

Abstract
HCMV is a widespread opportunistic pathogen that causes birth defects in newborns and severe disease in immunocompromised individuals. The broad tropism of HCMV infection suggests that it utilizes multiple receptors. We recently demonstrated that EGFR serves as a receptor for HCMV. Here we show that HCMV also uses integrin αvβ3 as a coreceptor. Upon infection, HCMV glycoproteins gB and gH independently bind to EGFR and αvβ3, respectively, to initiate viral entry and signaling. αvβ3 then translocates to lipid rafts where it interacts with EGFR to induce coordinated signaling. The coordination between EGFR and αvβ3 is essential for the early events of HCMV infection, including viral entry, RhoA downregulation, stress fiber disassembly, and viral nuclear trafficking. Our findings support a model where EGFR and αvβ3 work together as coreceptors for HCMV entry and signaling. This discovery is fundamental to understanding HCMV pathogenesis and developing treatment strategies targeted to viral receptors.
Human cytomegalovirus (HCMV) is a β-group herpesvirus that causes severe complications in immunocompromised individuals. HCMV is also a leading cause of virus-associated birth defects, is associated with atherosclerosis and coronary restenosis, and has been implicated as a cofactor in the progression of HIV-1 infection1–4. The oncogenic potential of HCMV has also been demonstrated5–7.
Identification of receptors for HCMV is essential for understanding HCMV pathogenesis, since these receptors are involved in mediating the immediate early events necessary for infection. Many cell surface components have been identified as virus receptors8, including signaling receptors such as chemokine receptor9, PDGFR10, FGFR11, EGFR12, TNF receptor family13, and integrin14. Recently, we demonstrated that EGFR is essential for HCMV binding, signaling, and entry, and the HCMV envelope glycoprotein, gB, is the ligand for EGFR15.
Herpesviruses utilize multiple receptors, which may explain their broad tropism of infection16. Like other herpesviruses, the entry of HCMV into host cells occurs through a cascade of events that require interactions between cellular and viral molecules16. Pathological analyses of infected individuals indicate that HCMV can infect most major cell types. Its infection also triggers a variety of signaling cascades to elicits potent biological effects on host cells15,17,18. Multiple receptors are likely involved in HCMV infection.
HCMV disrupts actin stress fibers to facilitate infection19. Moreover, HCMV induces quiescent cells to re-enter G1 phase, which benefits viral DNA replication20. The rapid kinetics of these events suggest that they are regulated by receptor-mediated signaling. These events appear to be jointly regulated by growth factor receptors and integrins21–23. Although EGFR is capable of mediating these events, integrin is recognized as the essential element that links extracellular stimuli to the cytoskeleton and cell cycle regulators21,23–25.
The interaction between different receptors on the plasma membrane is one mechanism responsible for coordination between signaling pathways25. Evidence suggests that lipid raft microdomains regulate the interaction between integrin and growth factor receptor26. These domains are docking sites for many signaling molecules, and are origins for coordinated signaling27–29. Many microbial pathogens, including viruses, use lipid rafts for entry, trafficking, and budding30,31. In the present study, we set out to determine if integrin coordinates with EGFR to facilitate HCMV infection and if lipid rafts are involved in this coordination.
RESULTS
Both EGFR and αvβ3 are required for HCMV infection
To determine whether integrins participate in HCMV infection, we performed infection-blocking experiments using function-blocking antibodies against various integrins. αvβ3-specific antibodies inhibited HCMV infection of HEL cells by 82%, (Fig. 1a), whereas other isotype-matched antibodies against α1, α2, α3, α4, α5, α6, β1, β2, β4, αvβ5, and αvβ6 had no significant effect on infection (Fig. 1a and data not shown). When HCMV was preincubated with soluble αvβ3 molecules prior to infection, infectivity was inhibited by 77%; however, no inhibition was observed when HCMV was preincubated with αvβ5 (Fig. 1a). The inhibitory effect of αvβ3 was neutralized by preincubation with αvβ3-specific antibody, suggesting specificity of inhibition (Fig. 1a, αvβ3+Ab). Preincubation of cells with natural ligands of αvβ3 (Fig. 1a) blocked HCMV infectivity by 54% (vitronectin) and 23% (fibronectin). An RGD peptide, GRGDSP, did not block HCMV infection (Fig. 1a), indicating that αvβ3-mediated infection depends on a ligand motif different from the typical tripeptide RGD motif. Consistent with our previous observation15, EGFR-specific antibody inhibited HCMV infection by 97% (Fig. 1a).
Figure 1.
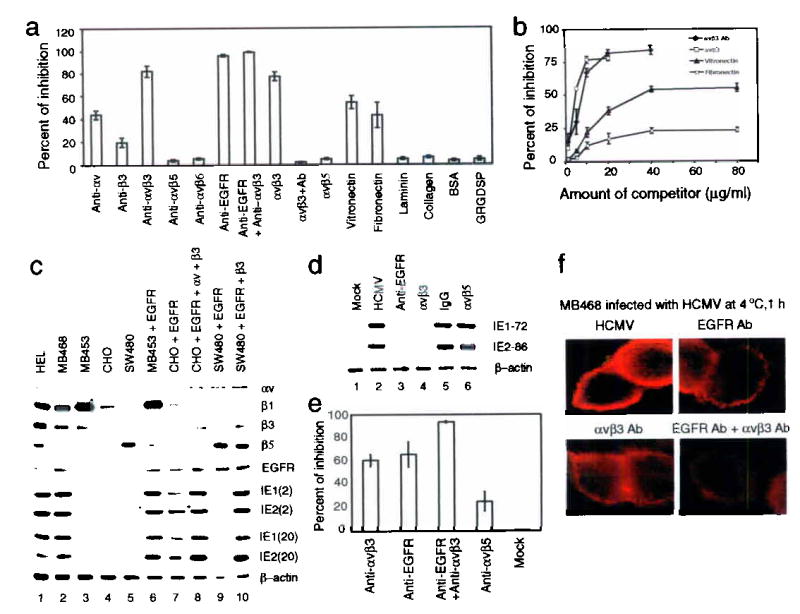
Both αvβ3 and EGFR are required for HCMV infection, and HCMV binds both EGFR and αvβ3 independently.
(a, b) HCMV infection blocking assays revealed that inhibition occurred when EGFR or αvβ3 was blocked (a), and the effects were dose-dependent (b). (c) IE gene products were detected in cells expressing both EGFR and αvβ3. (d) IE gene products decreased when HCMV-EGFR or HCMV-αvβ3 binding was inhibited, (e) EGFR- and αvβ3-specific antibodies inhibited HCMV binding, as determined by real-time PCR. (f) Immunofluorescence photomicrographs localizing UL94 show that binding of HCMV to MB468 cells was inhibited by blocking HCMV-EGFR or HCMV-αvβ3 interaction.
Inhibition of infectivity by αvβ3-specific antibody was dose-dependent (Fig. 1b). Similar dose-dependent inhibition was also seen when HCMV and cells were preincubated with soluble αvβ3 and vitronectin or fibronectin, respectively (Fig. 1b). These results suggest that integrin αvβ3 is involved in HCMV infection.
