

Abstract
Apoptosis of alveolar epithelial cells (AECs) has been implicated as a key event in the pathogenesis of lung fibrosis. Recent studies demonstrated a role for the synthesis and binding of angiotensin II to receptor AT1 in the induction of AEC apoptosis by bleomycin (BLEO) and other proapoptotic stimuli. On this basis we hypothesized that BLEO-induced apoptosis and lung fibrosis in mice would be inhibited by the AT1 antagonist losartan (LOS) or by targeted deletion of the AT1 gene. Lung fibrosis was induced by intratracheal administration of BLEO (1 U/kg) to wild-type C57BL/6J mice. Co-administration of LOS abrogated BLEO-induced increases in total lung caspase 3 activity detected 6 hours after in vivo administration and reduced by 57% BLEO-induced caspase 3 activity in blood-depleted lung explants exposed to BLEO ex vivo (both P < 0.05). Co-administration of LOS in vivo reduced DNA fragmentation and immunoreactive caspase 3 (active form) in AECs, measured at 14 days after intratracheal BLEO, by 66% and 74%, respectively (both P < 0.05). LOS also inhibited the accumulation of lung hydroxyproline by 45%. The same three measures of apoptosis and lung fibrosis were reduced by 89%, 85%, and 75%, respectively (all P < 0.01), in mice with a targeted disruption of the AT1a receptor gene (C57BL/6J-Agtr1atm1Unc). These data indicate an essential role for angiotensin receptor AT1a in the pathogenesis of BLEO-induced lung fibrosis in mice and suggest that AT1 receptor signaling is required for BLEO-induced apoptosis of AECs in mice as it is in rat and human AECs.
Idiopathic pulmonary fibrosis is a progressive and often fatal human disease characterized by infiltration of inflammatory cells into interstitial and alveolar spaces, ongoing damage to the lung parenchyma, fibroblast proliferation, and accumulation of interstitial collagens. 1 Ongoing evaluations of both older and newer data have lead to the recent characterization of idiopathic pulmonary fibrosis as a disease of abnormal wound repair, in which abnormalities in epithelial-mesenchymal interactions are of key importance. 1,2 This evolving theory about the pathogenesis of idiopathic pulmonary fibrosis is, in some respects, a revival of the hypothesis first put forth by Haschek and Witschi 3 and Adamson and colleagues 4 that the severity of the fibrogenic response in the lung is directly related to the severity of epithelial injury.
A growing body of evidence suggests that alveolar epithelial cell (AEC) death by apoptosis is a key event in the initiation and progression of lung fibrosis. In mice exposed to bleomycin (BLEO) by intratracheal instillation, up-regulation of the receptor Fas on lung epithelial cells and Fas ligand on infiltrating lymphocytes were associated with DNA fragmentation in epithelia and subsequent accumulation of collagens. 5 Intratracheal instillation of Fas-activating antibodies caused epithelial cell apoptosis and subsequent collagen accumulation, the severity of which was proportional to the amount of Fas-activating antibody instilled. 6 Other investigators have shown that BLEO itself induces apoptosis of AECs, which precedes the deposition of collagens. 7 More importantly, several groups have found that blockage of epithelial apoptosis with caspase inhibitors administered in vivo can prevent BLEO-induced lung cell apoptosis and the subsequent accumulation of lung collagens. 7,8
Recent work from this laboratory has shown that exposure of cultured AECs to Fas ligand, 9 tumor necrosis factor-α, 10 or BLEO 11 all induce expression of angiotensinogen mRNA and protein, and its cleavage to the peptide angiotensin II (ANGII). Moreover, apoptosis of cultured AECs in response to these apoptosis inducers was abrogated by antagonists of ANG receptor AT1, such as losartan (LOS) or L158809. 11-13 For all these reasons, it was hypothesized that angiotensin receptor AT1 is essential for AEC apoptosis and lung fibrosis in vivo. To test this theory, normal mice and mice deficient in ANG receptor AT1a, the AT1 subtype expressed in lung, 14 were subjected to intratracheal BLEO administration and quantitation of apoptosis and lung collagens. We report here the prevention of both BLEO-induced AEC apoptosis and lung collagen accumulation in mice by administration of the AT1-selective receptor antagonist LOS or by targeted deletion of the AT1a receptor gene.
Materials and Methods
Reagents and Materials
The AT1-selective antagonist LOS was obtained from Merck and Co., West Point, PA. Alkaline phosphatase-conjugated streptavidin, digoxigenin-labeled deoxyuridine trisphosphate (dig-dUTP), and biotinylated deoxyuridine trisphosphate (bio-dUTP) were obtained from Boehringer Mannheim, Indianapolis, IN. BLEO was obtained from Sigma Chemical Co., Saint Louis, MO. Reagents for detection of alkaline phosphatase and other secondary reagents for in situ end labeling (ISEL) of DNA or Western blotting were from sources described earlier. 7 All other materials were of reagent grade and were obtained from Sigma Chemical Co.
Animals, Induction of Pulmonary Fibrosis, and Surgical Procedures
All mice were obtained from The Jackson Laboratories, Bar Harbor, ME, and were housed in a satellite facility of University Laboratory Animal Resources, Michigan State University. Control animals were wild-type C57BL/6J mice used at 7 to 8 weeks of age. Some experiments also used mice of the same genetic background but with a targeted disruption in the ANG receptor AT1a gene (C57BL/6J-Agtr1atm1Unc) that removes a portion of the coding region sufficient to eliminate specific binding of AT1-selective agonists in all organs tested. 15 Heterozygous animals were used on the basis of availability at the same age and body weight as wild types.
