

Abstract
Transforming Growth Factor (TGF)-β is markedly induced and rapidly activated in the infarcted myocardium. However, understanding of the exact role of (TGF)-β signaling in the infarcted and remodeling heart has been hampered by the complex and unusual biology of TGF-β activation and by the diversity of its effects eliciting multiple, and often opposing cellular responses. Experimental studies suggest that TGF-β signaling may be crucial for repression of inflammatory gene synthesis in healing infarcts mediating resolution of the inflammatory infiltrate. In addition, TGF-β may play an important role in modulating fibroblast phenotype and gene expression, promoting extracellular matrix deposition in the infarct by upregulating collagen and fibronectin synthesis and by decreasing matrix degradation through induction of protease inhibitors. TGF-β is also a key mediator in the pathogenesis of hypertrophic and dilative ventricular remodeling by stimulating cardiomyocyte growth and by inducing interstitial fibrosis. In this review we summarize the current knowledge on the role of TGF-β in infarct healing and cardiac remodeling.
Keywords: fibrosis, growth factors, infarction, remodeling, cytokines
1. Introduction
Members of the TGF-β family are markedly induced in the infarcted myocardium and through their potent effects are capable of playing a central role in infarct healing, cardiac repair and left ventricular remodeling. However, understanding of the exact role of TGF-β in cardiac injury has been hampered by the complex and unusual biology of TGF-β activation and by the amazing diversity of its effects eliciting multiple, and often opposing cellular responses. Our review summarizes current knowledge on the expression, activation and functional role of TGF-β in the infarcted and remodeling myocardium. Evidence suggests that TGF-β is a central mediator involved in the inflammatory and fibrotic phase of healing and may critically modulate many cellular steps in post-infarction cardiac repair. In addition, TGF-β plays a key role in hypertrophic and fibrotic remodeling of the heart mediating cardiomyocyte growth, fibroblast activation and extracellular matrix deposition.
Three structurally similar isoforms of TGF-β (TGF-β1, β2 and β3), encoded by three distinct genes, have been identified in mammalian species [1]. These three isoforms signal through the same cell surface receptors and have similar cellular targets, although each isoform is expressed in a distinct pattern under control of a unique promoter [2]. TGF-β1 is the prevalent isoform and is found almost ubiquitously, whereas the other isoforms are expressed in a more limited spectrum of cells and tissues. Although the three isoforms have similar in vitro properties their in vivo effects are distinct. Knockout experiments in mice have suggested that each TGF-β isoform plays an independent role in embryonic development underlining their non-compensated functions.
TGF-β is produced by many cell types as a large latent complex, unable to associate with its receptors. The extracellular concentration of TGF-β activity is primarily regulated by conversion of latent TGF-β to active TGF-β. Most tissues contain significant amounts of latent TGF-β; activation of only a small fraction of this latent TGF-β generates maximal cellular response [3].
A variety of molecules have been described as TGF-β activators. Proteases including plasmin, Matrix metalloproteinase (MMP)-2 and MMP-9 are capable of activating TGF-β, coupling matrix degradation with activation of a molecule that has a primary role in maintaining matrix integrity and stability [5]. Thrombospondin (TSP)-1 is a key TGF β activator which acts by disrupting the non-covalent interactions between the Latency-associated Peptide (LAP) and the TGF-β molecule [6]. Reactive oxygen species generation and a mildly acidic environment are also capable of inducing TGF-β activation.
The members of the TGF-β superfamily transduce their signal from the membrane to the nucleus through distinct combinations of transmembrane type I and type II serine/threonine receptors and their downstream effectors, the Smad proteins [1], [8]. Active TGF-β binds to the constitutively active type II receptor (TβRII) at the cell surface. The complex subsequently interacts with, and transphosphorylates the cytoplasmic domain of the type I receptor (TβRI), also known as ALK5 [9]. Apart from the well-characterized ALK5, which is expressed by many different cell types, endothelial cells express a second type I TGF-β receptor, termed ALK1 [10]. Phosphorylation of the TβRI activates the type I receptor kinase domain, which then propagates downstream intracellular signals, through the Smad proteins, essential components in the signaling pathway of TGF-β [11] (Figure 1). Besides Smad-mediated transcription, TGF-β activates other signaling cascades, including extracellular signal-regulated kinase (Erk), c-Jun-N-terminal kinase (JNK), TGF-β-activated kinase 1 (TAK1), abelson nonreceptor tyrosine kinase (c-Abl) and p38 Mitogen-activated protein kinase (MAPK) pathways [12] (Figure 1).
Figure 1. Smad-dependent and Smad-independent pathways in TGF-β signaling.
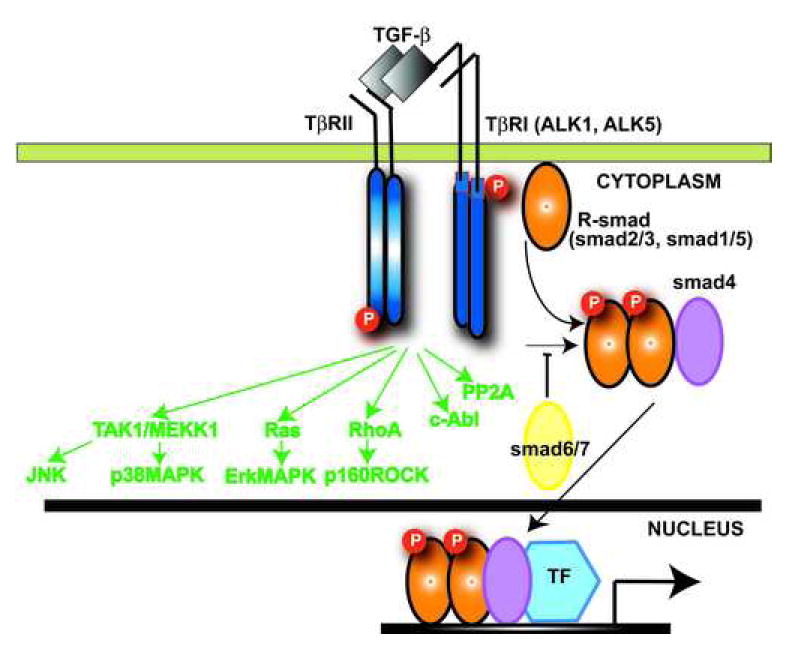
TGF-β binds a complex of transmembrane receptor serine/threonine kinases (type I and type II) inducing transphosphorylation of the GS segments in the type I receptor by the type II receptor kinases. The activated type I receptors phosphorylate selected Smads. Receptor-activated Smads (R-Smads) form a complex with the common Smad4. Subsequently the Smad complexes translocate into the nucleus, where they regulate transcription of target genes. The structurally divergent inhibitory Smads (Smad6 and Smad7), negatively regulate TGF-β signaling. In addition, TGF-β signals through Smad-independent cascades (green) activating Erk, JNK, p38MAPK, Protein Phosphatase 2A (PP2A) and RhoA pathways.