We looked for a correlation between viral gene expression and the presence of both EGFR and αvβ3 in various cell lines. We found that HCMV immediate-early (IE) proteins, IE1-72 and IE2-86, were expressed only in EGFR- and αvβ3-positive HEL and MB468 cells (Fig. 1c, lanes 1 and 2). There was no detectable IE protein expression in EGFR-negative cells, MB453 (lane 3) and CHO cells (lane 4), nor in β3-negative SW480 cells (lane 5). Transfection of an EGFR-expression construct into αvβ3-positive, EGFR-negative MB453 cells rendered these cells susceptible to infection (lane 6). High levels of IE protein expression were detected in CHO cells following co-transfection with EGFR-, αv-, and β3-expression plasmids (lane 8). SW480 cells, a β3-negative cell line which expresses a low level of EGFR32, were not susceptible to infection following transfection with the EGFR expression construct (lane 9). However, SW480 cells became susceptible to infection when EGFR- and β3-expression constructs were co-transfected (lane 10). The same results were obtained when infection was performed at two different M.O.I. (2 and 20 pfu/cell). In addition, levels of IE1-72 and IE2-86 were reduced dramatically when the interaction between HCMV-EGFR or HCMV-αvβ3 was blocked by EGFR-neutralizing antibody (Fig. 1d, lane 3) or soluble αvβ3 (lane 4), respectively. These data indicate that both EGFR and αvβ3 are required for HCMV infection.
EGFR and αvβ3 each interact with HCMV independently
To determine whether HCMV physically interacts with host cells via αvβ3, we incubated HEL cells with HCMV and quantified membrane-bound viruses by real-time DNA PCR33. Pretreatment of cells with EGFR- and αvβ3-specific antibodies inhibited HCMV binding by 65% and 60%, respectively (Fig. 1e). Ninety four percent of viral binding was inhibited when the cells were pretreated with both antibodies (Fig. 1e).
We used MB468 cells to monitor surface-bound particles by detecting HCMV capsid-associated protein UL94 via indirect immunofluorescence. Binding of HCMV to cells decreased when cells were pretreated with EGFR- or αvβ3-specific antibody (Fig. 1f). When cells were pretreated with the combination of both antibodies, inhibition of HCMV binding was nearly complete (Fig. 1f). Thus, HCMV can independently bind to target cells via both αvβ3 and EGFR.
HCMV activates PI3-K and Src signal pathways
To determine whether αvβ3 is activated upon HCMV infection, we assayed for phosphorylation of the β3 subunit in infected HEL cells (15 pfu/cell). HCMV induced tyrosine phosphorylation of β3 within 5 min post infection (p.i.) (Fig. 2a). As reported previously15, HCMV induced phosphorylation of EGFR within 5 min p.i. (Fig. 2a). Concurrently, PI3-K and Src, two downstream signaling molecules of EGFR and αvβ3, were activated following HCMV infection (Fig. 2a).
Figure 2.
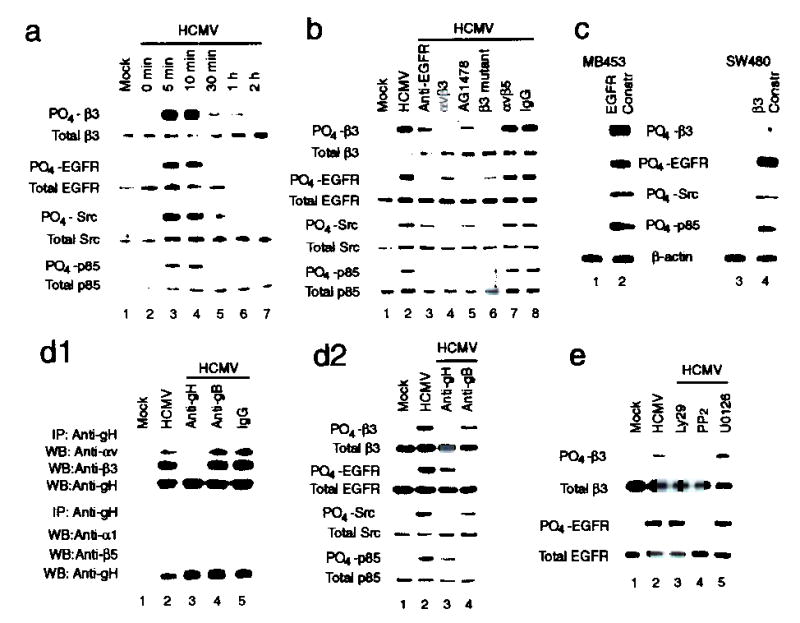
Coordinated signaling between EGFR and αvβ3 is generated when both αvβ3 and EGFR are activated via gH and gB, respectively.
(a) Both αvβ3 and EGFR were activated upon HCMV infection, concurrently with the activation of downstream Src and PI3-K signaling. (b,c) HCMV activated EGFR-mediated PI3-K and αvβ3-mediated Src signaling. Coordination between EGFR-and αvβ3-mediated signal occurred when both receptors were activated, (d1) HCMV interacted with αvβ3 via gH. (d2) HCMV-induced phosphorylation of β3 and Src were inhibited by blocking gH. (e) Crosstalk between EGFR and αvβ3 was inhibited by Ly29 or PP2 but not by U0126. PO4: phosphorylated; Constru: construct.
Phosphorylation of the β3 and Src were induced by HCMV binding to αvβ3, since the phosphorylation of β3 and Src were inhibited when cells were infected with HCMV pretreated with soluble αvβ3 (Fig. 2b, lane 4) or when cells were stably transfected with the β3-FF mutant plasmid (lane 6)34. Likewise, the phosphorylation of EGFR and p85 was inhibited when HCMV-induced EGFR activation was inhibited by EGFR-specific antibody (Fig. 2b, lane 3), or by AG1478 (100 nM), a specific inhibitor of EGFR kinase (lane 5). HCMV induced only PI3-K signaling in β3-negative, EGFR-positive SW480 cells (Fig. 2c, lane 3). Only Src signaling was induced in EGFR-negative, αvβ3-positive MB453 cells (lane 1). Both Src and PI3-K were activated when either MB453 or SW480 cells were converted to EGFR- and αvβ3-positive status (lanes 2 and 4, respectively). These results suggest that HCMV-induced PI3-K and Src signal pathways are mediated by EGFR and αvβ3, respectively.
HCMV gH is involved in the HCMV-αvβ3 interaction
The HCMV envelope protein gH has been implicated in signaling and fusion events during infection35,36. We found that gH interacted with αvβ3, since both αv and β3 subunits were co-immunoprecipitated with gH (Fig. 2d1, upper panel). The HCMV-αvβ3 interaction was disrupted by pretreating virus with gH-neutralizing antibody (lane 3). An isotype-matched antibody directed against the envelope protein gB - which we implicated in the interaction between HCMV and EGFR - could not disrupt this interaction (lane 4). Subunits α1 and β5 were not co-immunoprecipitated by gH-specific antibody (Fig. 2d1, lower panel). These data suggest that HCMV can specifically interact with αvβ3 via gH and that the gH-αvβ3 interaction is independent of the gB-EGFR interaction.
