table 1) was derived from exon microarray hybridization data using a platform designed to provide a comprehensive genome-wide analysis of annotated and predicted exons (see Methods section). In order to identify motifs that regulate tissue-specific alternative splicing, it is critical to identify a set of alternative exons having similar expression patterns indicative of regulation by a shared splicing program. Therefore, the group of exons studied here was carefully selected by analysis of exon microarray data from a panel of 16 normal adult human tissues. Probesets that exhibited gene-level NI that were significantly higher in heart and skeletal muscle, relative to 14 other tissues, were first identified. For this part of the analysis, we grouped the heart and skeletal muscle exon expression together to enhance the power of the statistical tests. Then a manual filtering process was performed so as to retain only probesets representing cassette exons, and to eliminate probesets corresponding to alternative first and last exons or to exon regions generated from alternative 5′ and 3′ splice sites. The final dataset consisted of 56 muscle-enriched, internal cassette exons. Most of these exons (∼80%) are integral multiples of 3 nt in length, with a median length of 84 nt, consistent with the notion that alternative exons are smaller than average constitutive exon length [∼145 nt (47,48)]. However, the genes with such alternative exons have a median size of 123 kb, much longer than the average gene length. To explore evolutionary conservation of candidate splicing regulatory elements, we also identified highly conserved orthologs for most of these human muscle-enriched exons in mouse, chicken and frog (Supplementary Table 1). It is important to note that while many of these exons show evidence of alternative splicing in Genbank, most were not previously known to exhibit muscle-enriched splicing and were not identified in the pilot study of muscle-enriched exons by Sugnet et al. (11). Therefore, analysis of this dataset should yield novel insights into the vertebrate muscle alternative splicing program, and should provide an opportunity to explore computationally the regulatory motifs that carry out this program.
Muscle-enriched splicing patterns for a random subset of these exons were validated experimentally in the human dataset by RT-PCR (Figure 1). Although splicing patterns were not absolutely muscle specific, in almost every case the efficiency of exon inclusion was highest in heart and skeletal muscle, confirming the predictions of the exon microarray. Importantly, mRNA and/or EST evidence from the genetic databases (data not shown) demonstrates that the majority of these exons are alternatively spliced in at least one of the other species examined (mouse, chicken or frog), suggesting that the incidence of conserved alternative exons in this specialized dataset is higher than the reported rate for general alternative exons (49). Taken together, these results indicate that the muscle-enriched exons constitute a special class of highly conserved alternative exons.
Figure 1.

Validation of microarray predictions of muscle-enriched alternative exons. RT-PCR confirmation of muscle-enriched alternative exon expression. Amplifications were performed using primers in the flanking constitutive exons. RNA sources used for amplification by lane number: 1, brain; 2, kidney; 3, liver; 4, stomach; 5, bone marrow; 6, testis; 7, heart; 8, skeletal muscle. Arrowheads indicate positions of the alternative exon inclusion products that are most enriched in heart and skeletal muscle.
Intron sequences flanking orthologous alternative exons in the mouse and human genomes tend to be evolutionarily conserved (24), consistent with the observation that cis-regulatory elements for tissue-specific alternative splicing are often located in those proximal intron regions. We used VISTA genome alignment tools to compare the proximal intron sequences in this muscle-enriched dataset and extended the evolutionary comparison to include chicken and frog. In the proximal 200-nt upstream (U200) and downstream (D200) introns, mouse sequences were highly similar to their human orthologs (median identity of 61 and 58%, respectively), while chicken and frog introns were much less homologous. The full quantitative data are shown in Supplementary Table 3 and representative alignments of exons with relatively high conservation (FXR1), or lower conservation restricted mainly to the exon (LRRFIP1), are displayed in Figure 2. The reduced overall homology of chicken and frog introns suggests that conserved motifs in these regions are likely conserved specifically for their function as cis-regulatory elements for muscle-specific alternative splicing, rather than being passively conserved as part of a larger conserved element.
Figure 2.

Conservation of intron sequences flanking muscle-enriched exons. Representative VISTA genome alignments of exon and flanking intron sequences from the mouse, chicken and frog genomes with the prototypical muscle-enriched exon from human. Exon boundaries are indicated by vertical lines. Shaded regions indicate sequences that exceed 75% identity, while curves above baseline indicate regions with >50% identity to the human sequence.
Frequent occurrence of muscle-enriched exons in genes encoding proteins with functions in cytoskeletal organization
Previous studies have demonstrated that the brain-specific alternative splicing factor, NOVA1, modulates the splicing of many components of the neuronal synapse (50). We hypothesized that the muscle alternative splicing program might similarly coordinate the expression of a particular class of genes that share a common pathway or cellular process. Using the method described previously (22,51), to examine the gene ontology (GO) terms associated with each parent gene for the muscle-enriched exons, we found a strong association with cytoskeleton organization and biogenesis, microtubule stabilization and muscle development (Supplementary Table 4). These associations were statistically significant (P < 0.001), suggesting that the muscle alternative splicing program is critical for proper expression of the unique cytoskeleton characteristic of vertebrate muscle.