Induction of Lung Injury and Fibrosis
Animals under pentobarbital anesthesia received a single intratracheal instillation of bleomycin sulfate (BLEO) at 1 U/ kg body weight, in 50 μl of sterile saline. The 50 μl dose was instilled at end-expiration, and the liquid was followed immediately by 300 μl of air to ensure delivery to the distal airways. Control animals were instilled with an equal volume of sterile saline. In some studies the AT1 receptor antagonist LOS was added to the intratracheal instillate at 20 μmol/L; the same animals also received daily intraperitoneal injections of LOS at 10 mg/kg in sterile saline throughout the test interval. Other treatment groups received daily intraperitoneal sham injections of the saline alone. LOS or sham injections were continued for 14 days after instillation of BLEO, at which point all animals were sacrificed for histology, detection of collagen or DNA fragmentation, and caspase 3 activation in epithelial cells.
Surgical Procedures
Immediately before sacrifice, animals were given intraperitoneal injections of sodium pentobarbital and the trachea was cannulated. The left lung was ligated at the hilus, excised distal to the ligation, and immediately frozen in liquid N2 for hydroxyproline assay of total collagen (see below). The remaining lung tissues were carefully removed and were instilled with 4% paraformaldehyde in phosphate-buffered saline (PBS) at 20 cm of H2O constant pressure, then immersed in the same fixative for 30 minutes followed by storage in 70% ethanol. The fixed tissues were washed with PBS three times for 15 minutes and were then embedded in paraffin. Five μm sections of lung were deparaffinized by passing through xylene, xylene:alcohol 1:1, 100% alcohol, and 70% alcohol for 10 minutes each. Ethanol was removing by rinsing with distilled water.
Lung Explant Culture
Explants of 1 mm2 were prepared by mincing of blood-depleted (PBS-perfused) mouse lung, and were cultured in Transwell polycarbonate inserts (3.0-μm pore; Costar, Corning, NY) under a thin layer (1 mm) of Dulbecco’s modified Eagle’s medium cell culture medium to facilitate gas exchange. 16 All explants were obtained from normal mouse lung that was PBS-perfused in situ before excision of the lungs. After excision of the lungs, treatment with BLEO or LOS was initiated ex vivo by intratracheal instillation of BLEO at 25 mU/ml in 300 μl of sterile Dulbecco’s modified Eagle’s medium (+/− LOS at 10−6 mol/L). The culture medium for explants also contained BLEO at 25 mU/ml, +/− LOS at 10−6 mol/L. Explants were harvested by transfer into liquid N2 and storage at −80°C until assay.
Identification and Quantitation of Apoptotic Cells and Total Lung Caspase 3 Activity
Localization of DNA Fragmentation
ISEL of fragmented DNA was conducted by a modification of the method of Mundle and colleagues. 17 Briefly, ethanol was removed from deparaffinized lung sections by rinsing in distilled water for at least 10 minutes. The slides were then placed in 3% hydrogen peroxide (Sigma Chemical Co.) for 30 minutes at 20°C, rinsed with PBS, and incubated with Proteinase K (Sigma) in standard saline citrate for 15 minutes at 37°C. Samples were rinsed once in water, three times in 0.15 mol/L PBS for 4 minutes each, and were then incubated in standard saline citrate (0.3 mol/L NaCl and 30 mmol/L sodium citrate in water, pH 7.0) at 80°C for 20 minutes. After four rinses in PBS and four rinses in buffer A (50 mmol/L Tris/HCl, 5 mmol/L MgCl, 10 mmol/L B-mercaptoethanol, and 0.005% bovine serum albumin in water, pH 7.5), the sections were incubated at 18°C for 2 hours with ISEL solution (0.001 mmol/L digoxigenin-dUTP; 20 U/ml DNA Polymerase I; and 0.01 mmol/L each of dATP, dCTP, and dGTP in buffer A). Afterward the sections were rinsed thoroughly five times with buffer A and three additional times in PBS. Detection of incorporated dUTP was achieved with by incubation for 2 hours at 37°C with AP-conjugated anti-digoxigenin (Boehringer Mannheim) at 1/400 dilution. Bound AP-antibody was then detected with the Fast Blue chromogen system and the sections were mounted with Fluoromount solution (Southern Biotechnology, Birmingham, AL).
Immunohistochemistry (IHC) for Activated Caspase 3
IHC was performed with an antibody that recognizes only the active form of the enzyme (BioVision, Mountain View, CA). Deparaffinized lung sections were blocked with a solution of 3% bovine serum albumin in PBS for 1 hour; the primary antibody was then applied overnight at 4°C in 3% bovine serum albumin/PBS. After washing in PBS, the antibody was detected with a biotin-conjugated secondary antibody and avidin-linked chromogen system. Type II pneumocytes were identified with the anti-cytokeratin antibody MNF116, an established marker of type II cells. 18 Detection of mouse lung antigens with this mouse monoclonal antibody was achieved with the Mouse-on-Mouse Iso-IHC kit (InnoGenex, San Ramon, CA), according to the manufacturer’s instructions.