The gH-specific antibody blocked both αvβ3 and Src activation in HCMV-infected HEL cells (Fig. 2d2). Conversely, gB-specific antibody blocked both EGFR and p85 activation. These data suggest that HCMV activates EGFR- and αvβ3-mediated signaling pathways via gB and gH, respectively.
Crosstalk between EGFR-and αvβ3-mediated signaling
As shown above, HCMV only infects cells expressing both EGFR and αvβ3, suggesting that cooperation between EGFR and αvβ3 is required. When HCMV-induced EGFR activation was inhibited by pretreating cells with EGFR-specific antibody (Fig. 2b, lane 3) or AG1478 (lane 5), the β3 phosphorylation was also negatively affected (Fig. 2b, compare lanes 3 and 5 with lane 2). Similarly, when activation of αvβ3 was inhibited by preincubating HCMV with soluble αvβ3 (lane 4) or by transfecting cells with a β3-FF construct (lane 6), the EGFR phosphorylation was also affected (compare lanes 4 and 6 with lane 2). Src activation was inhibited profoundly by blocking EGFR activation (lanes 3 and 5), and PI3-K activation was inhibited by blocking αvβ3 activation (lanes 4 and 6). Similar results were observed when the activation of either αvβ3 or EGFR was inhibited by blocking the αvβ3-gH or EGFR-gB interaction, respectively (Fig. 2d2, lanes 3 and 4). These data suggest that there is crosstalk between EGFR and αvβ3 when both receptors are activated,which generates coordinated signaling and results in synergistic increases in both EGFR- and αvβ3-mediated signaling (Fig. 2b, lane 2 and Fig. 2d2, lane 2).
Phosphorylation of β3 decreased substantially after pretreatment of cells with Ly294002 (Ly29, 20 μM), a specific PI3-K inhibitor (Fig. 2e, lane 3). The phosphorylation of EGFR decreased when cells were treated with PP2, a specific Src inhibitor (10 μM, lane 4). The MAP-kinase inhibitor U0126 had no effect on phosphorylation of either β3 or EGFR (lane 5). We conclude that EGFR-dependent PI3-K and αvβ3-dependent Src signaling pathways coordinate following HCMV infection, leading to synergistic increases in downstream signaling.
EGFR-αvβ3 interaction is essential for signaling crosstalk
To determine whether HCMV induces an interaction between EGFR and αvβ3 and whether this leads to signaling coordination, we infected HEL cells with HCMV at 15 pfu/cell. EGFR co-precipitated with αvβ3 immunoprecipitated by αvβ3-specific antibody from lysates prepared 5 and 10 min after infection (Fig. 3a, row 1, lanes 3 and 4). Conversely, αv and β3 subunits co-precipitated with EGFR immunoprecipitated with EGFR-specific antibody (Fig. 3a, rows 3 and 4, lanes 3 and 4). EGFR did not co-precipitate with αvβ5 (Fig. 3a, row 6). The interaction between EGFR and αvβ3 was disrupted by inhibiting HCMV-induced activation of either EGFR or αvβ3 (Fig. 3b, lanes 3–6), as well as by inhibition of Src by PP2 or PI3-K by Ly29 (Fig. 3b, lanes 7 and 8). In similar assays, herpes simplex virus (HSV-1) did not induce an EGFR-αvβ3 interaction (Supplementary Fig. 1 online), suggesting that this interaction is specific to HCMV infection.
Figure 3.
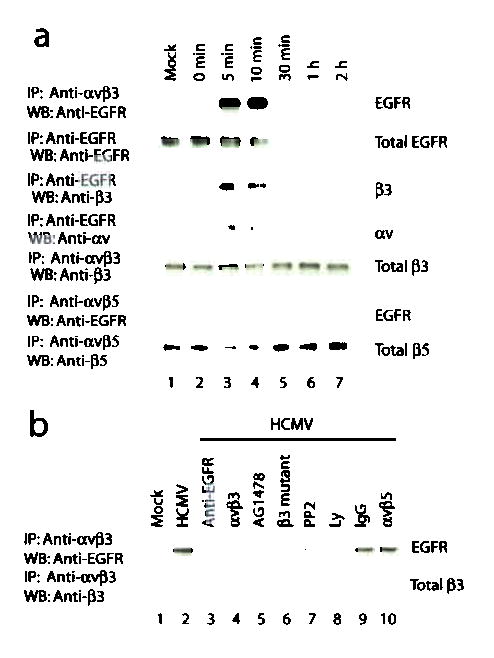
HCMV induces the formation of an EGFR-αvβ3 complex when both αvβ3 and EGFR are activated.
(a) EGFR co-precipitated with αvβ3 but not with αvβ5 in lysates from HCMV-infected HEL cells at 5 and 10 min p.i. (b) The interaction between EGFR and αvβ3 was disrupted by inhibiting the activation of EGFR, αvβ3, PI3-K, or Src. IP: immunoprecipitation; WB: western blot.
We compared complex formation data (shown in Fig. 3a,b) with signaling status (shown in Fig. 2a,b) and found that substantial phosphorylation of EGFR, β3, PI3-K and Src occurred at 5 and 10 min, the same times when EGFR-αvβ3 complexes were seen. Inhibiting the formation of EGFR-αvβ3 complex by blocking either EGFR or αvβ3 activation resulted in much lower levels of αvβ3- and EGFR-mediated signaling (Fig. 2b, d2). These data suggest that the HCMV-induced EGFR-αvβ3 interaction - which occurs only when both receptors are activated - is required for the coordination between the two receptors.
The lipid raft is involved in the signal coordination
We examined whether lipid rafts regulate the HCMV-induced interaction between EGFR and αvβ3. Prior to infection, αvβ3 was located in nonraft fractions (Total β3, Fig. 4a2, lane 1) and not in raft fractions (Fig. 4a1, lane 1). Exposure to HCMV induced movement of αvβ3 molecules into lipid rafts following αvβ3 activation (PO4-β3, Fig. 4a1). This translocation was not affected by EGFR-specific antibody or by AG1478 (data not shown), suggesting that the activation and translocation of αvβ3 to lipid rafts is independent of EGFR activation.
Figure 4.
