

Abstract
The checkpoint kinase ATM (ataxia telangiectasia mutated) transduces genomic stress signals to halt cell cycle progression and promote DNA repair in response to DNA damage. Here, we report the characterisation of an essential cofactor for ATM, ATMIN (ATM INteracting protein). ATMIN interacts with ATM through a C-terminal motif, which is also present in Nijmegen breakage syndrome (NBS)1. ATMIN and ATM colocalised in response to ATM activation by chloroquine and hypotonic stress, but not after induction of double-strand breaks by ionising radiation (IR). ATM/ATMIN complex disruption by IR was attenuated in cells with impaired NBS1 function, suggesting competition of NBS1 and ATMIN for ATM binding. ATMIN protein levels were reduced in ataxia telangiectasia cells and ATM protein levels were low in primary murine fibroblasts lacking ATMIN, indicating reciprocal stabilisation. Whereas phosphorylation of Smc1, Chk2 and p53 was normal after IR in ATMIN-deficient cells, basal ATM activity and ATM activation by hypotonic stress and inhibition of DNA replication was impaired. Thus, ATMIN defines a novel NBS1-independent pathway of ATM signalling.
Keywords: ATM, ATMIN, DNA damage, NBS1
Introduction
Ataxia telangiectasia mutated (ATM) is a member of the phosphatidylinositol kinase-related protein family that includes ATR and DNAPKcs. These kinases respond to the presence of DNA damage or replication blocks by activating cell cycle checkpoints and promotion of DNA repair (Shiloh, 2003; Bakkenist and Kastan, 2004). ATM is mutated in the genomic instability syndrome ataxia telangiectasia (A-T), which is characterised by problems in motor coordination, immunodeficiency and increased tumour incidence (McKinnon, 2004).
ATM is believed to form inactive homodimers in the absence of stimulation. In response to activating signals, the best-studied example being induction of double-strand breaks (DSBs), ATM undergoes rapid dimer dissociation and ATM kinase activity is strongly elevated. Active ATM phosphorylates numerous substrates including Smc1 and p53, at serine or threonine residues followed by glutamine (the ‘SQ/TQ' motif) (Kim et al, 1999; Shiloh, 2003).
ATM activation is accompanied by intermolecular ATM autophosphorylation. Serine 1981 was the first identified site of ATM autophosphorylation and antibodies specific for phosphorylated serine 1981 have been widely used as a biological marker for the active monomeric form of ATM (Bakkenist and Kastan, 2003). Subsequently, serines 367 and 1893 have been identified as additional ATM autophosphorylation sites (Kozlov et al, 2006). Nijmegen breakage syndrome (NBS) is a heritable human condition that shows some overlapping clinical features with A-T (Digweed and Sperling, 2004). The protein mutated in NBS, called NBS1 or nibrin, is a component of the MRN (Mre11/Rad50/NBS1) complex (D'Amours and Jackson, 2002; Stracker et al, 2004). Human NBS1 mutations are hypomorphic and thus retain some MRN activity (Maser et al, 2001; Digweed and Sperling, 2004), presumably because NBS1-null mutations are not compatible with survival (Zhu et al, 2001; Dumon-Jones et al, 2003). MRN associates with ATM after induction of DNA damage and is required for ATM activation and recruitment of ATM to DSBs (Uziel et al, 2003; Lee and Paull, 2005; Kim et al, 2006). NBS1 directly interacts with ATM, and a short C-terminal motif in NBS1 has been shown to significantly contribute to ATM/NBS1 binding (Falck et al, 2005).
Although the ATM kinase is best known as a master regulator of the cell cycle checkpoints and repair after DSBs, ATM also responds to other types of cellular stresses. UV light, hypotonic stress and inhibitors of DNA replication like hydroxyurea (HU) trigger ATM serine 1981 autophosphorylation and cause increased phosphorylation of ATM substrates without producing DSB (Bakkenist and Kastan, 2003). NBS1 is not required for ATM autophosphorylation by these other stimuli (Difilippantonio et al, 2005) and the molecular mechanism that underlies DSB-independent ATM activation has been enigmatic.
Our manuscript describes ATM INteracting protein (ATMIN) as a regulatory protein that controls the activity of the DNA damage checkpoint kinase ATM in the absence of DSBs. ATMIN and ATM reciprocally stabilise each other, suggesting an intimate functional connection. ATMIN colocalises with S1981-phosphorylated ATM and is required for ATM activity in basal conditions, after HU treatment and in response to hypotonic stress, but not after ionising radiation (IR). Our data demonstrate that ATMIN is essential for NBS1-independent ATM activation by these stimuli, thus defining a novel pathway of ATM signalling.
Results
ATMIN interacts with ATM
Recently, a novel ATM substrate protein called ASCIZ was described that contained multiple SQ/TQ sites and two N-terminal C2H2 zinc (Zn2+) fingers (McNees et al, 2005). However, analysis of several expressed sequence tags (ESTs) suggested that the published 667-amino-acid ASCIZ protein sequence is incomplete. Two additional upstream exons, each encoding one additional Zn2+ finger, add 156 amino acids upstream of the described methionine start codon (Supplementary Figure 1A). RT–PCR analysis confirmed that the full-length mRNA contained the additional exons (Supplementary Figure 1B). Thus, the human cDNA encodes a protein of 823 amino acids with a predicted molecular size of 88.3 kDa. Phylogenic comparison revealed significant amino-acid sequence similarity to ESTs in several metazoan species including mouse, chicken and zebrafish (Supplementary Figure 1C) and the full-length proteins of all species analysed contained four highly conserved Zn2+ fingers (Supplementary Figure 1C and Figure 1A). In addition, a putative PEST sequence, which has been implicated in ubiquitin-mediated protein degradation (Rogers et al, 1986), was predicted (Figure 1A). To discriminate the complete protein from the previously described truncated form, and due to its biological properties described below, we refer to the full-length protein as ATMIN.
Figure 1.