For quantitation of ISEL- or caspase 3-positive epithelial cells, the number of positive cells within the surfaces of the alveolar walls were counted in a minimum of six randomly selected ×400 microscopic fields per lung section. Positive cells within the alveolar airspaces, or otherwise clearly not within the surface of the alveolar wall, were not scored. The counts of positive nuclei per field were expressed as a percentage of the total number of nuclei in the same microscopic field. Sections from each of at least five mice per treatment group were analyzed by an investigator blinded to sample identity.
Caspase 3 Enzyme Activity
Assay of total lung caspase 3 enzyme activity was conducted with a commercially available kit (Molecular Probes, Eugene, OR). Fast-frozen lung was homogenized in assay kit buffer and was analyzed according to the manufacturer’s instructions on a Biotek FL600 fluorescence plate reader. In all samples, specificity of the reaction for caspase 3 was verified by abrogation of the signal with a caspase 3-selective irreversible inhibitor (data not shown).
Quantitation of Lung Collagen
For quantitation of total lung collagen, tissues frozen in liquid N2 were dried to constant weight in preweighed tubes at 80°C. The weighed dry tissue was hydrolyzed in 6 N HCl and was subjected to determination of hydroxyproline as described earlier by Woessner. 19 The efficiency of the hydrolysis was verified with rat tail collagen by comparison to standard hydroxyproline (Sigma Chemical Co.).
Results
Quantitation of Apoptosis and Lung Injury
On the basis of earlier work with rat models, 7,11 we hypothesized that BLEO would induce apoptosis of mouse lung AECs that might be detected in situ by end labeling of fragmented DNA or by IHC for the active form of caspase 3. Consistent with this expectation, intratracheal instillation of BLEO caused increased ISEL and positive caspase 3 IHC within cells in the surfaces of alveolar corners, the expected locations of type II pneumocytes (Figure 1 ▶ ; B, F, and G, arrows; see subsequent figures for quantitation). Many ISEL- or caspase 3-positive cells co-localized with positive immunoreactivity to monoclonal antibody MNF116 (Figure 1C) ▶ , an established marker of type II pneumocytes. 18 Interestingly, MNF116 immunoreactivity was observed in relatively normal regions of BLEO-exposed lung (Figure 1D ▶ , right) but not in more severely affected regions (Figure 1D ▶ , left), consistent with the elimination of type II cells in these areas. Co-administration of the AT1 receptor antagonist LOS with the BLEO (Figure 1H) ▶ significantly reduced caspase 3 IHC (see below for quantitation).
Figure 1.
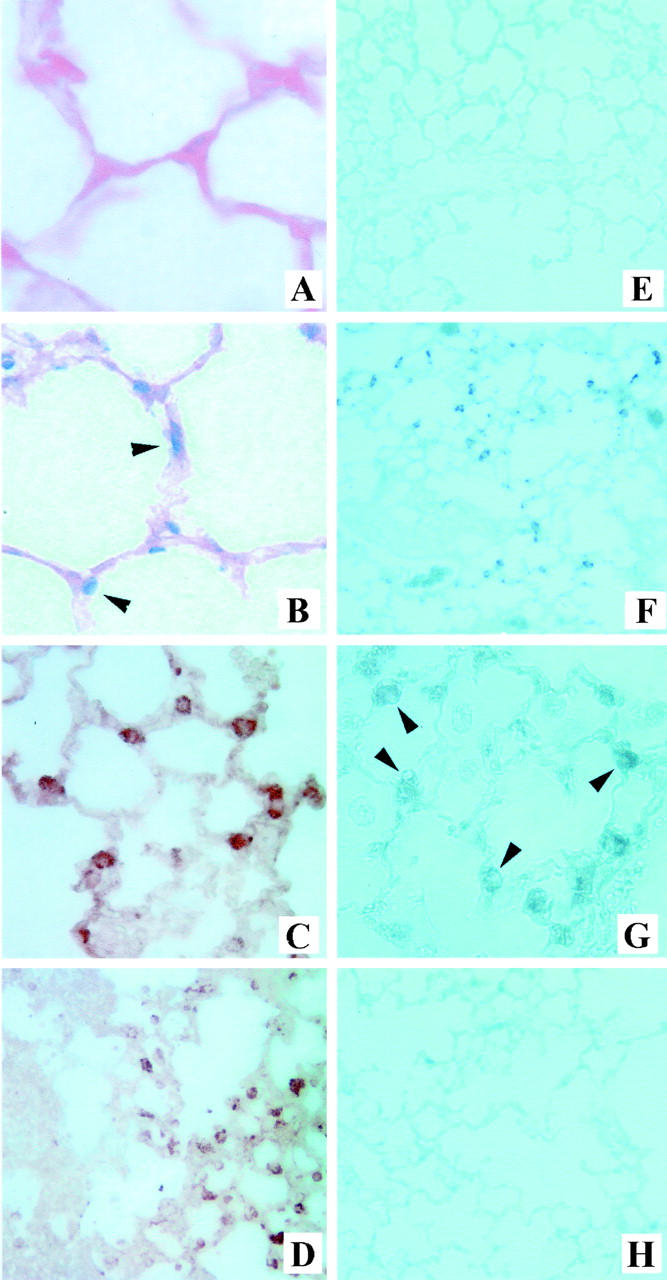
Detection of DNA fragmentation, activation of caspase 3, and alveolar type II pneumocytes in mouse lung. Deparaffinized lung sections were prepared from mice instilled intratracheally 14 days earlier with sterile saline (A, C, and E) or BLEO (B, D, F, and G). The sections were subjected to ISEL of fragmented DNA (A and B) or IHC with antibodies against the active form of caspase 3 (E–H) or with the type II cell-specific antibody MNF116 (18, C and D). G: Higher magnification of active caspase 3 labeling in F. H: Active caspase 3 labeling in mice treated with BLEO and LOS, an AT1 receptor antagonist. Note ISEL and active caspase 3 labeling in cells in the corners of alveolar walls in the lungs of BLEO-treated mice (B, F, and G, arrowheads) but not in saline-treated mice (A and E) or in mice treated with BLEO and LOS (H). Note also the co-localization of MNF116 (C) with anti-caspase 3 IHC (G) or ISEL (B) in BLEO-treated lungs. D reveals labeling of the type II cell marker MNF116 in relatively normal regions of BLEO-treated lung (D, right) but not in more severely affected regions (D, left). See text for details. Original magnifications: ×400 (A, B, C, G); ×200 (D); ×100 (E, F, H).