2. TGF-βs: Pleiotropic mediators with potent and diverse effects on cellular behavior
The TGF-βs are some of the most pleiotropic and multifunctional peptides known. They have potent and direct effects on many different cell types and are involved in a wide variety of biological processes such as embryonic development, cell growth and differentiation, cell proliferation and apoptosis, fibrous tissue deposition and regulation of the immune response. The cellular actions of TGF-β are not only dependent on the cell type but also on its state of differentiation and on the cytokine milieu [2]. Mice with genetic disruptions of the TGF-β genes have been generated and revealed the role of TGF-β isoforms in embryogenesis and immune regulation. Approximately 50% of TGF-β1 null mice die in utero due to defective yolk sac vasculogenesis and hematopoiesis [13]. The remaining mice develop to term and show no gross developmental abnormalities, but about 2-4 weeks after birth they succumb to a wasting syndrome associated with multifocal inflammation and massive lymphocyte and macrophage infiltration in many organs, but primarily in the heart and lungs [14]. The phenotype of TGF-β1 null mice suggests a prominent role for TGF-β1 in homeostatic regulation of immune cell proliferation and extravasation into the tissues [14]. In contrast to TGF-β1 -/- animals, TGF-β2 knockout mice exhibit perinatal mortality and a wide range of developmental defects. The developmental processes most commonly involved include epithelial-mesenchymal interactions, cell growth, and extracellular matrix production resulting in cardiac, pulmonary, craniofacial, spinal, ocular, inner ear, and urogenital defects [15]. TGF-β3 deficient animals also exhibit defective epithelial-mesenchymal interactions resulting in cleft palate and abnormal lung development [16]. The distinct phenotypes of the TGF-β knockout animals indicate numerous non-compensated functions of the three TGF-β isoforms.
TGF-β is expressed at high levels in the heart both during embryonic development and adult life [17]. In adult mouse hearts TGF-β is localized in both the cardiomyocytes and the extracellular matrix [17]. In embryonic development TGF-β has been implicated in cardiac valve morphogenesis [18]. TGF-β2 null mice exhibit multiple cardiac developmental abnormalities with varying penetrance, including double outlet right ventricle, atrial and ventricular septal defects and occasional thickened semilunar valves reflecting disturbances of looping, myocardialization and endocardial cushion differentiation [19]. Although the role of endogenous TGF-β expression in the normal adult heart remains unknown, it has been suggested that it may sustain the spontaneous beating rate of cardiomyocytes [20]. In addition, storage of latent TGF-β which can be activated following injury may play an important role in tissue repair by promoting new synthesis and deposition of extracellular matrix.
3.The role of the inflammatory response in infarct healing
Myocardial infarction triggers an inflammatory response that ultimately results in healing and formation of a scar [21], [22]. The cardiac repair process can be divided into three overlapping phases: the inflammatory phase, the proliferative phase and the maturation phase. During the inflammatory phase, cardiomyocyte death and hypoxia result in free radical generation, initiation of the complement cascade, and activation of Nuclear Factor (NF)-κB and Toll Like Receptor (TLR)-mediated signaling pathways. These events induce chemokine and cytokine synthesis and upregulate adhesion molecule expression in endothelial cells and leukocytes, resulting in infiltration of monocytes, lymphocytes and polymorphonuclear cells into the infarcted area. Neutrophils and macrophages clear the wound from dead cells and matrix debris. During the proliferative phase of healing, expression of inflammatory mediators is suppressed, while fibroblasts and endothelial cells infiltrate the wound. Although the mechanisms responsible for resolution of inflammation are poorly understood, timely repression of inflammatory mediator synthesis is important for the transition to fibrous tissue deposition. As most inflammatory cells undergo apoptotic death, activated myofibroblasts produce extracellular matrix proteins and an extensive microvascular network evolves. Maturation of the scar follows: fibroblasts and vascular cells undergo apoptosis and a scar containing cross-linked collagen bundles is formed. The cellular and molecular events associated with infarct healing directly influence left ventricular remodeling and affect prognosis in patients with myocardial infarction.
TGF-β may be a key mediator in regulating many of the events associated with infarct healing. TGF-β may play an important role in monocyte recruitment in the healing infarct, promoting granulation tissue formation. Activation of TGF-β signaling pathways may be important in suppressing expression of pro-inflammatory cytokines and chemokines in the infarcted myocardium resulting in resolution of the inflammatory infiltrate. Furthermore, TGF-β may critically regulate fibrous tissue deposition by mediating acquisition of the myofibroblastic phenotype, by inducing extracellular matrix protein synthesis, and by promoting matrix preservation through increased expression of Tissue Inhibitors of Metalloproteinases (TIMP). In order to better understand the role of β-mediated actions in infarct healing, we will briefly discuss the effects of TGF-β stimulation on the main cell types involved in cardiac repair.
3.1 The effects of TGF-β on inflammatory and immune cells (Figure 2)
Figure 2. The diverse, multifunctional, and pleiotropic effects of TGF-β on cell types involved in infarct healing.
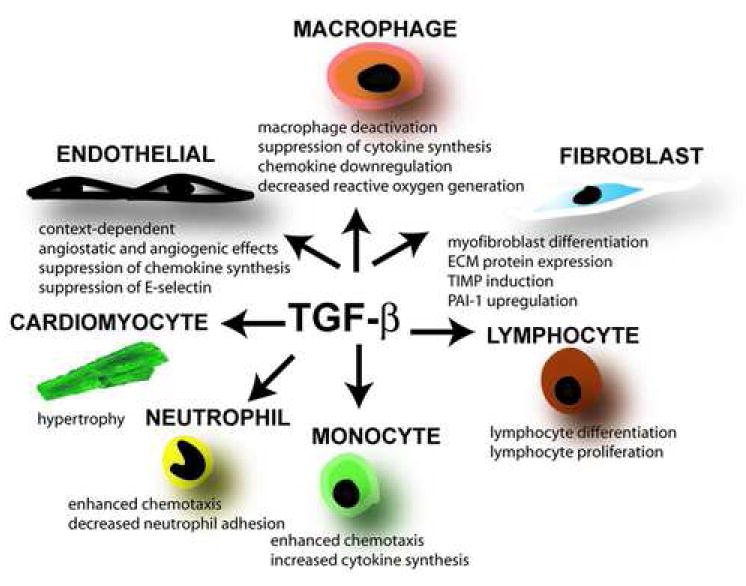
During the pro-inflammatory phase of healing TGF-β may induce mononuclear cell chemotaxis and modulate lymphocyte phenotype. The effects of TGF-β on mature macrophages are predominantly suppressive, inhibiting pro-inflammatory cytokine and chemokine synthesis and decreasing reactive oxygen generation. These actions may be important in regulating resolution of the post-infarction inflammatory response. TGF-β also induces acquisition of the myofibroblastic phenotype and promotes extracellular matrix deposition by increasing collagen and fibronectin synthesis, and by inhibiting matrix degradation through TIMP upregulation. Spared cardiomyocytes respond to TGF-β by undergoing hypertrophy. The effects of TGF-β on endothelial cells are complex and context-dependent; their importance in regulating infarct angiogenesis is unknown. TGF-β mediated effects on fibrous tissue deposition and cardiac hypertrophy may be important in the pathogenesis of left ventricular remodeling.
Femtomolar concentrations of TGF-β1 elicit a direct chemotactic response from neutrophils [23] and monocytes [26]; this effect may be important for movement of infiltrating leukocytes in inflamed tissues. Conversely, the actions of TGF-β1 on the endothelium result in decreased leukocyte adhesion and inhibition of neutrophil migration across the endothelial layer, in part through decreased surface expression of E-selectin [24]. The effects of TGF-β on lymphocytes, monocytes and macrophages can be either stimulatory or inhibitory, depending on the cytokine milieu, the state of differentiation and the tissue origin of the cells [25], highlighting the pleiotropic nature of the cytokine. Picomolar concentrations of TGF-β activate monocytes, stimulating synthesis of a variety of cytokines (such as Interleukin-1β and Tumor Necrosis Factor- α), chemokines (such as Monocyte Chemoattractant Protein-1), and growth factors (including basic Fibroblast Growth Factor and Platelet-Derived Growth Factor-BB) [2], [26] and increasing integrin expression. In contrast to the activating effects of TGF-β on peripheral blood monocytes, its actions on mature macrophages are predominantly suppressive. TGF-β has a deactivating effect on macrophages, suppressing pro-inflammatory cytokine and chemokine synthesis [27], [28] and decreasing reactive oxygen generation. The phenotype of the TGF-β1 null mouse suggests that the anti-inflammatory actions of the molecule on endothelial cells and macrophages are critical for suppression of the inflammatory response and for regulation of tissue homeostasis.