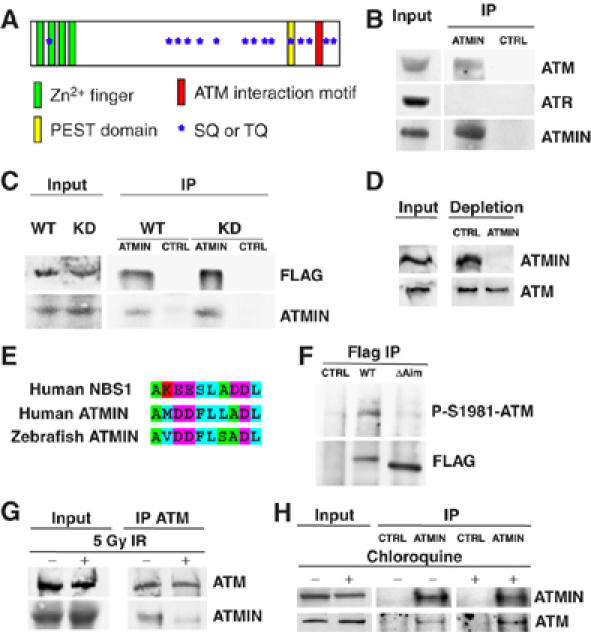
ATMIN interacts with ATM. (A) Schematic representation of the ATMIN protein. Approximate regions of ATMIN encoding Zn2+ fingers are indicated by green boxes, the PEST domain as a yellow box, the ATM interaction motif as a red box, SQ/TQ motifs as blue stars. (B) ATMIN immunoprecipitation (IP) was performed with HCT116 cell lysates followed by immunoblotting with antibodies specific for ATM, ATR and ATMIN. (C) HEK 293T cells were transfected with FLAG-tagged wild-type ATM (WT) or FLAG-tagged kinase-dead ATM (KD), ATMIN IP was performed, followed by immunoblotting with FLAG and ATMIN-specific antibodies. (D) HEK 293T cell protein extracts were depleted of ATMIN protein or control depleted, followed by immunoblotting with ATMIN- and ATM-specific antibodies. (E) Alignment of the ATM interaction motifs of human NBS1, human ATMIN and zebrafish ATMIN. Amino acids are coloured according to biochemical properties (hydrophobic, blue; acidic, purple; basic, red; polar, green). (F) FLAG IP was performed on HEK 293T cell lysates transfected with control vector, FLAG-tagged wild-type ATMIN or ATMINΔAim, followed by immunoblotting with FLAG- and P-S1981-ATM-specific antibodies. (G) HEK 293T cells were irradiated with 5 Gy or mock treated, ATM IP was performed, followed by immunoblotting with ATMIN and ATM antibodies. (H) HEK 293T cells were treated with 25 μg/ml chloroquine (4 h) or mock treated, ATMIN or control IP was performed, followed by immunoblotting with ATMIN and ATM antibodies.
To analyse ATMIN function, we produced a panel of ATMIN-specific monoclonal antibodies. Immunoprecipitation (IP) of endogenous ATMIN showed interaction with ATM, but not with the related kinase ATR (Figure 1B). ATMIN interacted to similar extents with ectopically expressed wild-type (WT) and kinase-dead (KD) ATM (Figure 1C). Complete depletion of ATMIN protein resulted in a small, but reproducible co-depletion of ATM (Figure 1D). Recently, a short motif has been identified in the C-terminus of NBS1 that mediates ATM interaction (Falck et al, 2005). Sequence alignment revealed a conserved region with similarity in the C-terminus of ATMIN (Figure 1E). A mutant ATMIN protein with eight alanine substitutions in the ATM-interaction motif (ATMINΔAim) showed reduced interaction with ATM (Figure 1F), indicating that the ATMIN C-terminus contributes to ATM binding. ATM is strongly activated by DSBs, but changes in chromatin structure induced by chloroquine or hypotonic salt have been shown to also stimulate ATM activation in the absence of DSBs (Bakkenist and Kastan, 2003). IP of endogenous ATM revealed interaction with ATMIN in untreated cells, but after IR treatment, ATM/ATMIN interaction was reduced (Figure 1G), whereas chloroquine treatment enhanced the interaction of ATMIN with ATM (Figure 1H).
Colocalisation of ATMIN with ATM, before, but not after DNA damage
Next, we determined ATMIN subcellular localisation using immunofluorescence (IF). Immunostaining with two different ATMIN-specific monoclonal antibodies revealed that ATMIN was localised in nuclear foci in IMR90 primary human fibroblasts and we observed a similar localisation in several other primary and tumour cells lines (Figure 2 and data not shown). ATM shows a dynamic localisation pattern during the cell cycle and re-localises to DSBs after cells have been treated with agents such as IR (Shiloh, 2003). In the absence of DNA damage, an antibody that recognises total ATM protein revealed IF staining, which was evenly distributed throughout the cell in IMR90 fibroblasts. However, often some regions of slightly more intense nuclear ATM staining were present and in these regions ATMIN and ATM showed partial colocalisation (Figure 2A). Treatment with IR resulted in accumulation of ATM in nuclear foci, presumably at DSBs. ATMIN was not recruited to DSBs, as ATM/ATMIN did not colocalise after IR (Figure 2B).
Figure 2.
