

Abstract
NSF is an AAA protein, broadly required for intracellular membrane fusion. NSF functions as a SNARE chaperone which binds, through SNAPs, to SNARE complexes and utilizes the energy of ATP hydrolysis to disassemble them thus facilitating SNARE recycling. While this is a major function of NSF, it does seem to interact with other proteins, such as the AMPA receptor subunit, GluR2, and β2-AR and is thought to affect their trafficking patterns. New data suggest that NSF may be regulated by transient post-translational modifications such as phosphorylation and nitrosylation. These new aspects of NSF function as well as its role in SNARE complex dynamics will be discussed.
Keywords: NSF, SNARE, α-SNAP, GluR2, β2-AR, post-translational modification
1. Introduction
Vesicular traffic is essential for cellular homeostasis. Cargo-containing transport vesicles bud from a donor compartment, then fuse with an appropriate acceptor compartment assuring a vectorial flow of membrane proteins/lipids and luminal contents through the cell. At its core, vesicular transport requires cargo selection and vesicle production through a budding process. The subsequent vesicle transport and targeting process concludes with specific membrane fusion of the vesicle to its target membrane. Early molecular analysis of vesicular trafficking between Golgi cisternae identified an essential N-ethylmaleimide Sensitive Factor (NSF) [1]. Subsequent studies identified NSF adaptors, SNAPs [2], and SNAP-binding receptors [3], SNAREs. The characterization of these proteins has lead to a detailed picture of the events leading to membrane fusion. Despite this knowledge, there are many unanswered questions regarding how SNAREs are regulated and how NSF itself might be regulated. In addition, new data suggest that NSF may have other cellular roles that require its ATPase-driven chaperone activity. This review will attempt to give an “NSF-centric” overview of some of these topics.
2. SNAREs
SNARE proteins are the minimal machinery for membrane fusion [4]. SNAREs are classified into vesicle (v) and target-membrane (t)-SNAREs according to their localization [3] or R (arginine) and Q (glutamine) -SNAREs based on a key residue in the center of their SNARE domains [5]. There are two types of t-SNAREs: syntaxin-type and SNAP-25 type. v-SNAREs and syntaxin-like t-SNAREs are type II integral membrane proteins with a single transmembrane domain (TMD). The TMDs not only anchor SNAREs to a membrane, but may contribute to complex assembly and fusion pore formation [6,7]. SNAP-25-like proteins lack a TMD and are generally anchored to the membrane through thioester-linked acyl groups [8]. All SNARE proteins characteristically contain conserved heptad repeats of approximately 60–70 residues termed the SNARE motif. SNAP-25-like SNAREs contain two such motifs. The SNARE motif forms an amphipathic α-helix where the hydrophobic residues are in register on the same face to form the core of the SNARE complexes [9].
In addition to the SNARE motif, many SNARE proteins have divergent N-terminal domains. The N-terminal half of syntaxin-1a contains an autonomously-folded, three-helix bundle, called the Habc-domain [10]. This domain forms a groove that accepts the SNARE motif generating a “closed” conformation of syntaxin-1a. The N-terminal domain of v-SNAREs is highly divergent and divides the family into “brevins” that contain short domains, and “longins” that contain longer structures [11]. For the brevins, the N-terminus attains a partially folded state when SNARE complexes are assembled and may play a role in controlling the stability of primed fusion complexes [12]. The longin domains appear to regulate v-SNARE sorting and have been shown to be important for spatially directed exocytosis in neurons [13]. From these examples, it seems possible that these N-terminal domains may be key structural elements to allow regulation of SNARE complex assembly and perhaps disassembly.
2.1. Assembly of SNARE Complexes
Structural information from neuronal SNARE complexes shows that the SNARE motifs assemble into parallel, twisted, coiled-coil, four-helix bundles by burying the hydrophobic residues inside the core (Figure 1). Three of the helices are contributed by t-SNAREs (1 from syntaxin and 2 from SNAP-25), with the other helix provided by the v-SNARE. The coiled-coil is composed of 15 hydrophobic layers arranged perpendicular to the axis of the helical bundle, and contains a central hydrophilic zero-layer with one R- and three Q-residues. The surface of the complex is highly grooved with several charged regions [9,14].
Figure 1. 20S Particle.
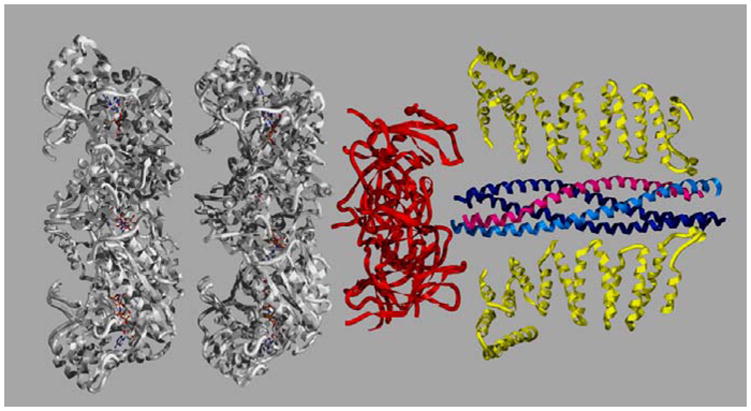
Depicted are the crystal structures for each element of the 20S particle. The two ATP-binding domains of NSF (D1 and D2) in white are modeled from the NSF-D2 structure (1D2N). A trimer of NSF-N domains, in red, is based on the three-in-three-out model of May et al. 1999 (1QDN). Only two yeast α-SNAPs (Sec17p, 1QQE ) are depicted in yellow. The coiled-coil SNARE complex is depicted with syntaxin-1a in light blue, synaptobrevin/VAMP-2, in magenta, and SNAP-25 in dark blue (2BUO). The images were created with Swiss PDB viewer and rendered with Pov-Ray.
Great focus has been put on the molecular steps of SNARE complex assembly. It is thought to start at the SNAREs’ N-termini and proceed in a zipper-like fashion toward the membrane anchor at the C-termini [15]. Before assembly, syntaxin-1a shows significant α-helicity, but SNAP-25 and synaptobrevin/VAMP-2 are largely unstructured. Upon ternary complex formation, SNAP-25 and synaptobrevin show dramatic increases in α-helicity. Considerable evidence [16] now implies that syntaxin-1a and SNAP-25 form a transient 1:1 complex, which serves as an “on-pathway” intermediate. After establishment of the t-SNARE heterodimer, synaptobrevin, from the opposing membrane, binds to form a high affinity complex, which spans the two fusing bilayers. The energy associated with SNARE complex assembly is thought to drive membranes into close apposition, which then directly or indirectly leads to fusion [16–18]. After membrane fusion, SNAREs remain as a complex in the same membrane. Disassembly of these cis complexes is achieved by the concerted action of α-SNAP and NSF.
3. α-SNAP
Clary et al. [2] determined that NSF required a peripheral membrane protein adaptor to bind Golgi membranes. This protein was called Soluble NSF Attachment Protein or SNAP. In mammals, there are three SNAPs, α, β, and γ; in yeast there is one, Sec17p. In mice, α-SNAP is encoded by the Napa gene and its deletion is embryonic lethal [19]. A G→A missense mutation in exon 4 of the Napa gene results in an M105I substitution that appears to be the cause of hydrocephalus with hop gait (hyh) in mice. Affected animals show a dome-shaped head with a small cerebral cortex. They die postnatally from a progressive enlargement of the ventricular system. Binding of the M105I mutant to SNARE complexes, however, was indistinguishable from that of the wild-type α-SNAP. The mutation also had no effect on NSF-mediated, SNARE complex disassembly. Intriguingly, both mRNA and protein levels of α-SNAP were decreased in hyh mice. Though the biochemical data do not yield a clear explanation of the defect in hyh mice, the animals’ phenotype indicates that the reduced availability of α-SNAP is detrimental to apical transport and cell fate regulation in neurons [19].
3.1. α-SNAP Binding to SNARE Complexes
α-SNAP does not bind synaptobrevin but can associate with the syntaxin/SNAP-25 heterodimer [20]. α-SNAP appears to first bind to syntaxin/SNAP-25. Association of synaptobrevin with this complex generates a third binding site for α-SNAP [20,21]. Consistently, the binding affinity of α-SNAP to syntaxin and SNAP-25 is weak but addition of synaptobrevin dramatically enhances binding [22]. Deletion mutagenesis suggests that the N-terminal 63 and C-terminal 37 residues of α-SNAP are important for binding to SNARE complexes. Binding of α-SNAP involves the C-terminal residues (194–243, part of the SNARE motif) of syntaxin-1 and the N-terminal residues (25–100) of SNAP-25. However, since there is large sequence variation among the SNARE family, it seems likely that α-SNAP primarily recognizes the overall shape of the coiled-coil structure and not specific residues.