Correlation with exon expression identifies splicing regulatory elements
Alternative splicing regulatory elements responsible for tissue-specific splicing are often located in proximal intron sequences (25). To search for candidate intronic regulatory motifs for the muscle-specific splicing program, we correlated the frequencies of hexamers in specific intronic regions with the logarithm of ratios of gene-normalized exon expression levels in skeletal muscle, across the 56 muscle-enriched exons in the human dataset. The ratio for any exon was enumerated against its average gene-normalized expression level across all the tissues. Thus, it is similar to the splicing index used above. The cis-elements exhibiting significant correlation with expression were considered potentially functional in regulating muscle-specific splicing. These were further examined for relative over-representation in introns of muscle-enriched exons, compared to a background set of introns flanking constitutive exons, using a hypergeometric distribution based on word counting in the oligonucleotide sequences. Finally, we examined their spatial conservation through vertebrate evolution (8) by testing whether the motif is over-represented in the other species using exactly the same statistical measures as used for humans.
Here we consider ugcaug in the downstream 200 nt (D200) of intron sequence as an example. This hexamer represents the binding site for mammalian Fox-1 and Fox-2 splicing factors (31), which have identical RRM domains. ugcaug has been reported as a common motif in proximal introns adjacent to tissue-specific exons. In a few cases, functional splicing assays have confirmed the importance of this motif in regulation of splicing (27–33). In the large group of muscle-enriched exons studied here, we found a highly significant correlation of ugcaug frequency with muscle expression (P = 6.8E−05). The distribution and the linear fit for a single iteration of correlation analysis (see Methods section) are shown in Figure 3A. Similar analysis shows that muscle expression does not correlate with ugcaug occurrences in the upstream intron, whereas in the downstream intron the magnitude of correlation decreases with distance from the exon as demonstrated by the increasing P-values (Figure 3B). These dependencies are further corroborated by strong over-representation of this motif in proximal downstream introns in human and other vertebrates (Figure 3C). Indeed, almost half of the muscle-enriched exons in all four datasets (23/56 in human, 21/54 in mouse, 20/43 in chicken and 19/36 in frog) possessed at least one ugcaug motif in the first 200 nt of the downstream proximal intron. Together, these results strongly support the hypothesis that ugcaug is potentially an important regulatory element for muscle-specific alternative splicing, as predicted by the correlation with expression analysis.
Figure 3.

Correlation with exon expression for the ugcaug regulatory element. (A) Linear fit between ratios of gene-normalized exon expression levels and counts of ugcaug in 200 nt of downstream intronic sequence across 356 exons (56 muscle-specific exons and 300 randomly selected exons) (P = 4.6E–06). (B) Dependence of correlational P-values of ugcaug count with distance. Filled circles indicate correlation with total motif count, while unfilled boxes indicate correlation with bin-wise count (bin size = 100 nt). E.g. for 300 nt, the filled circle reflects the strength of correlation with the count in 1–300 nt of intron, while the unfilled box reflects the correlation with the count in 201–300 nt of intron. Minus sign indicates upstream intron. (C) Contrast scores of ugcaug in upstream and downstream introns of human, mouse, chicken and frog. (‘E’ in Figure 3B and 3C indicates position of the exon).
Analysis of upstream and downstream intron sequences
We extended the above analysis to identify additional muscle-specific cis-elements in upstream and downstream intron sequences. In the downstream 200 nt sequence, we searched all possible hexamers and identified a total of 35 hexamers that were significantly correlated with expression (P⩽0.05) and also over-represented in the human dataset (P ⩽ 0.05, q⩽0.2); nine of these were also over-represented in at least one other species (Table 1A and Supplementary Table 5A). Several of these elements have been previously characterized experimentally as regulators of muscle-specific splicing. These motifs fell into three distinct classes: (i) the Fox1/2-binding motif ugcaug (P = 6.8E−05) and two closely related hexamers (gcaugg, uuugca); of note, in all four species the majority (58–76%) of GCAUG motifs in the D200 region occurred in the context of the full UGCAUG hexamer. (ii) ug-rich elements gugugu and uguguc (correlation P-values = 0.032 and 0.005, respectively), that resemble binding sites for the CELF family of splicing factors; and (iii) the novel motif acuaac (P = 0.0006) and related hexamers cuaacc (P = 0.004) and cacuaa (P = 0.04). The latter class is similar to the uacuaac element noted in a recent study of a small group of muscle-specific exons in mouse (11). The distribution of these elements in flanking introns of exons in the human, mouse, chicken and frog datasets is shown in Figure 4A. Importantly, this analysis revealed that ugcaug was the most over-represented hexamer in all four datasets, and both gugugu and acuaac were also consistently in the top ∼1% of the most over-represented hexamers in these species.