3.2 TGF-β modulates fibroblast phenotype and promotes fibrosis
TGF-β plays a key role in the development of tissue fibrosis [29]. TGF-β stimulation induces conversion of fibroblasts to myofibroblasts [30] and enhances extracellular matrix protein synthesis. In addition, TGF-β suppresses the activity of proteases that degrade extracellular matrix by inhibiting MMP expression and by inducing synthesis of protease inhibitors, such as Plasminogen Activator Inhibitor (PAI)-1 and TIMPs [1]. Activation of the Smad3 signaling pathway appears to be important in mediating TGF-β-induced extracellular matrix protein synthesis and TIMP upregulation [31]. In addition, recent evidence suggests that at least some of the pro-fibrotic effects of TGF-β may be in part mediated through Connective Tissue Growth Factor (CTGF) upregulation [32].
3.3. Effects of TGF-β on the angiogenic potential of endothelial cells
The effects of TGF-β on endothelial cells are complex and context-dependent being either stimulatory or inhibitory, depending on differentiation of the cells and on the presence of environmental cues [33]. TGF-β is involved in the development of the vascular system by modulating the function of both endothelial cells and pericytes [33]. The phenotype of gene knockouts of TGF-β, the TGF-β receptors and many of its downstream signaling proteins suggests an essential role for TGF-β signaling in vascular development. TGF-β has been described as being either angiogenic or angiostatic in vivo, depending on the nature of the assay used [33]. In addition, the in vitro effects of TGF-β on endothelial cell proliferation, migration and expression of proteases are dependent on the source of endothelial cells and the dose of TGF-β used to stimulate them. Recent investigations have suggested that TGF-β regulates the activation state of the endothelium via a fine balance between distinct signaling pathways involving the two TβRI receptors, ALK1 and ALK5. ALK1 activation stimulates endothelial cell proliferation and migration via Smad1/5 transcription factors, whereas ALK5 inhibits EC proliferation and migration through smad2/3-mediated interactions [37]. Endoglin, an accessory transmembrane receptor for TGF-β plays a pivotal role in modulating the balance between ALK1 and ALK5 signaling to regulate endothelial cell proliferation [38].
4.Expression and activation of TGF-β in myocardial infarcts
TGF-β is markedly upregulated in experimental models of myocardial infarction. TGF-β isoforms demonstrate distinct patterns of expression in the infarct: TGF-β1 and β2 are induced early, whereas TGF-β3 shows delayed and prolonged upregulation [39], [40]. TGF-β expression in the infarcted heart is attenuated by angiotensin-converting enzyme inhibitors and angiotensin receptor blockers [41], [42] suggesting that angiotensin II signaling plays an important role in stimulating TGF-β synthesis [43]. Extravasated platelets may be the main source of latent TGF-β in the early stages of infarct healing, whereas macrophages and fibroblasts may be responsible for the sustained upregulation of TGF-β during the proliferative phase of healing. TGF-β expression is predominantly localized in the infarct border zone [44]. Increased expression of the downstream effectors of TGF-β signaling is noted in the healing infarct. Smad2, 3, and 4 protein expression is significantly upregulated in the scar and border zone area [45]. In contrast, expression of the inhibitory Smad7 is decreased in myocardial scars; this may contribute to cardiac fibrosis in the remodeling myocardium [46]. Although evidence suggests that bioactive TGF-β is released in the cardiac extracellular fluids 3-5h following reperfused infarction [47], the mechanisms responsible for TGF-β activation in the infarcted heart are poorly understood. Our recent experiments suggested that TSP-1 induction in the infarct border zone may play an important role in activation of TGF-β signaling pathways in mouse and canine infarcts [48].
5.Effects of exogenous TGF-β on the ischemic heart
Lefer and colleagues first suggested a protective effect of exogenous TGF-β in the ischemic heart, demonstrating that TGF-β injection reduced ischemic myocardial injury presumably by attenuating the deleterious effects of pro-inflammatory cytokines such as TGF-β [49]. Intravenous administration of TGF-β1 prior to reperfusion attenuated myocardial necrosis and endothelial dysfunction and decreased neutrophil adherence to the ischemic coronary endothelium in a feline model of myocardial ischemia and reperfusion [50], [49]. Baxter and coworkers suggested that the protective effects of exogenous TGF-β1 on the ischemic myocardium may be mediated through attenuation of cardiomyocyte apoptosis [51]. Recent investigations have focused on the effects of exogenous TGF-β in myocardial regeneration following infarction. Both TGF-β and Bone Morphogenetic Protein (BMP)-2 increase the expression of cardiac transcription factors in embryonic stem cells directing them to differentiate into cardiomyocytes [52]. Implantation of TGF-β pre-programmed CD117+ stem cells into the infarcted myocardium results in myocardial regeneration and induces angiogenesis; these effects are not noted when untreated stem cells are implanted [53]. Although these studies provide insight into the potential actions of TGF-β in myocardial ischemia, the effects of exogenous TGF-β administration on the ischemic heart are likely to be complex and dependent on the dose, species, timing and route of administration.
6. The role of endogenous TGF-β in regulating infarct healing
6. 1.The role of TGF-β in the post-infarction inflammatory response
Due to its pleiotropic and multifunctional effects, TGF-β may play an important and complex role in regulating the post-infarction inflammatory response. Although TGF-β1, - β2 and -β2 exhibit distinct patterns of expression in healing infarcts, the specific role of these isoforms in cardiac repair remains unknown. Unfortunately, as described previously, mice with mutations for genes involved in the TGF-β signaling cascade exhibit developmental defects and diffuse inflammation preventing their use as tools to dissect the role of TGF-β in infarct healing. Thus, information on the in vivo actions of TGF-β in healing infarcts is predominantly derived from studies using TGF-β inhibition strategies.
Although TGF-β is a potent mononuclear cell chemoattractant, its role in regulating monocyte chemotaxis in the infarct remains unknown. In the post-ischemic canine cardiac lymph TGF-β appears to be an important factor mediating monocyte recruitment during the first five hours of reperfusion [47]. Complement activation, free radical generation and monocyte chemoattractant chemokines also play a crucial role in recruiting mononuclear cells in the infarcted myocardium [47], [54]. TGF-β critically regulates cytokine and chemokine expression by monocytes, macrophages and endothelial cells. Its effects on macrophages are primarily deactivating, suppressing chemokine and pro-inflammatory cytokine synthesis. In addition, TGF-β inhibits chemokine synthesis by cytokine-stimulated endothelial cells [55]. Inhibition of TGF-β signaling through gene transfection with the extracellular domain of TGF-β type II receptor (TβRII) into the limb skeletal muscles resulted in increased mortality, enhanced neutrophil infiltration and increased pro-inflammatory cytokine and chemokine gene expression when applied early (within 24h) following coronary occlusion [56]. These findings suggest an important role for TGF-β signaling in resolution of inflammation and repression of cytokine and chemokine gene synthesis. In addition, mice with disruption of TSP-1, a critical activator of TGF-β, exhibit enhanced and prolonged inflammation following myocardial infarction associated with impaired Smad2 phosphorylation [48]. TSP-1 null mice show extension of the inflammatory reaction into the non-infarcted area, suggesting that localized induction of TSP-1 in the infarct border zone may serve as a barrier limiting expansion of inflammation into the non-infarcted area by locally activating TGF-β. In the absence of TSP-1, expansion of the inflammatory infiltrate leads to enhanced adverse remodeling [48]. Although the experiments on TSP-1 -/-mice support the role of TGF-β in suppression and containment of the inflammatory response, TSP-1 also has non-TGF-β mediated actions that may be important in regulation of the post-infarction inflammatory response.