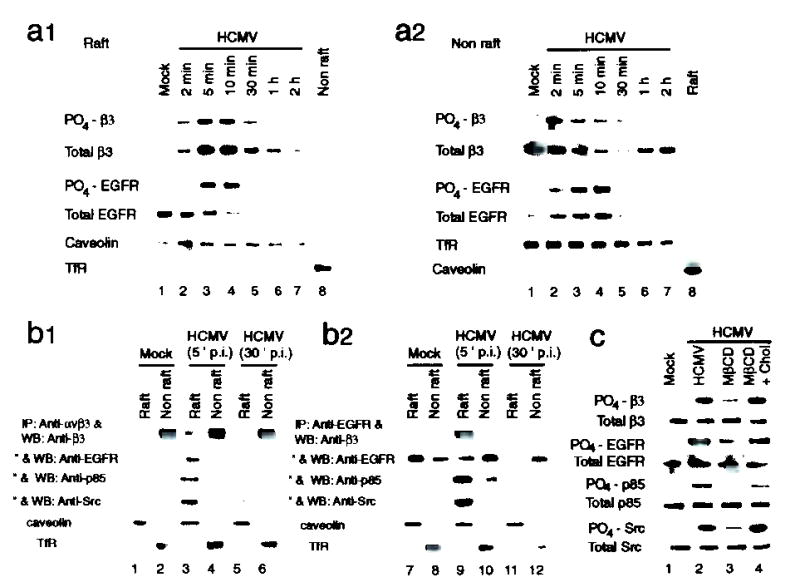
The lipid raft microdomain is involved in regulating HCMV-induced formation of EGFR-αvβ3 complex and coordination of signaling.
(a1, a2) Activated αvβ3 moved from non-raft fractions (a2) into lipid raft fractions (a1), where it colocalized with activated EGFR at 5 and 10 min p.i. (b1, b2) EGFR and αvβ3, together with p85 subunit of PI3-K and Src, formed multimeric complexes only when both receptors were co-localized in lipid raft microdomains. (c) Disruption of lipid rafts with MβCD reduced HCMV-induced signaling coordination. The effect of MβCD was reversed by cholesterol (Chol). TfR: transferrin receptor.
In uninfected cells, the majority of EGFR molecules were located in the raft fraction (compare Total EGFR, Fig. 4a1,a2, lane 1). By 10 min p.i., the total level of EGFR in lipid rafts decreased substantially (Fig. 4a1), with a corresponding increase in non-raft regions (Fig. 4a2). These events were not inhibited by blocking the HCMV-αvβ3 interaction (data not shown). Although HCMV induced movement of EGFR out of lipid rafts, a substantial level of phosphorylated EGFR localized with phosphorylated αvβ3 in the lipid raft fraction at 5 and 10 min p.i. (Fig. 4a1, lanes 3 and 4). HCMV-induced EGFR-αvβ3 complex formation was detected within this time frame (Fig. 3a, lanes 3 and 4), suggesting that lipid rafts regulate the interaction between EGFR and αvβ3.
EGFR was co-precipitated by αvβ3-specific antibody from raft fractions (5 min p.i.) but not from non-raft fractions (Fig. 4b1, lanes 3 and 4). p85 also co-precipitated with αvβ3 only from raft fractions (lanes 3 and 4). Similarly, both β3 and Src were precipitated by EGFR-specific antibody only from raft fractions (Fig. 4b2, lanes 9 and 10). However, when EGFR moved out of the lipid rafts (30 min p.i.), EGFR and p85 no longer co-precipitated with αvβ3 from raft fractions (b1, lanes 5 and 6). Conversely, neither β3 nor Src were pulled down by EGFR-specific antibody after EGFR moved into non-raft regions (b2, lanes 11 and 12). These data suggest that EGFR and αvβ3 interact with each other to form a multimeric complex only when both receptors are co-localized in lipid raft microdomains. Notably, the HCMV-induced phosphorylation of EGFR, β3, p85, and Src were all reduced when cells were pretreated with methyl-β-cyclodextrin (MβCD, 5 mM, Fig. 4c, lane 3), a cholesterol-depleting drug which disrupts the integrity of lipid rafts29. Inhibition did not occur after MβCD was removed and cells were incubated with media containing cholesterol (30 μg/ml) before infection (lane 4). We suggest that lipid rafts play an important role in the regulation of EGFR-αvβ3 interactions and, thereby, the coordination of EGFR- and αvβ3-dependent signaling pathways.
PI3-K and Src signaling are required for HCMV entry
To determine whether coordination of EGFR- and αvβ3-mediated signaling pathways is required for HCMV entry, we measured internalized viral DNA by real-time PCR. HCMV was internalized as early as 5 min p.i., and internalization reached a plateau at 30 min p.i. Internalization at 30 min p.i. was inhibited by 96% following Ly29 treatment, by 74% following PP2 treatment, and by 99% following MβCD treatment (Fig. 5a). HCMV entry was also studied by monitoring the localization of the capsid-associated protein UL94 in MB468 cells under fluorescence microscopy. Most of the UL94 was internalized and had translocated into nuclei by 2 hr p.i. (Fig. 5b). When cells were pretreated with Ly29, PP2, or MβCD, the majority of virus remained on the cell surface at 2 hr p.i. (Fig. 5b). Thus, HCMV entry can be blocked when either EGFR- or αvβ3-dependent signaling is inhibited or when the integrity of lipid rafts is disrupted.
Figure 5.
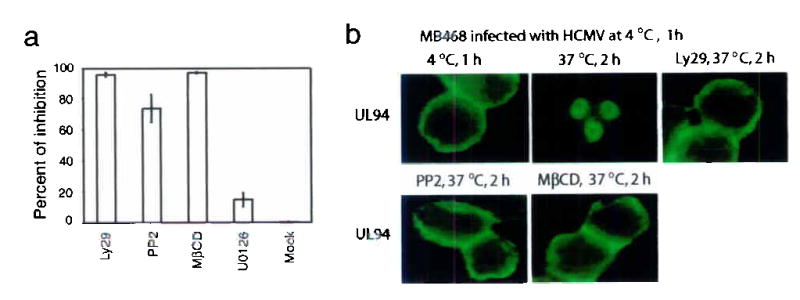
Coordination of PI3-K signaling and Src signaling within lipid rafts is required for HCMV entry.
(a) Ly29, PP2, and MβCD inhibited HCMV entry (as determined by real-time PCR) into HEL cells. Results are shown as percentages of inhibition, (b) Immunofluorescence photomicrographs localizing UL94 show that Ly29, PP2, and MβCD inhibited the internalization of HCMV into MB468 cells.
HCMV triggers downregulation of RhoA activity
Many viruses disrupt actin stress fibers to facilitate their nuclear translocation30,37. The RhoA/cofilin pathway is primary regulator of the generation and maintenance of stress fibers22,38. We found that levels of activated RhoA (GTP-RhoA) and total RhoA decreased dramatically following HCMV infection (Fig. 6a). Correspondingly, phosphorylation of cofilin, the downstream target of the RhoA22, decreased along with the decreased RhoA activity (Fig. 6a, lanes 3–7). HCMV-induced reductions of active RhoA (Fig. 6b, 5 min p.i.) and total RhoA (Fig. 6b, 1 hr p.i.) were inhibited when infection was inhibited with EGFR-specific antibody (I), soluble αvβ3 (II), Ly29, and PP2 (Fig. 6b, comparing lanes 3, 4, 6, and 7 with lane 2). Furthermore, when cells were treated with combinations of the inhibitors, I + II (lane 5) and Ly29 + PP2 (lane 8), there was negligible reduction of RhoA (comparing lanes 5 and 8 with lane 1). Phosphorylation of cofilin showed a similar pattern (Fig. 6b). These results suggest that signaling from both EGFR and αvβ3 are required for HCMV-induced downregulation of the RhoA/cofilin pathway.