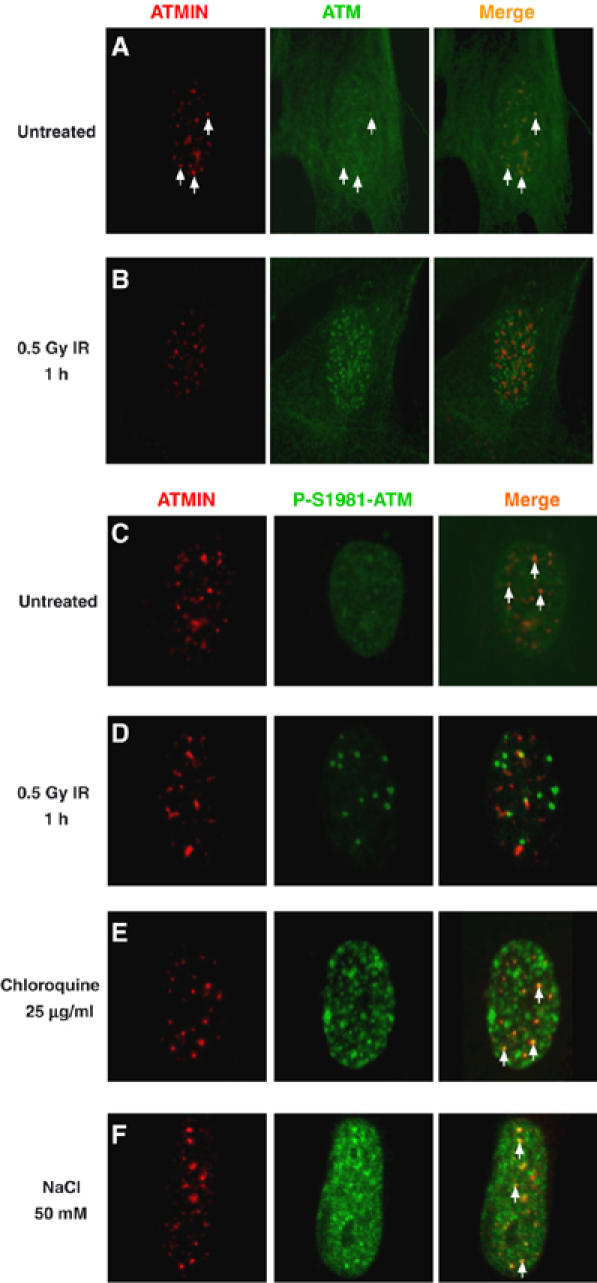
ATMIN colocalises with ATM after chloroquine and hypotonic treatment, but not after IR. (A, B) Double-immunostaining for ATMIN (red), ATM (green) and merge of ATM/ATMIN in either untreated IMR90 fibroblasts (A) or 1 h after treatment with 0.5 Gy IR (B). White arrows indicate sites of ATM/ATMIN colocalisation. (C–F) Double-immunostaining for ATMIN (red), phosphorylated ATM (P-S1981-ATM; green) and merge in untreated IMR90 primary fibroblasts (C), or 1 h after treatment with 0.5 Gy IR (D), after 4 h with 25 μg/ml chloroquine (E) or 1 h with 50 mM NaCl (F). White arrows indicate sites of P-S1981-ATM/ATMIN colocalisation.
Immunostaining for ATM phosphorylated at serine residue 1981 (P-S1981-ATM) was barely detectable in untreated primary human fibroblasts, but wherever regions with marginally increased P-S1981-ATM staining were present, these showed significant colocalisation with ATMIN (Figure 2C, white arrows). P-S1981-ATM staining was greatly augmented by IR treatment, but P-S1981-ATM and ATMIN did not colocalise after induction of DSBs (Figure 2D). In contrast, treatment of cells with reagents inducing changes in chromatin structure such as chloroquine or hypotonic salt resulted in ATM activation as judged by P-S1981-ATM IF staining and the majority of detectable ATMIN colocalised with P-S1981-ATM (Figure 2E and F, white arrows). Quantification revealed that 61% of ATMIN foci colocalised with P-ATM in untreated cells, but only 5% after IR. ATMIN/P-ATM colocalisation increased to 88% after chloroquine treatment and to 98% in response to hypotonic salt. Thus, ATMIN colocalised with ATM after chloroquine treatment and hypotonic shock, but not after IR.
NBS1 is required for ATM/ATMIN dissociation after IR
As ATMIN and NBS1 share a similar ATM interaction motif, we investigated whether NBS1 plays a role in the disruption of the ATMIN/ATM complex. After IR, in NBS1-mutant cells reconstituted with wild-type NBS1 (Cerosaletti et al, 2000) P-S1981-ATM was efficiently recruited to DSBs marked by phosphorylated histone H2AX (γH2AX) and ATMIN/P-S1981-ATM colocalisation was lost (Figure 3A and B; NBS1−/−+WT NBS1). In cells with compromised NBS1 function, the extent of ATM activation after IR was reduced and ATM recruitment to sites of DNA damage was less efficient (Figure 3C; NBS1−/−+vector). Moreover, colocalisation of P-S1981-ATM with ATMIN was greatly increased in NBS1-deficient cells compared to NBS1-proficient cells (90% ATMIN/P-ATM colocalisation in NBS1−/−+vector cells and 28% in NBS1−/−+WT NBS1 cells; Figure 3B and D, white arrows), indicating that NBS1 contributes to the disruption of the ATMIN/P-S1981-ATM complex by IR. It is noteworthy that the few intense P-S1981-ATM foci, likely to represent the small amount of active ATM recruited to DSB in NBS1−/−+vector cells (Figure 3D, yellow arrowheads), did not co-stain with ATMIN. There was still significant ATMIN/P-S1981-ATM colocalisation after IR in NBS1-mutant cells complemented with C-terminally truncated NBS1 (Figure 3E and F; NBS1−/−+ΔC NBS1), suggesting that the C-terminus of NBS1 contributes to ATMIN/ATM dissociation.
Figure 3.
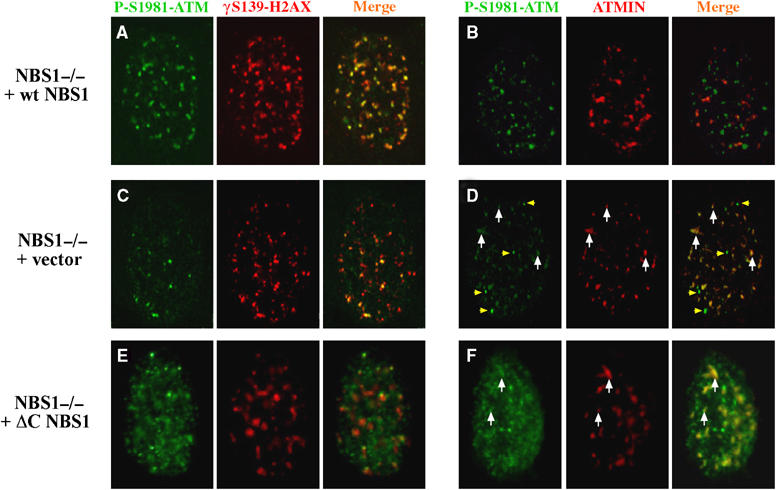
ATMIN/ATM colocalisation after IR in NBS1-deficient cells. (A–F), Double-immunostaining for phosphorylated ATM (P-S1981-ATM; green) with either phosphorylated histone H2AX (γH2AX; red) or ATMIN (red) was performed in NBS1-deficient cells complemented with wild-type NBS1 (NBS1−/− +wt NBS1; (A, B)), empty vector (NBS1−/− +vector; (C, D)) or a C-terminally truncated form a NBS1 (NBS1−/− +ΔC NBS1; (E, F)) after treatment with 0.2 Gy IR for 5 min. White arrows indicate sites of P-S1981-ATM/ATMIN colocalisation, yellow arrowheads in (D) indicate P-S1981-ATM staining not colocalising with ATMIN.