6.2. The role of TGF-β in fibrous tissue deposition in the healing infarct
TGF-β promotes matrix deposition by inducing extracellular matrix protein expression by fibroblasts and by inhibiting matrix degradation through upregulation of TIMPs and PAI-1. In addition, TGF-β is critically involved in phenotypic modulation of fibroblasts into myofibroblasts. In vitro experiments have demonstrated potent and consistent effects of TGF-β on cardiac fibroblasts. TGF-β markedly enhances collagen type I and type III synthesis [57], decreases collagenase expression, increases TIMP-1 synthesis [58], upregulates integrin expression and induces acquisition of the myofibroblastic phenotype in isolated cardiac fibroblasts [59].
In the healing infarct activated myofibroblasts are the main source of collagen [60], and participate in extracellular matrix remodeling through MMP synthesis and release. Two independent studies inhibiting TGF-β by using gene transfer of the extracellular domain of the TβRII receptor suggested an important role for TGF-β in fibrous tissue deposition following myocardial infarction [56], [61]. In both studies TGF-β inhibition after the inflammatory phase of infarct healing resulted in decreased fibrous tissue deposition in the infarcted area. Although anti-TGF-β treatment markedly decreased collagen deposition in the infarct, it also altered the qualitative characteristics of the wound, increasing the cellular content and the density of myofibroblasts [61]. The mechanisms responsible for these effects remain unknown.
7. The role of TGF-β in cardiac remodeling
The term “ventricular remodeling” describes the alterations in size, shape and function of the left ventricle in response to changes in hemodynamic loading conditions, neurohormonal activation, or induction of local mediators that alter the structural characteristics of the myocardium. Remodeling is a dynamic and complex process resulting from activation of cellular and molecular pathways involving the cardiomyocytes, fibroblasts and extracellular matrix. Cardiac remodeling can be physiologic (described in elite athletes) or pathologic. Pathologic remodeling occurs in three major patterns: a) concentric remodeling when pressure overload causes growth in cardiomyocyte thickness, b) eccentric remodeling resulting from a volume load that produces cardiomyocyte lengthening and c) post-infarction remodeling, which involves a combined pressure and volume load on the non-infarcted area as well as interactions with the cellular and matrix components of the cardiac scar [62], [63]. TGF-β may be a crucial regulator of cardiac remodeling through its direct and potent actions in cardiomyocyte hypertrophy and cardiac extracellular matrix metabolism.
7.1. The link between TGF-β and the renin-angiotensin system
Numerous studies have established the importance of the renin-angiotensin system (RAS) in cardiac remodeling and clinical trials documented the beneficial effects of angiotensin II inhibition in patients with myocardial infarction and heart failure [64]. The RAS is markedly activated in response to hemodynamic overload and local generation of angiotensin II directly induces cellular responses in both cardiomyocytes and interstitial cells [65]. Angiotensin II exerts growth promoting effects on cardiomyocytes [66] and stimulates fibroblast proliferation and expression of extracellular matrix proteins [65], through interactions involving the Angiotensin Type 1 (AT1) receptor. Extensive evidence suggests a direct functional association between the RAS system and the TGF- pathway, indicating that TGF-β1 acts downstream of Angiotensin II [65]. Angiotensin II stimulation induces TGF-β1 mRNA and protein expression by cardiomyocytes and cardiac fibroblasts [67], [68]. In addition, angiotensin II enhances expression of endoglin in cardiac fibroblasts increasing their responsiveness to the fibrogenic actions of TGF- β [69]. Treatment with Angiotensin Converting Enzyme inhibitors or AT1 receptor blockers markedly decreased TGF-β1 levels in hypertrophied [70] and infarcted hearts [42], [41] suggesting that TGF-β induction in the remodeling myocardium is at least in part mediated through angiotensin II signaling. In addition, there is direct proof that TGF-β1 plays a critical role in mediating the hypertrophic response due to angiotensin II [71].
7.2. TGF-β signaling and post-infarction ventricular remodeling (Figure 3)
Figure 3. Role of TGF-β signaling in infarct healing and post-infarction remodeling.
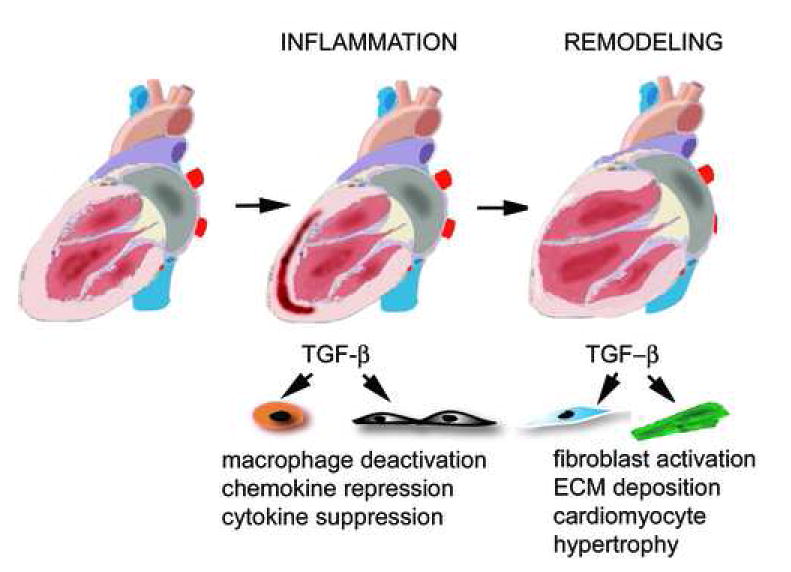
Myocardial infarction triggers an inflammatory reaction that ultimately results in formation of a scar. Infarct healing is associated with alterations in the geometric characteristics of the ventricle, dilation and hypetrophy; these changes are termed “ventricular remodeling”. In the early phases of infarct healing TGF-β may be important in resolution of the inflammatory response by deactivating macrophages and by suppressing endothelial cell chemokine and cytokine synthesis. At a later stage, TGF-β activates fibrogenic pathways by inducing extracellular matrix deposition and may contribute to the pathogenesis of left ventricular remodeling by promoting fibrosis and hypertrophy of the non-infarcted myocardium.
In the infarcted heart left ventricular remodeling begins within hours after the infarct and continues to progress over weeks or months [63]. Reparation of the necrotic area and formation of a scar is associated with profound changes in ventricular architecture and geometry of the ventricle leading to increased circumference and sphericity, and significantly increased left ventricular volume (Figure 3). Post-infarction remodeling is linked to heart failure progression and is associated with poor prognosis following myocardial infarction. Ventricular dilation following myocardial infarction is an important predictor of mortality [72] and adverse cardiac events [73], including the development of heart failure and ventricular arrhythmias. Although the pathways involved in remodeling remain poorly understood, it is clear that the pathologic and structural changes associated with infarct healing directly influence remodeling and affect prognosis in patients with myocardial infarction.
Because of its important role in regulating fibrous tissue deposition, composition of the extracellular matrix, and cardiac hypertrophy, TGF-β appears to be a crucial mediator in the pathogenesis of post-infarction remodeling. TGF-β inhibition during the proliferative phase of healing resulted in attenuated left ventricular remodeling decreasing cardiomyocyte hypertrophy and reducing interstitial fibrosis in the non-infarcted ventricle [56]. Anti-TGF-β therapy also had a significant effect on the geometry of the infarct, shortening and thickening the infarcted segment without affecting the absolute size of the infarct [61].