The crystal structure of Sec17p shows that it contains an N-terminal twisted sheet of α-helical hairpins with a protruding C-terminal α-helical globular bundle (Figure 1). One edge of the twisted sheet is longer than the other forming concave and convex faces in the structure. The concave face has a distribution of negative charges, which is most pronounced at the extreme C terminus. Both the concave face and the longer edge contain residues that are conserved among all SNAPs [23]. As outlined by Rice and Brunger (1999), there are two possible models of SNAP-SNARE interaction: “face-on” and “edge-on”. Mutagenesis data show SNAP-SNARE complex interactions involve positively charged α-SNAP residues distributed over the concave surface of its twisted sheet domain [24].
4. NSF
NSF is encoded by the Sec18 gene in yeast [25] and a neuronal specific version is encoded by the comatose gene (dNSF1) in Drosophila [26]. NSF is clearly important since conditional mutations in Sec18p and comatose both lead to cessation of membrane transport under restrictive temperatures. Each protomer of the homo-hexamer has three domains: the amino-terminal domain (NSF-N, 1–205) is required for SNAP-SNARE binding; the first ATP-binding domain (NSF-D1, 206–477), which provides ATPase activity, is required for SNARE complex disassembly; and the second ATP-binding domain (NSF-D2, 478–744) is required for hexamerization [27,28]. From sequence homologies of NSF-D1 and NSF–D2, NSF is a member of the AAA family of ATPases that generally uses ATP hydrolysis to alter the conformation of a substrate protein [29].
4.1 NSF Structure
From several studies using quick-freeze/deep-etch electron microscopy [30] or cryo-electron microscopy (cryo-EM) with single-particle averaging of NSF-α-SNAP-SNARE complexes [31], NSF clearly shows six-fold symmetry. It appears to be arranged like a barrel composed of two rings (presumably NSF-D1 and NSF–D2) with six knobs extending from the barrel (presumably NSF-N). The conformation depends on the nucleotide bound. ATP-charged NSF is a hollow cylinder: NSF-D1 and NSF-D2 form hexameric rings that are arranged in a double-layered barrel, while NSF-N domains emerge from the sides of the ring. The ADP-bound morphology is slightly wider, and does not possess outwardly flared NSF-N domains [30,31]. These nucleotide-state-dependent conformations may hold the secret to how NSF uses ATP hydrolysis to disassemble SNARE complexes.
NSF-N is a kidney-shaped domain composed of two subdomains: NA (a.a. 1–83) and NB (a.a. 87–201), which are joined by a short linker [32,33] (Figure 2A). NA is made of six β strands arranged in a barrel with two “ψ loops” containing short α helices (α1 and α2) extending over the top. NB is an α/β roll, where four β strands wrap around a single amphipathic α-helix. The interface between NA and NB is predominantly hydrophilic with two small hydrophobic clusters at the edges of the interface. The surface of NSF-N possesses an overall positive charge and contains three grooves, all large enough to accommodate an α-helix [32,33]. At this stage, each could be important for SNAP-SNARE complex binding, though mutagenesis experiments suggest that groove 3 is the likely binding site. Groove 3 contains the first NA ψ loop in the double ψ loop beta barrel motif, which consists of strand β5 and the loop between strands α1 and α2 [32]. Mutation of one highly conserved residue (R67E) in this surface completely eliminates binding [34]. Sec18p-N (yeast NSF) has the same overall fold [35] with a conserved groove 3.
Figure 2. Post-translational modifications of NSF.
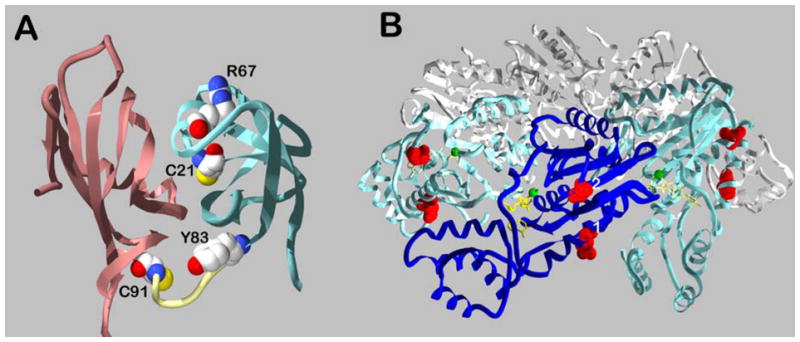
Panel A, crystal structure of the amino-terminal domain of NSF, showing the locations of the modified cysteine and tyrosine residues. The NA subdomain is in rose and the NB subdomain is in aqua. The image is based on 1QDN and was generated using Swiss PDB viewer and rendered with Pov-Ray. Panel B, crystal structure of the NSF-D2 hexamer. Three subunits are shown in white, two in aqua and one in blue. The modified residues are indicated by red. Position 1 is predicted to be equivalent to the phosphorylated Ser237 in the NSF-D1 domain. Position 2 is Ser569, phosphorylated by Pctaire1. The image is based on 1D2N and was created with Swiss PDB viewer and rendered with Pov-Ray.
The structure of NSF-D2 was the first AAA domain to be determined [36,37] (Figure 2B). Overall, it consists of two subdomains: an N-terminal nucleotide-binding subdomain (residues 505–676), and a C-terminal α-helical subdomain (residues 677–750). The N-terminal subdomain (which contains the Walker A and B box motifs) has the wedge shape with a central five-stranded parallel β-sheet. The C-terminal subdomain is composed of four α helices. This subdomain lies above the nucleotide-binding domain and contributes several residues to nucleotide binding.
Although the structure of NSF-D1 has not been determined, its sequence homology to NSF-D2 indicates that the NSF-D2’s structure can be used as a guide to understand the mechanism of the catalytically active NSF-D1. Both domains contain nucleotide-binding sites with the classical Walker A and B box motifs. The conserved lysine residues (266, 549) in the two Walker A boxes are crucial to ATP-binding [38,39]. The aspartic acid in the DEXX sequence of the Walker B box is thought to coordinate a Mg2+ ion that is needed for ATP hydrolysis, whereas the glutamate is required to activate water for the hydrolysis reaction. Mutation of Glu329 (E329Q) in NSF-D1 results in a dominant negative form of NSF that can bind but not disassemble SNAP-SNARE complexes [28,40].
Within the N-terminal subdomain of both ATP-binding domains, there is a highly conserved region called Second Region of Homology (SRH), which is unique to AAA proteins. At the C-terminus of SRH are two highly conserved arginine residues called Arginine Fingers. Both are important for nucleotide hydrolysis in other AAA proteins and are thought to allow for inter-subunit communication between the protomers of the hexameric ATPases [41]. Mutations of these residues in NSF only have a limited effect on ATPase activity but almost eliminate SNARE complex disassembly [34]. Sensor 1 is present at the N terminus of the SRH. A conserved polar residue in Sensor 1 is in close proximity to the phosphates of the bound nucleotide. Mutation of this residue (T394P) in Sec18p eliminates ATPase activity [42]. The Sensor 2 comes from the C-terminal helical subdomain. The residues in Sensor 2 are in close proximity to the bound nucleotide and have been proposed to monitor the state of the bound nucleotide. This could propagate nucleotide-dependent conformational change to the outer regions of NSF [41].
4.2. NSF Binding to the α-SNAP-SNARE Complexes
NSF and α-SNAP do not interact in solution. The interaction of NSF with α-SNAP occurs when α-SNAP binds to membranes, to a plastic surface, or alternatively when trimerized [43]. Cross-linking between α-SNAP and NSF in the membrane-bound complex further confirms this interaction [44]. The deletion mutants show that the C-terminal 45 amino acids of α-SNAP are crucial for interaction with NSF [45]. Consistently when this domain was grafted onto the trimerization domain of thrombospondin, the resulting recombinant protein bound NSF in the absence of SNAREs [43]. Point mutations in this domain, specifically of the penultimate leucine residue (Leu294), yield an α-SNAP that can bind NSF but cannot activate its ATPase activity [46].