The instillation of BLEO also resulted in histological changes typical of BLEO-induced lung fibrosis by day 14 after instillation (Figure 2) ▶ . These include the infiltration of inflammatory cells, thickening of alveolar walls, and collagen accumulation (Figure 2B) ▶ ; for quantitation, see Figures 5 and 7 ▶ below. Again, the co-administration of LOS with the BLEO (Figure 2C) ▶ significantly reduced the alterations in lung morphology and collagen deposition (see below).
Figure 2.
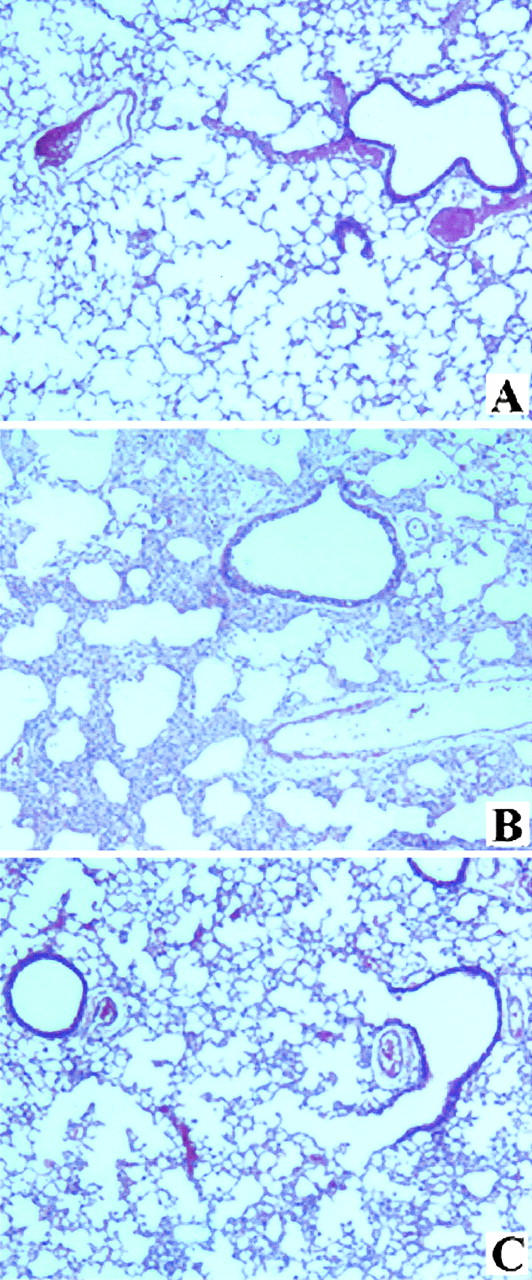
Histology of mouse lungs at 14 days after instillation of BLEO. A–C: Hematoxylin and eosin preparations of mouse lung instilled intratracheally 14 days earlier with sterile saline (A), BLEO (B), or BLEO and LOS (C). See text and Materials and Methods section for details. Original magnifications, ×200.
Figure 5.

AT1 receptor blockade inhibits lung collagen accumulation at 14 days after BLEO instillation. Normal mice were administered BLEO in the presence or absence of LOS as described in Figure 3 ▶ . Fourteen days later, total lung collagen was determined by assay of total hydroxyproline (HP) in hydrolyzed lung tissue (see Materials and Methods). Bars are the means ± SEM of n = 6; *, P < 0.05 versus control (CTL) by analysis of variance and Student-Newman-Keul’s test.
Inhibition of Apoptosis and Collagen Deposition by an AT1 Antagonist
Apoptosis also was detected as an increase in the total activity of caspase 3 in lung tissue, measured by enzyme assay of lung homogenates. As early as 6 hours after instillation of BLEO intratracheally (Figure 3A) ▶ , lung caspase 3 activity was increased 100%, but the increase was prevented by co-administration of the AT1 receptor antagonist LOS) (see Materials and Methods). In Figure 3B ▶ , BLEO also increased caspase 3 activity when applied in vitro at 25 mU/ml to mouse lung explants that were depleted of blood before explant culture. Application of BLEO in vitro increased caspase 3 activity in the explants by nearly 100% in 24 hours (P < 0.05), but LOS (10−6 mol/L) inhibited the increase by 57%.
Figure 3.