ATM is required for ATMIN stabilisation
The in silico prediction of a PEST sequence indicated that ATMIN protein stability might be regulated by the ubiquitin/proteasome system. However, treatment with proteasome inhibitor only led to a minor increase in ATMIN protein levels (Figure 4A). In contrast, ectopically overexpressed ATMIN was very unstable, and proteasome inhibition caused significant ATMIN stabilisation (Figure 4B). Whereas endogenous ATMIN protein forms nuclear foci (Figure 2), overexpressed ATMIN was found throughout the nucleoplasm (data not shown), a mislocalisation also observed previously that may contribute to instability (McNees et al, 2005). Inhibition of de novo protein synthesis by anisomycin treatment led to a reduction of overexpressed ATMIN protein levels, but deletion of the C-terminus containing the predicted PEST sequence resulted in higher steady-state levels and increased ATMIN stability (Figure 4C).
Figure 4.
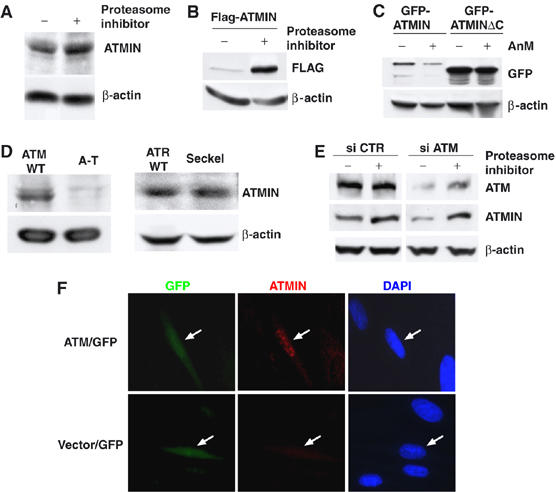
ATM augments ATMIN protein levels. (A) Protein lysates isolated from HEK 293T cells treated with proteasome inhibitor or mock treated for 6 h were analysed by immunoblotting with ATMIN and β-actin-specific antibodies. (B) HEK 293T cells were transfected with FLAG-tagged ATMIN, treated with proteasome inhibitor for 6 h or mock treated, followed by immunoblotting with FLAG-tag and β-actin-specific antibodies. (C) HEK 293T cells were transfected with GFP-tagged ATMIN or a C-terminal truncation of ATMIN (GFP-ATMINΔC), treated with anisomycin (5 μM) for 6 h or mock treated, followed by immunoblotting with GFP and β-actin-specific antibodies. (D) Protein lysates isolated from A-T cells, Seckel syndrome cells and respective control cells were analysed by immunoblotting with ATMIN and β-actin-specific antibodies. (E) HEK 293T cells were transfected with siRNA pools directed against ATM or control pools, proteasome inhibitor or mock treated, and ATM, ATMIN and β-actin protein levels were analysed. (F) A-T cells were co-transfected with GFP expression vector and FLAG-tagged ATM (ATM/GFP) or with GFP and empty control vector, (vector/GFP). GFP fluorescence, ATMIN immunostaining (red) and DAPI staining is shown. White arrows indicate transfected cells.
The discrepancy in stability between endogenous and overexpressed ATMIN could be explained if another protein was necessary to stabilise ATMIN. Therefore, we investigated the role of ATM in ATMIN stability. Western blot and IF analysis revealed that ATMIN protein levels were reduced in ATM-deficient A-T cells, but normal in cells derived from Seckel syndrome patients, which express significantly diminished levels of ATR (Alderton et al, 2004) (Figure 4D). Reduction of ATM protein levels by transfection of siRNAs led to a decrease in ATMIN levels, which was reversed by proteasome inhibition (Figure 4E). Re-expression of ATM in A-T cells resulted in increased levels of ATMIN protein (Figure 4F). Thus ATMIN stability is regulated by ATM.
ATMIN is required for efficient ATM activation in the absence of DNA damage
To elucidate the functional significance of ATMIN for ATM signalling, we analysed the consequences of ATMIN depletion using RNAi in human cells. Co-transfection of plasmids directing expression of siRNAs targeting ATMIN (si-ATMIN) with GFP into IMR90 fibroblasts resulted in diminished ATMIN protein levels in GFP-positive transfected cells (Figure 5A, white arrowheads), but not in GFP-negative untransfected cells or in cells expressing a mismatched control siRNA (si-mmCTR). IF analysis also revealed reduced staining for P-S1981-ATM in IR-treated si-ATMIN cells (Figure 5B). To investigate the reason for the apparent lack of P-S1981-ATM in ATMIN-depleted cells, we analysed the effects of ATMIN knockdown biochemically. ATMIN protein levels were significantly decreased by si-ATMIN expression, which was accompanied by a small reduction in ATM protein levels. The levels of S1981-phosphorylated ATM after chloroquine treatment was slightly reduced in ATMIN-depleted cells and this decrease was more noticeable after IR (Figure 5C), indicating that ATMIN contributes to both ATM protein expression and S1981 autophosphorylation.
Figure 5.
