

Abstract
Monocyte-macrophage activation by interferon (IFN)-γ is a key initiating event in inflammation. Usually, the macrophage response is self-limiting and inflammation resolves. Here, we describe a mechanism by which IFN-γ contributes to inflammation resolution by suppressing expression of vascular endothelial growth factor-A (VEGF-A), a macrophage product that stimulates angiogenesis during chronic inflammation and tumorigenesis. VEGF-A was identified as a candidate target of the IFN-γ-activated inhibitor of translation (GAIT) complex by bioinformatic analysis, and experimentally validated by messenger RNA–protein interaction studies. Although IFN-γ induced persistent VEGF-A mRNA expression, translation was suppressed by delayed binding of the GAIT complex to a specific element delineated in the 3′UTR. Translational silencing resulted in decreased VEGF-A synthesis and angiogenic activity. Our results describe a unique anti-inflammatory pathway in which IFN-γ-dependent induction of VEGF-A mRNA is translationally silenced by the same stimulus, and they suggest the GAIT system directs a post-transcriptional operon that contributes to inflammation resolution.
Keywords: inflammation, interferon-gamma, RNA, translational control, VEGF-A
Introduction
Monocyte/macrophages respond to infection or injury by activating an ensemble of cytokine- and growth factor-inducible programs of gene expression that targets invading microbes and infected cells. The macrophage inflammatory response must be tightly regulated to balance destruction of foreign agents with protection of host tissue (Nathan, 2002). Remarkably, the macrophage also exhibits anti-inflammatory functions that participate in tissue-healing processes after removal or neutralization of the initial trauma. Thus, macrophages must integrate a diversity of inputs to determine the appropriate pro- or anti-inflammatory response in a particular situation. During the resolution phase, macrophage expression of inflammatory genes is often switched off at the transcriptional level (Ohmori and Hamilton, 1994). However, recent attention has focused on post-transcriptional mechanisms that reduce inflammatory gene expression by dynamic interactions between cis-elements in messenger RNAs and specific RNA-binding protein(s), which cause mRNA destabilization or translational silencing (Wilusz et al, 2001; Kracht and Saklatvala, 2002).
Interferon (IFN)-γ is an inflammatory cytokine released by T-lymphocytes and natural killer cells that activate multiple host-defense responses in macrophages (Schroder et al, 2004). IFN-γ induces the formation and activation of IFN-γ-activated inhibitor of translation (GAIT), a heterotetrameric RNA-binding complex in monocytes/macrophages (Sampath et al, 2004). The GAIT complex inhibits translation of ceruloplasmin (Cp), an acute phase inflammatory protein secreted systemically into plasma by hepatocytes, and locally in sites of inflammation by cytokine-stimulated macrophages (Mazumder and Fox, 1999). IFN-γ induces Cp mRNA transcription and protein expression in peripheral blood monocytes and U937 cells, a human pro-monocytic cell line; however, synthesis of the protein stops after about 16 h despite the presence of abundant Cp mRNA (Mazumder et al, 1997; Mazumder and Fox, 1999). The GAIT complex forms about 16 h after cell activation by IFN-γ. It binds to a bipartite stem–loop element (GAIT element) in the Cp mRNA 3′UTR, inhibiting its translation and causing complete cessation of Cp synthesis by about 24 h (Sampath et al, 2003). The GAIT complex consists of ribosomal protein L13a, glutamyl-prolyl tRNA synthetase (EPRS), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and NS1-associated protein 1 (NSAP1) (Sampath et al, 2004). L13a and EPRS are mobilized into the GAIT complex after release from their host complexes, that is, the ribosome and the tRNA multisynthetase complex, respectively (Mazumder et al, 2003; Sampath et al, 2004). Delayed translational silencing of Cp expression may be necessary to limit Cp accumulation in the cellular microenvironment and prevent oxidative injury (Mukhopadhyay et al, 1997).
Cp is not likely to be the sole GAIT pathway target. The very high cellular abundance of the four GAIT complex components and the near-stoichiometric release of L13a and EPRS from their parent complexes indicate that the complex is in marked excess of Cp mRNA (Mazumder et al, 2003; Sampath et al, 2004). Thus, Cp may belong to a family of transcripts regulated by the GAIT complex. Coordinate, post-transcriptional regulation of functionally related genes has been shown for mRNAs containing common structural elements in their UTRs, recognized by specific RNA-binding, trans-acting factor(s) (Brown et al, 2001; López de Silanes et al, 2005). This control system has been termed a ‘post-transcriptional operon' (Keene and Tenenbaum, 2002), and examples include targets of fragile X mental retardation protein, ELAV/Hu proteins, and the Pumilio/Puf family of proteins that interact with specific structural elements in mRNAs and coordinately regulate their stability or translation (Tenenbaum et al, 2000; Brown et al, 2001; Gerber et al, 2006).
To discover potential target transcripts of the GAIT complex, we used a pattern-searching algorithm to query a 3′UTR database. Among the transcripts predicted to contain GAIT element-like RNA structures, vascular endothelial growth factor-A (VEGF-A) was of particular interest. It is produced by inflammatory macrophages and it increases vascular permeability, stimulates angiogenesis, and enhances monocyte recruitment, all hallmarks of inflammation (Ferrara and Davis-Smyth, 1997; Weis and Cheresh, 2005; Zittermann and Issekutz, 2006). Macrophages play a key role in inflammatory and tumor angiogenesis, and they regulate the angiogenic switch in a mouse model of breast cancer (Sunderkötter et al, 1994; Lin et al, 2006). VEGF-A synthesis is induced in macrophages activated by pro-inflammatory agonists, for example, IFN-γ plus bacterial lipopolysaccharide (Xiong et al, 1998; Ramanathan et al, 2003). VEGF-A expression is regulated by hypoxia and cytokines at the levels of transcription, mRNA stability, and translation (Pages and Pouyssegur, 2005; Hua et al, 2006). Stimulus-dependent activation of any of these regulatory mechanisms increases VEGF-A expression. Overproduction of VEGF-A can cause excessive neovascularization and vessel permeability, events associated with tumorigenesis and chronic inflammatory conditions (Tammela et al, 2005). Because negative regulatory mechanisms for VEGF-A expression under inflammatory conditions have not been identified, GAIT element-mediated translational repression of VEGF-A after inflammatory stimulation might be particularly important.
Here, we show the GAIT complex suppresses VEGF-A expression in IFN-γ-activated monocytic cells by inhibiting its translation. We conclude that VEGF-A expression is subject to negative regulation after inflammatory stimulation, and that the GAIT regulatory system directs a post-transcriptional operon that limits the response to inflammatory stimuli, and may contribute to the resolution of chronic inflammation.
Results
VEGF-A mRNA is a binding target of the GAIT complex in IFN-γ-treated monocytes
To date, a functional GAIT element has been identified only in Cp mRNA (Sampath et al, 2003). We utilized a consensus GAIT element pattern, based on the sequence and secondary structure of the human Cp GAIT element, to query a nonredundant 3′UTR database containing 217 135 sequences using PatSearch, a folding energy-independent, pattern-matching algorithm that searches for nucleotide sequences as well as secondary structures, that is, stems and loops of defined length (Grillo et al, 2003) (Figure 1A). PatSearch predicted putative GAIT elements in the 3′UTRs of 190 eukaryotic transcripts, including 93 mammalian and 52 human mRNAs (Supplementary Table S1). In contrast, query of a 5′UTR database containing 190 432 sequences showed GAIT-like elements in only five mammalian transcripts, suggesting the element is a bona fide 3′UTR regulatory element. As expected, the human Cp GAIT element was identified (Figure 1B). A gene ontology (GO)-based literature search (using the GoPubmed server) of the human transcripts for which GO annotation was available, showed that more than 50% (19/39) were associated with the inflammatory response or induced by inflammatory agonists (Doms and Schroeder, 2005). Prediction of a GAIT element in the 3′UTR of VEGF-A (Figure 1B) was notable, because it is a key mediator of inflammatory angiogenesis, and its expression is induced by multiple stimuli. GAIT element-mediated translational repression of VEGF-A would be particularly significant, as negative regulatory mechanisms for VEGF-A expression have not been identified.
Figure 1.