The AT1-selective receptor antagonist LOS inhibits BLEO-induced activation of caspase 3. A: BLEO was instilled intratracheally into normal mice with and without LOS in the intratracheal instillate (see Materials and Methods). Six hours later, the lungs were perfused to remove blood, excised, and the enzymatic activity of caspase 3 was measured in lung homogenates. B: Lung explants were prepared from normal mouse lung tissue perfused before excision (see Materials and Methods). BLEO (25 mU/ml) was applied in serum-free culture medium for 24 hours in the presence or absence of LOS (10−6 mol/L). Bars are the means ± SEM of n = 6; *, P < 0.05 versus control (CTL); **, P < 0.05 versus BLEO+LOS by analysis of variance and Student-Newman-Keul’s test.
The ability of LOS to inhibit lung epithelial apoptosis was also observed in vivo at 14 days after BLEO administration. In Figure 4A ▶ , intratracheal BLEO increased the abundance of ISEL-positive cells by 11-fold (P < 0.01), but LOS inhibited the increase by 66% (P < 0.05). Similarly, intratracheal BLEO increased the number of caspase 3-positive cells by 25-fold (Figure 4B ▶ , P < 0.01), but LOS blocked the increase by 74% (P < 0.05). Measurement of lung collagen accumulation in the same animals by hydroxyproline assay (Figure 5) ▶ revealed an increase in total lung collagen of 56% by 14 days after intratracheal BLEO, but LOS reduced the increase by 45%, to a value not significantly different from the control (CTL).
Figure 4.

AT1 receptor blockade inhibits DNA fragmentation and caspase 3 activation in lung epithelial cells 14 days after BLEO instillation. Normal mice were given a single intratracheal instillation of BLEO in the presence or absence of LOS in the instillate. LOS also was administered thereafter daily intraperitoneally. Fourteen days later, lung sections were prepared and labeled by ISEL (A) or by IHC for the active form of caspase 3 (B). Labeling was quantitated in cells within the alveolar surfaces (see Figure 1C ▶ ). Bars are the means ± SEM of n = 6; *, P < 0.01 versus control (CTL); **, P < 0.05 versus BLEO by analysis of variance and Student-Newman-Keul’s test.
Inhibition of Apoptosis and Collagen Deposition by AT1 Gene Deletion
Receptor subtype AT1a is the AT1 isoform expressed in the lungs of mice. 14 To test the hypothesis that angiotensin receptor AT1 is essential for BLEO-induced epithelial apoptosis and lung fibrogenesis, heterozygous AT1a-null mice were exposed to intratracheal BLEO in the same manner as wild-type mice of the same genetic background. In Figure 6 ▶ , the deletion of one allele of the AT1a gene (+/−) reduced BLEO-induced ISEL by 89% (Figure 6A) ▶ and inhibited BLEO-induced caspase 3 IHC by 85% (Figure 6B) ▶ , both relative to the response in wild-type mice (**, P < 0.01).
Figure 6.

Mice deficient in angiotensin receptor AT1a exhibit reduced DNA fragmentation and caspase 3 activation in lung epithelial cells 14 days after BLEO instillation.. Normal [wild type (w.t.)] or heterozygous AT1a knockout mice (+/−) were administered BLEO intratracheally as in Figure 3 ▶ . Fourteen days later, lung sections were prepared and labeled by ISEL (A) or by IHC for the active form of caspase 3 (B), which were quantitated as described in Figure 4 ▶ and Materials and Methods. Bars are the means ± SEM of n = 5; *, P < 0.001 versus wild-type unchallenged (w.t. − BLEO); **, P < 0.01 versus wild-type challenged (w.t. + BLEO) by analysis of variance and Student-Newman-Keul’s test.
When the susceptibility of the same mice to BLEO-induced fibrosis was measured, heterozygous AT1a-null mice did not exhibit a statistically significant increase in lung hydroxyproline at 14 days after intratracheal BLEO (Figure 7A) ▶ , in contrast to wild-type mice. Expression of the hydroxyproline data as the absolute amount of collagen per left lung (Figure 7B) ▶ suggested that unchallenged AT1a +/− mice, of the same age and body weight as the wild types, have more total collagen per left lung at baseline relative to wild-type mice, but the difference was not statistically significant.
Figure 7.

Mice deficient in angiotensin receptor AT1a exhibit reduced lung collagen accumulation in response to BLEO instillation. Normal [wild type (w.t.)] or heterozygous AT1a knockout mice (+/−) were administered BLEO intratracheally as in Figure 3 ▶ . Fourteen days later lung tissue was fast-frozen, hydrolyzed, and total collagen was measured by hydroxyproline assay (HP) as described in Materials and Methods. A: HP data are expressed as a percentage of the corresponding control (− BLEO). B: Data are expressed as the absolute amount of HP per left lung. Bars are the means ± SEM of n = 5; *, P < 0.01 versus untreated (− BLEO) by analysis of variance and Student-Newman-Keul’s test.
Discussion
Inhibitors of angiotensin-converting enzyme (ACE) or antagonists of ANG receptor AT1 have been shown to have anti-apoptotic and anti-fibrotic effects in the heart, 20 kidney, 21 and liver. 22 The first reports of anti-fibrotic actions in the lung by the ACE inhibitor captopril, published many years ago, 23,24 were recently extended by the demonstration that the AT1 antagonists LOS and L158809 have even more potent anti-fibrotic potential in the lungs than do ACE inhibitors. 25 The present work extends those observations by showing that at least one of the mechanisms by which AT1 antagonists act is through the inhibition of apoptosis in AECs.