NSF-N is required for binding SNAP-SNARE complexes [28,39,43]. Consistently, the Sec17p-(yeast α-SNAP)-binding-defective Sec18-1 allele maps to a 120-amino acid region in the N domain (G89D) [47,48]. Only ATP-charged NSF is binding competent. The ATP-binding mutant, K266A, shows weak binding to the SNAP-SNARE complexes, suggesting that binding of ATP by NSF-D1 most likely induces conformational changes in the D1 and N domains that are crucial to the interaction of NSF with SNAP-SNARE complexes. The neighboring D1 or D2 also possibly participates in binding to α-SNAP because the monomeric mutant N-D1 and hexameric N-D2 can bind but monomeric N domain cannot [28]. This implies that two elements are potentially important to the binding interaction: oligomerization of NSF-N and proximity to an ATP binding domain.
NSF does not bind to SNARE complexes in the absence of α-SNAP [21,49]. Based on this, NSF and α-SNAP are thought to bind in a sequential manner, giving rise to the 20S particle, named for its sedimentation coefficient [50]. In the presence of non-hydrolysable ATP, the 20S particle is stable and can be purified. The SNARE complex in this particle is perhaps reflective of the cis configuration that would occur post-fusion. The 20S particle is disassembled in a process coupled to ATP hydrolysis, which probably reflects a major role of NSF in vivo [3,50]. NSF, however, can dissociate NSF-α-SNAP-syntaxin and NSF-α-SNAP-syntaxin-SNAP-25 complexes in vitro, suggesting that it could be a general SNARE chaperone affecting the conformations of multiple SNARE-containing complexes [51].
Electron microscopy studies of the 20S particle show that it has a spark-plug shape [30], with the SNARE complex at the thinner end and the two rings of NSF clearly visible at the wider end (Figure 1). Three α-SNAP molecules coat the rod-like SNARE complex along its length. While NSF binds to one end, the membrane-spanning regions of VAMP and syntaxin appear located at the other end, adjacent to the amino terminus of α-SNAP [52]. A cyro-EM structure of the 20S particle, suggests that the rings of the two ATP binding domains, NSF-D1 and NSF-D2, form a double-layered barrel, arranged in an anti-parallel orientation (A similar conclusion was made about the p97/VCP protein until crystallography proved it incorrect [53,54]). Six protrusions, thought to be the NSF N domains, appear to extend sideways from one end of the barrel. Near these protrusions, a cap-like density corresponding to α-SNAP and SNAREs is clearly visible [31]. These data suggest a mechanism by which NSF could untwist the SNARE complexes.
4.3. NSF’s ATPase Activity
The intrinsic ATPase activity of NSF is very low [27]. NSF-N has been proposed to exert some control over NSF’s ATPase activity because antibodies to it cause a 2-fold increase in hydrolytic activity [55]. Binding to immobilized α-SNAP stimulates the ATPase activity [56]; however, maximal stimulation of ATPase activity is achieved when both α-SNAP and SNARE complexes are included [51]. The penultimate leucine of α-SNAP is critical for this activity [45,46]. Consistently, the L294A mutation of α-SNAP is unable to mediate 20S particle disassembly. A direct interaction between NSF and Leu294 (or adjacent residues) seems likely but is as yet unproven.
NSF-D1 accounts for the majority of basal and SNAP-stimulated ATPase activity [38,57]. Mutations in the ATP-binding site of domain D1 (K266A and E329Q) cause a 70–80% decrease in ATPase activity relative to wild type NSF [38]. The ATP-binding mutant (K266A) disrupts NSF ability to bind to the SNAP-SNARE complexes. The hydrolysis mutant (E329Q) fails to dissociate the SNAP-SNARE complexes [28]. Mutations in the NSF-D2 decrease ATPase activity but only minimally [38]. The ATPase activity of Sec18p is stimulated by Sec17p. The hydrolysis mutant, E350Q in Sec18p-D1, shows no basal or stimulated ATPase activity [58]. The temperature-sensitive, paralytic, mutation comatosest17 (G274E) in Drosophila NSF1 is near the D1 ATP-binding site [59]. When this mutation was engineered into mammalian NSF (G282E), the resulting mutant protein had no ATPase activity [60]. These data demonstrate the importance of the D1 domain to NSF’s ATPase activity and thus to its function in membrane trafficking events.
5. Binding of NSF to Other Substrates
The well-documented role of NSF in vesicular transport is logically based on its interactions with SNAREs and SNAPs; however, several studies point to additional roles for NSF [61]. Specifically, NSF has been shown to be associated with a number of non-SNARE proteins, which can be divided into two classes (Table 1). The first are the C-terminal, cytoplasmic domains of a number of cell-surface signaling receptors. The second class of interactions is more diverse and includes peripheral and soluble cellular proteins as well as cytoskeletal elements. Examples of these interactions will be discussed below.
Table 1.
| NSF Interaction | Reference |
|---|---|
| NSF/Receptor | |
| GluR2 | Osten et al., 1998; Nishimune et al., 1998; Song et al., 1998 |
| β2-AR | Cong et al., 2001 |
| CRLR-RAMP3 | Bomberger et al., 2005 |
| GABAA receptor β subunit | Goto et al., 2005 |
| GABAB receptor 2 | Pontier et al., 2006 |
| D1-like dopamine receptor | Heydorn et al., 2004 |
| Dopamine D2 receptor | Zou et al., 2005 |
| Muscarinic M1, M3, M4 and M5 receptors | Heydorn et al., 2004 |
| Tachykinin NK1 and NK2 receptors | |
| Somatostatin SST1 receptor | |
| DOP receptor | |
| Chemokine receptors US27 and US28 | |
| NSF/Rab family | |
| Rab 3, 4, 6 | Han et al., 2000 |
| Rab 11-containing complex(γ-SNAP/Rip11/Gaf-1) | Tani et al., 2003 |
| Rab 11-FIP3 | Martin et al., 2006 |
| Rab 5 containing complex (EEA-1, Rabaptin-5, Rabex-5, syntaxin 13) | McBride et al., 1999 |
| NSF/others | |
| β-arrestin 1 | McDonald et al., 1999 |
| GATE-16 | Sagiv et al., 2000 |
| LMA1 | Xu et al., 1998 |
| βPIX | Martin et al., 2006 |
| GABARAP | Kittler et al., 2001 |
| PTP-MTG2 | Huynh et al., 2004 |
| Pctaire 1 | Liu et al., 2006 |
| GEC1 | Chen et al., 2006 |
5.1. NSF Binding to Membrane Receptors
The direct interactions between NSF and the cytoplasmic tails of several cell surface receptors have been reported: e.g. the AMPA receptor, the β2-AR receptor, the dopaminergic receptor, the adrenomedulin (AM) receptor and the γ-amino-butyric acid (GABA) receptor [62–68]. NSF binding is proposed to modulate the trafficking of these receptors between the plasma membrane and the endosome.
NSF is shown to directly bind to the C-terminal tail of the GluR2 subunit of the AMPA receptor in a SNAP-independent manner. The stable binding is nucleotide-dependent and requires all three domains of NSF. The minimal NSF binding domain of GluR2 is located between Lys844 and Gln853 [62–64]. Further mutagenesis of this segment showed that deletion of the last 5 amino acids abolishes NSF binding [69]. The NSF-GluR2 interaction is proposed to play a role in the stabilization of surface AMPA receptors on the postsynaptic membrane since a peptide that disrupts the NSF/GluR2-interaction causes a rapid decrease in the size of synaptic currents [70].
A two-hybrid approach was used to demonstrate that the C-terminus of β2-AR mediates binding to NSF. Mutations at any of the last three residues (S411A, L412A, or L413A) ablate NSF binding [65]. The addition of a single alanine residue at the end of the β2-AR tail also abrogates NSF binding. Binding to the β2-AR, however, is not affected by the addition of α-SNAP [71]. The binding of NSF to the β2-AR is critical to allow the receptor to undergo rapid recycling, which might also involve another interaction of NSF with β-arrestin1. β-arrestins play an important role of desensitization of many G-protein coupled receptors (GPCR) [72]. Over-expression of NSF in HEK 293 cells significantly enhances agonist-induced β2-AR clearance and rescues the inhibition of β2-AR internalization mediated by the phosphomimetic mutant of β-arrestin 1(βarr1S412D) [73].
There are five dopamine receptor subtypes, which are divided into two classes: D1-like and D2-like. NSF is found to bind to C-terminal tails of the D1 and D5 receptors fused to glutathione S-transferase (GST), suggesting that NSF could possibly be involved in the recycling of D1-like receptors [66].