TGF-β1 induces cardiomyocyte hypertrophy through activation of TAK1, a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family [74]. The TGF-β1/TAK1-p38MAPK pathway is activated in spared cardiomyocytes following myocardial infarction and may play an important role in the development of hypertrophy in the remodeling myocardium [74]. In addition, TGF-β may critically modulate the composition and spatial localization of the extracellular matrix network in the infarcted heart. Activation of TGF-β signaling appears to be predominantly localized in the infarct border zone and may enhance extracellular matrix protein expression by local fibroblasts. Furthermore TGF-β enhances fibroblast TIMP synthesis promoting matrix deposition in the viable myocardium of the border zone. Enhanced deposition of matrix in the non-infarcted myocardium may induce dysfunction resulting in dilative remodeling of the infarcted ventricle.
7.3 TGF-β and hypertrophic remodeling (Figure 4)
Figure 4. TGF-β in hypetrophic and dilative remodeling.
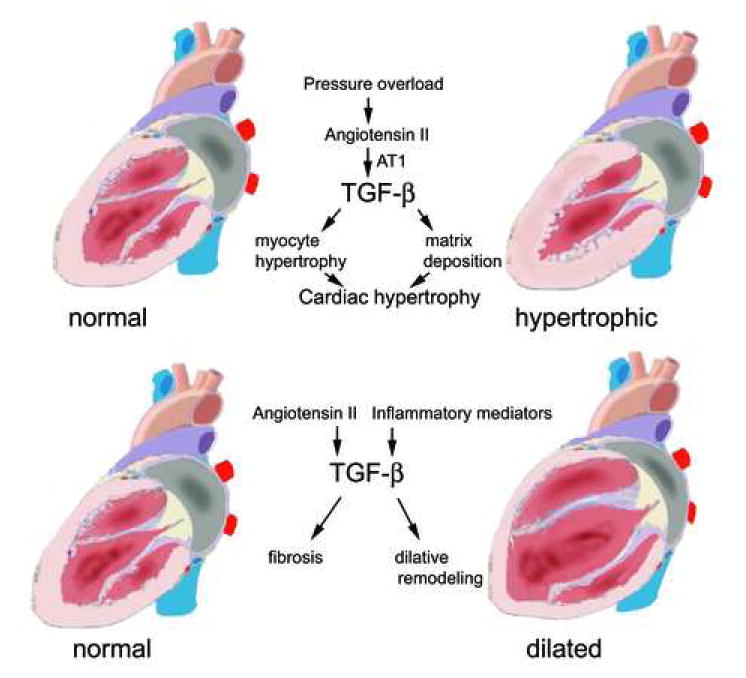
Evidence suggests a central role for TGF-β in the pathogenesis of hypertrophic cardiac remodeling. Pressure overload activates the Renin Angiotensin System generating angiotensin II. The hypertrophic effects of Angiotensin II are mediated through activation of TGF-β signaling pathways. TGF-β induces both cardiomyocyte hypertrophy and interstitial fibrosis. In contrast, the role of TGF-β in dilated cardiomyopathy is less clearly established. TGF-β induction mediated through Angiotensin II generation or via activation of inflammatory pathways may result in cardiac fibrosis playing a role in dilative remodeling.
Cardiac hypertrophy due to hemodynamic overload is associated with increased deposition of extracellular matrix, proliferation of cardiac fibroblasts and hypertrophic growth of cardiac myocytes. Several lines of evidence suggest an important role for TGF-β in regulating hypertrophic cardiac remodeling [75]. First, in vitro experiments demonstrated that TGF-β1 stimulation alters the program of differentiated gene expression in isolated cardiac myocytes promoting synthesis of fetal contractile proteins, characteristic of pressure-overload hypertrophy [76]. Second, TGF-β1 is markedly induced in the hypertrophied myocardium [77] and its expression correlates with fibrosis in the pressure-overloaded human heart [65]. Third, overexpression of TGF-β in transgenic mice results in cardiac hypertrophy which is characterized by both interstitial fibrosis and hypertrophic growth of cardiac myocytes [78]. Finally, there is direct evidence that TGF-β1 plays a critical role in angiotensin II-mediated cardiac hypertrophy. Schultz and co-workers demonstrated that TGF-β1 -/- mice bred in an immune compromised Rag1 deficient background (in order to overcome the lethal phenotype of TGF-β1 null animals, which develop diffuse multiorgan inflammatory lesions) were protected from the development of cardiac hypertrophy in response to subpressor doses of Angiotensin II [71]. These findings indicated that TGF-β1 acts downstream of Angiotensin II promoting cardiomyocyte growth in the heart.
Limited information is available on the signaling pathways responsible for the TGF-β- mediated hypertrophic response. A recent study demonstrated that cardiac hypertrophy due to aortic banding is associated with TAK1 activation. Constitutively activated TAK1 in the adult mouse heart mimicked hypertrophic TGF-β responses resulting in cardiomyocyte hypertrophy, interstitial fibrosis, “fetal” cardiac gene induction and early demise [79]. Although these experiments suggest that TAK1 activation is sufficient to provoke hypertrophy and heart failure in the mouse myocardium, direct proof of the role of this pathway in TGF-β mediated hypertrophic response is lacking.
7.4 The role of TGF-β in fibrotic remodeling of the cardiomyopathic ventricle
Development of interstitial fibrosis plays an important role in the pathobiology of cardiomyopathy and contributes to diastolic and systolic dysfunction by increasing both passive and active stiffness of the ventricle [80]. In addition, fibrotic remodeling impairs anisotropic conduction allowing the generation of re-entry circuits and promoting arrhythmias [81]. Marked fibrosis is noted in the hypertrophied heart, as well as in hearts with dilated cardiomyopathy [82]. Furthermore, extensive deposition of interstitial collagen in the absence of a completed infarction is often found in dysfunctional myocardial segments from patients with ischemic cardiomyopathy [83] and correlates inversely with contractile reserve [84]. TGF-β may play an important role in fibrotic remodeling of the cardiomyopathic ventricle by modulating fibroblast phenotype and gene expression and by promoting extracellular matrix deposition through upregulation of TIMP synthesis. We have recently demonstrated that in patients with chronic ischemic cardiomyopathy, dysfunctional myocardial segments with recovery of function following surgical revascularization had increased inflammatory leukocyte recruitment and MCP-1 expression, compared with irreversibly dysfunctional segments [85]. In contrast, myocardial segments with persistent dysfunction exhibited established interstitial fibrosis [83]. These findings suggest that chronic ischemic cardiomyopathy is a continuous process. At an early stage induction of inflammatory mediators by brief ischemic insults that do not result in cardiomyocyte necrosis leads to recruitment of leukocytes in the myocardium. Acute inflammation may activate endogenous inhibitory factors, such as TGF-β, which may suppress the inflammatory process, but may also stimulate extracellular matrix protein expression, leading to fibrous tissue deposition and irreversible dysfunction.