Table 1.
Significant words identified by linear correlation analysis. Top 10 words in (Panel A) downstream and (Panel B) upstream introns (length = 200 nt).
| Word | Correlation analysis P-value | Over-representation analysis | Contrast score | Phylogenetically conserved? | Putative trans-factors | |
|---|---|---|---|---|---|---|
| P-value | q-value | |||||
| Panel A: D200 | ||||||
| UGCAUG | 6.8E−05 | 1.7E−15 | 6.1E−12 | 0.0024 | Frog, mouse, chicken | Fox-1 |
| ACUAAC | 0.0006 | 2.5E−08 | 2.3E−05 | 0.0008 | Frog, mouse, chicken | * |
| GCAUGG | 0.0006 | 3.6E−05 | 0.004 | 0.0009 | Mouse | Fox-1 |
| CGUGUG | 0.0007 | 0.009 | 0.12 | 0.0005 | CELF | |
| GCAUGA | 0.002 | 0.002 | 0.04 | 0.0006 | Fox-1 | |
| AGCAUG | 0.002 | 0.0007 | 0.02 | 0.0007 | Fox-1 | |
| UAACCC | 0.003 | 9.6E−05 | 0.008 | 0.0006 | ||
| CUAACC | 0.004 | 2.9E−05 | 0.004 | 0.0007 | Frog | * |
| CACCAA | 0.005 | 0.005 | 0.08 | 0.0004 | ||
| UGUGUC | 0.005 | 0.006 | 0.09 | 0.0007 | Chicken | CELF |
| Panel B: U200 | ||||||
| CCCCUU | 0.002 | 9.8E−05 | 0.004 | 0.0009 | ||
| UUUCCA | 0.002 | 0.0006 | 0.02 | 0.0009 | PTB | |
| UCCUCC | 0.002 | 8.3E−05 | 0.004 | 0.0007 | ||
| UCUCCA | 0.002 | 0.0002 | 0.007 | 0.0006 | ||
| AUCUCC | 0.003 | 0.02 | 0.19 | 0.0002 | ||
| CCCCCU | 0.003 | 0.03 | 0.2 | 0.0004 | Frog | PTB |
| UCUUUC | 0.004 | 1.1E−07 | 1.8E−05 | 0.0020 | ||
| CUCCUC | 0.006 | 0.003 | 0.05 | 0.0004 | ||
| UCAUCU | 0.007 | 0.001 | 0.02 | 0.0005 | ||
| AAAUCU | 0.009 | 0.003 | 0.05 | 0.0005 | ||
Asterisk indicates the previously identified, but as yet uncharacterized, novel element acuaac. q-value indicates multiple testing correction using the false discovery rate (FDR) method (43). Phylogenetic conservation was assessed by examining relative over-representation of each word in each species and employing the same P-value cutoffs as in human (P⩽0.05, q⩽0.2). Complete list of significant words is shown in Supplementary Table 5.
Figure 4.

Phylogenetic conservation of regulatory motifs in the proximal intron sequences. Bar graphs show the over-representation of the indicated cis-regulatory motifs in the proximal intron sequences for muscle-enriched exons in four vertebrate species. (A) Enrichment of selected regulatory motifs in 1-kb flanking intron regions. The highest abundance of ugcaug, ugugug and acuaac elements is consistently within the proximal downstream region of ∼200 nt, while cucucu and ucuu elements were enriched in the proximal upstream intron. The representative hnRNP A1-binding site uaggg was not over-represented near muscle-enriched exons. (B) Analysis of the putative CELF-binding site ugc in 0.5-kb flanking introns. ugc is enriched in the D50 region of all four species. Vertical axis, contrast score, i.e. difference in motif frequency between muscle datasets and control datasets of constitutive exons, occurrences/nt × 103; horizontal axis, nt range relative to the alternative exon; E indicates position of the muscle-enriched exon.
For upstream intron sequences (200 nt), we found a total of 27 hexamers that were significantly correlated with expression and also over-represented in human, of which three were over-represented in at least one other species (Table 1B and Supplementary Table 5B). Many such elements are strongly pyrimidine-rich, characteristic of binding sites for PTB protein, an inhibitor of splicing for many alternative exons (35). In all four species, the muscle-enriched datasets showed strong over-representation of the reported PTB-binding sites, cucucu and ucuu, in the proximal upstream intron (Figure 4). cucucu was concentrated mainly in the U200 region. ucuu was focused even more tightly in the U100 region (Figure 4B), where it was consistently among the top five over-represented tetramers in all four species. Lesser over-representation of ucuu motifs over a broad area of downstream intron sequences was also noted, perhaps consistent with previous findings that optimal splicing repression by PTB requires binding sites both upstream and downstream of the regulated exon (52,53).