Other potential mechanisms by which AT1 antagonists might act on the lung in vivo, a topic recently reviewed by Marshall, 26 include decreased vascular tone, decreased vascular permeability, and altered fibroblast activity. At least some of these actions could be envisioned to be related to the ability of ACE inhibitors or AT1 antagonists to reduce blood pressure. Indeed, the AT1-null mice used here were shown earlier to exhibit reductions in systemic blood pressure of ∼12 mm Hg for heterozygous animals at baseline. 15 Thus, it is possible that some of the anti-fibrotic actions of LOS or AT1a deletion might be related to lowered systemic or pulmonary hydrostatic pressures, and the data herein do not strictly exclude that possibility.
On the other hand, the ability of LOS to prevent caspase 3 activation by BLEO in cultured lung explants (Figure 3B) ▶ argues against the involvement of decreased blood pressure in the inhibition of apoptosis because no hydrostatic pressure changes occur in explants manipulated ex vivo. Moreover, the induction of LOS-inhibitable caspase 3 activity in lung explants, depleted of blood by previous PBS perfusion, argues against a primary role for blood-derived cells in the initiation of the apoptosis. This experiment also supports the theory that the inhibitory effect of LOS on apoptosis was not mediated by an indirect action on infiltrating inflammatory cells.
The initial protocol for the reported in vivo studies was designed to determine whether blockade or deletion of the AT1 receptor, throughout the time course of the 14-day BLEO model, was capable of inhibiting or blocking the apoptotic and fibrotic responses. The success of this strategy, particularly in light of the many known functions of angiotensin discussed in preceding paragraphs, raises the interesting question of whether the blockage of fibrogenesis was due to the acute or delayed consequences of AT1 blockade. Although a time course study of various LOS administration protocols was not performed, quantitation of the number of erythrocytes reaching the alveolar airspaces by 6 hours after BLEO (a crude index of lung barrier collapse) suggested that LOS did not prevent acute, transient barrier collapse (data not shown) despite its ability to reduce caspase 3 activation at the same sampling time (Figure 3) ▶ . This observation, although very preliminary, is consistent with the theory that the blockage of apoptosis in AECs is key to the subsequent blockade of collagen deposition. In contrast, blockage of receptor AT1 may also inhibit mitosis of lung fibroblasts 27 and reduce collagen synthesis by the same cells, 28 relatively delayed effects that might be independent of AEC apoptosis at early time points. Moreover, endothelial cells also express receptor AT1 and undergo apoptosis in response to angiotensin, albeit at relatively high concentrations, 29,30 and other cell types resident in the lung are known to respond to angiotensin in ways currently under intense study. 31 Thus, it is possible that the acute early effects of AT1 blockade on AEC apoptosis are not necessary for inhibition of collagen deposition at later time points, and this study does not exclude that possibility.
On the other hand, earlier work has shown that AECs undergoing apoptosis in response to Fas ligand, tumor necrosis factor-α, or BLEO begin secreting angiotensin into the extracellular space within hours of exposure, 9-11 at least in vitro. Those studies also showed that the autocrine production of angiotensin and its binding to receptor AT1 on AECs were required for apoptosis in response to these agents; this mechanism can explain the ability of LOS to block AEC apoptosis in vivo in the present study. Moreover, previous studies with apoptosis inhibitors support the contention that the acute apoptotic response is a pivotal event in the BLEO model. Wang and colleagues 7 showed that the ACE inhibitor captopril or the caspase inhibitor ZVAD-fmk had essentially equal ability to block the appearance of apoptotic epithelial cells in rats exposed to intratracheal BLEO and to prevent subsequent collagen deposition. 7 That report, which was confirmed by Kuwano and colleagues 8 in studies of mice exposed to BLEO and/or ZVADfmk, suggested that the blockade of fibrogenesis by captopril was indeed related to inhibition of apoptosis, rather than the many other effects of ACE inhibition in vivo. 26 Later work confirmed that the ZVAD compound had no inhibitory effect on angiotensin-converting enzyme itself. 13 Thus, the present data are consistent with the ability of ACE inhibition by captopril to block both AEC apoptosis and collagen deposition in rats, 7 and extend this concept to angiotensin receptor blockade in mice. The data herein also are in agreement with recent reports that LOS inhibits BLEO-induced collagen deposition in rat lung 32 and that AT1a-null mice show reduced liver fibrosis in response to carbon tetrachloride. 33
The AT1 receptor is expressed as two isoforms, AT1a and AT1b, for which no selective antagonists have yet been developed. 34 Subtype AT1a is known to be expressed in the lungs of mice, but AT1b was not detected in mouse lung by reverse transcriptase-polymerase chain reaction. 14 Although it is possible that cells of minor abundance in the lung, such as type II cells, might express AT1b in quantities not detected in earlier studies, the primary isolates of type II pneumocytes from Wistar rats did not reveal AT1b expression by reverse transcriptase-polymerase chain reaction despite the use of two different primer sets and high-amplification cycle numbers (data not shown). In any case, the finding that deletion of only one allele of the AT1a gene significantly reduced BLEO-induced apoptosis and collagen accumulation supports the notion that AT1a is the only active AT1 receptor subtype on the alveolar epithelium of mice.