8. TGF-β as a therapeutic target in the infarcted and remodeling heart
Experimental studies suggest that the effects of anti-TGF-β strategies are dependent on the timing of the intervention [61], [86]. Anti-TGF gene therapy within 24h following infarction enhanced cytokine and chemokine synthesis and increased neutrophil infiltration resulting in exacerbated left ventricular dysfunction and increased mortality [56]. In contrast, late TGF-β inhibition attenuated cardiac hypertrophy and decreased interstitial fibrosis in the remodeling heart reducing left ventricular dilatation and dysfunction. Thus TGF-β inhibition during the inflammatory phase of infarct healing is detrimental and results in an exacerbated and prolonged local inflammatory response. Late TGF-β inhibition, on the other hand, does not interfere with the mechanisms that mediate resolution of the inflammatory infiltrate and has beneficial actions through attenuation of fibrotic and hypertrophic remodeling. Although this view is somewhat oversimplified, it can serve as an initial guide to frame future studies exploring the mechanistic basis of the TGF-β mediated effects in the healing infarct. Several important questions need to be answered:
-
What are the effects of TGF-β on various cell types involved in infarct healing and how do these effects influence the remodeling process?
All cell types involved in healing of myocardial infarcts express TGF-β receptors and respond to TGF-β stimulation. Generation of mutant mice with selective disruption of the TGF-β response in specific cell types may provide important information on the mechanisms of TGF-β mediated effects in healing infarcts. Understanding the in vivo significance of the actions of TGF-β on specific cell types may allow us to predict the consequences of TGF-β inhibition in patients with myocardial infarction.
-
What are the signaling mechanisms responsible for the TGF-β mediated effects in healing infarcts?
In the healing infarct, the anti-inflammatory and pro-fibrotic actions of TGF-β may involve activation of distinct signaling pathways. Dissecting the signaling pathways responsible for distinct TGF-β mediated effects in the infarcted myocardium is important in order to design strategies selectively targeting specific responses. Agents that target specific pathways downstream of the TGF-β receptor are more likely to have the desired effects while avoiding unwanted complications [87]. Recently, several agents targeting Smad-dependent and Smad-independent pathways have been developed. Halofuginone, a low molecular weight plant alkaloid that inhibits Smad3 activation and rapidly enhances Smad7 expression, has potent antifibrotic properties [88]. In addition, the c-abl kinase inhibitor imatinib ameliorates renal [89] and pulmonary [90] fibrosis by interfering with a Smad-independent TGF-β signaling pathway. Understanding the role of Smad-dependent and Smad-independent signaling in infarct healing may allow us to predict the consequences of various approaches targeting the TGF-β signaling cascade in patients with myocardial infarction.
-
Does TGF-β have disparate effects on infarct healing and left ventricular remodeling?
TGF-β may play distinct roles in regulating events involved in infarct healing and left ventricular remodeling. Activation of TGF-β signaling pathways in the infarct may be crucial for the formation and maintenance of a collagenous scar, increasing tensile strength and preventing left ventricular dilation [91]. In contrast, TGF-β-mediated effects in the non-infarcted remodeling myocardium may result in inappropriate fibrosis and dysfunction. Thus, the consequences of TGF-β inhibition during the fibrotic phase of infarct healing may depend on the topographic localization of the inhibitory agent.
-
What are the mechanisms responsible for TGF-β activation?
Although TGF-β activation plays a crucial role in the biology of TGF-β, little is known on the mechanisms of TGF-β activation in the infarcted heart. TSP-1 appears to be critically involved in mediating TGF-β activation in the infarct border zone. However, the significance of other TGF-β activating mechanisms such as proteases, integrins, and free radical generation remains unknown. The contribution of the various TGF-β activating factors may depend on their spatial distribution and the time course of their expression. Thus, TSP-1 may be important for TGF-β activation in the infarct border zone, whereas free radical generation may play a more limited role in activating readily available TGF-β stores immediately after injury.
-
What is the role of specific TGF-β isoforms in infarct healing and cardiac remodeling?
The differential expression of TGF-β1, β2 and β3 in the healing infarct may indicate distinct roles of the TGF-β isoforms in the infarcted heart. TGF-β1 is the most abundant isoform, the predominant isoform secreted by macrophages and fibroblasts, and the only isoform found in human platelets. Cutaneous wounds treated with either TGF-β1 or with TGF-β2 had increased extracellular matrix deposition in the early stages of wound healing. In contrast, application of TGF-β3 decreased monocyte recruitment and extracellular matrix deposition in healing wounds [93] and reduced scarring. Investigations exploring the role of specific TGF-β isoforms in infarct healing have not been performed.
-
What is the role of TGF-β signaling in high-risk populations, such as the diabetics and the elderly?
Evidence suggests that aging and diabetes significantly alter the cellular responses to TGF-β. Senescent animals with ischemic dermal ulcers [94], and diabetic rats with incisional wounds [95] exhibit impaired responses to TGF-β stimulation. Furthermore, dermal fibroblasts from senescent subjects show diminished expression of TGF-β receptors and blunted responsiveness to growth factors and fibrogenic substances [96]. The relevance of these findings in healing myocardial infarcts is unknown. Understanding the role of TGF-β signaling in elderly and diabetic patients with myocardial infarction is important in order to design optimal therapeutic strategies for these high-risk groups.
8. Conclusions
TGF-β is induced and activated in healing infarcts and, through its pleiotropic and multifunctional effects may regulate a wide range of cellular responses critical to cardiac repair. Unfortunately, the complex and often contradictory biological properties of TGF-β have hampered understanding of its role in infarct healing and cardiac remodeling, TGF-β may be involved in recruitment of mononuclear cells, repression of inflammatory gene synthesis, resolution of the inflammatory infiltrate, deposition of fibrous tissue and neovessel formation following myocardial infarction playing a crucial role in orchestrating the post-infarction inflammatory response. Understanding the timing, localization and signaling mechanisms responsible for TGF-β mediated effects is important in order to design novel therapeutic strategies targeting the TGF-β signaling cascade in the infarcted and remodeling heart.
Acknowledgments
This work was supported by NIH grant R01 HL76246, a grant-in-aid from the American Heart Association Texas affiliate and the Methodist Hospital Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication.As a service to our customers we are providing this early version of the manuscript.The manuscript will undergo copyediting, typesetting, and review of the resulting proofbefore it is published in its final citable form. Please note that during the productionprocess errorsmaybe discovered which could affect the content, and all legal disclaimersthat apply to the journal pertain.