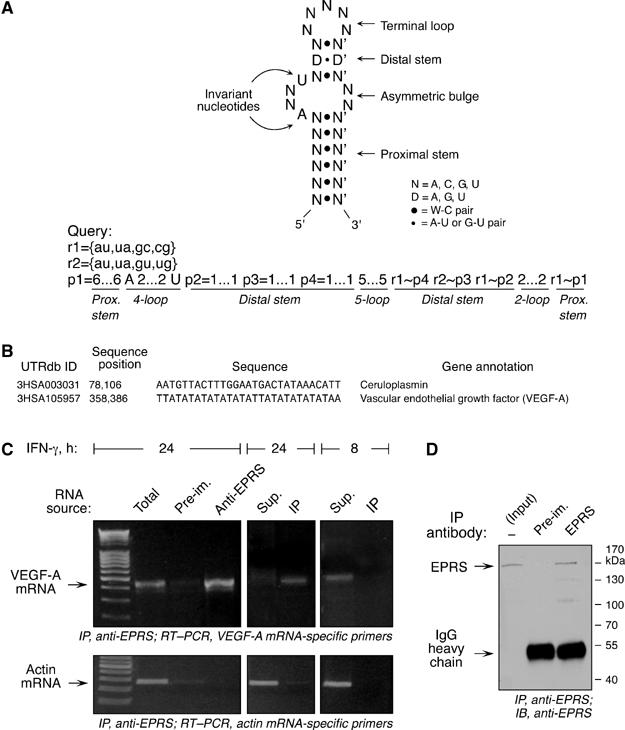
VEGF-A mRNA interacts with the GAIT complex. (A) Secondary structure and sequence features of the human Cp GAIT element (top panel). The query pattern, based on the secondary structure and sequence features of the Cp GAIT element, was used to search a nonredundant 3′UTR database using the PatSearch program (bottom panel). Following the syntax of the PatSearch algorithm, allowed base-pairs are represented by rnumber and patterns defined by pnumber. The GAIT element-specific stems and loops are shown below. (B) PatSearch result predicted the presence of GAIT elements in Cp and VEGF-A 3′UTR. UTRdb ID refers to the sequence entry in the UTR database, and sequence position refers to the 3′UTR position of the sequence encoding the predicted GAIT element. (C) To show VEGF-A mRNA interaction with the GAIT complex in vivo, U937 cells were treated with IFN-γ for 8 or 24 h, and lysates were immunoprecipitated (IP) with anti-EPRS antibody to isolate GAIT complex, or with control pre-immune (Pre-im.) serum. RNA associated with the GAIT complex, or present in the non-immunoprecipitated supernatant (Sup.), was subjected to RT–PCR using primers specific for VEGF-A or β-actin mRNA, and products were resolved in 1.6% agarose gels. (D) To verify antibody specificity, lysate from U937 cells treated with IFN-γ for 24 h was immunoprecipitated with polyclonal anti-human EPRS antibody and immunoblotted with the same antibody, or with pre-immune serum as control.
To assess GAIT complex binding to VEGF-A mRNA, we used an anti-EPRS antibody to immunoprecipitate the GAIT complex from lysates of cells treated with IFN-γ for 24 h. EPRS is the only component of the heterotetrameric GAIT complex that directly interacts with the Cp GAIT element, and co-immunoprecipitates with the other GAIT complex components (Sampath et al, 2004). RNA associated with the isolated GAIT complex was amplified by RT–PCR using VEGF-A-specific primers and revealed abundant, bound VEGF-A mRNA (Figure 1C). RT–PCR using actin-specific primers showed specificity of the interaction. Quantitative RT–PCR, normalized to β-actin mRNA, showed a 13.3-fold enrichment of VEGF-A mRNA in the anti-EPRS immunoprecipitate compared to total cellular RNA, and a 180-fold enrichment in the anti-EPRS immunoprecipitate compared with the pre-immune IgG immunoprecipitate. VEGF-A mRNA was specifically present in the immunoprecipitated fraction from cells treated with IFN-γ for 24 h, but not for 8 h, showing that VEGF-A mRNA interacts with EPRS in the functional GAIT complex induced after 24-h treatment with IFN-γ, but not with EPRS in the nonfunctional pre-GAIT complex formed after 8-h. Anti-EPRS antibody specificity and immunoprecipitation efficiency were shown by immunoblot analysis of anti-EPRS immunoprecipitate from 8- and 24-h lysates from IFN-γ-treated U937 cells (Figure 1D and Supplementary Figure S1). Thus, VEGF-A mRNA is a likely target for GAIT complex-mediated translational repression.
Translational silencing of VEGF-A expression in IFN-γ-treated monocytic cells
We examined translational regulation of endogenous, cellular VEGF-A gene expression by IFN-γ. U937 cells exhibited low, basal expression of VEGF-A mRNA, but a robust induction by IFN-γ was seen after 8 h, and still higher expression after 24 h (Figure 2A). Quantitative RT–PCR, normalized to β-actin mRNA, showed a 3.2- and 9.7-fold increase in VEGF-A mRNA after 8 and 24 h of IFN-γ treatment, respectively. Likewise, cellular VEGF-A protein increased markedly 8 h after IFN-γ treatment. However, VEGF-A was reduced to nearly the basal level after 24 h despite abundant transcript, a finding consistent with post-transcriptional repression (Figure 2B). Human peripheral blood mononuclear cells (PBMC) treated with IFN-γ also showed reduced VEGF-A protein after 24 h despite the presence of abundant VEGF-A mRNA, indicating an active silencing mechanism in primary monocytes (Figure 2C and D). IFN-γ treatment for 8 h increased the rate of de novo synthesis of VEGF-A in both U937 cell lysates and conditioned media, as measured by metabolic labeling with 35S-Met/Cys, followed by immunoprecipitation with anti-VEGF-A antibody (Figure 2E, top panel). However, synthesis declined after 16 h of IFN-γ treatment and was almost completely inhibited after 24 h. Labeling of protein in conditioned medium and lysate not subjected to immunoprecipitation was unaffected by IFN-γ, indicating global protein synthesis was not inhibited and delayed silencing of VEGF-A synthesis was transcript-selective (Figure 2E, bottom panel). To eliminate the possibility of increased proteolytic degradation of VEGF-A protein after 24-h IFN-γ treatment, recombinant VEGF-A protein was incubated with lysate and conditioned medium from cells treated with IFN-γ for 8 and 24 h. No significant degradation of recombinant VEGF-A was observed, suggesting the low amount of VEGF-A protein in the 24-h cell lysate and conditioned medium was not due to induction of proteolytic activity (Supplementary Figure S2).
Figure 2.
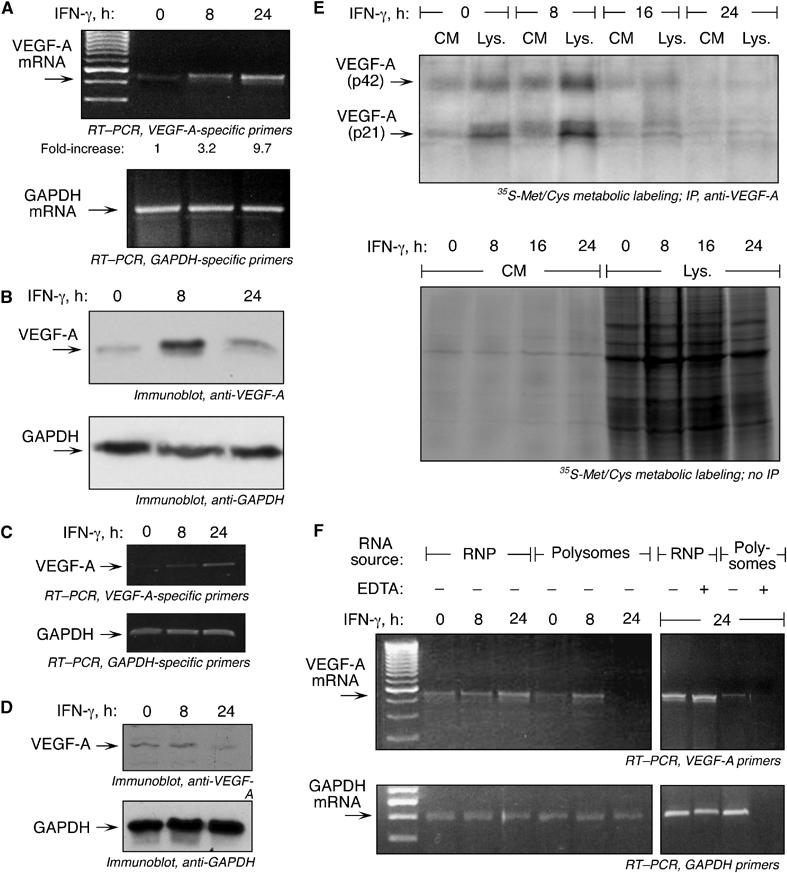
Translational silencing of VEGF-A expression in vivo. (A) RT–PCR analysis of total RNA from U937 cells treated with IFN-γ for 0, 8, or 24 h. RT–PCR was done using primers specific for VEGF-A (top panel) and GAPDH (bottom panel). Real-time PCR results indicating the increase in VEGF-A mRNA expression in IFN-γ-treated cells compared to untreated cells are included below the top panel (expressed as fold-increase normalized to β-actin). (B) Cell lysates from U937 cells treated with IFN-γ for 0, 8, or 24 h were processed in Laemlli gel-loading buffer in absence of reducing agent. Lysates were subjected to immunoblotting with anti-VEGF-A (top) and anti-GAPDH (bottom panel) antibodies. (C) RT–PCR analysis of total RNA from human PBMCs treated with IFN-γ for 0, 8, or 24 h. RT–PCR was performed using primers specific for VEGF-A (top panel) and GAPDH (bottom panel). (D) Cell lysates from PBMCs treated with IFN-γ for 0, 8, or 24 h were processed in Laemlli gel-loading buffer in absence of reducing agent. Lysates were subjected to immunoblotting with anti-VEGF-A (top) and anti-GAPDH (bottom panel) antibodies. (E) U937 cells were treated with IFN-γ for up to 24 h. At the end of each interval, cells were metabolically labeled with [35S]Met/Cys for 1 h. Conditioned media and cell lysates were immunoprecipitated with anti-VEGF-A antibody and resolved by electrophoresis on SDS–10% polyacrylamide gel (top panel). Monomeric and dimeric VEGF-A forms are indicated by arrows. The same samples were subjected to electrophoresis without immunoprecipitation (bottom). (F) U937 cells were treated with IFN-γ for 8 or 24 h and cytosolic extracts were fractionated into polysomal and non-polysomal, RNP fractions by ultracentrifugation on a 20% sucrose cushion in the presence or absence of 10 mM EDTA. RNA associated with each fraction was isolated and subjected to RT–PCR using primers specific for VEGF-A (top panel) and GAPDH (bottom panel).