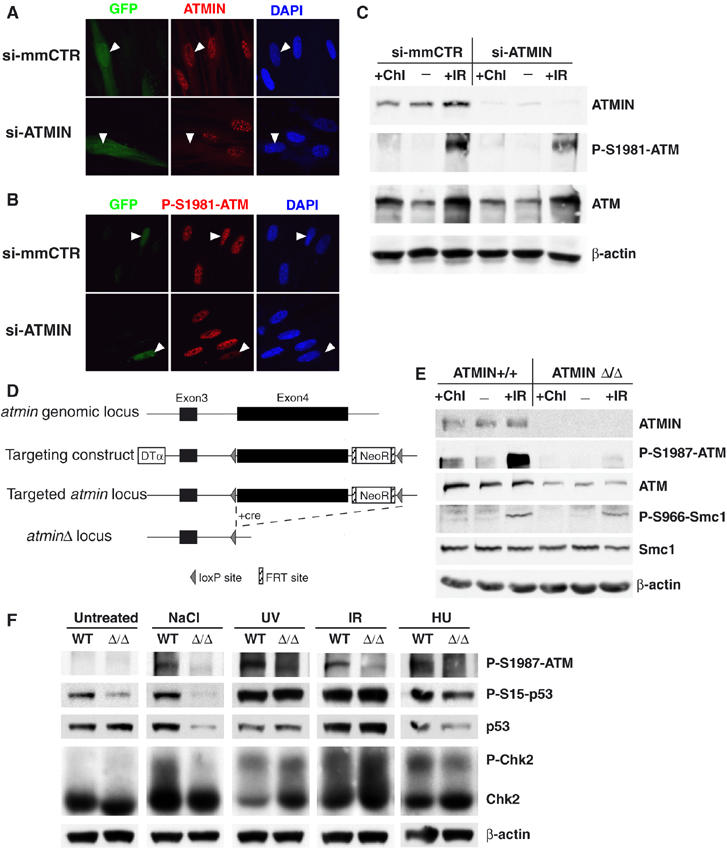
ATMIN is required for ATM protein expression and function. (A, B) IMR90 cells were co-transfected with GFP and a pSUPER vector expressing a shRNA specific for ATMIN (si-ATMIN) or with GFP and a pSUPER vector expressing a mismatched shRNA (si-mmCTR), and processed for IF 30 min after treatment with 5 Gy IR. Two different siRNAs targeting ATMIN gave similar results. Immunostaining for ATMIN (A), P-S1981-ATM (B), is shown together with GFP fluorescence. White arrowheads indicate transfected cells. (C) HEK 293T cells were transfected with vectors expressing either si-ATMIN or si-mmCTR. 48 h after transfection cells were treated with 5 Gy IR (1 h), 25 μg/ml chloroquine (Chl; 4 h) or mock treated and expression of the indicated protein was determined by Western blot analysis. (D) Schematic representation of the atmin genomic locus, the targeting construct, the targeted atmin locus, and the targeted atmin locus after cre-mediated recombination (atminΔ). Exons are represented by black rectangles, intronic DNA is shown as a black line. LoxP sites are indicated by triangles, FRT sites as hatched rectangles. DTα, diphtheria toxin α chain; NeoR, Neomycin resistance gene. (E) atmin+/+ and atminΔ/Δ MEFs were treated with 5 Gy IR (1 h), 25 μg/ml chloroquine (Chl; 4 h) or mock treated and expression of the indicated protein was determined by Western blot analysis. (F) atmin+/+ and atminΔ/Δ MEFs were treated with 2 mM HU for 2 h, hypotonic salt (50 mM NaCl) for 1 h, 50 J m−2 UV (harvested after 3 h), 1 Gy IR (harvested after 15 min) or mock treated and expression of the indicated protein was determined by Western blot analysis.
To test whether the residual ATMIN protein present in ATMIN-knockdown cells plays a role in ATM activation, we generated a targeted null mutation in the murine atmin gene (atminΔ). Exon 4 of the atmin locus, which encodes 615 of the 823 amino acids of ATMIN, was flanked by loxP sites and subsequently deleted (Figure 5D). Heterozygous atmin+/Δ mice were viable and fertile. atmin+/Δ mice were intercrossed and primary mouse embryonic fibroblast (MEFs) were isolated from E12.5 embryos. ATMIN protein was detected in atmin+/+ control MEFs, but was absent in homozygous mutant atminΔ/Δ cells (Figure 5E). ATM protein levels were lower in atminΔ/Δ MEFs. Moreover, ATM S1987 phosphorylation (the corresponding residue to human S1981) was significantly reduced in untreated cells and after IR treatment. Smc1 phosphorylation was augmented after IR and ATMIN deficiency caused only a marginal reduction, in agreement with recent observations that ATM S1987 phosphorylation is not absolutely required for IR-induced ATM activity in murine cells (Pellegrini et al, 2006). ATMIN deficiency also impaired stimulation of ATM S1987 phosphorylation by chloroquine (Figure 5E), suggesting that ATM/ATMIN colocalisation after chloroquine treatment is functionally relevant. The effect of atmin inactivation on ATM is thus stronger than, but in agreement with, the consequences of ATMIN knockdown in human cells.
Untreated atminΔ/Δ MEFs showed reduced phosphorylation of p53 at serine 15, suggesting that ATMIN contributes to basal ATM activity (Figure 5F), consistent with the ATMIN/ATM colocalisation we observed in untreated cells (Figure 2). ATMIN was also required for efficient ATM S1987 phosphorylation, p53 phosphorylation, p53 stabilisation and Chk2 phosphorylation in response to hypotonic stress. DNA-damage signalling triggered by replication stress is believed to be largely dependent on ATR (Shechter et al, 2004), but NBS1-independent ATM autophosphorylation is also stimulated by inhibitors of DNA replication (Bakkenist and Kastan, 2003; Difilippantonio et al, 2005). ATMIN was required for efficient ATM S1987 phosphorylation induced by HU treatment, and ATMIN-deficient cells showed a small reduction in p53 and Chk2 phosphorylation. In contrast, IR and UV induced phosphorylation of all ATM substrates analysed was unaffected (Figure 5F). Therefore, ATMIN appears to be required for ATM activity in a stimulus-dependent manner.