Figure 6.
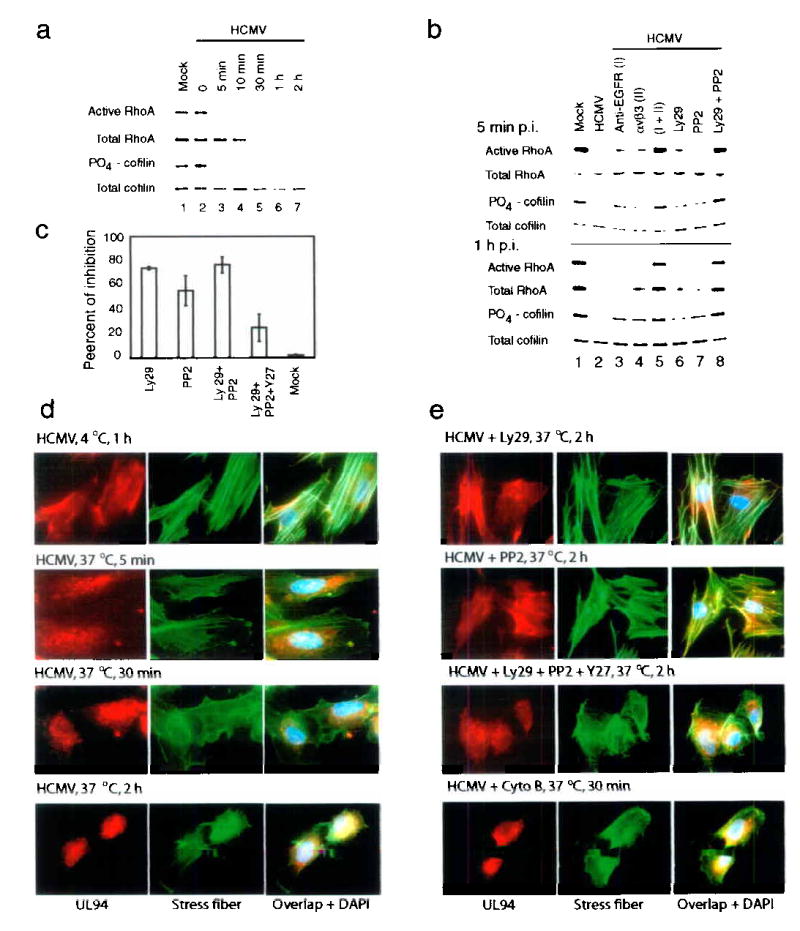
Coordination between EGFR- and αvβ3-mediated signaling is required for HCMV-induced downregulation of RhoA activity, disruption of stress fibers, and virus nuclear translocation.
(a) HCMV induced downregulation of the RhoA/cofilin pathway, (b) RhoA/cofilin downregulation was inhibited by blocking EGFR- or αvβ3-mediated signaling, (c) Ly29 or PP2 inhibited HCMV nuclear translocation (determined by real-time PCR), which was relieved by Y-27632. (d) HCMV induced stress fiber disassembly, which correlated with UL94 nuclear translocation. (e) Stress fiber disassembly and UL94 nuclear translocation were inhibited by blocking PI3-K or Src signaling. The inhibition was relieved by Y-27632. Cytochalasin B (Cyto B) facilitated HCMV nuclear translocation.
Stress fiber disruption and viral nuclear translocation
To determine if actin stress fibers change structurally following HCMV-induced RhoA downregulation, we used Alexa Fluor 488 labeled phalloidin to visually monitor actin in infected HEL cells. By 5 and 30 min p.i., the number and intensity of stress fibers had dramatically decreased (Fig 6d, rows 2 and 3), correlating with RhoA/cofilin downregulation (Fig. 6a). Movement of UL94 was also monitored via indirect immunofluorescence. During this period, UL94 entered cells and distributed throughout the cytoplasm (Fig. 6d, rows 2 and 3). At 2 h p.i., stress fibers were disrupted drastically, concomitantly with nuclear translocation of UL94 (Fig. 6d, row 4). These events were substantially inhibited by the treatment of cells with either Ly29 or PP2 (Fig. 6e, rows 1 and 2). The inhibition by Ly29 and PP2 could be reversed by addition of the RhoA inhibitor Y-27632 (Y27, 10 μM), as treatment of cells with the combination of Ly29, PP2, and Y27 led to both disruption of stress fibers and induction of nuclear translocation of UL94 (Fig. 6e, row 3). There was a close temporal relationship between the disruption of stress fibers and nuclear translocation of UL94 (Fig. 6d,e). Treatment of cells with cytochalasin B (5 μM), an actin-disrupting compound, led to a more rapid translocation of UL94, as early as 30 min p.i. (Fig. 6e, row 4).
Real-time PCR was performed to quantify nuclear translocation of viral DNA at 2 h p.i. Addition of inhibitors reduced viral nuclear translocation (Fig. 6c) by 73% (Ly29), 55% (PP2), and 76% (Ly29 + PP2). The inhibition by Ly29 and PP2 could be relieved by the addition of Y-27632 (Ly29 + PP2 + Y27). These data suggest that both HCMV-induced RhoA downregulation and disruption of stress fibers are essential for the nuclear translocation of HCMV.
DISCUSSION
Many viruses require more than one receptor to facilitate infection16,39–41. These receptors and their viral ligands usually form higher-order molecular complexes to generate coordinated signaling essential for the early events of infection, such as virus entry and trafficking16,42–44. Receptors known to be involved in coordinated signaling include growth factor receptor and integrin25. The coordination between receptors is essential for the signaling required for various cellular events, including cell proliferation, differentiation, and cell mobility23. In the present study, HCMV simultaneously and independently bound to EGFR (via gB) and αvβ3 (via gH) and induced both EGFR-dependent PI3-K and αvβ3-dependent Src signaling. Coordination between these two signaling pathways existed, as there was increased activation of each individual pathway when both pathways were activated. An HCMV-induced interaction between EGFR and αvβ3 within lipid rafts was required for the coordinated signaling. Disruption of the EGFR-αvβ3 interaction and specific inhibition of the coordination between P3-K and Src inhibited viral entry. Furthermore, coordination between PI3-K and Src was needed for HCMV-induced RhoA downregulation, which was required for disruption of actin stress fibers and viral nuclear translocation. Interestingly, RhoA activity did not seem to be essential for early stages of viral entry (Supplementary Fig. 2 online).
HCMV activates distinct signaling pathways via αvβ3 and EGFR. Various cellular processes - such as embryonic development, tissue homeostasis and cell cycle progression of normal cells - are tightly regulated by both integrin-mediated adhesion to the extracellular matrix and the binding of growth factors to their receptors25. Therefore, it is likely that each distinct receptor has its own predominant means of mediating cognate signaling pathways, each with specific biological activities. A significant observation from our studies is that HCMV appears induce coordination between EGFR- and αvβ3-mediated signaling pathways, and this coordination appears to be critical for successful viral infection.