To verify that IFN-γ inhibited VEGF-A mRNA translation, we examined VEGF-A mRNA association with polysomes. Inhibition of translation initiation is accompanied by mRNA translocation from the high-density, polysomal pool to a low-density, ribosome-poor ribonucleoprotein (RNP) pool. Cytosolic extracts of IFN-γ-treated cells were fractionated into polysome and RNP fractions and VEGF-A mRNA determined by RT–PCR. After IFN-γ treatment for 8 h, when VEGF-A protein synthesis is high, abundant VEGF-A mRNA was polysome-associated (Figure 2F, top panel). However, after 24 h, VEGF-A mRNA was absent from the polysome fraction, indicating VEGF-A translation was blocked. As a control, RT–PCR with GAPDH-specific primers showed continuous association of GAPDH mRNA with polysomes, indicating translation repression was selective for VEGF-A mRNA (Figure 2F, bottom panel). To confirm that mRNA in the polysome fraction was not the result of nonspecific interactions or formation of heavy mRNP aggregates, the cell lysates were treated with 10 mM EDTA to disrupt ribosome assembly on mRNA. EDTA entirely shifted the GAPDH message from the polysomal to the RNP fraction, indicating authentic polysome association.
The putative GAIT element in the VEGF-A 3′UTR mediates translation inhibition
To begin to investigate the role of the putative 3′UTR GAIT element, we examined whether the VEGF-A mRNA 3′UTR confers translational silencing to a heterologous reporter RNA in an in vitro translation system. A segment of the 1942-nt VEGF-A 3′UTR containing nucleotides 11–900 (3′UTR11–900) was inserted downstream of the firefly luciferase (FLuc) open reading frame (Figure 3A, top panel). The segment excludes the distal AU-rich elements (AREs) that confer RNA instability (Levy et al, 1996). A capped, poly(A)-tailed RNA was transcribed in vitro and used as template for translation in a rabbit reticulocyte lysate (RRL) system. Lysates from cells treated with IFN-γ for 16 or 24 h strongly repressed FLuc-VEGF-A 3′UTR(11–900)-A30 translation (Figure 3A, middle panel). In contrast, lysates from untreated cells or cells treated with IFN-γ for 8 h did not inhibit reporter translation. The time course of translation silencing by cell lysates is consistent with the binding of VEGF-A mRNA to the GAIT complex, and with the time course of GAIT complex activation (Mazumder and Fox, 1999). Capped Renilla luciferase (RLuc) RNA lacking the VEGF-A 3′UTR, used as a control template, was not inhibited. To confirm the specific role of the VEGF-A 3′UTR in translation inhibition, we added an excess of VEGF-A 3′UTR(11–900)-A30 RNA as a decoy. The decoy restored translation of reporter RNA, indicating VEGF-A mRNA 3′UTR is responsible for the activity (Figure 3A, bottom panel). To more precisely localize the translation-silencing region of VEGF-A 3′UTR, a shorter segment (nt 324–455), containing the predicted GAIT element (nt 358–386), was inserted downstream of FLuc. In vitro translation of FLuc-VEGF-A 3′UTR(324–455)-A30 RNA was inhibited by lysates from 16- and 24-h IFN-γ-treated U937 cells (Figure 3B, top panel), but translation of a reporter RNA containing an adjacent segment of the VEGF-A 3′UTR not encompassing the putative GAIT element (FLuc-VEGF-A 3′UTR(441–560)-A30) was unaffected (Figure 3B, bottom panel).
Figure 3.
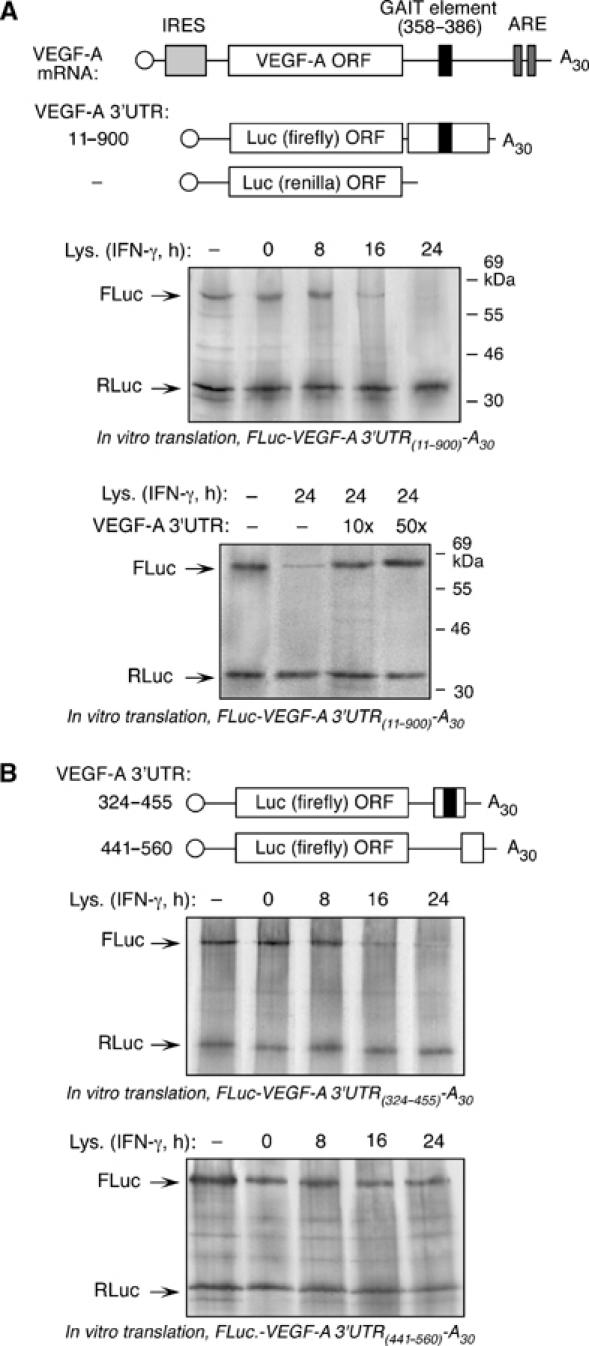
The 3′UTR of VEGF-A mRNA mediates translation inhibition. (A) Schematic of VEGF-A mRNA and chimeric luciferase constructs used for in vitro translation (top panel). The m7G cap is indicated by an open circle, the IRES by a light gray rectangle, the putative GAIT element by a black rectangle, and the AREs by dark gray rectangles. Capped, FLuc-VEGF-A 3′UTR(11–900)-A30 RNA was translated in RRL containing [35S]Met, and in absence or presence of cytosolic extracts from U937 cells treated with IFN-γ for up to 24 h (middle panel). Capped, RLuc RNA lacking the GAIT element was co-translated in each reaction as control. Translation reactions were resolved on SDS–10% polyacrylamide gel. The same RNAs were translated in the presence of cytosolic extract from 24-h, IFN-γ-treated U937 cells, and in the presence of 10- and 50-fold molar excess of in vitro transcribed VEGF-A 3′UTR RNA as competitor (bottom panel). (B) Schematic of chimeric luciferase constructs used for in vitro translation (top panel). In vitro translation, in presence of IFN-γ-treated U937 cytosolic extracts, of capped FLuc-VEGF-A 3′UTR(324–455)-A30 encompassing the putative GAIT element (middle panel), and FLuc-VEGF-A 3′UTR(441–560)-A30 (bottom panel). RLuc RNA was co-translated in each reaction.