References
- 1.Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–61. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 3.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 4.Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol. 1988;106:1659–65. doi: 10.1083/jcb.106.5.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–45. [PubMed] [Google Scholar]
- 6.Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 7.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 8.Piek E, Heldin CH, Ten Dijke P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13:2105–24. [PubMed] [Google Scholar]
- 9.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–78. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 10.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. Embo J. 2002;21:1743–53. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 12.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 13.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121:1845–54. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 14.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–4. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–70. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–14. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson NL, Flanders KC, Smith JM, Ellingsworth LR, Roberts AB, Sporn MB. Expression of transforming growth factor-beta 1 in specific cells and tissues of adult and neonatal mice. J Cell Biol. 1989;108:661–9. doi: 10.1083/jcb.108.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heine U, Munoz EF, Flanders KC, Ellingsworth LR, Lam HY, Thompson NL, et al. Role of transforming growth factor-beta in the development of the mouse embryo. J Cell Biol. 1987;105:2861–76. doi: 10.1083/jcb.105.6.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, et al. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation. 2001;103:2745–52. doi: 10.1161/01.cir.103.22.2745. [DOI] [PubMed] [Google Scholar]
- 20.Roberts AB, Roche NS, Winokur TS, Burmester JK, Sporn MB. Role of transforming growth factor-beta in maintenance of function of cultured neonatal cardiac myocytes. Autocrine action and reversal of damaging effects of interleukin-1. J Clin Invest. 1992;90:2056–62. doi: 10.1172/JCI116087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res. 2005;66:22–32. doi: 10.1016/j.cardiores.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 22.Frangogiannis NG. Targeting the inflammatory response in healing myocardial infarcts. Curr Med Chem. 2006;13:1877–93. doi: 10.2174/092986706777585086. [DOI] [PubMed] [Google Scholar]
- 23.Fava RA, Olsen NJ, Postlethwaite AE, Broadley KN, Davidson JM, Nanney LB, et al. Transforming growth factor beta 1 (TGF-beta 1) induced neutrophil recruitment to synovial tissues: implications for TGF-beta-driven synovial inflammation and hyperplasia. J Exp Med. 1991;173:1121–32. doi: 10.1084/jem.173.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith WB, Noack L, Khew-Goodall Y, Isenmann S, Vadas MA, Gamble JR. Transforming growth factor-beta 1 inhibits the production of IL-8 and the transmigration of neutrophils through activated endothelium. J Immunol. 1996;157:360–8. [PubMed] [Google Scholar]
- 25.Celada A, Maki RA. Transforming growth factor-beta enhances the M-CSF and GM-CSF-stimulated proliferation of macrophages. J Immunol. 1992;148:1102–5. [PubMed] [Google Scholar]
- 26.Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, et al. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci U S A. 1987;84:5788–92. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Werner F, Jain MK, Feinberg MW, Sibinga NE, Pellacani A, Wiesel P, et al. Transforming growth factor-beta 1 inhibition of macrophage activation is mediated via Smad3. J Biol Chem. 2000;275:36653–8. doi: 10.1074/jbc.M004536200. [DOI] [PubMed] [Google Scholar]
- 28.Feinberg MW, Shimizu K, Lebedeva M, Haspel R, Takayama K, Chen Z, et al. Essential role for Smad3 in regulating MCP-1 expression and vascular inflammation. Circ Res. 2004;94:601–8. doi: 10.1161/01.RES.0000119170.70818.4F. [DOI] [PubMed] [Google Scholar]
- 29.Lijnen PJ, Petrov VV, Fagard RH. Induction of cardiac fibrosis by transforming growth factor-beta(1) Mol Genet Metab. 2000;71:418–35. doi: 10.1006/mgme.2000.3032. [DOI] [PubMed] [Google Scholar]
- 30.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–11. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001;276:17058–62. doi: 10.1074/jbc.M100754200. [DOI] [PubMed] [Google Scholar]
- 32.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–27. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 33.Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8:21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 34.Oshima M, Oshima H, Taketo MM. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol. 1996;179:297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- 35.Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development. 1999;126:1631–42. doi: 10.1242/dev.126.8.1631. [DOI] [PubMed] [Google Scholar]
- 36.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626–31. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goumans MJ, Lebrin F, Valdimarsdottir G. Controlling the angiogenic switch: a balance between two distinct TGF-b receptor signaling pathways. Trends Cardiovasc Med. 2003;13:301–7. doi: 10.1016/s1050-1738(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 38.Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, et al. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. Embo J. 2004;23:4018–28. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deten A, Holzl A, Leicht M, Barth W, Zimmer HG. Changes in extracellular matrix and in transforming growth factor beta isoforms after coronary artery ligation in rats. J Mol Cell Cardiol. 2001;33:1191–207. doi: 10.1006/jmcc.2001.1383. [DOI] [PubMed] [Google Scholar]
- 40.Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–77. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu CM, Tipoe GL, Wing-Hon Lai K, Lau CP. Effects of combination of angiotensin-converting enzyme inhibitor and angiotensin receptor antagonist on inflammatory cellular infiltration and myocardial interstitial fibrosis after acute myocardial infarction. J Am Coll Cardiol. 2001;38:1207–15. doi: 10.1016/s0735-1097(01)01518-2. [DOI] [PubMed] [Google Scholar]
- 42.Sun Y, Zhang JQ, Zhang J, Ramires FJ. Angiotensin II, transforming growth factor-beta1 and repair in the infarcted heart. J Mol Cell Cardiol. 1998;30:1559–69. doi: 10.1006/jmcc.1998.0721. [DOI] [PubMed] [Google Scholar]
- 43.Hao J, Wang B, Jones SC, Jassal DS, Dixon IM. Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro. Am J Physiol Heart Circ Physiol. 2000;279:H3020–30. doi: 10.1152/ajpheart.2000.279.6.H3020. [DOI] [PubMed] [Google Scholar]
- 44.Dean RG, Balding LC, Candido R, Burns WC, Cao Z, Twigg SM, et al. Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem. 2005;53:1245–56. doi: 10.1369/jhc.4A6560.2005. [DOI] [PubMed] [Google Scholar]
- 45.Hao J, Ju H, Zhao S, Junaid A, Scammell-La Fleur T, Dixon IM. Elevation of expression of Smads 2, 3, and 4, decorin and TGF-beta in the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol. 1999;31:667–78. doi: 10.1006/jmcc.1998.0902. [DOI] [PubMed] [Google Scholar]
- 46.Wang B, Hao J, Jones SC, Yee MS, Roth JC, Dixon IM. Decreased Smad 7 expression contributes to cardiac fibrosis in the infarcted rat heart. Am J Physiol Heart Circ Physiol. 2002;282:H1685–96. doi: 10.1152/ajpheart.00266.2001. [DOI] [PubMed] [Google Scholar]
- 47.Birdsall HH, Green DM, Trial J, Youker KA, Burns AR, MacKay CR, et al. Complement C5a, TGF-beta 1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation. 1997;95:684–92. doi: 10.1161/01.cir.95.3.684. [DOI] [PubMed] [Google Scholar]
- 48.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, et al. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 49.Lefer AM, Tsao P, Aoki N, Palladino MA., Jr Mediation of cardioprotection by transforming growth factor-beta. Science. 1990;249:61–4. doi: 10.1126/science.2164258. [DOI] [PubMed] [Google Scholar]
- 50.Lefer AM, Ma XL, Weyrich AS, Scalia R. Mechanism of the cardioprotective effect of transforming growth factor beta 1 in feline myocardial ischemia and reperfusion. Proc Natl Acad Sci U S A. 1993;90:1018–22. doi: 10.1073/pnas.90.3.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baxter GF, Mocanu MM, Brar BK, Latchman DS, Yellon DM. Cardioprotective effects of transforming growth factor-beta1 during early reoxygenation or reperfusion are mediated by p42/p44 MAPK. J Cardiovasc Pharmacol. 2001;38:930–9. doi: 10.1097/00005344-200112000-00015. [DOI] [PubMed] [Google Scholar]
- 52.Behfar A, Zingman LV, Hodgson DM, Rauzier JM, Kane GC, Terzic A, et al. Stem cell differentiation requires a paracrine pathway in the heart. Faseb J. 2002;16:1558–66. doi: 10.1096/fj.02-0072com. [DOI] [PubMed] [Google Scholar]