In an earlier report describing the mechanisms by which angiotensin induces apoptosis in primary cultures of AECs, Papp and colleagues 35 showed that blockage of AT1 signaling through protein kinase C (PKC) with the specific PKC inhibitor chelerythrin could attenuate the apoptotic response to angiotensin. This finding is consistent with the known role of PKC in AT1 signaling in a variety of cell types, 30 but the pathways from PKC to the effector caspase 3, which is also required for this response, 35 are currently unknown. Given that AEC apoptosis in response to Fas ligand, tumor necrosis factor-α, or BLEO all require the autocrine production and binding of angiotensin to AT1, 9-11 the report of Papp and colleagues 35 suggests that PKC inhibitors would also block AEC apoptosis in response to these agents in vivo as well. This prediction was not tested in the present study, but will be an interesting topic for future inquiry.
In summary, BLEO-induced apoptosis of lung epithelial cells in mice was significantly inhibited by the AT1-selective angiotensin receptor antagonist LOS or by targeted deletion of the gene for angiotensin receptor subtype AT1a. Both methods of reducing AT1 action also reduced or abrogated lung collagen accumulation in response to BLEO challenge. These data agree with earlier demonstrations of the anti-fibrotic action of ACE inhibitors and AT1-selective antagonists in rat models of lung fibrosis, and with in vitro studies showing a role for receptor AT1 in mediating apoptosis of AECs. They also suggest the possibility that AT1 antagonists may hold potential for the treatment of lung fibrosis in humans; this possibility is supported by the recent finding that patients with pulmonary fibrosis have a higher frequency of the D allele of angiotensin-converting enzyme, 36 a deletion polymorphism that confers higher levels of ACE.
Footnotes
Address reprint requests to Bruce D. Uhal, Ph.D, Department of Physiology, Michigan State University, 3185 Biomedical and Physical Sciences Building, East Lansing, MI 48824-3320. E-mail: uhal@msu.edu.
Supported by the United States Public Health Service (grant HL-45136), the American Heart Association (grant-in-aid 0250269N), and the Michigan State University Foundation.
References
- 1.Selman M, King T, Pardo A: Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001, 134:136-151 [DOI] [PubMed] [Google Scholar]
- 2.Gauldie J, Kolb M, Sime P: A new direction in the pathogenesis of pulmonary fibrosis? Respir Res 2002, 3:1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haschek WM, Witschi HP: Pulmonary fibrosis: a possible mechanism. Toxicol Appl Pharmacol 1979, 51:475-487 [DOI] [PubMed] [Google Scholar]
- 4.Adamson IYR, Young L, Bowden DH: Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol 1988, 130:377-383 [PMC free article] [PubMed] [Google Scholar]
- 5.Hagimoto N, Kuwano K, Nomoto Y, Kunitake R, Hara N: Apoptosis and expression of FAS/FAS ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol 1997, 16:91-101 [DOI] [PubMed] [Google Scholar]
- 6.Hagimoto N, Kuwano K, Miyazaki H, Kunitake R, Fujita M, Kawasaki M, Kanika Y, Hara N: Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of FAS antigen. Am J Respir Cell Mol Biol 1997, 17:272-278 [DOI] [PubMed] [Google Scholar]
- 7.Wang R, Ibarra-Sunga O, Pick R, Uhal BD: Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am J Physiol 2000, 279:L143-L151 [DOI] [PubMed] [Google Scholar]
- 8.Kuwano K, Kunitak R, Maeyama T, Hagimoto N, Kawasaki M, Matsuba T, Yoshimi M, Inoshima I, Yoshid K, Hara N: Attenuation of bleomycin-induced pneumopathy in mice by a caspase inhibitor. Am J Physiol 2001, 280:L316-L325 [DOI] [PubMed] [Google Scholar]
- 9.Wang R, Zagariya A, Ang E, Ibarra-Sunga O, Uhal BD: Fas-induced apoptosis of alveolar epithelial cells requires angiotensin II generation and receptor interaction. Am J Physiol 1999, 277:L1245-L1250 [DOI] [PubMed] [Google Scholar]
- 10.Wang R, Alam G, Zagariya A, Gidea C, Pinillos H, Lalude O, Choudhary G, Uhal BD: Apoptosis of lung epithelial cells in response to TNF-alpha requires angiotensin II generation de novo. J Cell Physiol 2000, 185:253-259 [DOI] [PubMed] [Google Scholar]
- 11.Li X, Zhuang H, Soledad-Conrad V, Zhang J, Uhal BD: Bleomycin-induced apoptosis of alveolar epithelial cells requires angiotensin synthesis de novo. Am J Physiol 2003, 284:L501-L507 [DOI] [PubMed] [Google Scholar]
- 12.Uhal BD, Wang R, Laukka J, Zhaung J, Soledad-Conrad V, Filippatos G: Inhibition of amiodarone-induced lung fibrosis but not alveolitis by angiotensin system antagonists. Pharmacol Toxicol 2003, 92:81-87 [DOI] [PubMed] [Google Scholar]
- 13.Filippatos G, Uhal BD: Blockade of apoptosis by ACE inhibitors and angiotensin receptor antagonists. Curr Pharm Design 2003, 9:707-714 [DOI] [PubMed] [Google Scholar]