We then focused specifically on the predicted GAIT element in the VEGF-A 3′UTR. The Mfold algorithm (under base-pairing constraints) predicts a bipartite stem–loop structure similar to the Cp GAIT element (Figure 4A), containing A7 and U10 residues in the internal bulge required for Cp GAIT element activity (Sampath et al, 2003). The wild-type 29-nt VEGF-A GAIT element and an element containing a U10C mutation (GAITmut) were inserted downstream of FLuc to generate capped and poly(A)-tailed Luc-VEGF-A GAIT-A30 and Luc-VEGF-A GAITmut-A30 RNAs. In vitro translation of Luc-VEGF-A GAIT-A30 RNA, but not Luc-VEGF-A-GAITmut-A30, was repressed by 16- and 24-h IFN-γ-treated U937 cell lysates (Figure 4B). Human PBMCs exhibited similar inhibitory activity; lysates from cells treated with IFN-γ for 24 h inhibited translation of Luc reporter RNA bearing the VEGF-A GAIT element by more than 50% (Figure 4C). Thus, the 29-nt sequence in the VEGF-A 3′UTR acts as a bona fide GAIT element and inhibits translation of an upstream reporter in the presence of lysates from IFN-γ-treated monocytes. The translational silencing ability of the VEGF-A GAIT element was tested in vivo. CMV-driven plasmids expressing FLuc upstream of the 29-nt wild-type and U10C mutant VEGF-A GAIT elements were transfected into U937 cells. A plasmid containing RLuc under SV40 promoter was co-transfected as control. IFN-γ treatment for 24 h inhibited CMV-FLuc-GAIT-A30 expression by about 65% (Figure 4D). No inhibition was observed after 8-h treatment. Expression of FLuc from the plasmid containing the mutant GAIT element, or no insert, was not inhibited by IFN-γ treatment. These results recapitulated the in vitro observations and confirmed translational silencing activity of the VEGF-A GAIT element in vivo.
Figure 4.
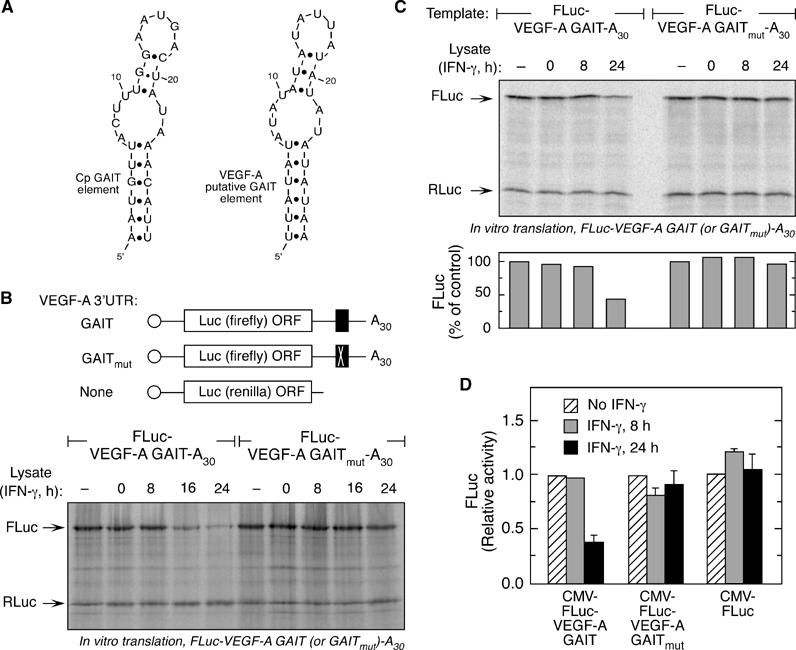
Functional identification of the VEGF-A 3′UTR GAIT element. (A) Folding structures of the Cp (nt 78–106) and the putative VEGF-A GAIT (nt 358–386) elements as predicted by the Mfold algorithm. Base pairing between A7:U23 and U8:A22 was disallowed while folding the VEGF-A GAIT element. (B) Chimeric luciferase constructs containing wild-type or mutant VEGF-A 3′UTR GAIT elements. Capped and poly-A tailed RNAs, containing the putative VEGF-A GAIT element (FLuc-VEGF-A GAIT-A30) or a mutant (U10C) GAIT element (FLuc-VEGF-A GAITmut-A30), downstream of FLuc (top panel), were subjected to in vitro translation in presence of cytosolic extracts from IFN-γ-treated U937 cells (bottom panel). RLuc RNA was co-translated in each reaction. (C) Capped and poly-A tailed RNAs, containing the putative VEGF-A GAIT element or a mutant GAIT element as in (B), were subjected to in vitro translation in presence of cytosolic extracts from IFN-γ-treated human PBMC (top panel). RLuc RNA was co-translated in each reaction. Fluc was quantified by densitometry, normalized to Rluc, and expressed as per cent of control condition without cell lysate (bottom). (D) U937 cells were transfected with eukaryotic, CMV-driven expression vectors containing the FLuc gene upstream of either wild-type (CMV-FLuc-VEGF-A GAIT-A30) or mutant VEGF-A GAIT element (CMV-FLuc-VEGF-A GAITmut-A30) or lacking any GAIT element (CMV-FLuc). Cells were co-transfected with a vector containing RLuc gene under the SV40 promoter. Following transfection, cells were treated with IFN-γ for 8 (gray bars) or 24 h (black bars), or with medium alone (hatched bars). Luciferase activity in cell lysates was measured by dual luciferase assay. Results show mean and standard deviation of values from three independent experiments.
The GAIT complex binds VEGF-A GAIT element and causes translational silencing
We investigated the role of the GAIT complex, consisting of EPRS, ribosomal protein L13a, NSAP1 and GAPDH, in binding the putative VEGF-A GAIT element and silencing translation. Protein binding to an [α-32P]UTP-labeled VEGF-A GAIT element riboprobe was assessed by electrophoretic mobility shift assay (EMSA). Lysates from U937 cells treated with IFN-γ for 16 or 24 h showed a protein or complex that bound the VEGF-A GAIT element with a binding pattern similar to that of the Cp GAIT element (Figure 5A). The interaction was not observed in lysates from cells treated with IFN-γ for 8 h or less. Antibodies against all four GAIT complex proteins supershifted the nucleoprotein complex formed by the VEGF-A GAIT element with a lysate from 24-h IFN-γ-treated cells, confirming the interaction with the mature, heterotetrameric GAIT complex (Figure 5B). We investigated whether interaction with the GAIT complex was responsible for translational silencing activity of the VEGF-A 3′UTR. The GAIT complex was immunodepleted from a lysate from IFN-γ-treated cells using anti-EPRS antibody (Figure 5C). The depleted lysate failed to inhibit in vitro translation of FLuc-VEGF-A 3′UTR(11–900)-A30 RNA (Figure 5D). Finally, we examined whether ablation of GAIT complex activity, by RNAi-mediated knockdown of L13a, an essential component of the GAIT complex, restored VEGF-A production in vivo. U937 cells stably transfected with a plasmid expressing a short hairpin RNA directed against L13a (U937-L13a-SHR) showed strong inhibition of L13a expression compared to a control cell line stably transfected with the plasmid alone (U937-pSUPER) (Figure 6A). VEGF-A protein synthesis was suppressed upon 24-h IFN-γ treatment in the control cells despite the presence of abundant VEGF-A mRNA, but the L13a-deficient cell line showed little or no translational suppression (Figure 6B and C). Consistent with the lack of translational silencing in U937-L13a-SHR cells, the GAIT complex immunoprecipitated from these cells treated with IFN-γ for 24-h showed substantially reduced association of VEGF-A mRNA compared with control cells (Figure 6D). Together, these results show the GAIT complex binds the VEGF-A GAIT element, and is responsible for the translational silencing mediated by the VEGF-A 3′UTR.
Figure 5.
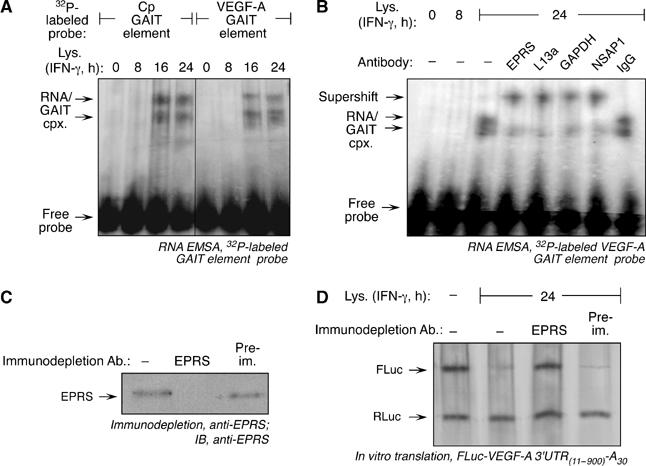
The GAIT complex binds the VEGF-A GAIT element and causes translational silencing. (A) RNA EMSA using 32P-labeled Cp and VEGF-A GAIT element probes. The riboprobes were incubated with cytosolic extracts from U937 cells treated with IFN-γ for up to 24 h. RNA–protein complexes were resolved by electrophoresis on a nondenaturing 5% polyacrylamide gel. (B) RNA–protein complexes formed between 32P-labeled VEGF-A GAIT element RNA and lysates from 24-h, IFN-γ-treated U937 cells were supershifted with antibodies against GAIT complex components. The cell lysate was incubated with the respective antibodies or non-immune IgG before incubation with the riboprobe. (C) Lysate from U937 cells treated with IFN-γ for 24 h was incubated with protein-A Sepharose beads coupled to anti-EPRS antibody (or to pre-immune serum, Pre-im.) to immunodeplete the GAIT complex. The beads were pelleted, and the supernatant subjected to immunoblotting with anti-EPRS antibody to verify effective immunodepletion. (D) At 24-h, IFN-γ-treated U937 cell lysates, immunodepleted with anti-EPRS antibody or pre-immune serum, were added to in vitro translation reactions containing FLuc-VEGF-A 3′UTR(11–900)-A30 and RLuc RNAs.