Many of the remaining significant hexamers, both for upstream and downstream introns, have low similarity to the previously discovered elements. Although these may represent novel elements, given that splicing elements are often degenerate, they can also be specific examples of known degenerate motifs. Our analysis using degenerate motifs presented below suggests that the latter possibility is more likely. Finally, for some of the major splicing regulatory elements described above, we observed that the profiles of positional over-representation have been conserved through vertebrate evolution: mouse, chicken and frog. This is displayed in Figure 4 for Fox, CELF and PTB-binding sites. Such strong positional conservation of motifs lends additional support to our findings using correlation with expression.
The Fox-binding site, ugcaug, was previously shown to be over-represented downstream of brain-enriched alternative exons (7,8), raising the possibility that the brain- and muscle-specific alternative splicing programs might exhibit functional similarities by sharing related components of the splicing machinery. To determine which of the candidate muscle cis-regulatory elements might be shared with brain-specific alternative splicing, and which are unique to the muscle program, we compared the frequency of several key cis-regulatory motifs in muscle (this study) and brain (8) datasets. Two elements, acuaac and ugugug, were clearly muscle specific since their frequencies were consistently higher in the D200–D300 region adjacent to muscle-enriched exons compared with the intronic region downstream of brain-enriched exons (Supplementary Figure 1; positive contrast scores). These motifs were also not over-represented in brain relative to control exons (data not shown). In contrast, the motifs ugcaug and cucucu occurred at even higher frequencies in the proximal introns of the brain-enriched dataset than they did in the muscle-enriched dataset (Supplementary Figure 1; negative contrast scores for ugcaug in the D200–D400 region, and for cucucu in the U100 region). Essentially equivalent distribution patterns were observed in the mouse, chicken and frog datasets (data not shown). These results strongly suggest that tissue-specific alternative splicing programs may utilize a combination of unique and shared cis-regulatory motifs that will require much additional analysis in the future.
Motifs identified via PWM analysis are consistent with word analyses
Because many splicing factors bind degenerate oligonucleotide sequences in RNA, we extended our analyses to include degenerate motifs through the use of PWMs (3,54). PWMs are probabilistic representations of degenerate binding sites. Over-represented PWMs in introns of 56 muscle-specific exons in the human dataset were obtained using the DME algorithm (44,45). We scanned multiple parameter settings of DME in order to obtain a large number of PWMs and reduce bias from DME. To identify the functional PWMs, we assessed their correlation with muscle expression using linear splines (19,22). Linear splines are among the simplest non-linear variants of linear models. In contrast to many other approaches, they facilitate adaptively learning the cutoff scores of PWMs that discriminate true targets from false targets of trans-factors. Previous regression approaches have used either maximum score of the PWM (22) or a global average of PWM scores for all potential binding sites (20) on an input sequence as the predictor variable. However, realistically, a small number of sites, sometimes >1, are bound by the corresponding trans-factor. Here we overcame this limitation by including both strength of PWM and the number of putative binding sites in our linear splines approach (see Methods section).
Degenerate 6-nt and 4-nt sequences that were over-represented in the proximal downstream intron sequence are shown in Table 2A and B and Supplementary Table 6A and B. Notably, all of the top 10 over-expressed PWM hexamer motifs in the D200 region are consistent with the major over-expressed unique motifs identified above. Among these, the six most statistically significant motifs represent close matches to the Fox-binding site, ugcaug; two (nhcuaa and hcuaan) are very similar to the novel acuaac element; and the remaining motifs (sukugs and cugysr) resemble ug-rich-binding site for CELF proteins. Analysis of over-expressed 4-mers in the D50 region revealed that the top-scoring motif is ugcm. While this motif is included in the Fox recognition sequence, other considerations suggest that it would not be sufficient for Fox binding. Instead, ugc likely represents the cug-rich sequences, characteristic of some CELF-binding sites.
Table 2.
Significant position weight matrices (PWMs) identified by correlation analysis using linear splines.
 |
 |
Panel A: Top 10 PWMs of width 6 nt in proximal downstream intron sequences (length = 200 nt).
Panel B: All significant PWMs of width 4 nt in proximal downstream intron sequences (length = 50 nt).
Panel C: Top 10 PWMs of width 6 nt in upstream intron sequences (length = 200 nt). Complete list of significant PWMs in downstream and upstream 200 nt introns is shown in Supplementary Table 6.
In the U200 region, all of the statistically over-represented motifs were quite pyrimidine-rich relative to the control group (Table 1B and 2C). Further investigation will be required to determine whether these elements are primarily bound by the PTB protein or by additional splicing factor(s). All remaining PWMs that have high significance in upstream and downstream introns exhibit at least partial similarity to the above three elements. For PWMs with only partial similarity, the similarity is observed either at the 5′ or at the 3′ end of the motif, indicating that the remainder of the motif most probably represents the flanking region. For example, for the PWM rrwgca, the last four bases match the 5′ end of the ugcaug, and hence, the first two bases are presumably the flanking region of this putative Fox-binding motif.