- 53.Li TS, Hayashi M, Ito H, Furutani A, Murata T, Matsuzaki M, et al. Regeneration of infarcted myocardium by intramyocardial implantation of ex vivo transforming growth factor-beta-preprogrammed bone marrow stem cells. Circulation. 2005;111:2438–45. doi: 10.1161/01.CIR.0000167553.49133.81. [DOI] [PubMed] [Google Scholar]
- 54.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–9. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 55.Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J. 2001;15:1428–30. doi: 10.1096/fj.00-0745fje. [DOI] [PubMed] [Google Scholar]
- 56.Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, et al. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res. 2004;64:526–35. doi: 10.1016/j.cardiores.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 57.Eghbali M, Tomek R, Sukhatme VP, Woods C, Bhambi B. Differential effects of transforming growth factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts. Regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ Res. 1991;69:483–90. doi: 10.1161/01.res.69.2.483. [DOI] [PubMed] [Google Scholar]
- 58.Chua CC, Chua BH, Zhao ZY, Krebs C, Diglio C, Perrin E. Effect of growth factors on collagen metabolism in cultured human heart fibroblasts. Connect Tissue Res. 1991;26:271–81. doi: 10.3109/03008209109152444. [DOI] [PubMed] [Google Scholar]
- 59.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–7. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 60.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–38. [PMC free article] [PubMed] [Google Scholar]
- 61.Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430–7. doi: 10.1161/01.CIR.0000165066.71481.8E. [DOI] [PubMed] [Google Scholar]
- 62.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–67. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 63.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–82. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 64.Pfeffer JM, Fischer TA, Pfeffer MA. Angiotensin-converting enzyme inhibition and ventricular remodeling after myocardial infarction. Annu Rev Physiol. 1995;57:805–26. doi: 10.1146/annurev.ph.57.030195.004105. [DOI] [PubMed] [Google Scholar]
- 65.Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–32. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 66.Sadoshima J, Izumo S. Molecular characterization of angiotensin II--induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. 1993;73:413–23. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- 67.Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–63. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 68.Campbell SE, Katwa LC. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol. 1997;29:1947–58. doi: 10.1006/jmcc.1997.0435. [DOI] [PubMed] [Google Scholar]
- 69.Chen K, Mehta JL, Li D, Joseph L, Joseph J. Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ Res. 2004;95:1167–73. doi: 10.1161/01.RES.0000150369.68826.2f. [DOI] [PubMed] [Google Scholar]
- 70.Kim S, Ohta K, Hamaguchi A, Yukimura T, Miura K, Iwao H. Effects of an AT1 receptor antagonist, an ACE inhibitor and a calcium channel antagonist on cardiac gene expressions in hypertensive rats. Br J Pharmacol. 1996;118:549–56. doi: 10.1111/j.1476-5381.1996.tb15437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787–96. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51. doi: 10.1161/01.cir.76.1.44. [DOI] [PubMed] [Google Scholar]
- 73.St John Sutton M, Pfeffer MA, Plappert T, Rouleau JL, Moye LA, Dagenais GR, et al. Quantitative two-dimensional echocardiographic measurements are major predictors of adverse cardiovascular events after acute myocardial infarction. The protective effects of captopril. Circulation. 1994;89:68–75. doi: 10.1161/01.cir.89.1.68. [DOI] [PubMed] [Google Scholar]
- 74.Matsumoto-Ida M, Takimoto Y, Aoyama T, Akao M, Takeda T, Kita T. Activation of TGF-{beta}1-TAK1-p38 MAPK pathway in spared cardiomyocytes is involved in left ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2005 doi: 10.1152/ajpheart.00186.2005. [DOI] [PubMed] [Google Scholar]
- 75.Azhar M, Schultz Jel J, Grupp I, Dorn GW, 2nd, Meneton P, Molin DG, et al. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391–407. doi: 10.1016/s1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parker TG, Packer SE, Schneider MD. Peptide growth factors can provoke "fetal" contractile protein gene expression in rat cardiac myocytes. J Clin Invest. 1990;85:507–14. doi: 10.1172/JCI114466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Villarreal FJ, Dillmann WH. Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am J Physiol. 1992;262:H1861–6. doi: 10.1152/ajpheart.1992.262.6.H1861. [DOI] [PubMed] [Google Scholar]
- 78.Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1) Am J Physiol Heart Circ Physiol. 2002;283:H1253–62. doi: 10.1152/ajpheart.00578.2001. [DOI] [PubMed] [Google Scholar]
- 79.Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, et al. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556–63. doi: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 80.Sun Y, Weber KT. Animal models of cardiac fibrosis. Methods Mol Med. 2005;117:273–90. doi: 10.1385/1-59259-940-0:273. [DOI] [PubMed] [Google Scholar]
- 81.Khan R, Sheppard R. Fibrosis in heart disease: understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology. 2006;118:10–24. doi: 10.1111/j.1365-2567.2006.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brooks A, Schinde V, Bateman AC, Gallagher PJ. Interstitial fibrosis in the dilated non-ischaemic myocardium. Heart. 2003;89:1255–6. doi: 10.1136/heart.89.10.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frangogiannis NG, Shimoni S, Chang SM, Ren G, Dewald O, Gersch C, et al. Active interstitial remodeling: an important process in the hibernating human myocardium. J Am Coll Cardiol. 2002;39:1468–74. doi: 10.1016/s0735-1097(02)01792-8. [DOI] [PubMed] [Google Scholar]
- 84.Nagueh SF, Mikati I, Weilbaecher D, Reardon MJ, Al-Zaghrini GJ, Cacela D, et al. Relation of the contractile reserve of hibernating myocardium to myocardial structure in humans. Circulation. 1999;100:490–6. doi: 10.1161/01.cir.100.5.490. [DOI] [PubMed] [Google Scholar]
- 85.Frangogiannis NG, Shimoni S, Chang SM, Ren G, Shan K, Aggeli C, et al. Evidence for an active inflammatory process in the hibernating human myocardium. Am J Pathol. 2002;160:1425–33. doi: 10.1016/S0002-9440(10)62568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liao R. Yin and Yang of myocardial transforming growth factor-beta 1: timing is everything. Circulation. 2005;111:2416–7. doi: 10.1161/01.CIR.0000167557.59069.D9. [DOI] [PubMed] [Google Scholar]
- 87.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xavier S, Piek E, Fujii M, Javelaud D, Mauviel A, Flanders KC, et al. Amelioration of radiation-induced fibrosis: inhibition of transforming growth factor-beta signaling by halofuginone. J Biol Chem. 2004;279:15167–76. doi: 10.1074/jbc.M309798200. [DOI] [PubMed] [Google Scholar]
- 89.Wang S, Wilkes MC, Leof EB, Hirschberg R. Imatinib mesylate blocks a non-Smad TGF-beta pathway and reduces renal fibrogenesis in vivo. Faseb J. 2005;19:1–11. doi: 10.1096/fj.04-2370com. [DOI] [PubMed] [Google Scholar]
- 90.Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–16. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Holmes JW, Borg TK, Covell JW. Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng. 2005;7:223–53. doi: 10.1146/annurev.bioeng.7.060804.100453. [DOI] [PubMed] [Google Scholar]
- 92.Koch RM, Roche NS, Parks WT, Ashcroft GS, Letterio JJ, Roberts AB. Incisional wound healing in transforming growth factor-beta1 null mice. Wound Repair Regen. 2000;8:179–91. doi: 10.1046/j.1524-475x.2000.00179.x. [DOI] [PubMed] [Google Scholar]
- 93.Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J Cell Sci. 1995;108(Pt 3):985–1002. doi: 10.1242/jcs.108.3.985. [DOI] [PubMed] [Google Scholar]
- 94.Wu L, Xia YP, Roth SI, Gruskin E, Mustoe TA. Transforming growth factor-beta1 fails to stimulate wound healing and impairs its signal transduction in an aged ischemic ulcer model: importance of oxygen and age. Am J Pathol. 1999;154:301–9. doi: 10.1016/s0002-9440(10)65276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Broadley KN, Aquino AM, Hicks B, Ditesheim JA, McGee GS, Demetriou AA, et al. The diabetic rat as an impaired wound healing model: stimulatory effects of transforming growth factor-beta and basic fibroblast growth factor. Biotechnol Ther. 1989;1:55–68. [PubMed] [Google Scholar]
- 96.Mogford JE, Tawil N, Chen A, Gies D, Xia Y, Mustoe TA. Effect of age and hypoxia on TGFbeta1 receptor expression and signal transduction in human dermal fibroblasts: mpact on cell migration. J Cell Physiol. 2002;190:259–65. doi: 10.1002/jcp.10060. [DOI] [PubMed] [Google Scholar]