Figure 6.
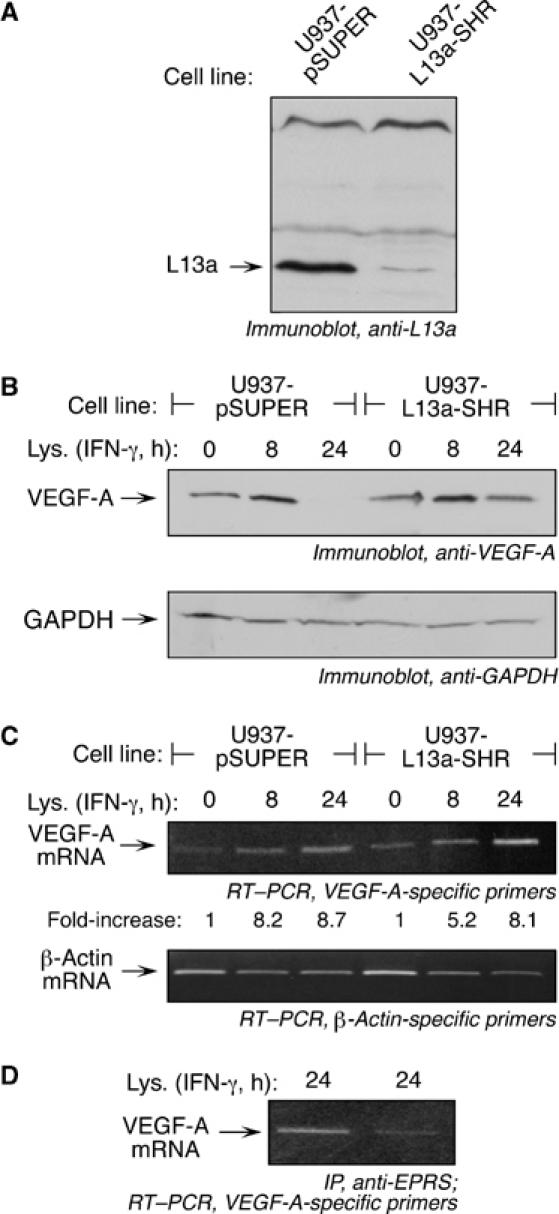
Ablation of the GAIT complex in vivo prevents translational silencing of VEGF-A. (A) Lysates from U937 cells stably transfected with pSUPER vector (U937-pSUPER) or pSUPER encoding a short hairpin RNA targeting L13a (U937-L13a-SHR) were immunoblotted with anti-L13a antibody. (B) Lysates from the stably transfected cell lines in (A) were treated with IFN-γ for 0, 8, or 24 h and processed in Laemlli gel-loading buffer in absence of reducing agent. Lysates were subjected to immunoblotting with anti-VEGF-A (top panel) and anti-GAPDH (bottom panel) antibodies. (C) Total RNA was isolated from the stably transfected cell lines treated with IFN-γ for 0, 8, or 24 h, and analyzed by RT–PCR using primers specific for VEGF-A (top panel) and β-actin (bottom panel). Real-time PCR results indicating increased VEGF-A mRNA expression in IFN-γ-treated cells compared to untreated cells (expressed as fold-increase normalized to β-actin) are inserted below the top panel. (D) The cell lines described in (A) were treated with IFN-γ for 24 h and lysates immunoprecipitated with anti-EPRS antibody, followed by RT–PCR with VEGF-A-specific primers.
Silencing of VEGF-A translation in monocytic cells inhibits angiogenic activity
Macrophage-derived VEGF-A induces angiogenesis during inflammation and tumor growth. We tested whether silencing VEGF-A translation in IFN-γ-stimulated U937 cells influences their pro-angiogenic activity measured as endothelial cell (EC) proliferation and in vitro tube formation. Subconfluent ECs were incubated with medium conditioned by IFN-γ-treated U937 cells and proliferation quantified by MTT assay. Medium conditioned by untreated U937 cells increased EC proliferation, consistent with basal production of VEGF-A (Figure 7A). Medium from cells treated with IFN-γ for 8 or 12 h increased proliferation further, but activity was reduced to basal level or less by medium conditioned by cells treated with IFN-γ for 16 or 24 h. Neutralizing anti-VEGF-A antibody blocked the activity in conditioned media, confirming the specific role of VEGF-A. A maximal dose of recombinant human VEGF-A, used as positive control, increased proliferation to about the same extent as the 12-h conditioned medium. Because ECs form capillary-like tubes on a matrigel-coated surface in response to VEGF-A, we examined whether IFN-γ treatment of U937 cells regulates their induction of EC tube formation. Conditioned medium from untreated cells induced tube formation, which was enhanced nearly 2.5-fold by medium from cells treated by IFN-γ for 8 h (Figure 7B and C). Tube formation returned to basal level when 16- or 24-h medium was used, consistent with decreased production of VEGF-A during this period. Both assays show that angiogenic activity of IFN-γ-treated U937 cells is blocked by GAIT-mediated translational silencing of VEGF-A. Thus, the GAIT pathway causes delayed inhibition of the angiogenic function of monocytic cells after inflammatory stimulus.
Figure 7.
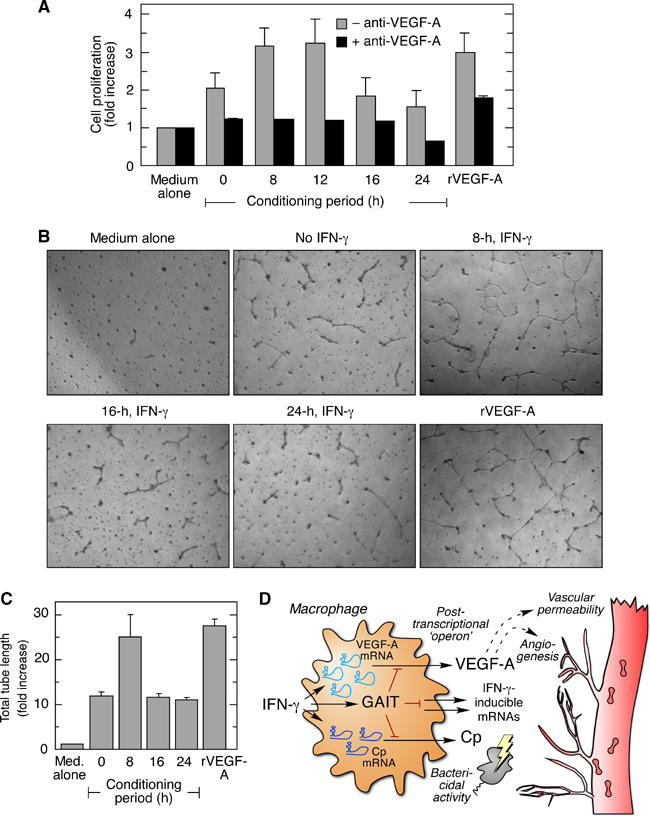
Silencing of VEGF-A translation in monocytic cells inhibits angiogenic activity. (A) EC proliferation was measured in presence of medium conditioned by IFN-γ-treated U937 cells. U937 cells were pre-treated with IFN-γ for up to 24 h, and then fresh medium was added for an additional 2 h. The conditioned medium was added to 50% confluent ECs, and proliferation measured by MTT assay. Cells were treated with recombinant VEGF-A (rVEGF-A, 10 ng/ml) as a positive control. Stimulation of proliferation was expressed as fold-increase compared to cells treated with medium alone (gray bars). Parallel wells contained conditioned medium pre-incubated with anti-VEGF-A antibody (black bars). Shown are the mean and standard deviation from three independent experiments. (B) Tube-formation by ECs on growth factor-depleted matrigel was determined after 12 h in presence of conditioned medium from U937 cells treated with IFN-γ for 8, 16, or 24 h, or with recombinant human VEGF-A (10 ng/ml). (C) EC tube formation was quantitated by computer-assisted tracing. Shown are the mean and standard deviation from three representative fields, for three independent experiments. (D) IFN-γ activates the transcription of VEGF-A, Cp, and other pro-inflammatory genes in macrophages at the site of chronic inflammation. Subsequently, IFN-γ activates the GAIT complex that binds to the GAIT element in the 3′UTR of VEGF-A, Cp, and possibly other transcripts, and silences their translation. This mechanism prevents persistent expression of these inflammatory proteins and reduces or resolves chronic inflammation and tissue injury.