Furthermore, in contrast to previous work (22), the new formulation of linear splines used here allowed us to obtain not only the potential target exons, but also the binding sites of the above splicing factors (see Methods section). The results for a representative set of motifs are summarized in the Supplementary Table 7. For the putative Fox-binding motif wgcauk, we find ugcaug as the most frequently occurring oligonucleotide sequence, as expected of Fox-binding sites. We have observed similar accuracy in binding site prediction in the context of transcriptional regulation (Das,D., unpublished data). Interestingly, we notice that not all possible combinations of nucleotides of a degenerate PWM are realized in the set of 56 muscle-specific exons. For example, for the candidate CELF-binding motif, cugysr, only cuguga is predicted as the binding site. These are consistent with the previous observations made in the context of transcriptional regulation (55,56).
DISCUSSION
In this study we have demonstrated that the correlation with expression approach, applied to global exon expression profiles, represents a powerful new tool for identification of cis-regulatory motifs for alternative splicing. Using a dataset of high-confidence muscle-enriched alternative exons extracted from human exon microarray data, we correlated motif occurrences in the flanking introns with the splicing index measure of relative muscle enrichment to identify candidate regulatory motifs for the muscle-splicing program. The logic of this strategy is supported by many studies of transcriptional regulation, and a few of splicing regulation (57), showing that functional response often correlates with regulatory motif copy number. The analysis presented here demonstrates that the number of Fox splicing factor binding sites (ugcaug) correlates strongly with the muscle splicing index (Figure 3A), consistent with previous reports that Fox proteins can regulate various tissue-specific alternative splicing events. The validity of correlation results were further supported by over-representation analysis, by comparative genomics showing that top scoring correlation motifs are phylogenetically conserved among vertebrate genomes, and by previous experimental studies implicating most of the same motifs in regulation of muscle-specific exon(s). Since tissue-specific alternative splicing is rarely an all or nothing phenomenon (e.g. Figure 1), correlation with expression may offer an attractive approach toward understanding complex tissue-specific patterns of alternative splicing. This approach may be particularly effective when PWMs are utilized in the splines-based framework to account simultaneously for both relative affinity and number of motif occurrences, providing insight into both the target exons and binding sites associated with a given motif.
Our immediate goal here in this proof of concept study was to examine whether the correlation with expression method can be used to identify splicing regulatory motifs, and consider muscle-specific alternative splicing program as an example of this application. This analysis strongly implicated several classes of known regulatory factors including Fox (ugcaug), CELF (gugugu and ucugug), PTB (cucucu and ucuu) and putative KH-type splicing factor (acuaac) as important mediators of muscle-enriched splicing. The current study thus confirms and substantially extends earlier reports that these factors can regulate one or a few muscle-enriched exons by providing significant new computational evidence that they correlate with muscle exons in a much larger dataset. Interestingly, there was a notable lack of novel cis-elements in the proximal flanking introns that strongly correlate with muscle expression across the entire dataset. This could indicate that much of the fundamental machinery for regulation of generalized muscle-enriched splicing has been identified or, more likely, that additional features need to be incorporated in the algorithms to identify the remaining components. Such features may include wider motifs and motifs located more distally from the regulated exons. It is also possible that there are weaker elements, which may only be revealed when combinatorial interactions among motifs are included in the regression models, or which may be required for spatially or temporally distinct subsets of muscle-enriched exons. To obtain an initial estimate as to which of these factors may be most influential, we extended our study to include PWMs of width 5–7 nt. The results are displayed on our website (http://vision.lbl.gov/People/ddas/NAR_SPLICE1/). We observe that most motifs have similarities to the known motifs as identified above. There is one motif in D200, GGSYVYW, which seems novel. But since it has much higher P-value than others (P = 0.01), it is not readily clear if it is truly functional. Hence, we suspect that inclusion of combinatorial interactions among motifs may be most effective in revealing the novel motifs. One question that needs to be addressed in future studies, as improved measures of binding specificity become available, is the importance of additional splicing factors such as the muscleblind proteins that are already known to influence the splicing of at least a few muscle-specific alternative exons (51).
A working model that summarizes these findings is presented in Figure 5. Fox, CELF and acuaac-binding factors are proposed as positive regulators of muscle-enriched exons via their binding to the downstream proximal intron. The distribution of binding motifs among individual introns suggests that these factors function independently in some cases, and collaboratively in others, to specify muscle-enriched splicing. For Fox proteins an especially widespread role is suggested by the high absolute abundance of ugcaug-binding motifs: almost half of the muscle-enriched exons in datasets of all four species possess at least one ugcaug motif in the D200 intronic region, and some of those lacking a proximal ugcaug have phylogenetically conserved distal ugcaug motif(s) (data not shown) analogous to the myosin II heavy chain-B neural specific exon (58). It will be interesting in the future to explore how coordination among these and other factors ultimately determines the spatial and temporal details of muscle-enriched splicing events. Based on studies in other systems, PTB is predicted as a negative regulator of splicing, functioning primarily from upstream intronic sites to prevent inappropriate inclusion in non-muscle cell types (35,59,60). Finally, it is important to note that variations of this general model likely pertain to individual exons; in particular, Fox and CELF proteins can also have a negative role in the regulation of exons that are skipped in muscle (27,61–63). Future experimental analysis of these splicing factors, using functional splicing assays and targeted disruption of splicing factor activity in vivo (64), will be required to more fully test the predictions of this model.