Adrenomedullin receptors are comprised of receptor activity-modifying proteins (RAMP2 or RAMP3) and calcitonin receptor-like receptor (CRLR), which is the GPCR. RAMPs (1–3) are single transmembrane accessory proteins indispensable to the determination of receptor phenotype. Co-expression of RAMP1 with CRLR generates a calcitonin gene-related peptide-1(CGRP-1) receptor, while co-expression of RAMP2 or RAMP3 with CRLR yields adrenomedullin receptors, AM-1 and AM-2 receptors, respectively [74]. Upon stimulation with AM, the CRLR-RAMP receptor complex is internalized and undergoes degradation or recycling dependent on the cell type. NSF might be involved in altering the intracellular trafficking of the CRLR-RAMP3 receptor complex. When NSF is co-expressed with CRLR-RAMP3 complexes in HEK 293 cells, CRLR-RAMP3 is sorted for recycling rather than undergoing degradation. N-ethylmaleimide (NEM) treatment blocks the resensitization/recycling of the receptor after agonist-stimulated desensitization in the rat mesangial cells. RAMP3 is thought to interact with NSF through its C-terminal, type-I PDZ motif (-DTLL). Deletion of the PDZ motif significantly affects the resensitization and recycling of the CRLR-RAMP3 receptor complex in the presence of NSF. Mutagenesis of the DTLL sequence indicates that Asp145, Thr146 and Leu148 are the critical amino acids in the PDZ motif that regulate the RAMP3/NSF interaction [67]
Using full-length C-terminal tails of the metabotropic GABAB receptor 1 (GBR1) and GBR2 as bait in the yeast two-hybrid screen, NSF is revealed as a binding partner of GBR2. This was confirmed by using the GST-GBR2 fusion protein and purified recombinant NSF. The interaction of GBR2 and NSF also occurs in CHO cells and is disrupted upon agonist stimulation. The binding region has been narrowed to the 27 amino acids (residues 799–825; Pep-27) at the C-terminus of GBR2. Inhibition of NSF binding to GBR2 with a TAT-Pep27 fusion peptide blocks the agonist-promoted desensitization of GBR in hippocampal slices and CHO cells. TAT-Pep27 also blocks the GABA-induced protein kinase C (PKC) recruitment and GBR phosphorylation in CHO cells. Given that GBR does not undergo agonist-stimulated internalization in any of the systems tested, the author suggested that NSF regulates GABAB receptor signaling efficacy in a different way from other receptors. The preassociation of NSF with GBR2 primes the receptor and promotes PKC recruitment and phosphorylation of the GBR2 receptor, resulting in agonist-promoted desensitization of the receptor [68].
5.2. Other NSF-Binding Interactions
NSF has also been shown to interact with a diverse array of proteins including small GTP-binding proteins of the Rab family, GABA receptor associated protein (GABARAP)/Golgi-associated ATPase enhancer of 16 kDa (GATE-16)/low molecular weight activity 1 (LMA1) family members, βPIX, protein tyrosine phosphatase (PTP)-MEG2, and Pctaire1. The functional significance of most of these interactions is still largely speculative though provocative. In many cases, the chaperone function of NSF may play a role in the assembly/disassembly cycle of complexes containing these proteins.
Han et al. (2000) used the NSF-N-D1 truncation as bait and recovered Rab 6 as a binding partner by two-hybrid screen. This interaction requires the C-terminal 30 amino acids of Rab 6 and can stimulate the ATPase of NSF [75]. Two other Rabs (3 and 4) appear to bind and to stimulate NSF’s ATPase activity. NSF also interacts with Rab-containing complexes. NSF binds to a Rab 11-containing complex made up of γ-SNAP and Rab interacting protein 11/γ-SNAP associated factor 1 (Rip11/Gaf-1) [76]. Two-hybrid analysis revealed that γ-SNAP interacts directly with NSF via its extreme C-terminus and requires the penultimate leucine (Leu312). Intact NSF is required to interact with γ-SNAP. Both the N-terminal and C-terminal regions of γ-SNAP also mediate binding to the C-terminal domain of Gaf-1/Rip11, a Rab 11 effector. The complex comprising γ-SNAP and Gaf-1/Rip11 is disassembled by NSF in an ATPase-dependent manner. A yeast two-hybrid screen identified Rab 11-FIP3, another Rab 11-binding protein, as a NSF binding partner [77]. NSF has been detected in a complex with Rab 5 effectors: early endosome antigen 1 (EEA-1), Rabaptin-5, Rabex-5, and the SNARE syntaxin-13 [78]. Despite this wide range of interactions, their physiological relevance is still not certain. NSF could be involved in control of the assembly/disassembly cycles of these Rab-containing complexes. Alternatively, these complexes could play a role in targeting NSF to membrane subdomains involved in high levels of membrane fusion. Further experiments are clearly required.
GATE-16 functions in the secretory system [79] and is a member of a ubiquitin-like family of proteins that contain GABARAP and LMA1 [80]. GATE-16 binds to NSF as demonstrated by in vitro studies and by co-immunoprecipitation from cell extracts. Since ubiquitin binds to the N-terminal domain of p97/VCP (a domain similar to NSF-N)[81], GATE-16’s similarity to ubiquitin may be useful in analyzing the NSF-GATE-16 interactions. GATE-16 stimulates NSF’s ATPase activity. It also interacts with Golgi v-SNARE GOS28 in an NSF- and SNAP-dependent manner leading the authors to propose that GATE-16 is transferred to GOS28 from NSF, stabilizing GOS28 in a “primed” conformation [82]
Using a truncated form of NSF (Δ-1-9) as bait, Martin et al. (2006) detected four NSF interacting proteins in a yeast two-hybrid screen: Rab 11-FIP3 (discussed above), αCOP, Mink2 and βPIX. βPIX, a Pak-binding Rho guanine exchange factor, was of specific interest due to its apparent role in the control of post-synaptic structure. Co-immunoprecipitation experiments confirmed the interaction and GST pull-down assays with βPIX fragments showed that the leucine zipper motif in the C-terminus of βPIX was important for binding. Subsequent studies failed to demonstrate a physiological role for the interaction but the authors suggest that, since βPIX can form higher molecular weight complexes, NSF may play a role in their assembly/disassembly cycle.
6. NSF and the Cytoskeleton
Genetic studies in Drosophila have linked NSF to cytoskeletal dynamics [83]. Drosophila expresses two NSF isoforms: dNSF1, which is the dominant isoform in the adult central nervous system and dNSF2, which shows a much broader distribution [84]. dNSF1 null flies die as pharate adults but dNSF2 deletion is lethal at or before the first instar [85]. Over expression of the dominant negative mutant of dNSF2 (E326Q) in neurons results in a very interesting phenotype: the overgrowth and hypersprouting of neuromuscular junctions (NMJ). Reversal of this phenotype served as the basis of a screen which identified a number of different classes of proteins involved in such varied processes as transcription and ubiquitin-mediated degradation. Interestingly, several actin-binding proteins were identified (i.e. moesin, jaguar (a myosin VI), and quail (a villin-like protein) [83,86]). β-tubulin was also found to suppress the dNSF2E/Q-induced phenotype. Such data suggest that NSF could play a role in controlling synaptic structure through an effect on the cytoskeleton. Such an effect would be consistent with the loss of cell polarity seen in Dictyostelium amoebae expressing defective NSF mutants [87]. However, what is the nature of this connection between NSF and the cytoskeleton? The authors of the Drosophila studies suggest that neurotransmitter release (or lack thereof) is not responsible since NMJ overgrowth is not seen in the syntaxin or synaptobrevin null mutants. However, these SNAREs represent only one membrane trafficking pathway in neurons. Additionally, overexpression of α-SNAP reverses the phenotype, suggesting some connection between NMJ overgrowth and membrane trafficking [83]. Perhaps, the effect of the dominant-negative dNSF2E/Q mutant is through disruption of a membrane trafficking pathway that is required to localize cytoskeletal binding proteins to specific regions of the presynaptic or post-synaptic plasma membrane. Further analysis will be required to dissect the mechanistic basis of this interesting effect.
7. NSF regulation
Initially, NSF was thought to be a “house-keeping protein”; constitutively active and not subjected to any regulation. Perhaps this misconception grew out of the fact that regulation of secretion appears to be largely prefusion and directed to SNARE complex assembly. However, recent studies show that NSF activity is not uniform and can be regulated by several different mechanisms, including the reversible inactivation by S-nitrosylation and phosphorylation.
7.1. Phosphorylation
NSF has been shown to be phosphorylated in rat brain synaptosomes in a depolarization-induced, calcium-dependent manner and this event correlates with glutamate release from synaptosomes. PKC appears to be responsible and it phosphorylates NSF on Ser237 in NSF-D1. Mutation of this residue to alanine eliminates in vitro phosphorylation and mutation to glutamic acid attenuates NSF binding to SNAP-SNARE complexes [88]. Structurally, the effect of phosphorylation at Ser237 can only be discussed based on the structure of NSF-D2 (Figure 2B). Ser237 would be in the middle of α1 and well within the reach of the adjacent subunit, particularly the loop between the α8 and α9. The phosphorylation of this residue could restrict the movement of the α8 by charge-charge interaction and thereby affect the conformational changes in Sensor 2 associated with ATP hydrolysis.