Discussion
Our studies reveal a novel, negative regulatory mechanism of VEGF-A gene expression in which VEGF-A mRNA, after induction by IFN-γ, is translationally inhibited by binding of the heterotetrameric GAIT complex to a defined element in its 3′UTR. The 29-nt regulatory element was predicted by a ‘pattern-matching' search of a 3′UTR database against the Cp GAIT element, and verified by conferring translational silencing to a heterologous reporter in vitro and in vivo. The utility of the pattern-matching approach has been demonstrated by the recent identification of an iron-response element in CDC14A mRNA that was missed by previous search algorithms based on sequence and folding energy criteria (Sanchez et al, 2006). Secondary structure analysis (by Mfold) of the VEGF-A GAIT element indicates that the functionally essential 5- or 6-bp proximal stem (Sampath et al, 2003) is conserved in primates, cow, pig, and dog, but not in rodents. The limited conservation suggests GAIT-mediated regulation of VEGF-A translation might have evolved in higher mammals, or possibly lost in rodents. The Cp mRNA GAIT element exhibits a similar, limited conservation among species. Interestingly, many genes involved in inflammation and inflammatory angiogenesis, including interleukin-8, CXCR1, MPIF-1, and PARC, are absent in mice but present in humans (Mestas and Hughes, 2004). This disparity, together with the differential response of mouse and human macrophages to agonists such as IFN-γ and lipopolysaccharide, indicates significant differences in innate immunity between mice and humans. Such differences are not unexpected given the divergence of the species about 65–75 million years ago and their evolution in dissimilar ecological niches.
VEGF-A expression is tightly controlled under normal and pathological conditions by a multiplicity of stimuli and by diverse mechanisms. VEGF-A is essential during embryonic development, and disruption of a single allele results in abnormal vascular development and embryonic lethality (Carmeliet et al, 1996; Ferrara et al, 1996). In contrast, VEGF-A expression is stringently regulated in the adult, and synthesis occurs only under specific conditions such as wound healing, periodic reconstitution of the uterine endometrium, and inflammation. Uncontrolled VEGF-A expression leads to aberrant and persistent neovascularization, increased vessel permeability, and enhanced leukocyte recruitment, and is a characteristic of many pathological conditions, including cancer, atherosclerosis, and chronic inflammatory diseases (Ferrara and Davis-Smyth, 1997). Negative regulation of VEGF-A gene expression offers the potential benefits of fine-tuning of the steady-state level and rapid downregulation to basal level, following stimulus-induced upregulation. VEGF-A synthesis is suppressed under normal conditions by von Hippel-Landau (VHL) tumor suppressor protein, which ubiquitinates HIF-1α, the major transcriptional activator of VEGF-A, causing its degradation (Shweiki et al, 1992; Tanimoto et al, 2000). Multiple stimuli, for example, hypoxia, cytokines, and female hormones, induce VEGF-A expression, by enhancing transcription, mRNA stability, or translation; however, negative regulatory processes that act post-induction have not been reported (Pages and Pouyssegur, 2005). To date, GAIT-mediated translational silencing of the VEGF-A mRNA in IFN-γ-treated monocyte/macrophages is the only negative regulatory mechanism of VEGF-A gene expression under inflammatory conditions. Thus, IFN-γ has a dual regulatory role in VEGF-A gene expression; it enhances VEGF-A mRNA and protein levels as an early response, but subsequently inhibits VEGF-A protein synthesis in a negative regulatory manner by activation of the GAIT system (Figure 7D). Also, the involvement of EPRS, a tRNA synthetase, in the regulation of expression of a key angiogenic protein is interesting, as two other tRNA synthetases, tyrosyl- and tryptophanyl tRNA synthetases, are induced by inflammatory agents and exhibit noncanonical activities related to angiogenesis (Otani et al, 2002; Wakasugi et al, 2002). The relationship between the three tRNA synthetases and angiogenesis-related activity is unclear, but multiple tRNA synthetases appear to have noncanonical functions related to inflammation (Lee et al, 2004).
Inflammation is a highly complex, self-limiting response, where ‘go' signals can trigger subsequent ‘stop' signals, and molecules responsible for inducing the inflammatory response, for example, tumor necrosis factor-α (TNF-α), prostaglandin E2, transforming growth factor-β1 (TGF-β1), and IFN-γ, can switch from pro- to anti-inflammatory activities depending on timing and context (Nathan, 2002). Transcriptional upregulation and delayed translational silencing of macrophage VEGF-A and Cp suggests IFN-γ is a binary inflammatory signal in which its action switches from pro-inflammatory during onset to anti-inflammatory in later stages. This view is consistent with accumulating evidence that IFN-γ expresses anti-inflammatory activities, especially during late stages of the macrophage inflammatory response (Ohmori and Hamilton, 1994; Hodge-Dufour et al, 1998; Mühl and Pfeilschifter, 2003). Likewise, IFN-γ can inhibit inflammatory angiogenesis by inducing macrophage expression of the angiostatic chemokines monokine induced by IFN-γ (Mig) and IFN-inducible protein 10 (IP-10) (Arenberg et al, 1996; Sgadari et al, 1997). The GAIT system joins other post-transcriptional pathways that downregulate inflammation (Kracht and Saklatvala, 2002). Post-transcriptional inhibitory pathways have the advantage of reversibility, and they provide a rapid and regulatable switch from pro- to anti-inflammatory activities. GAIT-mediated translational silencing may provide a mechanism by which IFN-γ-driven ‘go' signals are switched off, and the pathway may play an important role in the resolution of chronic inflammation.
Coordinated translational silencing of VEGF-A and Cp indicates that the GAIT system constitutes a post-transcriptional operon inhibiting the expression of related inflammatory genes (Figure 7D). The pattern-matching algorithm predicts GAIT-like elements in multiple mRNAs, suggesting that additional genes may be targets. GO analysis of predicted human target mRNAs shows that about 50% are associated with the inflammatory response or inflammatory cell activities, such as cell adhesion, motility, phagocytosis, pathogen recognition, and generation of reactive oxygen species. Association of two other mRNAs with the GAIT complex has been verified by immunoprecipitation of 24-h, IFN-γ-treated U937 cell lysates, followed by RT–PCR (Supplementary Figure S3). Death-associated protein kinase 1 is a serine/threonine kinase that mediates IFN-γ-induced cell death, and GLUT10 is a facilitative glucose transporter, which when mutated causes aberrant angiogenesis and vasculopathy (Deiss et al, 1995; Coucke et al, 2006). However, functional GAIT elements in transcripts other than VEGF-A and Cp have not been experimentally validated. The finding of several inflammatory and IFN-γ-inducible genes as possible GAIT targets is consistent with the postulated role of the GAIT pathway in anti-inflammatory processes. For example, both heparan sulfate 2-O-sulfotransferase and heparan glucosaminyl-N-deacetylase/N-sulfotransferase are involved in the biosynthesis of heparan sulfate, an important mediator of leukocyte–EC interaction in inflammation (Wang et al, 2005). Likewise, glucose transporters are hypoxia-inducible genes, whose expression, like that of VEGF-A, increases during wound healing and in the presence of lipopolysaccharide (Blouin et al, 2004). Post-transcriptional operons may be particularly important for controlling expression of macrophage inflammatory genes, as at least three other mechanisms have been described. Tristetraprolin, a zinc-finger protein, binds to AREs in TNF-α, GM-CSF, and interleukin-3 mRNAs in macrophages, promoting mRNA de-adenylation and degradation (Carballo et al, 2000). A microarray-based analysis has shown that T-cell intracellular antigen 1 (TIA-1), a macrophage protein that represses translation of inflammatory transcripts, interacts with about 300 mRNAs (López de Silanes et al, 2005). Finally, HuR acts as a negative post-transcriptional modulator of inflammation by inhibiting translation of inflammatory mRNAs, including TNF-α, cyclooxygenase-2, and TGF-β1 (Katsanou et al, 2005). Post-transcriptional operons, operating at the level of mRNA degradation or translational silencing, may have evolved as mechanisms to rapidly and coordinately suppress multiple inflammatory genes and downregulate the inflammatory response.
Materials and methods
Cell culture and plasmid construction
Human U937 monocytic cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS). Human PBMCs were isolated by leukapheresis, followed by countercurrent centrifugal elutriation (Czerniecki et al, 1997), under an Institutional Review Board-approved protocol that adhered to American Association of Blood Bank guidelines. PBMCs and U937 cells were treated with 500 U/ml of human IFN-γ (R&D Systems, Minneapolis, MN, USA) for up to 24 h. Bovine aortic ECs were cultured in Ham's F-12/DME (1:1) medium containing 10% FBS.
The VEGF-A 3′UTR (nt 11–900) was amplified from total RNA obtained from U937 cells, and cloned into pGEM-T vector (Promega, Madison, WI, USA). The cloned sequence was identical to the reported human VEGF-A 3′UTR sequence (Levy et al, 1997). This DNA segment was inserted downstream of FLuc, after releasing the Cp 3′UTR, in the vector pSP64 FLuc-Cp 3′UTR-A30 to generate pSP64-FLuc-VEGF-A 3′UTR(11–900)-A30. Sequences corresponding to nucleotides 324–455 and 441–560 of the VEGF-A 3′UTR were similarly inserted downstream of FLuc. A synthetic, 29-nt DNA, corresponding to nucleotides 358–386 of the VEGF-A 3′UTR, was inserted into the same plasmid to generate pSP64-FLuc-VEGF-A GAIT-A30. Likewise, the same 29-nt sequence containing a point mutation (U10C) was used to generate pSP64-FLuc-VEGF-A GAITmut-A30. Synthetic wild-type and mutant VEGF-A GAIT element sequences were also inserted downstream of FLuc cloned in the eukaryotic expression vector pcDNA3 (Invitrogen, Gaithersburg, MD, USA) to generate pCD-FLuc-VEGF-A GAIT and pCD-FLuc-VEGF-A GAITmut.