Our studies show that HCMV induces translocation of αvβ3 into lipid rafts, thus enabling a physical interaction between αvβ3 and EGFR. In fact, the EGFR-αvβ3 interaction - as well as the formation of a multimeric complex composed of EGFR, αvβ3, and their downstream signal molecules, Src and the p85 subunit of PI3-K - occurs only in lipid rafts. We conclude that the lipid rafts are involved in the regulation of the EGFR-αvβ3 interaction and, consequently, coordinated signaling.
Based on our findings, we propose a new model of HCMV infection (Supplementary Fig. 3 online) in which HCMV membrane glycoproteins gB and gH bind EGFR and αvβ3 and trigger simultaneous activation of EGFR, αvβ3, and their respective signaling pathways. Activated EGFR and αvβ3 co-localize in lipid rafts, where they interact to form multimeric complexes that generate coordinated signaling. By this mechanism, HCMV is able to trigger potent, cooperative biological effects in target cells necessary for successful viral infection. We propose that HCMV entry occurs specifically within lipid rafts and not at random sites
Feire et al recently demonstrated that the integrin β1 subunit is also involved in HCMV infection45. Our preliminary data also indicates that β1 cooperates with EGFR to facilitate infection, but its efficiency is less than that of β3 (Supplementary Fig. 4 online). Whether β1 uses the same mechanism as αvβ3 to enhance infection needs to be investigated.
According to our model, the broad tropism of HCMV is due primarily to its ability to bind both EGFR and αvβ3. The ability to activate both EGFR and integrin may explain how HCMV elicits potent mitogenic activity. The coordinated signaling may also contribute to HCMV-induced cell migration, inflammatory responses, and fibrosis that, for example, lead to restenosis2. In the case of our current study, identifying coreceptors that act synergistically will surely influence future treatment strategies for combating HCMV infection.
METHODS
Cells and Virus
Human embryonic lung fibroblasts (HEL), human colorectal adenocarcinoma cells (SW480), human breast cancer cells (MDA-MB453 and MDA-MB468), and Chinese hamster ovary cells (CHO) were obtained from American Type Culture Collection and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum15. HEL cells transfected with pRcRSV-β3-FF plasmid were selected using G418. Towne strain HCMV (p38–40) was passaged and purified46 and analyzed by electron microscopy and SDS-PAGE (Supplementary Fig. 5 online). We used HCMV virions purified through a sucrose for biological assays and virions purified through a CsCl gradient for binding and trafficking assays.
Plasmids, Antibodies and Reagents
See Supplementary Methods online.
Infection Blocking Assays
We preincubated HEL cells with function-blocking antibodies against various integrins (20 μg/ml) and EGFR (1 μg/ml), integrin ligands (40 μg/ml), or GRGDSP (200 μg/ml) in DMEM at 4°C for 1 h. The cells were washed and infected with virus at M.O.I. of 2 pfu/cell. We also infected cells with HCMV preincubated with soluble integrins (10 μg/ml), with or without neutralizing antibody treatment at 3μg/ml. The cells were washed and covered with 1% methylcellulose in DMEM supplemented with 10% FBS and incubated at 37°C until microplaques were visible. We fixed and stained monolayers with 0.8% Crystal violet in 50% ethanol. We counted plaques under an inverted microscope. Results are shown as percentages of inhibition as compared to that of control cells. The data were from three independent experiments performed in duplicate. For determination of viral gene expressions, cells were pretreated with neutralizing antibodies or left untreated for 1 h before infection, and infected with HCMV (M.O.I. 2 pfu/cell) and harvested at 48 h p.i. In addition, we infected different cell lines as well as cells transfected with different plasmids expressing EGFR, αv subunit, or β3 subunit with HCMV at M.O.I. of 2 and 20 pfu/cell, respectively. Viral IE proteins were detected by immunoblotting.
Virus Binding Assays
The binding assays were performed by a method adapted from studies of EBV47. HEL cells were fixed briefly with ice-cold 0.1% paraformaldehyde and incubated with HCMV virions for 1hr at 4°C. Cells were washed and DNA molecules in HCMV particles remaining on the cell surface were released with lysis buffer (50 mM Tris-HCL, pH 8.0,120 mM NaCL, 0.5% NP40). We determined the copy numbers of DNA in the lysates by real-time PCR33,48. To block the binding of HCMV to cells, we preincubated the cells with neutralizing antibodies at 4°C for 1h.
Real-time DNA PCR
Real-time PCR was performed according to published procedures48. We used a pair of PCR primers (5′-GCTCCCCGAGCTCATTTTCA-3′ and 5′- CACAGCACGATGCCCACCA-3′) and a fluorescent probe FAM (5′-CACATACCCTACCGCCACGGCC-3′) TAMRA to determine the quantities of HCMV UL123 (IE 1-72) gene in samples. Samples with known copy numbers of HCMV DNA were used as standards.
Virus Entry Assays
We incubated HEL cells with HCMV at 4°C for 1 hr. Cells were washed and internalization was initiated by transferring the cells to 37°C. HCMV remaining on the cell surface was removed by incubation with trypsin (100 μg/ml) for 5 min at 37°C. The quantity of internalized viral DNA was measured by real-time PCR. Viral entry was blocked by preincubating cells with different compounds at 4°C for 1 hr.
Measurement of Viral DNA in Nuclei
After HCMV internalization, we isolated nuclei from HEL cells18 after virions remaining on the surface were removed with trypsin. We quantified viral DNA in nuclei by real-time PCR. For trafficking blocking assays, we incubated cells with HCMV at 37°C for 5 min. The membrane-bound viruses were removed with trypsin. Cells were re-cultured with or without inhibitors at 37°C for 2 hr. Nuclei were isolated and viral DNA in the nuclei was measured. We verified the purity of nuclei by immunoblotting with GRP78-specific antibody49.
Isolation of Caveolae/Rafts
We isolated raft using a published method50. Briefly, cells were lysed in TNE buffer (25 mM Tris-HCl, pH 7.5, 150mM NaCl, 5 mM EDTA) containing 1% Triton X-100 and adjusted to 40% sucrose using TNE-containing 80% sucrose. We placed the lysates in an ultracentrifuge tube and overlaid with 5–6 ml of TNE containing 38% sucrose and then with 2–3 ml of TNE containing 5% sucrose. The lysates were subjected to ultracentrifugation for 20 hr at 35,000 rpm at 4° C. We collected eight fractions and harvested the raft (fraction 3) and non-raft (fraction 8) fractions.