NSF also can be phosphorylated by the serine/threonine kinase, Pctaire1. Pctaire1 phosphorylates NSF on Ser569 in NSF-D2 and affects NSF oligomerization [89] (Figure 2B). Mutation of Ser569 to alanine (S569A) abolishes phosphorylation and stabilizes the NSF oligomer. The S569E mutant caused a defect in oligomerization. Inhibition of Pctaire1 activity by overexpression of its kinase-inactive mutant (Pctaire1-KD) also enhances the ability of NSF to hexamerize. Consistently, overexpression of Pctaire1-KD or NSF-S569A in PC12 cells significantly enhances high potassium-stimulated growth hormone release from dense core vesicles, suggesting phosphorylation of NSF by Pctaire1 plays a role in regulating the calcium-dependent exocytosis [89]. In the crystal structure of NSF-D2, Ser569 is located on the interface between monomers. Phosphorylation of this residue could impact the hexamerization of NSF.
Tyrosine phosphorylation of NSF was shown by Huynh et al. [90]. Tyrosine kinases Fes and Fer phosphorylate NSF on Tyr83 and the tyrosine phosphatase, PTP-MEG2, specifically removes the phosphate (Figure 2A). Phosphorylation at Tyr83 increases NSF’s ATPase activity but prevents α-SNAP binding. This suggests that tyrosine phosphorylated-NSF would be functionally inactive and lead to an accumulation of dead-end cis-SNARE complexes, thereby inhibiting membrane fusion. The hypothesis is supported in Jurkat T cells transfected with a NSF Y83F mutant or with PTP-MEG2. These cells contained enlarged secretory vesicles which were fragmented into smaller vesicles when the cells were treated with the PTP inhibitor, pervanadate. This indicates that tyrosine phosphorylation/dephosporylation of NSF can regulate a dynamic cycle of vesicle fusion. In such a model, PTP-MEG2 functions as a positive regulator of NSF to promote secretory vesicle fusion.
As mentioned previously, NSF-N contains two subdomains: NA and NB. Tyr83 is located on the loop connecting these two subdomains and is involved in stabilization of this loop by forming a hydrogen bond with an adjacent amino acid (Gln90). Gln90 also forms a hydrogen bond with Lys87, further stabilizing the loop. Addition of a negative charge to Tyr83 might affect these interactions and disrupt the interface between the subdomains thus perturbing α-SNAP binding.
7.2. S-Nitrosylation and Oxidation
Nitric Oxide (NO) is a second messenger in the cardiovascular system that limits vascular inflammation and thrombosis by, in part, affecting endothelial cell and platelet exocytosis. Given that the inhibition of NSF by NEM is based on the sensitivity of specific cysteines to alkylation, NSF would seem a viable target for regulation by S-nitrosylation. The nitrosylation of NSF was first demonstrated, in endothelial cells, by Lowenstein and colleagues [91]. They showed that exogenous NSF could rescue the NO-induced inhibition of von Willebrand Factor release from permeabilized endothelial cells. Surprisingly, treatment of NSF with the NO donor 2-(N, N-diethylamino)-diazenolate-2-oxide (DEA-NONOate) did not inhibit NSF’s ATPase activity but did block NSF’s ability to disassemble SNARE complexes. DTT restored the activity of nitrosylated-NSF, consistent with the reversible nature of the modification. Mutagenesis of NSF’s nine cysteines indicates that NSF-N domain Cys21 and 91 (Figure 2A) and D1-domain Cys264 are likely sites of S-nitrosylation. Mutations of those residues to alanine all partially decreased NSF’s ATPase activity. Mutation of Cys21 blocks the ability of NSF to interact with SNARE complexes and mutations of Cys91 and Cys264 act as dominant negatives. Cys21 is located on the interface between the two subdomains of NSF-N, which is proposed to be the binding sites between NSF and α-SNAP. Mutation of this residue could affect α-SNAP binding and abolish the interaction between NSF and SNARE complexes. Cys91 is located on the similar position as Tyr83. Its mutation may restrict the conformational change of N domain induced by ATP hydrolysis in NSF-D1 and affect the ability of NSF to disassemble the SNARE complexes. Cys264 is located in the Walker A motif of NSF-D1, which is critical to ATP binding. Mutation of this residue may affect the nucleotide binding and inactivate the NSF.
The nitrosylation of NSF not only occurs in endothelial cells, but is also seen in platelets. Lowenstein and colleagues demonstrated that NO inhibits platelet granule exocytosis by S-nitrosylation of NSF and adding NSF to the platelets restores secretion. S-nitrosylation of NSF on Cys91 also regulates its binding to the AMPAR GluR2 subunit [92]. Substantial augmentation of NSF-GluR2 binding is observed by treatment with the NO donors. NSF-GluR2 binding is diminished in eNOS null mice. NO donors elicit a rapid, time-dependent surface insertion of GluR2, which is markedly reduced by disrupting the interaction between GluR2 and NSF. It appears NSF S-nitrosylation could be a physiologic mediator of the receptor’s surface expression during NO-induced synaptic plasticity. It should be noted that there are many potentially nitrosylatable proteins in a cell and it is perhaps overly optimistic to believe the NSF is the only substrate that counts.
Recently, it has been shown that NSF can be regulated by hydrogen peroxide. H2O2 inhibits exocytosis from thrombin-stimulated endothelial cells. This inhibition can be reversed by adding NSF. H2O2 is thought to inactivate NSF through oxidation of the Cys264 in NSF-D1. Consistently, mutation of Cys264 to threonine eliminates the sensitivity of NSF to H2O2 [93]. While this might suggest that NSF could be a redox sensor in the cell, whose activity is decreased when the oxidation state of the cytosol increases, it seems that, as with nitrosylation, NSF may not be the only relevant target of this level of regulation.
8. Summary
Since its discovery, NSF has been linked to the secretory pathway. Subsequent work has established a role for NSF in the assembly/disassembly cycle of the SNARE proteins, a function essential for cell survival. More recent work suggests that NSF may serve as a structural chaperone for other, non-SNARE, complexes. As work progresses on these interactions, one should be guarded to discriminate between truly new functions for NSF and logical sequellae of defective membrane trafficking.
The appreciation of NSF regulation has also been a recent development and reflects our increased understanding of the complexity of membrane trafficking mechanisms. Phosphorylation, nitrosylation, and oxidation may all play roles in controlling NSF activity and thus membrane trafficking. These modifications are generally transient and offer an ideal mechanism for acute but reversible regulation of membrane trafficking. However, care should be taken to establish the uniqueness of the effect on NSF. Since multiple proteins can be modified by nitrosylation or oxidation, the challenge is to establish that their inactivating effects on NSF are truly the cause of the defect in membrane trafficking observed.
In the years since NSF’s discovery, much has been learned, however several questions still remain and new ones have surfaced. It is still unclear, at a molecular level, how NSF uses ATP hydrolysis to disassemble the SNARE complexes. It is now clear that NSF is regulated in novel ways, but what are the physiological ramifications of these events? This is a particularly interesting question given the reduction in NSF levels seen associated with epilepsy [94–96]. Finally, what role(s) does NSF play in the non-SNARE complexes? Is it a chaperone affecting their disassembly or is it along for the ride to where it is needed for membrane trafficking? Future studies will address these and many more questions.
Acknowledgments
We apologize to the excellent, but alas uncited, researchers who have contributed to our understanding of NSF. Due to space constraints we could not discuss all of the published studies of NSF function. The authors would like to thank the Whiteheart laboratory for their care in reading this manuscript. We specifically thank Dr. Elena A. Matveeva whose efforts have made much of the work discussed possible. We are indebted to Dr. Phyllis Hanson (Washington University) for her insightful comments over the years and to Dr. Elizabeth Kubalek-Wilson (Scripps Research Institute) for her perseverance in our studies of NSF’s structure. This work is supported by grants from the National Institutes of Health, NS046242 (to SWW) and from the Department of Veterans Affairs (to JTS).