Bioinformatic analysis
Structure and sequence information from the Cp GAIT element was utilized to generate a consensus query pattern for the GAIT element using the PatSearch syntax (Grillo et al, 2003). The query pattern was matched against nonredundant 3′UTR and 5′UTR sequence databases; nucleotide mismatches and mispairings were not permitted. RNA secondary structure predictions were performed using the Mfold program incorporating base-pairing constraints. GO-based literature search for functional attributes of PatSearch results was performed using the GOPubmed server (Doms and Schroeder, 2005).
Determination of in vivo interaction between GAIT complex and VEGF-A mRNA
Lysates from IFN-γ-treated U937 cells were immunoprecipitated with 4 μg of rabbit polyclonal anti-EPRS antibody (raised against bacterially expressed, His-tagged human EPRS-linker domain, amino acids 681–884; IgG fraction purified by peptide-affinity chromatography) using Seize primary immunoprecipitation kit (Pierce, Rockford, IL, USA). mRNAs associated with immunoprecipitated mRNP complexes were isolated by phenol:chloroform extraction and ethanol precipitation. The isolated RNA was further purified using RNeasy kit (Qiagen, Valencia, CA, USA), and subjected to reverse transcription using oligo-dT primers and PCR amplification using gene-specific primers.
Quantitative RT–PCR
RNA isolated from cells or from immunoprecipitated mRNP complexes was reverse-transcribed using oligo(dT) primers. cDNA was subjected to real-time PCR using SYBR-green PCR master-mix in an ABI Prism 7000 Sequence Detection System. PCR-amplified VEGF-A was normalized to amplified β-actin cDNA.
In vitro transcription
pSP64 plasmid constructs containing VEGF-A 3′UTR or GAIT element sequences downstream of FLuc were linearized and transcribed in vitro using the mMessage mMachine transcription system (Ambion, Austin, TX, USA) to generate capped poly(A)-tailed RNAs. Capped RLuc RNA was similarly transcribed from the plasmid pRL-SV40 (Promega). The pGEM-T-VEGF-A 3′UTR was linearized and transcribed using the Megascript transcription system (Ambion) to generate VEGF-A 3′UTR RNA. [α-32P]UTP-labeled VEGF-A and Cp GAIT element RNAs were transcribed using the T7-riboprobe system (Promega) from a synthetic oligonucleotide template having a T7 promoter-adapter.
In vitro translation
Capped poly(A)-tailed template RNAs were translated in RRL (Promega) in the presence of a methionine-free amino-acid mixture and translation-grade [35S]methionine (Perkin Elmer, Boston, MA, USA). Cytosolic extract (500 ng of protein) from untreated or IFN-γ-treated U937 cells were added to translation reactions. Excess (10- or 50-fold) VEGF-A 3′UTR RNA was used in decoy experiments. Reactions were resolved by SDS–PAGE (10% polyacrylamide) and visualized by phosphorimaging.
RNA electrophoretic mobility shift assay
[α-32P]UTP-labeled VEGF-A or Cp GAIT element RNA was incubated with cytosolic extracts from U937 cells incubated with IFN-γ for up to 24 h. For supershift assays, the lysate was pre-incubated with affinity-purified anti-EPRS, anti-L13a (Mazumder et al, 2003), anti-GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-NSAP (Anaspec, San Jose, CA, USA) polyclonal antibodies, or with IgG purified from pre-immune rabbit serum. RNA–protein complexes were resolved by native gel electrophoresis and visualized by phosphorimaging.
Cell transfection
U937 cells were transiently transfected with 6 μg of pCD-FLuc-VEGF-A GAIT, pCD-FLuc-VEGF-A GAITmut, and pCD-FLuc DNAs using human dendritic cell nucleofection kit V (Amaxa biosystems, Cologne, Germany). RLuc-expressing vector pRL-SV40 (1 μg, Promega) was co-transfected for normalization of transfection efficiency. After 12 h, transfected cells were incubated with IFN-γ, lysed, and luciferase activity was measured using dual luciferase assay kit (Promega).
Immunodepletion of GAIT complex
U937 cell lysates were incubated with polyclonal anti-EPRS antibody or pre-immune IgG coupled to protein-A Sepharose CL beads (Sigma) in RIPA buffer. The beads were pelleted and the process was repeated twice with supernatants. The supernatants were immunoblotted with anti-EPRS antibody to verify immunodepletion.
Stable knockdown of L13a expression
U937 cells were subjected to nucleofection with either pSUPER (Oligoengine, Seattle, WA, USA) encoding a small hairpin RNA targeted to human L13a (kind gift from Barsanjit Mazumder, Cleveland State University) or empty pSUPER vector. Puromycin-resistant clones of transfected cells were selected and expanded to form stably transfected cell lines showing knockdown of L13a expression.
Immunoblot analysis
Cell lysates were denatured under nonreducing conditions, and resolved on SDS–12% PAGE. After transfer, the blot was probed with anti-human VEGF-A antibody and HRP-conjugated anti-rabbit secondary antibody, and detected by ECL (Amersham, Arlington Heights, IL, USA). Immunoblotting was also performed using anti-GAPDH antibody to ensure equal loading.
Metabolic labeling
U937 cells were treated with IFN-γ for up to 24 h, then washed and resuspended in RPMI 1640 medium (minus Met/Cys, Sigma). The cells were metabolically labeled for 1 h with [35S]Met/Cys, and conditioned media and cell lysates were subjected to immunoprecipitation with anti-VEGF-A antibody (Santa Cruz) coupled to protein A-Sepharose CL (Sigma) in RIPA buffer. Immunoprecipitated proteins, cell lysates, and conditioned media were resolved on SDS–12% PAGE, followed by visualization by phosphorimage analysis.
Isolation of polysome-associated mRNA
U937 cells were homogenized in polysome lysis buffer containing cycloheximide (0.1 mg/ml). Cytosolic extract was obtained by centrifugation at 10 000 g for 20 min. The extract was overlayed on a 20% (w/v) sucrose cushion and centrifuged at 150 000 g for 2 h. The polysome-containing pellet and the nonpolysomal supernatant were collected, subjected to proteinase K digestion, and associated mRNA isolated by phenol-chloroform extraction and ethanol precipitation. For polysome release experiments, cells were lysed in polysome lysis buffer containing 10 mM EDTA, and centrifuged on a 20% sucrose cushion in the same buffer.
Cell proliferation
Bovine aortic ECs (5 × 105 cells/well) were seeded in serum-free DMEM, and serum-starved for 18 h. U937 cells were treated with IFN-γ for up to 24 h and then harvested, washed, and placed in fresh serum-free RPMI medium for 2 h. The conditioned medium was filtered (0.45 μm filter), concentrated, and added to EC cultures for 24 h. A 10 ng/ml portion of recombinant human VEGF-A (R&D Systems) was used as positive control. In other wells, conditioned media was incubated with 2 μg/ml of anti-VEGF-A antibody for 1 h. After 24 h, MTT assay (Sigma) was done according to manufacturer's protocol.
Endothelial cell tube formation
ECs were seeded on growth factor-reduced matrigel (BD Biosciences, Bedford, MA, USA) in serum-free Ham's F12:DME medium. After 4 h, conditioned medium from IFN-γ-treated U937 cells was overlaid on the cells and incubated for 24 h. Recombinant human VEGF-A (10 ng/ml) was used as positive control. Micrographic images of EC tubes in each well were captured, manually traced (ACD Canvas), and the total length of traced lines quantified (Adobe Photoshop).
Supplementary Material
Supplementary Table S1
Supplementary Figures
Acknowledgments
We are grateful to Barsanjit Mazumder for the generous gift of a plasmid encoding a small hairpin RNA targeted to human L13a. This work was supported by NIH grants HL29582, HL67725, and HL76491 (to PLF). The authors do not have any conflicts of interest related to the work described in this article.