Figure 5.

A candidate model showing splicing factors implicated in regulation of conserved muscle-enriched alternative exons. Based on the conserved distribution of splicing factor binding sites across multiple vertebrate orders, the positive correlation with muscle-specific splicing and the high absolute abundance of ugcaug motifs, Fox proteins are proposed to play a major role in promoting inclusion of muscle-enriched exons. The distribution of binding motifs among individual introns suggests that CELF proteins and by KH-type splicing factor(s) function independently in some cases, and together with Fox proteins in others, to specify muscle-enriched splicing. In contrast, the enrichment of candidate PTB-binding sites in the proximal upstream intron suggests a role in preventing inappropriate inclusion of muscle-specific exons in other cell types.
Some of the cis-regulatory elements associated with muscle-enriched alternative exons have previously been observed flanking brain-enriched exons: ugcaug was the most over-represented motif in proximal downstream intron (7,8,11), and cucucu was the second most over-represented motif in the U100 region upstream of brain-enriched exons (7). These observations suggest general roles for Fox- and PTB-related proteins in regulating tissue-specific splicing, at least for muscle and brain, but raise the question as to how tissue specificity is ultimately determined. Several mechanisms may contribute to determination of temporal and spatial pattern of splicing switches, including tissue-specific differences in transcription and/or alternative splicing of Fox and PTB paralogs (28). Differential expression of additional RNA-binding proteins, such CELF proteins and KH-type acuaac-binding proteins in muscle, or NOVA1-related proteins in brain, likely also play a role, as may non-RNA-binding co-factors that preferentially interact with paralogs/isoforms of the primary RNA-binding proteins.
In summary, normal metazoan development requires not only a transcriptional program, but also an alternative pre-mRNA splicing program to ensure that each gene encodes specific protein isoforms in the appropriate spatial and temporal patterns. Enrichment within the muscle dataset of genes with functions in cytoskeleton organization, microtubule stabilization and muscle development supports the notion that this splicing program is essential for proper expression of the unique muscle cytoskeleton. The exon microarray employed in this study will enhance our ability to track the expression of individual exons during development and differentiation. As we have demonstrated here, this experimental approach is well complemented by the computational approach based on correlation with expression. We anticipate that correlation with exon expression will provide valuable insights into the cis-regulation of alternative splicing as additional datasets of tissue-specific exons become available for analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thank Charles Sugnet for use of the dataset of human cassette alternative exons in correlation studies. This work was supported by DE AC03 76SF00098, the National Institutes of Health NIH grant HL45182, National Aeronautics and Space Administration Grant T6275W and by the Director, Office of Biological and Environmental Research, US Department of Energy under contract DE-AC03-76SF00098. Funding to pay the Open Access publication charges for this article was provided by NIH grant HL45182.
Conflict of interest statement. None declared.