- 14.Burson J, Aguilera G, Gross K, Sigmond C: Differential expression of angiotensin receptor 1A and 1B in mouse. Am J Physiol 1994, 267:E260-E267 [DOI] [PubMed] [Google Scholar]
- 15.Ito M, Oliverio M, Mannon P, Best C, Maeda N, Smithies O, Coffman T: Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc Natl Acad Sci 1995, 92:3521-3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor B, Stoops T, Everett A: Protein phosphatase inhibitors arrest cell cycle and reduce branching morphogenesis in fetal rat lung cultures. Am J Physiol 2000, 278:L1062-L1070 [DOI] [PubMed] [Google Scholar]
- 17.Mundle S, Iftikhar A, Shetty V, Alvi S, Dameron S, Gregory S, Marcus B, Khan S, Raza A: In situ end labeling of DNA to detect apoptotic cell death in a variety of human tumors. Cell Death Differ 1994, 1:117-122 [PubMed] [Google Scholar]
- 18.Fehrenbach H, Kasper M, Koslowski R, Pan T, Schuh D, Muller M, Mason RJ: Alveolar epithelial type II cell apoptosis in vivo during resolution of keratinocyte growth factor-induced hyperplasia in the rat. Histochem Cell Biol 2000, 114:49-61 [DOI] [PubMed] [Google Scholar]
- 19.Woessner J: The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys 1961, 93:440-447 [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Weber TK: Cardiac remodelling by fibrous tissue: role of local factors and circulating hormones. Ann Med 1998, 1:S3-S8 [PubMed] [Google Scholar]
- 21.Mezzano S, Ruiz-Ortega M, Egido J: Angiotensin II and renal fibrosis. Hypertension 2001, 38:635-638 [DOI] [PubMed] [Google Scholar]
- 22.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, Tsujinoue H, Fukui H: Angiotensin-II type I receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology 2001, 34:745-750 [DOI] [PubMed] [Google Scholar]
- 23.Molteni A, Ward W, Ts’ao C, Solliday N, Dunne M: Monocrotaline-induced pulmonary fibrosis in rats: amelioration by captopril and penicillamine. Proc Soc Exp Biol Med 1985, 180:112-120 [DOI] [PubMed] [Google Scholar]
- 24.Ward W, Molteni A, Ts’ao C, Hinz J: Captopril reduces collagen and mast cell accumulation in irradiated rat lung. Int J Radiat Oncol Biol Phys 1990, 19:1405-1409 [DOI] [PubMed] [Google Scholar]
- 25.Molteni A, Moulder JE, Cohen E, Ward W, Fish B, Taylor J, Wolfe L, Brizio-Molteni L, Veno P: Control of radiation-induced pneumopathy and lung fibrosis by angiotensin-converting enzyme inhibitors and an angiotensin II type 1 receptor blocker. Int J Radiat Biol 2000, 76:523-532 [DOI] [PubMed] [Google Scholar]
- 26.Marshall RP: The pulmonary renin-angiotensin system. Curr Pharm Design 2003, 9:715-722 [DOI] [PubMed] [Google Scholar]
- 27.Marshall R, McNulty R, Laurent G: Angiotensin II is mitogenic for human lung fibroblasts via activation of the type I receptor. Am J Respir Crit Care Med 2000, 161:1999-2004 [DOI] [PubMed] [Google Scholar]
- 28.Marshall RP, Gohlke P, Chambers RC, Howell DC, Bottoms S, Unger T, McAnulty RJ, Laurent GJ: Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol, in press [DOI] [PubMed]
- 29.Dimmeler S, Rippmann V, Weiland U, Haendeler J, Zeiher AM: Angiotensin II induces apoptosis of human endothelial cells. Protective effect of nitric oxide. Circ Res 1997, 81:970-976 [DOI] [PubMed] [Google Scholar]
- 30.Li D, Yang B, Philips MI, Mehta JL: Proapoptotic effects of ANG II in human coronary artery endothelial cells: role of AT1 receptor and PKC activation. Am J Physiol 1999, 276:H786-H792 [DOI] [PubMed] [Google Scholar]
- 31.Harrison DG, Cai H, Landmesser U, Griendling KK: Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J Renin Angiotensin Aldosterone Syst 2003, 4:51-61 [DOI] [PubMed] [Google Scholar]
- 32.Fang X, Zhu Y, Hu X, Liu Y: Losartan in the rat model of bleomycin-induced pulmonary fibrosis and its impact on the expression of monocyte chemoattractant protein-1 and basic fibroblast growth factor. Zhonghua Jie He He Hu Xi Za Zhi 2002, 25:268-272 [PubMed] [Google Scholar]
- 33.Kanno K, Tazuma S, Chayama K: AT1A-deficient mice show less severe progression of liver fibrosis induced by CCl(4). Biochem Biophys Res Commun 2003, 308:177-183 [DOI] [PubMed] [Google Scholar]
- 34.Filippatos G, Tilak M, Pinillos H, Uhal BD: Regulation of apoptosis by angiotensin II in the heart and lungs. Int J Mol Med 2001, 7:273-280 [DOI] [PubMed] [Google Scholar]
- 35.Papp M, Li X, Zhuang J, Wang R, Uhal BD: Angiotensin receptor subtype AT1 mediates alveolar epithelial cell apoptosis in response to ANGII. Am J Physiol 2002, 282:L713-L718 [DOI] [PubMed] [Google Scholar]
- 36.Morrison C, Papp A, Hejmanowski A, Addis V, Prior T: Increased D allele frequency of the angiotensin converting enzyme gene in pulmonary fibrosis. Hum Pathol 2001, 32:521-528 [DOI] [PubMed] [Google Scholar]