References
- Arenberg DA, Kunkel SL, Polverini PJ, Morris SB, Burdick MD, Glass MC, Taub DT, Iannettoni MD, Whyte RI, Strieter RM (1996) Interferon-γ-inducible protein 10 (IP-10) is an angiostatic factor that inhibits human non-small cell lung cancer (NSCLC) tumorigenesis and spontaneous metastases. J Exp Med 184: 981–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin CC, Pagé EL, Soucy GM, Richard DE (2004) Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1α. Blood 103: 1124–1130 [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST (2001) Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile × syndrome. Cell 107: 477–487 [DOI] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ (2000) Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood 95: 1891–1899 [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380: 435–439 [DOI] [PubMed] [Google Scholar]
- Coucke PJ, Willaert A, Wessels MW, Callewaert B, Zoppi N, De Backer J, Fox JE, Mancini GM, Kambouris M, Gardella R, Facchetti F, Willems PJ, Forsyth R, Dietz HC, Barlati S, Colombi M, Loeys B, De Paepe A (2006) Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat Genet 38: 452–457 [DOI] [PubMed] [Google Scholar]
- Czerniecki BJ, Carter C, Rivoltini L, Koski GK, Kim HI, Weng DE, Roros JG, Hijazi YM, Xu S, Rosenberg SA, Cohen PA (1997) Calcium ionophore-treated peripheral blood monocytes and dendritic cells rapidly display characteristics of activated dendritic cells. J Immunol 159: 3823–3837 [PubMed] [Google Scholar]
- Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A (1995) Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev 9: 15–30 [DOI] [PubMed] [Google Scholar]
- Doms A, Schroeder M (2005) GoPubMed: exploring PubMed with the Gene Ontology. Nucleic Acids Res 33: W783–W786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW (1996) Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380: 439–442 [DOI] [PubMed] [Google Scholar]
- Ferrara N, Davis-Smyth T (1997) The biology of vascular endothelial growth factor. Endocr Rev 18: 4–25 [DOI] [PubMed] [Google Scholar]
- Gerber AP, Luschnig S, Krasnow MA, Brown PO, Herschlag D (2006) Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc Natl Acad Sci USA 103: 4487–4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo G, Licciulli F, Liuni S, Sbisa E, Pesole G (2003) PatSearch: a program for the detection of patterns and structural motifs in nucleotide sequences. Nucleic Acids Res 31: 3608–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge-Dufour J, Marino MW, Horton MR, Jungbluth A, Burdick MD, Strieter RM, Noble PW, Hunter CA, Pure E (1998) Inhibition of interferon γ induced interleukin 12 production: a potential mechanism for the anti-inflammatory activities of tumor necrosis factor. Proc Natl Acad Sci USA 95: 13806–13811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, Ji Y, Zhao C, Wang J, Yang BB, Zhang Y (2006) miRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE 1: e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsanou V, Papadaki O, Milatos S, Blackshear PJ, Anderson P, Kollias G, Kontoyiannis DL (2005) HuR as a negative posttranscriptional modulator in inflammation. Mol Cell 19: 777–789 [DOI] [PubMed] [Google Scholar]
- Keene JD, Tenenbaum SA (2002) Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell 9: 1161–1167 [DOI] [PubMed] [Google Scholar]
- Kracht M, Saklatvala J (2002) Transcriptional and post-transcriptional control of gene expression in inflammation. Cytokine 20: 91–106 [DOI] [PubMed] [Google Scholar]
- Lee SW, Cho BH, Park SG, Kim S (2004) Aminoacyl-tRNA synthetase complexes: beyond translation. J Cell Sci 117: 3725–3734 [DOI] [PubMed] [Google Scholar]
- Levy AP, Levy NS, Goldberg MA (1996) Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem 271: 2746–2753 [DOI] [PubMed] [Google Scholar]
- Levy NS, Goldberg MA, Levy AP (1997) Sequencing of the human vascular endothelial growth factor (VEGF) 3′ untranslated region (UTR): conservation of five hypoxia-inducible RNA-protein binding sites. Biochim Biophys Acta 1352: 167–173 [DOI] [PubMed] [Google Scholar]
- Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW (2006) Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 66: 11238–11246 [DOI] [PubMed] [Google Scholar]
- López de Silanes I, Galbán S, Martindale JL, Yang X, Mazan-Mamczarz K, Indig FE, Falco G, Zhan M, Gorospe M (2005) Identification and functional outcome of mRNAs associated with RNA-binding protein TIA-1. Mol Cell Biol 25: 9520–9531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder B, Fox PL (1999) Delayed translational silencing of ceruloplasmin transcript in gamma interferon-activated U937 monocytic cells: role of the 3′ untranslated region. Mol Cell Biol 19: 6898–6905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder B, Mukhopadhyay CK, Prok A, Cathcart MK, Fox PL (1997) Induction of ceruloplasmin synthesis by IFN-γ in human monocytic cells. J Immunol 159: 1938–1944 [PubMed] [Google Scholar]
- Mazumder B, Sampath P, Seshadri V, Maitra RK, DiCorleto P, Fox PL (2003) Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell 115: 187–198 [DOI] [PubMed] [Google Scholar]
- Mestas J, Hughes CC (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172: 2731–2738 [DOI] [PubMed] [Google Scholar]
- Mühl H, Pfeilschifter J (2003) Anti-inflammatory properties of pro-inflammatory interferon-γ. Int Immunopharmacol 3: 1247–1255 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay CK, Mazumder B, Lindley PF, Fox PL (1997) Identification of the prooxidant site of human ceruloplasmin: a model for oxidative damage by copper bound to protein surfaces. Proc Natl Acad Sci USA 94: 11546–11551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C (2002) Points of control in inflammation. Nature 420: 846–852 [DOI] [PubMed] [Google Scholar]
- Ohmori Y, Hamilton TA (1994) IFN-γ selectively inhibits lipopolysaccharide-inducible JE/monocyte chemoattractant protein-1 and KC/GRO/melanoma growth-stimulating activity gene expression in mouse peritoneal macrophages. J Immunol 153: 2204–2212 [PubMed] [Google Scholar]
- Otani A, Slike BM, Dorrell MI, Hood J, Kinder K, Ewalt KL, Cheresh D, Schimmel P, Friedlander M (2002) A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc Natl Acad Sci USA 99: 178–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages G, Pouyssegur J (2005) Transcriptional regulation of the vascular endothelial growth factor gene—a concert of activating factors. Cardiovasc Res 65: 564–573 [DOI] [PubMed] [Google Scholar]
- Ramanathan M, Giladi A, Leibovich SJ (2003) Regulation of vascular endothelial growth factor gene expression in murine macrophages by nitric oxide and hypoxia. Exp Biol Med (Maywood) 228: 697–705 [DOI] [PubMed] [Google Scholar]
- Sampath P, Mazumder B, Seshadri V, Fox PL (2003) Transcript-selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3′ untranslated region. Mol Cell Biol 23: 1509–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath P, Mazumder B, Seshadri V, Gerber CA, Chavatte L, Kinter M, Ting SM, Dignam JD, Kim S, Driscoll DM, Fox PL (2004) Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell 119: 195–208 [DOI] [PubMed] [Google Scholar]
- Sanchez M, Galy B, Dandekar T, Bengert P, Vainshtein Y, Stollte J, Muckenthaler MU, Hentze MW (2006) Iron regulation and the cell cycle: Identification of an iron-responsive element in the 3′ untranslated region of human cell division cycle 14A mRNA by a refined microarray-based screening strategy. J Biol Chem 281: 22865–22874 [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon-γ: an overview of signals, mechanisms and functions. J Leukoc Biol 75: 163–189 [DOI] [PubMed] [Google Scholar]
- Sgadari C, Farber JM, Angiolillo AL, Liao F, Teruya-Feldstein J, Burd PR, Yao L, Gupta G, Kanegane C, Tosato G (1997) Mig, the monokine induced by interferon-gamma, promotes tumor necrosis in vivo. Blood 89: 2635–2643 [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E (1992) Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 359: 843–845 [DOI] [PubMed] [Google Scholar]
- Sunderkötter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C (1994) Macrophages and angiogenesis. J Leukoc Biol 55: 410–422 [DOI] [PubMed] [Google Scholar]
- Tammela T, Enholm B, Alitalo K, Paavonen K (2005) The biology of vascular endothelial growth factors. Cardiovasc Res 65: 550–563 [DOI] [PubMed] [Google Scholar]
- Tanimoto K, Makino Y, Pereira T, Poellinger L (2000) Mechanism of regulation of the hypoxia-inducible factor-1α by the von Hippel-Lindau tumor suppressor protein. EMBO J 19: 4298–4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenbaum SA, Carson CC, Lager PJ, Keene JD (2000) Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc Natl Acad Sci USA 97: 14085–14090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakasugi K, Slike BM, Hood J, Otani A, Ewalt KL, Friedlander M, Cheresh DA, Schimmel P (2002) A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc Natl Acad Sci USA 99: 173–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Fuster M, Sriramarao P, Esko JD (2005) Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol 6: 902–910 [DOI] [PubMed] [Google Scholar]
- Weis SM, Cheresh DA (2005) Pathophysiological consequences of VEGF-induced vascular permeability. Nature 437: 497–504 [DOI] [PubMed] [Google Scholar]
- Wilusz CJ, Wormington M, Peltz SW (2001) The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol 2: 237–246 [DOI] [PubMed] [Google Scholar]
- Xiong M, Elson G, Legarda D, Leibovich SJ (1998) Production of vascular endothelial growth factor by murine macrophages: regulation by hypoxia, lactate, and the inducible nitric oxide synthase pathway. Am J Pathol 153: 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zittermann SI, Issekutz AC (2006) Endothelial growth factors VEGF and bFGF differentially enhance monocyte and neutrophil recruitment to inflammation. J Leuk Biol 80: 244–254 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1
Supplementary Figures