Abbreviations
- NSF
N-ethylmaleimide sensitive factor
- AAA
ATPases associated with various cellular activities
- SNAP
soluble NSF attachment protein
- SNARE
SNAP receptor
- VAMP
vesicle associated membrane protein
- TMD
transmembrane domain
- AMPAR
α-amino-5-hydroxy-3-methyl-4-isoxazole propionic acid receptor
- GluR2
glutamate receptor 2
- GABA
γ-amino-butyric acid
- GBR
GABAB receptor
- β2-AR
β2-adrenergic receptor
- GPCR
G-protein coupled receptor
- CRLR
calcitonin receptor-like receptor
- RAMP
receptor activity-modifying proteins
- AM
adrenomedullin
- NMJ
neuromuscular junction
- PKC
protein kinase C
- PTP
protein tyrosine phosphatase
- GATE-16
Golgi-associated ATPase enhancer of 16 kDa
- VCP
valosin containing protein
- Rip11/Gaf-1
Rab interaction protein 11/γ-SNAP associated factor 1
- EEA-1
early endosome antigen 1
- SRH
second region of homology
- LMA1
low molecular weight activity 1
- GEC1
glandular epithelial cell 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci U S A. 1988;85:7852–6. doi: 10.1073/pnas.85.21.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clary DO, Griff IC, Rothman JE. SNAPs, a family of NSF attachment proteins involved in intracellular membrane fusion in animals and yeast. Cell. 1990;61:709–21. doi: 10.1016/0092-8674(90)90482-t. [DOI] [PubMed] [Google Scholar]
- 3.Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–24. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 4.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–72. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 5.Fasshauer D, Sutton RB, Brunger AT, Jahn R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci U S A. 1998;95:15781–6. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han X, Wang CT, Bai J, Chapman ER, Jackson MB. Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science. 2004;304:289–92. doi: 10.1126/science.1095801. [DOI] [PubMed] [Google Scholar]
- 7.Xu Y, Zhang F, Su Z, McNew JA, Shin YK. Hemifusion in SNARE-mediated membrane fusion. Nat Struct Mol Biol. 2005;12:417–22. doi: 10.1038/nsmb921. [DOI] [PubMed] [Google Scholar]
- 8.Ungermann C, Langosch D. Functions of SNAREs in intracellular membrane fusion and lipid bilayer mixing. J Cell Sci. 2005;118:3819–28. doi: 10.1242/jcs.02561. [DOI] [PubMed] [Google Scholar]
- 9.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–53. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 10.Margittai M, Fasshauer D, Jahn R, Langen R. The Habc domain and the SNARE core complex are connected by a highly flexible linker. Biochemistry. 2003;42:4009–14. doi: 10.1021/bi027437z. [DOI] [PubMed] [Google Scholar]
- 11.Filippini F, Rossi V, Galli T, Budillon A, D’Urso M, D’Esposito M. Longins: a new evolutionary conserved VAMP family sharing a novel SNARE domain. Trends Biochem Sci. 2001;26:407–9. doi: 10.1016/s0968-0004(01)01861-8. [DOI] [PubMed] [Google Scholar]
- 12.Borisovska M, et al. v-SNAREs control exocytosis of vesicles from priming to fusion. Embo J. 2005;24:2114–26. doi: 10.1038/sj.emboj.7600696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossi V, Banfield DK, Vacca M, Dietrich LE, Ungermann C, D’Esposito M, Galli T, Filippini F. Longins and their longin domains: regulated SNAREs and multifunctional SNARE regulators. Trends Biochem Sci. 2004;29:682–8. doi: 10.1016/j.tibs.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Antonin W, Fasshauer D, Becker S, Jahn R, Schneider TR. Crystal structure of the endosomal SNARE complex reveals common structural principles of all SNAREs. Nat Struct Biol. 2002;9:107–11. doi: 10.1038/nsb746. [DOI] [PubMed] [Google Scholar]
- 15.Hanson PI, Heuser JE, Jahn R. Neurotransmitter release - four years of SNARE complexes. Curr Opin Neurobiol. 1997;7:310–5. doi: 10.1016/s0959-4388(97)80057-8. [DOI] [PubMed] [Google Scholar]
- 16.Fasshauer D, Margittai M. A transient N-terminal interaction of SNAP-25 and syntaxin nucleates SNARE assembly. J Biol Chem. 2004;279:7613–21. doi: 10.1074/jbc.M312064200. [DOI] [PubMed] [Google Scholar]
- 17.Fasshauer D, Otto H, Eliason WK, Jahn R, Brunger AT. Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J Biol Chem. 1997;272:28036–41. doi: 10.1074/jbc.272.44.28036. [DOI] [PubMed] [Google Scholar]
- 18.Sorensen JB, Wiederhold K, Muller EM, Milosevic I, Nagy G, de Groot BL, Grubmuller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. Embo J. 2006;25:955–66. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chae TH, Kim S, Marz KE, Hanson PI, Walsh CA. The hyh mutation uncovers roles for alpha Snap in apical protein localization and control of neural cell fate. Nat Genet. 2004;36:264–70. doi: 10.1038/ng1302. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi T, Yamasaki S, Nauenburg S, Binz T, Niemann H. Disassembly of the reconstituted synaptic vesicle membrane fusion complex in vitro. Embo J. 1995;14:2317–25. doi: 10.1002/j.1460-2075.1995.tb07226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanson PI, Otto H, Barton N, Jahn R. The N-ethylmaleimide-sensitive fusion protein and alpha-SNAP induce a conformational change in syntaxin. J Biol Chem. 1995;270:16955–61. doi: 10.1074/jbc.270.28.16955. [DOI] [PubMed] [Google Scholar]
- 22.McMahon HT, Sudhof TC. Synaptic core complex of synaptobrevin, syntaxin, and SNAP25 forms high affinity alpha-SNAP binding site. J Biol Chem. 1995;270:2213–7. doi: 10.1074/jbc.270.5.2213. [DOI] [PubMed] [Google Scholar]
- 23.Rice LM, Brunger AT. Crystal structure of the vesicular transport protein Sec17: implications for SNAP function in SNARE complex disassembly. Mol Cell. 1999;4:85–95. doi: 10.1016/s1097-2765(00)80190-2. [DOI] [PubMed] [Google Scholar]
- 24.Marz KE, Lauer JM, Hanson PI. Defining the SNARE complex binding surface of alpha-SNAP: implications for SNARE complex disassembly. J Biol Chem. 2003;278:27000–8. doi: 10.1074/jbc.M302003200. [DOI] [PubMed] [Google Scholar]
- 25.Novick P, Field C, Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–15. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- 26.Siddiqi O, Benzer S. Neurophysiological defects in temperature-sensitive paralytic mutants of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1976;73:3253–7. doi: 10.1073/pnas.73.9.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tagaya M, Wilson DW, Brunner M, Arango N, Rothman JE. Domain structure of an N-ethylmaleimide-sensitive fusion protein involved in vesicular transport. J Biol Chem. 1993;268:2662–6. [PubMed] [Google Scholar]
- 28.Nagiec EE, Bernstein A, Whiteheart SW. Each domain of the N-ethylmaleimide-sensitive fusion protein contributes to its transport activity. J Biol Chem. 1995;270:29182–8. doi: 10.1074/jbc.270.49.29182. [DOI] [PubMed] [Google Scholar]
- 29.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–29. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 30.Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90:523–35. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- 31.Furst J, Sutton RB, Chen J, Brunger AT, Grigorieff N. Electron cryomicroscopy structure of N-ethyl maleimide sensitive factor at 11 A resolution. Embo J. 2003;22:4365–74. doi: 10.1093/emboj/cdg420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu RC, Jahn R, Brunger AT. NSF N-terminal domain crystal structure: models of NSF function. Mol Cell. 1999;4:97–107. doi: 10.1016/s1097-2765(00)80191-4. [DOI] [PubMed] [Google Scholar]
- 33.May AP, Misura KM, Whiteheart SW, Weis WI. Crystal structure of the amino-terminal domain of N-ethylmaleimide-sensitive fusion protein. Nat Cell Biol. 1999;1:175–82. doi: 10.1038/11097. [DOI] [PubMed] [Google Scholar]
- 34.Matveeva EA, May AP, He P, Whiteheart SW. Uncoupling the ATPase activity of the N-ethylmaleimide sensitive factor (NSF) from 20S complex disassembly. Biochemistry. 2002;41:530–6. doi: 10.1021/bi015632s. [DOI] [PubMed] [Google Scholar]