Immunofluorescence Microscopy
We fixed cells in 4% paraformaldehyde and permeabilized them in 0.1 % Triton X-100. The localization of UL94 was determined by incubation with anti-UL94 antibody, followed by incubation with FITC-or Rhodamine-labeled secondary antibody. For staining of actin stress fibers, we incubated samples with Alexa Fluor 488 phalloidin (Molecular Probes) according to the manufacturer’s instructions. Nuclei were stained with 0.1% DAPI in PBS. The cells were then mounted in Mowiol (Aldrich) and examined under an epifluorescence microscope (Zeiss Axioskop). We analyzed the images using the Openlab 3.0.7 Zeiss imaging system.
Supplementary Material
Supplementary Methods
Plasmids, Antibodies and Reagents
Constructs expressing human β3 integrin subunit (pRcRSV-β3) and mutated β3 with phenylalanine substitutions for Tyr747 and 759 (pRcRSV-β3-FF) were obtained from D, Clemmons. Full-length human αv subunit cDNA was obtained by RT-PCR sing a pair of primers (5′-GCACTTCGGCGATGGCTTTTCCG-3′ and 5′-TTAAGTTTCTGAGTTTCCTTCACC-3′) and ligated directly into the pTARGET mammalian expression vector (Promega). Full-length soluble integrin αvβ3 and αvβ5 used for functional studies, as well as the polyclonal antibodies against αvβ3, αv, β3, αvβ5 and β5, and function-blocking monoclonal antibodies specific to selected integrins (α1, α2, α3, α4, α5, α6, αv, β1, β2, β3, β4, αvβ3, αvβ5 and αvβ3 ) were purchased from Chemicon International. Anti-gB and anti-gH antibodies (IgG1) were purchased from Advanced Biotechnologies and ViroStat. Anti-PI3-K p85 antibody and anti-EGFR neutralizing antibody were obtained from Upstate Biotechnology. Antibodies against viral proteins (IE1-72, IE2-86, and UL94) were generated in our laboratory. Antibodies directed against EGFR, GRP 78, Src, caveolin, and transferrin receptor, as well as normal mouse IgG and FITC- and Rhodamine-labeled secondary antibodies were obtained from Santa Cruz. RhoA activation assay kits were purchased from Cytoskeleton. Antibody against activated Src (anti-phospho-tyrosine 416) was obtained from Cell Signaling. Vitronectin, fibronectin, laminin, collagen type I, cytochalasin B, methyl-β-cyclodextrin (MβCD), soluble cholesterol and 4′, 6′-diamidino-2-phenylindole (DAPI) were obtained from Sigma. Ly294006, PP2, U0126, and Y-27632 were purchased from Calbiochem. RGD peptide (GRGDSP) was obtained from GIBCO-BRL.
RhoA Activity Assays
The assay was performed using a RhoA activation assay kit (Cytoskeleton) according to the manufacturer’s instructions. Briefly, GTP-bound RhoA (active RhoA) was precipitated from cell lysates based on the capability of GTP-RhoA to bind to GST-rhotekin which was associated with glutathione-agarose beads. The bound RhoA on the beads was resolved by SDS-PAGE and detected by immunoblotting. The total Rho A in the same cell lysates was also measured by immunoblotting.
Immunoprecipitation and Western immunoblotting
HEL cells were serum-starved for 24 h prior to infection with HCMV purified by sucrose gradient at 15 pfu/cell. At different time points, cells were lysed with buffer containing 50 mM Tris-HCL, pH 8.0, 120 mM NaCL, 0.5% NP40, 100 mM NaF, 0.2 mM Na-orthovanadate, 1 mM PMSF, 20 μM leupeptin and 1.5 μM aprotinin. The protein concentrations of cell lysates were determined using a Bio-Rad protein assay kit. αvβ3, EGFR and p85 were immunoprecipitated from lysates with their corresponding antibodies. Tyrosine-phosphorylated β3, EGFR and p85 were detected by immunoblotting with anti-phosphotyrosine antibody, while activated Src was detected by anti-phospho-Src (Tyr416) antibody. The level of total Src in lysates and levels of immunoprecipitated β3, EGFR, and p85 were detected by immunoblotting after stripping the same membranes.
Acknowledgments
We are very grateful to David Clemmons for β3 and mutated β3 plasmids. We thank N. Raab-Traub, J. S. Pagano, and D. Evers for helpful discussion and critical review of this manuscript. This study was supported by Public Health Service research grants AI47468 from the National Institute of Allergy and Infectious Diseases and CA 19014 from the National Cancer Institute, National Institute of Health.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
References
- 1.Huang, E.-S. & Kowalik, T.F. The pathogenicity of human cytomegalovirus: An overview. in Molecular Aspects of Human Cytomegalovirus Diseases (eds. Becker, Y., Darai, G. & Huang, E.S.) 1–45 (Springer-Verlag, Berlin, 1993).
- 2.Speir E, et al. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science. 1994;265:391–4. doi: 10.1126/science.8023160. [DOI] [PubMed] [Google Scholar]
- 3.Mocarski, E.S., and Courcelle C. T. Cytomegaloviruses and Their Replication, in Fields Virology, Vol. 2 (ed. Knipe, D.M., and Howley, P. M.) 2629–2673 (Lippincott William & Wilkins, Philadelphia, 2001).
- 4.Macher AM, et al. Death in the AIDS patient: role of cytomegalovirus. N Engl J Med. 1983;309:1454. doi: 10.1056/NEJM198312083092312. [DOI] [PubMed] [Google Scholar]
- 5.Shen Y, Zhu H, Shenk T. Human cytomagalovirus IE1 and IE2 proteins are mutagenic and mediate “hit-and-run” oncogenic transformation in cooperation with the adenovirus E1A proteins. Proc Natl Acad Sci U S A. 1997;94:3341–5. doi: 10.1073/pnas.94.7.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukac DM, Alwine JC. Effects of human cytomegalovirus major immediate-early proteins in controlling the cell cycle and inhibiting apoptosis: studies with ts13 cells. J Virol. 1999;73:2825–31. doi: 10.1128/jvi.73.4.2825-2831.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cinatl J, Scholz M, Kotchetkov R, Vogel JU, Doerr HW. Molecular mechanisms of the modulatory effects of HCMV infection in tumor cell biology. Trends Mol Med. 2004;10:19–23. doi: 10.1016/j.molmed.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Flint, S.J., Enquist, L.W., Krug, M.R., Racaniello, V.R. & Skalka, A.M. Chapter 4 Virus Attachemnt to Host cell. in Principles of Virology: molecular biology, pathogenesis, and control 101–131 (AWSM press, Washington, D.C., 2000).