In response to IR, an ATM-dependent DNA-damage response arrests cells at the G2–M transition (Zhou et al, 2000). However, atminΔ/Δ cells did nor show a defect in the IR-induced G2/M checkpoint (Figure 6A). Moreover, ATMIN deficiency did not result in radiosensitivity (Figure 6B), but resulted in increased cell death in response to chloroquine treatment (Figure 6C). Thus, ATMIN in dispensable for ATM functions triggered by DSBs, but protects cells from chloroquine-induced cell death.
Figure 6.
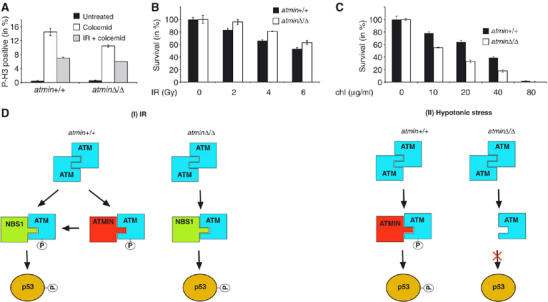
Models illustrating interaction of ATMIN and NBS1 with ATM. (A) atmin+/+ and atminΔ/Δ MEFs were treated with colcimid (1 μg/ml), and were exposed to 10 Gy of IR or mock treated. At 20 h after irradiation, the percentage of cells that were in M phase was determined by staining for phospho-histone H3. (B) atmin+/+ and atminΔ/Δ MEFs were treated with the indicated doses of IR and cell viability was determined by trypan blue exclusion. (C) atmin+/+ and atminΔ/Δ MEFs were treated with the indicated doses of chloroquine and cell viability was determined by trypan blue exclusion. (D) Model of NBS1 and ATMIN functions in response to ATM activation by IR and hypotonic shock.
Discussion
This study has identified ATMIN as a novel regulator of ATM signalling. The role of NBS1 and the MRN complex in ATM activation and the recruitment of ATM to DSBs, in response to irradiation, is well established, but the molecular mechanism controlling basal ATM activity and ATM activation triggered by other stresses has been unknown. We propose that ATMIN is an essential component of the signalling pathway regulating ATM function in response to stimuli such as hypotonic stress.
ATMIN localisation
It is noteworthy that the localisation of endogenous ATMIN to intranuclear foci that we describe here is in disagreement with the previously published localisation pattern of ectopically expressed GFP-tagged ASCIZ in U2OS cells (McNees et al, 2005). Ubiquitous fluorescence of ectopically expressed GFP-ASCIZ was observed, and we have also made similar observations with ectopically expressed GFP-ATMIN (data not shown). It is likely that either the GFP moiety of overexpressed GFP-ATMIN interferes with normal ATMIN localisation or that overexpression per se alters correct localisation. We have observed endogenous ATMIN intranuclear foci in all primary and tumour cell lines and under all conditions we have studied to date, and it appears that this localisation is important for ATMIN's function in the ATM-signalling pathway.
Mutual stabilisation of ATMIN and ATM
The significance of ATMIN/ATM interaction is illustrated by their mutual dependence for stabilisation. Reciprocal stabilisation is commonly observed in protein complexes, an example being the interaction of ATR with its cofactor ATRIP (Cortez et al, 2001), and also in the case of ATMIN and ATM this argues for an intimate functional connection. The degree of dependence on the other protein differs between ATMIN and ATM. ATMIN levels are completely dependent on ATM, as ATMIN protein is almost undetectable in A-T cells (Figure 4D and F). In contrast, ATM protein is reduced, but readily detectable in ATMIN-mutant cells (Figure 5E). This observation is in agreement with the localisation data: after chloroquine treatment or hypotonic stress, the majority of ATMIN colocalises with ATM, but only a subpool of ATM is associated with ATMIN (Figure 2E and F). The proteasome-dependent degradation of ATMIN is mediated by the C-terminus, which contains a predicted PEST sequence. The proximity of the PEST sequence with the NBS1-like ATM interaction motif suggests a possible mechanism of ATMIN stabilisation by ATM, because binding of ATM to ATMIN may shield the PEST sequence, thereby impeding ubiquitin attachment and ATMIN degradation.
Regulation of ATM S1981 autophosphorylation by ATMIN
ATM activation is accompanied by autophosphorylation at several residues. Phosphorylation at serine 1981 is a hallmark of active ATM, but whether this phosphorylation is also necessary for activation is unclear. Whereas a mutant form of ATM with serine 1981 substituted for alanine failed to rescue ATM function in A-T cells, a BAC transgenic expressing murine ATM with a mutation of serine 1987 to alanine complemented ATM deficiency in ATM-knockout mice (Kozlov et al, 2006; Pellegrini et al, 2006).
ATMIN plays an important role in the stimulation of S1981/1987 autophosphorylation by ATM. In both human and murine cells, loss of ATMIN function reduced S1981/1987 autophosphorylation in response to all stimuli tested. However, there was no correlation between the reduction of ATM S1981/1987 autophosphorylation and downstream signalling. The absence of ATMIN significantly impaired ATM autophosphorylation after IR, but the phosphorylation of ATM substrates occurred normally. Our data are thus consistent with the notion that ATM S1981/1987 autophosphorylation is not an absolute requirement for ATM substrate phosphorylation in murine cells. It is conceivable that a transient interaction of ATM and ATMIN after IR treatment occurs, which is important for ATM 1981/1987 autophosphorylation (Figure 6D). The significant ATMIN/P-S1981-ATM colocalisation after IR in NBS1-deficient cells (Figure 3D) may thus represent a ‘frozen' intermediate stage of ATM activation by IR. In contrast, under basal conditions and in response to hypotonic shock ATMIN associates with ATM, regulates ATM S1981/1987 autophosphorylation and is also required for subsequent signalling (Figure 6D). ATMIN-deficient cells show several hallmarks of ATM-deficient cells (Xu and Baltimore, 1996), including proliferation defects and premature senescence (our unpublished observations). Thus, the regulation of ATM function by ATMIN in the absence of DNA damage appears to be of significance.