REFERENCES
- 1.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 2.Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stamm S, Zhang MQ, Marr TG, Helfman DM. A sequence compilation and comparison of exons that are alternatively spliced in neurons. Nucleic Acids Res. 1994;22:1515–1526. doi: 10.1093/nar/22.9.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang XH, Kangsamaksin T, Chao MS, Banerjee JK, Chasin LA. Exon inclusion is dependent on predictable exonic splicing enhancers. Mol. Cell. Biol. 2005;25:7323–7332. doi: 10.1128/MCB.25.16.7323-7332.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brudno M, Gelfand MS, Spengler S, Zorn M, Dubchak I, Conboy JG. Computational analysis of candidate intron regulatory elements for tissue-specific alternative pre-mRNA splicing. Nucleic Acids Res. 2001;29:2338–2348. doi: 10.1093/nar/29.11.2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minovitsky S, Gee SL, Schokrpur S, Dubchak I, Conboy JG. The splicing regulatory element, UGCAUG, is phylogenetically and spatially conserved in introns that flank tissue-specific alternative exons. Nucleic Acids Res. 2005;33:714–724. doi: 10.1093/nar/gki210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hui J, Hung LH, Heiner M, Schreiner S, Neumuller N, Reither G, Haas SA, Bindereif A. Intronic CA-repeat and CA-rich elements: a new class of regulators of mammalian alternative splicing. EMBO J. 2005;24:1988–1998. doi: 10.1038/sj.emboj.7600677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miriami E, Margalit H, Sperling R. Conserved sequence elements associated with exon skipping. Nucleic Acids Res. 2003;31:1974–1983. doi: 10.1093/nar/gkg279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugnet CW, Srinivasan K, Clark TA, O’Brien G, Cline MS, Wang H, Williams A, Kulp D, Blume JE, et al. Unusual Intron Conservation near Tissue-Regulated Exons Found by Splicing Microarrays. PLoS Comput. Biol. 2006;2:e4. doi: 10.1371/journal.pcbi.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeo G, Hoon S, Venkatesh B, Burge CB. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc. Natl Acad. Sci. USA. 2004;101:15700–15705. doi: 10.1073/pnas.0404901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang XH, Heller KA, Hefter I, Leslie CS, Chasin LA. Sequence information for the splicing of human pre-mRNA identified by support vector machine classification. Genome Res. 2003;13:2637–2650. doi: 10.1101/gr.1679003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark TA, Sugnet CW, Ares M., Jr Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science. 2002;296:907–910. doi: 10.1126/science.1069415. [DOI] [PubMed] [Google Scholar]
- 15.Frey BJ, Mohammad N, Morris QD, Zhang W, Robinson MD, Mnaimneh S, Chang R, Pan Q, Sat E, et al. Genome-wide analysis of mouse transcripts using exon microarrays and factor graphs. Nat. Genet. 2005;37:991–996. doi: 10.1038/ng1630. [DOI] [PubMed] [Google Scholar]
- 16.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 17.Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- 18.Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nat. Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- 19.Das D, Banerjee N, Zhang MQ. Interacting models of cooperative gene regulation. Proc. Natl Acad. Sci. USA. 2004;101:16234–16239. doi: 10.1073/pnas.0407365101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conlon EM, Liu XS, Lieb JD, Liu JS. Integrating regulatory motif discovery and genome-wide expression analysis. Proc. Natl Acad. Sci. USA. 2003;100:3339–3344. doi: 10.1073/pnas.0630591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keles S, van der Laan M, Eisen MB. Identification of regulatory elements using a feature selection method. Bioinformatics. 2002;18:1167–1175. doi: 10.1093/bioinformatics/18.9.1167. [DOI] [PubMed] [Google Scholar]
- 22.Das D, Nahle Z, Zhang MQ. Adaptively inferring human transcriptional subnetworks. Mol. Syst. Biol. 2006;2:2006–0029. doi: 10.1038/msb4100067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Cherry JM, Botstein D, Li H. A systematic approach to reconstructing transcription networks in Saccharomycescerevisiae. Proc. Natl Acad. Sci. USA. 2002;99:16893–16898. doi: 10.1073/pnas.252638199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorek R, Ast G. Intronic sequences flanking alternatively spliced exons are conserved between human and mouse. Genome Res. 2003;13:1631–1637. doi: 10.1101/gr.1208803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 26.Clark TA, Schweitzer AC, Chen TX, Staples MK, Lu G, Wang H, Williams A, Blume JE. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol. 2007;8:R64. doi: 10.1186/gb-2007-8-4-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin Y, Suzuki H, Maegawa S, Endo H, Sugano S, Hashimoto K, Yasuda K, Inoue K. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 2003;22:905–912. doi: 10.1093/emboj/cdg089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakahata S, Kawamoto S. Tissue-dependent isoforms of mammalian Fox-1 homologs are associated with tissue-specific splicing activities. Nucleic Acids Res. 2005;33:2078–2089. doi: 10.1093/nar/gki338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol. Cell. Biol. 2005;25:10005–10016. doi: 10.1128/MCB.25.22.10005-10016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baraniak AP, Chen JR, Garcia-Blanco MA. Fox-2 mediates epithelial cell-specific fibroblast growth factor receptor 2 exon choice. Mol. Cell. Biol. 2006;26:1209–1222. doi: 10.1128/MCB.26.4.1209-1222.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ponthier JL, Schluepen C, Chen W, Lersch RA, Gee SL, Hou VC, Lo AJ, Short SA, Chasis JA, et al. Fox-2 splicing factor binds to a conserved intron motif to promote inclusion of protein 4.1R alternative exon 16. J. Biol. Chem. 2006;281:12468–12474. doi: 10.1074/jbc.M511556200. [DOI] [PubMed] [Google Scholar]