- 35.Babor SM, Fass D. Crystal structure of the Sec18p N-terminal domain. Proc Natl Acad Sci U S A. 1999;96:14759–64. doi: 10.1073/pnas.96.26.14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu RC, Hanson PI, Jahn R, Brunger AT. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat Struct Biol. 1998;5:803–11. doi: 10.1038/1843. [DOI] [PubMed] [Google Scholar]
- 37.Lenzen CU, Steinmann D, Whiteheart SW, Weis WI. Crystal structure of the hexamerization domain of N-ethylmaleimide-sensitive fusion protein. Cell. 1998;94:525–36. doi: 10.1016/s0092-8674(00)81593-7. [DOI] [PubMed] [Google Scholar]
- 38.Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE. N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol. 1994;126:945–54. doi: 10.1083/jcb.126.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matveeva EA, He P, Whiteheart SW. N-Ethylmaleimide-sensitive fusion protein contains high and low affinity ATP-binding sites that are functionally distinct. J Biol Chem. 1997;272:26413–8. doi: 10.1074/jbc.272.42.26413. [DOI] [PubMed] [Google Scholar]
- 40.Dalal S, Rosser MF, Cyr DM, Hanson PI. Distinct roles for the AAA ATPases NSF and p97 in the secretory pathway. Mol Biol Cell. 2004;15:637–48. doi: 10.1091/mbc.E03-02-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ogura T, Whiteheart SW, Wilkinson AJ. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J Struct Biol. 2004;146:106–12. doi: 10.1016/j.jsb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 42.Steel GJ, Harley C, Boyd A, Morgan A. A screen for dominant negative mutants of SEC18 reveals a role for the AAA protein consensus sequence in ATP hydrolysis. Mol Biol Cell. 2000;11:1345–56. doi: 10.1091/mbc.11.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wimmer C, Hohl TM, Hughes CA, Muller SA, Sollner TH, Engel A, Rothman JE. Molecular mass, stoichiometry, and assembly of 20 S particles. J Biol Chem. 2001;276:29091–7. doi: 10.1074/jbc.M011292200. [DOI] [PubMed] [Google Scholar]
- 44.Whiteheart SW, Brunner M, Wilson DW, Wiedmann M, Rothman JE. Soluble N-ethylmaleimide-sensitive fusion attachment proteins (SNAPs) bind to a multi-SNAP receptor complex in Golgi membranes. J Biol Chem. 1992;267:12239–43. [PubMed] [Google Scholar]
- 45.Barnard RJ, Morgan A, Burgoyne RD. Domains of alpha-SNAP required for the stimulation of exocytosis and for N-ethylmalemide-sensitive fusion protein (NSF) binding and activation. Mol Biol Cell. 1996;7:693–701. doi: 10.1091/mbc.7.5.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnard RJ, Morgan A, Burgoyne RD. Stimulation of NSF ATPase activity by alpha-SNAP is required for SNARE complex disassembly and exocytosis. J Cell Biol. 1997;139:875–83. doi: 10.1083/jcb.139.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eakle KA, Bernstein M, Emr SD. Characterization of a component of the yeast secretion machinery: identification of the SEC18 gene product. Mol Cell Biol. 1988;8:4098–109. doi: 10.1128/mcb.8.10.4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horsnell WG, Steel GJ, Morgan A. Analysis of NSF mutants reveals residues involved in SNAP binding and ATPase stimulation. Biochemistry. 2002;41:5230–5. doi: 10.1021/bi0160359. [DOI] [PubMed] [Google Scholar]
- 49.Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–18. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 50.Wilson DW, Whiteheart SW, Wiedmann M, Brunner M, Rothman JE. A multisubunit particle implicated in membrane fusion. J Cell Biol. 1992;117:531–8. doi: 10.1083/jcb.117.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matveeva E, Whiteheart SW. The effects of SNAP/SNARE complexes on the ATPase of NSF. FEBS Lett. 1998;435:211–4. doi: 10.1016/s0014-5793(98)01071-0. [DOI] [PubMed] [Google Scholar]
- 52.Hohl TM, Parlati F, Wimmer C, Rothman JE, Sollner TH, Engelhardt H. Arrangement of subunits in 20 S particles consisting of NSF, SNAPs, and SNARE complexes. Mol Cell. 1998;2:539–48. doi: 10.1016/s1097-2765(00)80153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X, et al. Structure of the AAA ATPase p97. Mol Cell. 2000;6:1473–84. doi: 10.1016/s1097-2765(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 54.Huyton T, et al. The crystal structure of murine p97/VCP at 3.6A. J Struct Biol. 2003;144:337–48. doi: 10.1016/j.jsb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 55.Sumida M, Hong RM, Tagaya M. Role of two nucleotide-binding regions in an N-ethylmaleimide-sensitive factor involved in vesicle-mediated protein transport. J Biol Chem. 1994;269:20636–41. [PubMed] [Google Scholar]
- 56.Morgan A, Dimaline R, Burgoyne RD. The ATPase activity of N-ethylmaleimide-sensitive fusion protein (NSF) is regulated by soluble NSF attachment proteins. J Biol Chem. 1994;269:29347–50. [PubMed] [Google Scholar]
- 57.Steel GJ, Morgan A. Selective stimulation of the D1 ATPase domain of N-ethylmaleimide-sensitive fusion protein (NSF) by soluble NSF attachment proteins. FEBS Lett. 1998;423:113–6. doi: 10.1016/s0014-5793(98)00072-6. [DOI] [PubMed] [Google Scholar]
- 58.Steel GJ, Laude AJ, Boojawan A, Harvey DJ, Morgan A. Biochemical analysis of the Saccharomyces cerevisiae SEC18 gene product: implications for the molecular mechanism of membrane fusion. Biochemistry. 1999;38:7764–72. doi: 10.1021/bi990315v. [DOI] [PubMed] [Google Scholar]
- 59.Pallanck L, Ordway RW, Ganetzky B. A Drosophila NSF mutant. Nature. 1995;376:25. doi: 10.1038/376025a0. [DOI] [PubMed] [Google Scholar]
- 60.Muller JM, Rabouille C, Newman R, Shorter J, Freemont P, Schiavo G, Warren G, Shima DT. An NSF function distinct from ATPase-dependent SNARE disassembly is essential for Golgi membrane fusion. Nat Cell Biol. 1999;1:335–40. doi: 10.1038/14025. [DOI] [PubMed] [Google Scholar]
- 61.Whiteheart SW, Matveeva EA. Multiple binding proteins suggest diverse functions for the N-ethylmaleimide sensitive factor. J Struct Biol. 2004;146:32–43. doi: 10.1016/j.jsb.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 62.Osten P, et al. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron. 1998;21:99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- 63.Nishimune A, et al. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21:87–97. doi: 10.1016/s0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- 64.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 65.Cong M, Perry SJ, Hu LA, Hanson PI, Claing A, Lefkowitz RJ. Binding of the beta2 adrenergic receptor to N-ethylmaleimide-sensitive factor regulates receptor recycling. J Biol Chem. 2001;276:45145–52. doi: 10.1074/jbc.M106087200. [DOI] [PubMed] [Google Scholar]
- 66.Heydorn A, Sondergaard BP, Hadrup N, Holst B, Haft CR, Schwartz TW. Distinct in vitro interaction pattern of dopamine receptor subtypes with adaptor proteins involved in post-endocytotic receptor targeting. FEBS Lett. 2004;556:276–80. doi: 10.1016/s0014-5793(03)01431-5. [DOI] [PubMed] [Google Scholar]
- 67.Bomberger JM, Parameswaran N, Hall CS, Aiyar N, Spielman WS. Novel function for receptor activity-modifying proteins (RAMPs) in post-endocytic receptor trafficking. J Biol Chem. 2005;280:9297–307. doi: 10.1074/jbc.M413786200. [DOI] [PubMed] [Google Scholar]
- 68.Pontier SM, et al. Coordinated action of NSF and PKC regulates GABAB receptor signaling efficacy. Embo J. 2006;25:2698–709. doi: 10.1038/sj.emboj.7601157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–74. doi: 10.1016/s0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- 70.Kim CH, Lisman JE. A labile component of AMPA receptor-mediated synaptic transmission is dependent on microtubule motors, actin, and N-ethylmaleimide-sensitive factor. J Neurosci. 2001;21:4188–94. doi: 10.1523/JNEUROSCI.21-12-04188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gage RM, Matveeva EA, Whiteheart SW, von Zastrow M. Type I PDZ ligands are sufficient to promote rapid recycling of G Protein-coupled receptors independent of binding to N-ethylmaleimide-sensitive factor. J Biol Chem. 2005;280:3305–13. doi: 10.1074/jbc.M406934200. [DOI] [PubMed] [Google Scholar]