Functional similarities between NBS1 and ATMIN
Several aspects of ATMIN function are analogous to NBS1: ATMIN interacts with ATM using a similar motif as NBS1 and ATMIN associates with ATM in a stimulus-dependent manner, as does NBS1. ATMIN colocalises with S1981-phosphorylated ATM in basal conditions and after chloroquine treatment and hypotonic stress, but not after IR. Conversely, NBS1 specifically associates with ATM in response to IR and recruits ATM to DSBs. Their stimulus-dependent association mirrors the relative importance of ATMIN and NBS1 for ATM-mediated substrate phosphorylation. Whereas NBS1 is essential for ATM function in response to IR, ATM activation in response to hypotonic stress or inhibitors of DNA replication is normal in NBS1-deficient cells (Difilippantonio et al, 2005). Our data demonstrate that ATMIN is required for ATM activity in response to these stimuli. Therefore, NBS1 and ATMIN might be viewed as two ATM cofactors that control ATM activation is response to distinct signals.
Materials and methods
Cell culture and treatment
HEK 293T, HCT116, IMR90, AT fibroblasts (AG04405), NBS-deficient and reconstituted cells, atmin+/+ and atminΔ/Δ cells were cultured in DMEM supplemented with 10% FCS. Seckel and control lymphoblastoid cells were maintained in RPMI supplemented with 10% FCS. In some cases, cells were treated with proteasome inhibitors (25 μM PI-1, Calbiochem), 5 μM anisomycin, 2 mM HU or 25 μg/ml chloroquine (all purchased from Sigma). Hypotonic swelling was performed using 50 mM NaCl in 1% FBS/PBS supplemented with 0.45% glucose. Cells were transfected using Lipofectamine 2000 (Invitrogen) and IR experiments were performed using a Cs137 Gamma Irradiator at 2.1 Gy/min and UV was applied using a Hoefer UVC 500 crosslinker. ATM and control siRNA pools were purchased from Dharmacon. For the G2–M checkpoint assay, cells were irradiated with 10 Gy IR or mock treated followed by incubation with or without 1 μg/ml colcemid for 20 h. Cells were stained with phospho-histone H3 antibody and the percentage of mitotic cells determined by FACS analysis. For the radio- and chloroquine-sensitivity analysis of the MEFs, cells were exposed to varying doses of IR (and harvested after 4 days) or chloroquine (for 12 h). Cell viability was determined by trypan blue exclusion.
Mice and MEFs
The atmin gene was targeted and disrupted using standard procedures (Behrens et al, 2002). After germ-line transmission of the targeted atmin locus, the floxed atmin exon 4 was removed using germ-line-deleting PGK-cre transgenic mice. Heterozygous atmin+/Δ mice are normal and fertile. MEFs were derived from E12.5 embryos resulting from heterozygous atmin+/Δ intercrosses as described previously (Behrens et al, 1999). Details of atmin targeting and the characterisation of the atmin-knockout phenotype will be described elsewhere.
Antibodies
The following antibodies were used for Western blotting and IF: S1981ATM and P-S1981ATM (Rockland); ATM 2C1, ATR H-300, P-S15-p53 (Santa Cruz); FLAG M2, β-actin (Sigma); P-S966-SMC1, SMC1 (Chemicon); γH2AX, Chk2 (Upstate); ATMIN, p53 and GFP (made in-house at CR-UK), HRP-conjugated goat anti-mouse/rabbit IgG (Sigma); FITC/cy3-conjugated goat anti-mouse/rabbit IgG (H&L) (Jackson).
Western blot analysis
Western blots were performed using standard procedures (Nateri et al, 2005). For Western blots, protein samples were separated by SDS–PAGE, and subsequently transferred onto PVDF membranes (Amersham). All primary antibodies were used at 1:1000 dilution and secondary antibodies at 1:2000.
Immunofluorescence
IMR90 cells were washed twice with cold PBS and fixed with cold methanol/acetone at −20°C for at least 1 h. All subsequent steps were performed at room temperature. Cells were washed with PBS and blocked with 10% FCS/PBS/0.1% Triton X100 before addition of primary antibodies diluted 1:400 in blocking buffer. Cells were washed three times for 5 min with blocking buffer followed by the addition of secondary antibodies diluted 1:400 in blocking buffer for 1 h. Cells were washed with PBS and incubated with DAPI before mounting in Mowiol (Calbiochem cat. no. 475904). Cells were visualised on a Zeiss Axioplan 2 upright microscope using AxioVision 4.1 imaging software.
Immunoprecipitations
Cells were washed with cold PBS and subsequently lysed for 30 min at 4°C with 150 mM NaCl, 80 mM Tris–HCl (pH 7.2), 0.2% NP-40, 10% Glycerol and complete protease inhibitor cocktail (Sigma). Lysates were cleared by centrifugation for 15 min at 4°C. Proteins were immunoprecipitated overnight with the indicated antibody. Immunocomplexes were captured by rotating for 2–3 h with Protein G (Sigma). In some cases, conjugated agarose beads were used. Immunoprecipitates were washed four times with lysis buffer and beads were resuspended in Laemmli sample buffer.
DNA constructs, RT–PCR and GenBank accession numbers
The murine full-length cDNA is derived from IMAGE clone 6847850 (BC060631) and the human cDNA is derived from two overlapping ESTs (BC002701, IMAGE clone 3612984 and CV808328, IMAGE clone 7501219). The chicken and fugu cDNAs were derived from a combination of partial ESTs and genomic alignments, the 5′ end of the chicken ATMIN cDNAs are covered by BM489446 (clone pgm2n.pk010.07) and BU216096 (clone ChEST673i4), the 5′ end of zebrafish ATMIN is on IMAGE clone 7427677 (CO936814).