- 9.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–7. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 10.Di Pasquale G, et al. Identification of PDGFR as a receptor for AAV-5 transduction. Nat Med. 2003;9:1306–12. doi: 10.1038/nm929. [DOI] [PubMed] [Google Scholar]
- 11.Qing K, et al. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat Med. 1999;5:71–7. doi: 10.1038/4758. [DOI] [PubMed] [Google Scholar]
- 12.Eppstein DA, et al. Epidermal growth factor receptor occupancy inhibits vaccinia virus infection. Nature. 1985;318:663–5. doi: 10.1038/318663a0. [DOI] [PubMed] [Google Scholar]
- 13.Terry-Allison T, et al. HveA (herpesvirus entry mediator A), a coreceptor for herpes simplex virus entry, also participates in virus-induced cell fusion. J Virol. 1998;72:5802–10. doi: 10.1128/jvi.72.7.5802-5810.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nemerow GR, Cheresh DA. Herpesvirus hijacks an integrin. Nat Cell Biol. 2002;4:E69–71. doi: 10.1038/ncb0402-e69. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature. 2003;424:456–61. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 16.Spear PO, Longnecker R. Herpesvirus entry: an update. J Virol. 2003;77:10179–85. doi: 10.1128/JVI.77.19.10179-10185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fortunato EA, McElroy AK, Sanchez I, Spector DH. Exploitation of cellular signaling and regulatory pathways by human cytomegalovirus. Trends Microbiol. 2000;8:111–9. doi: 10.1016/s0966-842x(00)01699-1. [DOI] [PubMed] [Google Scholar]
- 18.Johnson RA, Wang X, Ma XL, Huong SM, Huang ES. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J Virol. 2001;75:6022–32. doi: 10.1128/JVI.75.13.6022-6032.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones NL, Lewis JC, Kilpatrick BA. Cytoskeletal disruption during human cytomegalovirus infection of human lung fibroblasts. Eur J Cell Biol. 1986;41:304–12. [PubMed] [Google Scholar]
- 20.Kalejta RF, Shenk T. Manipulation of the cell cycle by human cytomegalovirus. Front Biosci. 2002;7:d295–306. doi: 10.2741/kalejta. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt A, Hall MN. Signaling to the actin cytoskeleton. Annu Rev Cell Dev Biol. 1998;14:305–38. doi: 10.1146/annurev.cellbio.14.1.305. [DOI] [PubMed] [Google Scholar]
- 22.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz MA, Ginsberg MH. Networks and crosstalk: integrin signalling spreads. Nat Cell Biol. 2002;4:E65–8. doi: 10.1038/ncb0402-e65. [DOI] [PubMed] [Google Scholar]
- 24.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 25.Giancotti FG, Tarone G. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu Rev Cell Dev Biol. 2003;19:173–206. doi: 10.1146/annurev.cellbio.19.031103.133334. [DOI] [PubMed] [Google Scholar]
- 26.Baron W, Decker L, Colognato H, ffrench-Constant C. Regulation of integrin growth factor interactions in oligodendrocytes by lipid raft microdomains. Curr Biol. 2003;13:151–5. doi: 10.1016/s0960-9822(02)01437-9. [DOI] [PubMed] [Google Scholar]
- 27.Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- 28.Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–67. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 30.Pelkmans L, Puntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science. 2002;296:535–9. doi: 10.1126/science.1069784. [DOI] [PubMed] [Google Scholar]
- 31.Manes S, del Real G, Martinez AC. Pathogens: raft hijackers. Nat Rev Immunol. 2003;3:557–68. doi: 10.1038/nri1129. [DOI] [PubMed] [Google Scholar]
- 32.Li E, et al. Association of p130CAS with phosphatidylinositol-3-OH kinase mediates adenovirus cell entry. J Biol Chem. 2000;275:14729–35. doi: 10.1074/jbc.275.19.14729. [DOI] [PubMed] [Google Scholar]
- 33.Wu E, et al. Membrane cofactor protein is a receptor for adenoviruses associated with epidemic keratoconjunctivitis. J Virol. 2004;78:3897–905. doi: 10.1128/JVI.78.8.3897-3905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ling Y, Maile LA, Clemmons DR. Tyrosine phosphorylation of the beta3-subunit of the alphaVbeta3 integrin is required for embrane association of the tyrosine phosphatase SHP-2 and its further recruitment to the insulin-like growth factor I receptor. Mol Endocrinol. 2003;17:1824–33. doi: 10.1210/me.2003-0143. [DOI] [PubMed] [Google Scholar]
- 35.Yurochko AD, et al. The human cytomegalovirus UL55 (gB) and UL75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF-kappaB during infection. J Virol. 1997;71:5051–9. doi: 10.1128/jvi.71.7.5051-5059.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keay S, Baldwin B. The human fibroblast receptor for gp86 of human cytomegalovirus is a phosphorylated glycoprotein. J Virol. 1992;66:4834–8. doi: 10.1128/jvi.66.8.4834-4838.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cudmore S, Reckmann I, Way M. Viral manipulations of the actin cytoskeleton. Trends Microbiol. 1997;5:142–8. doi: 10.1016/S0966-842X(97)01011-1. [DOI] [PubMed] [Google Scholar]
- 38.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. Embo J. 1999;18:578–85. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dimitrov DS. Virus entry: molecular mechanisms and biomedical applications. Nat Rev Microbiol. 2004;2:109–22. doi: 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider-Schaulies J. Cellular receptors for viruses: links to tropism and pathogenesis. J Gen Virol. 2000;81:1413–29. doi: 10.1099/0022-1317-81-6-1413. [DOI] [PubMed] [Google Scholar]
- 41.Smith AE, Helenius A. How viruses enter animal cells. Science. 2004;304:237–42. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- 42.Fuller AO, Perez-Romero P. Mechanisms of DNA virus infection: entry and early events. Front Biosci. 2002;7:d390–406. doi: 10.2741/A783. [DOI] [PubMed] [Google Scholar]
- 43.Greber UF. Signalling in viral entry. Cell Mol Life Sci. 2002;59:608–26. doi: 10.1007/s00018-002-8453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore JP, Doms RW. The entry of entry inhibitors: a fusion of science and medicine. Proc Natl Acad Sci U S A. 2003;100:10598–602. doi: 10.1073/pnas.1932511100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feire AL, Koss H, Compton T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc Natl Acad Sci U S A. 2004;101:15470–5. doi: 10.1073/pnas.0406821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang ES, Chen ST, Pagano JS. Human cytomegalovirus. I. Purification and characterization of viral DNA. J Virol. 1973;12:1473–81. doi: 10.1128/jvi.12.6.1473-1481.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller N, Hutt-Fletcher LM. A monoclonal antibody to glycoprotein gp85 inhibits fusion but not attachment of Epstein-Barr virus. J Virol. 1988;62:2366–72. doi: 10.1128/jvi.62.7.2366-2372.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim JH, Saito K, Yokoyama S. Chimeric receptor analyses of the interactions of the ectodomains of ErbB-1 with epidermal growth factor and of those of ErbB-4 with neuregulin. Eur J Biochem. 2002;269:2323–9. doi: 10.1046/j.1432-1033.2002.02877.x. [DOI] [PubMed] [Google Scholar]
- 49.Tomlinson CC, Damania B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J Virol. 2004;78:1918–27. doi: 10.1128/JVI.78.4.1918-1927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arreaza G, Melkonian KA, LaFevre-Bernt M, Brown DA. Triton X-100-resistant membrane complexes from cultured kidney epithelial cells contain the Src family protein tyrosine kinase p62yes. J Biol Chem. 1994;269:19123–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.