- 32.Kabat JL, Barberan-Soler S, McKenna P, Clawson H, Farrer T, Zahler AM. Intronic alternative splicing regulators identified by comparative genomics in nematodes. PLoS Comput. Biol. 2006;2:e86. doi: 10.1371/journal.pcbi.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou HL, Baraniak AP, Lou H. Role for Fox-1/Fox-2 in mediating the neuronal pathway of calcitonin/calcitonin gene-related peptide alternative RNA processing. Mol. Cell. Biol. 2007;27:830–841. doi: 10.1128/MCB.01015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ladd AN, Charlet N, Cooper TA. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol. Cell. Biol. 2001;21:1285–1296. doi: 10.1128/MCB.21.4.1285-1296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spellman R, Smith CW. Novel modes of splicing repression by PTB. Trends Biochem. Sci. 2006;31:73–76. doi: 10.1016/j.tibs.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Gardina PJ, Clark TA, Shimada B, Staples MK, Yang Q, Veitch J, Schweitzer A, Awad T, Sugnet C, et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics. 2006;7:325. doi: 10.1186/1471-2164-7-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Srinivasan K, Shiue L, Hayes JD, Centers R, Fitzwater S, Loewen R, Edmondson LR, Bryant J, Smith M, et al. Detection and measurement of alternative splicing using splicing-sensitive microarrays. Methods. 2005;37:345–359. doi: 10.1016/j.ymeth.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Kent WJ. BLAT – the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Program NCS, Green ED, Sidow A, Batzoglou S. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003;13:721–731. doi: 10.1101/gr.926603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hastie T, Tibshirani R, Friedman JH. The Elements of Statistical Learning. New York, USA, pp. 46–47: Springer Verlag; 2001. [Google Scholar]
- 41.Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. J. Mol. Biol. 1987;193:723–750. doi: 10.1016/0022-2836(87)90354-8. [DOI] [PubMed] [Google Scholar]
- 43.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith AD, Sumazin P, Das D, Zhang MQ. Mining ChIP-chip data for transcription factor and cofactor binding sites. Bioinformatics. 2005;21(Suppl. 1):i403–i412. doi: 10.1093/bioinformatics/bti1043. [DOI] [PubMed] [Google Scholar]
- 45.Smith AD, Sumazin P, Zhang MQ. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc. Natl Acad. Sci. USA. 2005;102:1560–1565. doi: 10.1073/pnas.0406123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schones DE, Sumazin P, Zhang MQ. Similarity of position frequency matrices for transcription factor binding sites. Bioinformatics. 2005;21:307–313. doi: 10.1093/bioinformatics/bth480. [DOI] [PubMed] [Google Scholar]
- 47.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 48.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 49.Nurtdinov RN, Artamonova, Mironov AA, Gelfand MS. Low conservation of alternative splicing patterns in the human and mouse genomes. Hum. Mol. Genet. 2003;12:1313–1320. doi: 10.1093/hmg/ddg137. [DOI] [PubMed] [Google Scholar]
- 50.Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, et al. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005;37:844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 51.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amir-Ahmady B, Boutz PL, Markovtsov V, Phillips ML, Black DL. Exon repression by polypyrimidine tract binding protein. RNA. 2005;11:699–716. doi: 10.1261/rna.2250405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner EJ, Baraniak AP, Sessions OM, Mauger D, Moskowitz E, Garcia-Blanco MA. Characterization of the intronic splicing silencers flanking FGFR2 exon IIIb. J. Biol. Chem. 2005;280:14017–14027. doi: 10.1074/jbc.M414492200. [DOI] [PubMed] [Google Scholar]
- 54.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 55.Friedman N, Barash Y, Elidan G, Kaplan T. Proceedings of the Seventh Annual International Conference on Computational Molecular Biology (RECOMB); Berlin, Germany: 2003. pp. 28–37. [Google Scholar]
- 56.Zhou Q, Liu JS. Modeling within-motif dependence for transcription factor binding site predictions. Bioinformatics. 2004;20:909–916. doi: 10.1093/bioinformatics/bth006. [DOI] [PubMed] [Google Scholar]
- 57.Cooper TA. Muscle-specific splicing of a heterologous exon mediated by a single muscle-specific splicing enhancer from the cardiac troponin T gene. Mol. Cell. Biol. 1998;18:4519–4525. doi: 10.1128/mcb.18.8.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawamoto S. Neuron-specific alternative splicing of nonmuscle myosin II heavy chain- B pre-mRNA requires a cis-acting intron sequence. J. Biol. Chem. 1996;271:17613–17616. [PubMed] [Google Scholar]
- 59.Wagner EJ, Garcia-Blanco MA. Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell. Biol. 2001;21:3281–3288. doi: 10.1128/MCB.21.10.3281-3288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharma S, Falick AM, Black DL. Polypyrimidine tract binding protein blocks the 5′ splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol. Cell. 2005;19:485–496. doi: 10.1016/j.molcel.2005.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 62.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 63.Zhang W, Liu H, Han K, Grabowski PJ. Region-specific alternative splicing in the nervous system: implications for regulation by the RNA-binding protein NAPOR. RNA. 2002;8:671–685. doi: 10.1017/s1355838202027036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu X, Fu XD. Conditional knockout mice to study alternative splicing in vivo. Methods. 2005;37:387–392. doi: 10.1016/j.ymeth.2005.07.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.