Full-length ATMIN cDNA was cloned by PCR from a murine brain cDNA library and verified by sequencing. In silico domain prediction was carried out using PFAM (http://www.sanger.ac.uk/Software/Pfam/search.shtml). PEST domain prediction was performed using the PESTfind algorithm (https://emb1.bcc.univie.ac.at/toolbox/pestfind/). The various ATMIN expression constructs and mutants were generated using standard cloning procedures. The GFP-ATMIN fusion was constructed by cloning the mouse cDNA into the C-terminal MCS of pEGFP-C3 (Clontech). The GFP-ATMINΔC was derived by removal of a 506 bp Sac1 fragment, which deletes the last 169 amino acids of ATMIN. The Aim motif was replaced with eight alanine substitutions by PCR mutagenesis. The FLAG-tag expression constructs were similarly made in vector pIRES2-EGFP (Clontech). siRNA experiments targeting ATM were performed using siRNA pools purchased from Dharmacon. ATMIN knockdown was performed using the pSuper expression plasmids (Brummelkamp et al, 2002). Mismatched oligos with 6 bp changes were used as controls. The sequences of the RNAi oligos were as follows:
- si-ATMINa
5′-GTC TCA CAT CTA CCG AAC T-3′
5′-AAC TCA GAC AGC AAT GGA T-3′
5′-GTA TCG CAC CTG CCT AAT T-3′
5′-AAT TCG GAT AGT AAC GGC T-3′
- si-ATMINb
5′-AAC TCA GAC AGC AAT GGA T-3′
5′-GTA TCG CAC CTG CCT AAT T-3′
5′-AAT TCG GAT AGT AAC GGC T-3′
- si-mmCTRa
5′-GTA TCG CAC CTG CCT AAT T-3′
5′-AAT TCG GAT AGT AAC GGC T-3′
- si-mmCTRb
5′-AAT TCG GAT AGT AAC GGC T-3′
mRNA was isolated from mouse cells using a Qiagen RNeasy Micro Kit and cDNA was synthesised using oligo dT primers. RT-PCR was performed using the following oligos:
- P1:
ATG GGG CCC ACG GAG GCG GCG GCG GCC GAT TCT
CGG GGC TGC TTG GTC GCT CAG TGG TTC AG
GAG GAT CAG GGC TCC TAC CGA CAG A
- P2:
CGG GGC TGC TTG GTC GCT CAG TGG TTC AG
GAG GAT CAG GGC TCC TAC CGA CAG A
- P3:
GAG GAT CAG GGC TCC TAC CGA CAG A
Supplementary Material
Supplementary Figure 1
Acknowledgments
We are grateful to P Concannon, C Da Costa, J Cronshaw, S Jackson, M Kastan, M Mitchell, C Morrison, AS Nateri and M Weitzman for providing reagents and advice. We thank V Constanzo and F Uhlmanr for critical reading of the manuscript. The London Research Institute is funded by CR-UK.
References
- Alderton GK, Joenje H, Varon R, Borglum AD, Jeggo PA, O'Driscoll M (2004) Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum Mol Genet 13: 3127–3138 [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2004) Initiating cellular stress responses. Cell 118: 9–17 [DOI] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, David JP, Mohle-Steinlein U, Tronche F, Schutz G, Wagner EF (2002) Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. EMBO J 21: 1782–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, Wagner EF (1999) Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet 21: 326–329 [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 [DOI] [PubMed] [Google Scholar]
- Cerosaletti KM, Desai-Mehta A, Yeo TC, Kraakman-Van Der Zwet M, Zdzienicka MZ, Concannon P (2000) Retroviral expression of the NBS1 gene in cultured Nijmegen breakage syndrome cells restores normal radiation sensitivity and nuclear focus formation. Mutagenesis 15: 281–286 [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716 [DOI] [PubMed] [Google Scholar]
- D'Amours D, Jackson SP (2002) The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 3: 317–327 [DOI] [PubMed] [Google Scholar]
- Difilippantonio S, Celeste A, Fernandez-Capetillo O, Chen HT, Reina San Martin B, Van Laethem F, Yang YP, Petukhova GV, Eckhaus M, Feigenbaum L, Manova K, Kruhlak M, Camerini-Otero RD, Sharan S, Nussenzweig M, Nussenzweig A (2005) Role of Nbs1 in the activation of the ATM kinase revealed in humanized mouse models. Nat Cell Biol 7: 675–685 [DOI] [PubMed] [Google Scholar]
- Digweed M, Sperling K (2004) Nijmegen breakage syndrome: clinical manifestation of defective response to DNA double-strand breaks. DNA Repair (Amst) 3: 1207–1217 [DOI] [PubMed] [Google Scholar]
- Dumon-Jones V, Frappart PO, Tong WM, Sajithlal G, Hulla W, Schmid G, Herceg Z, Digweed M, Wang ZQ (2003) Nbn heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res 63: 7263–7269 [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Kim JE, Minter-Dykhouse K, Chen J (2006) Signaling networks controlled by the MRN complex and MDC1 during early DNA damage responses. Mol Carcinog 45: 403–408 [DOI] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, Kastan MB (1999) Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 274: 37538–37543 [DOI] [PubMed] [Google Scholar]
- Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF (2006) Involvement of novel autophosphorylation sites in ATM activation. EMBO J 25: 3504–3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Paull TT (2005) ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308: 551–554 [DOI] [PubMed] [Google Scholar]
- Maser RS, Zinkel R, Petrini JH (2001) An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat Genet 27: 417–421 [DOI] [PubMed] [Google Scholar]
- McKinnon PJ (2004) ATM and ataxia telangiectasia. EMBO Rep 5: 772–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNees CJ, Conlan LA, Tenis N, Heierhorst J (2005) ASCIZ regulates lesion-specific Rad51 focus formation and apoptosis after methylating DNA damage. EMBO J 24: 2447–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nateri AS, Spencer-Dene B, Behrens A (2005) Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 437: 281–285 [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, Nussenzweig A (2006) Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature 443: 222–225 [DOI] [PubMed] [Google Scholar]
- Rogers S, Wells R, Rechsteiner M (1986) Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234: 364–368 [DOI] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J (2004) Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst) 3: 901–908 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Stracker TH, Theunissen JW, Morales M, Petrini JH (2004) The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair (Amst) 3: 845–854 [DOI] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y (2003) Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22: 5612–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Baltimore D (1996) Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev 10: 2401–2410 [DOI] [PubMed] [Google Scholar]
- Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, Mattern MR, Winkler JD, Khanna KK (2000) Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem 275: 10342–10348 [DOI] [PubMed] [Google Scholar]
- Zhu J, Petersen S, Tessarollo L, Nussenzweig A (2001) Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol 11: 105–109 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1