- 72.Lefkowitz RJ. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J Biol Chem. 1998;273:18677–80. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- 73.McDonald PH, Cote NL, Lin FT, Premont RT, Pitcher JA, Lefkowitz RJ. Identification of NSF as a beta-arrestin1-binding protein. Implications for beta2-adrenergic receptor regulation. J Biol Chem. 1999;274:10677–80. doi: 10.1074/jbc.274.16.10677. [DOI] [PubMed] [Google Scholar]
- 74.Kamitani S, Asakawa M, Shimekake Y, Kuwasako K, Nakahara K, Sakata T. The RAMP2/CRLR complex is a functional adrenomedullin receptor in human endothelial and vascular smooth muscle cells. FEBS Lett. 1999;448:111–4. doi: 10.1016/s0014-5793(99)00358-0. [DOI] [PubMed] [Google Scholar]
- 75.Han SY, Park DY, Park SD, Hong SH. Identification of Rab6 as an N-ethylmaleimide-sensitive fusion protein-binding protein. Biochem J. 2000;352(Pt 1):165–73. [PMC free article] [PubMed] [Google Scholar]
- 76.Tani K, Shibata M, Kawase K, Kawashima H, Hatsuzawa K, Nagahama M, Tagaya M. Mapping of functional domains of gamma-SNAP. J Biol Chem. 2003;278:13531–8. doi: 10.1074/jbc.M213205200. [DOI] [PubMed] [Google Scholar]
- 77.Martin HG, Henley JM, Meyer G. Novel putative targets of N-ethylmaleimide sensitive fusion protein (NSF) and alpha/beta soluble NSF attachment proteins (SNAPs) include the Pak-binding nucleotide exchange factor betaPIX. J Cell Biochem. 2006;99:1203–15. doi: 10.1002/jcb.20998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McBride HM, Rybin V, Murphy C, Giner A, Teasdale R, Zerial M. Oligomeric complexes link Rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell. 1999;98:377–86. doi: 10.1016/s0092-8674(00)81966-2. [DOI] [PubMed] [Google Scholar]
- 79.Legesse-Miller A, Sagiv Y, Porat A, Elazar Z. Isolation and characterization of a novel low molecular weight protein involved in intra-Golgi traffic. J Biol Chem. 1998;273:3105–9. doi: 10.1074/jbc.273.5.3105. [DOI] [PubMed] [Google Scholar]
- 80.Elazar Z, Scherz-Shouval R, Shorer H. Involvement of LMA1 and GATE-16 family members in intracellular membrane dynamics. Biochim Biophys Acta. 2003;1641:145–56. doi: 10.1016/s0167-4889(03)00086-7. [DOI] [PubMed] [Google Scholar]
- 81.Halawani D, Latterich M. p97: The cell’s molecular purgatory? Mol Cell. 2006;22:713–7. doi: 10.1016/j.molcel.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 82.Sagiv Y, Legesse-Miller A, Porat A, Elazar Z. GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. Embo J. 2000;19:1494–504. doi: 10.1093/emboj/19.7.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laviolette MJ, Nunes P, Peyre JB, Aigaki T, Stewart BA. A genetic screen for suppressors of Drosophila NSF2 neuromuscular junction overgrowth. Genetics. 2005;170:779–92. doi: 10.1534/genetics.104.035691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boulianne GL, Trimble WS. Identification of a second homolog of N-ethylmaleimide-sensitive fusion protein that is expressed in the nervous system and secretory tissues of Drosophila. Proc Natl Acad Sci U S A. 1995;92:7095–9. doi: 10.1073/pnas.92.15.7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Golby JA, Tolar LA, Pallanck L. Partitioning of N-ethylmaleimide-sensitive fusion (NSF) protein function in Drosophila melanogaster: dNSF1 is required in the nervous system, and dNSF2 is required in mesoderm. Genetics. 2001;158:265–78. doi: 10.1093/genetics/158.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peyre JB, Seabrooke S, Randlett O, Kisiel M, Aigaki T, Stewart BA. Interaction of cytoskeleton genes with NSF2-induced neuromuscular junction overgrowth. Genesis. 2006;44:595–600. doi: 10.1002/dvg.20254. [DOI] [PubMed] [Google Scholar]
- 87.Thompson CR, Bretscher MS. Cell polarity and locomotion, as well as endocytosis, depend on NSF. Development. 2002;129:4185–92. doi: 10.1242/dev.129.18.4185. [DOI] [PubMed] [Google Scholar]
- 88.Matveeva EA, Whiteheart SW, Vanaman TC, Slevin JT. Phosphorylation of the N-ethylmaleimide-sensitive factor is associated with depolarization-dependent neurotransmitter release from synaptosomes. J Biol Chem. 2001;276:12174–81. doi: 10.1074/jbc.M007394200. [DOI] [PubMed] [Google Scholar]
- 89.Liu Y, Cheng K, Gong K, Fu AK, Ip NY. Pctaire1 phosphorylates N-ethylmaleimide-sensitive fusion protein: implications in the regulation of its hexamerization and exocytosis. J Biol Chem. 2006;281:9852–8. doi: 10.1074/jbc.M513496200. [DOI] [PubMed] [Google Scholar]
- 90.Huynh H, et al. Control of vesicle fusion by a tyrosine phosphatase. Nat Cell Biol. 2004;6:831–9. doi: 10.1038/ncb1164. [DOI] [PubMed] [Google Scholar]
- 91.Matsushita K, et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139–50. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang Y, et al. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–40. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 93.Matsushita K, Morrell CN, Mason RJ, Yamakuchi M, Khanday FA, Irani K, Lowenstein CJ. Hydrogen peroxide regulation of endothelial exocytosis by inhibition of N-ethylmaleimide sensitive factor. J Cell Biol. 2005;170:73–9. doi: 10.1083/jcb.200502031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guan Z, et al. A spontaneous recurrent seizure-related Rattus NSF gene identified by linker capture subtraction. Brain Res Mol Brain Res. 2001;87:117–23. doi: 10.1016/s0169-328x(00)00286-2. [DOI] [PubMed] [Google Scholar]
- 95.Yu F, Guan Z, Zhuo M, Sun L, Zou W, Zheng Z, Liu X. Further identification of NSF* as an epilepsy related gene. Brain Res Mol Brain Res. 2002;99:141–4. doi: 10.1016/s0169-328x(01)00345-x. [DOI] [PubMed] [Google Scholar]
- 96.Matveeva EA, Vanaman TC, Whiteheart SW, Slevin JT. Asymmetric accumulation of hippocampal 7S SNARE complexes occurs regardless of kindling paradigm. Epilepsy Res. 2006 doi: 10.1016/j.eplepsyres.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goto H, Terunuma M, Kanematsu T, Misumi Y, Moss SJ, Hirata M. Direct interaction of N-ethylmaleimide-sensitive factor with GABA(A) receptor beta subunits. Mol Cell Neurosci. 2005;30:197–206. doi: 10.1016/j.mcn.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 98.Zou S, Li L, Pei L, Vukusic B, Van Tol HH, Lee FJ, Wan Q, Liu F. Protein-protein coupling/uncoupling enables dopamine D2 receptor regulation of AMPA receptor-mediated excitotoxicity. J Neurosci. 2005;25:4385–95. doi: 10.1523/JNEUROSCI.5099-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Heydorn A, Sondergaard BP, Ersboll B, Holst B, Nielsen FC, Haft CR, Whistler J, Schwartz TW. A library of 7TM receptor C-terminal tails. Interactions with the proposed post-endocytic sorting proteins ERM-binding phosphoprotein 50 (EBP50), N-ethylmaleimide-sensitive factor (NSF), sorting nexin 1 (SNX1), and G protein-coupled receptor-associated sorting protein (GASP) J Biol Chem. 2004;279:54291–303. doi: 10.1074/jbc.M406169200. [DOI] [PubMed] [Google Scholar]
- 100.Xu Z, Sato K, Wickner W. LMA1 binds to vacuoles at Sec18p (NSF), transfers upon ATP hydrolysis to a t-SNARE (Vam3p) complex, and is released during fusion. Cell. 1998;93:1125–34. doi: 10.1016/s0092-8674(00)81457-9. [DOI] [PubMed] [Google Scholar]
- 101.Kittler JT, Rostaing P, Schiavo G, Fritschy JM, Olsen R, Triller A, Moss SJ. The subcellular distribution of GABARAP and its ability to interact with NSF suggest a role for this protein in the intracellular transport of GABA(A) receptors. Mol Cell Neurosci. 2001;18:13–25. doi: 10.1006/mcne.2001.1005. [DOI] [PubMed] [Google Scholar]
- 102.Chen C, Li JG, Chen Y, Huang P, Wang Y, Liu-Chen LY. GEC1 interacts with the kappa opioid receptor and enhances expression of the receptor. J Biol Chem. 2006;281:7983–93. doi: 10.1074/jbc.M509805200. [DOI] [PubMed] [Google Scholar]