

Abstract
β-Glucan is one of the most abundant polysaccharides in fungal pathogens, yet its importance in antifungal immunity is unclear. Here we show that deficiency of dectin-1, the myeloid receptor for β-glucan, rendered mice susceptible to infection with Candida albicans. Dectin-1-deficient leukocytes demonstrated significantly impaired responses to fungi even in the presence of opsonins. Impaired leukocyte responses were manifested in vivo by reduced inflammatory cell recruitment after fungal infection, resulting in substantially increased fungal burdens and enhanced fungal dissemination. Our results establish a fundamental function for β-glucan recognition by dectin-1 in antifungal immunity and demonstrate a signaling non–Toll-like pattern-recognition receptor required for the induction of protective immune responses.
Infections with normally nonpathogenic fungi such as Candida albicans are an emerging problem resulting from modern medical interventions and the increasing prevalence of acquired immunodeficiency1. The high incidence of morbidity and mortality associated with such diseases, especially once the fungus has disseminated, demonstrates deficiencies in both present antifungal therapies and understanding of the host immune response. Protection against such organisms is mediated mainly by phagocytic cells that recognize, ingest and kill the invading pathogen, inducing a T helper type 1 immune response, which in turn activates fungicidal effector mechanisms, such as the respiratory burst1. Although cells such as neutrophils and macrophages are thought to be crucial in that process, the mechanism underlying the recognition and initiation of the protective responses to these pathogens remains unclear.
The cell walls of fungi consist mainly of carbohydrates, including mannose-based structures (the mannoproteins), β-glucan and chitin. For immune systems of infected hosts, such polysaccharides serve as pathogen-associated molecular patterns (PAMPs) that can be recognized by a variety of host-expressed pattern-recognition receptors, including the Toll-like receptors (TLRs), although the precise functions of each of the myriad receptors that can respond to these pathogens and contribute to the induction of protective responses have not been fully elucidated.
Historically, the cell walls of fungi were shown to be covered by a layer of mannoproteins, which prompted much interest in mannose-based recognition systems2. Subsequent evidence has suggested that this model may be too simplistic and that other PAMPs, particularly β-glucans, are exposed on the cell surface and therefore are potentially important in immune recognition3. In fungi such as C. albicans and Saccharomyces cerevisiae, β-glucans can comprise up to 50% of the dry weight of the fungal cell wall and are essential structural components that provide elasticity and mechanical strength2. In isolated form, β-glucans are known to stimulate immune function, having a variety of beneficial effects, including protection against tumor development or infection with fungal, bacterial, viral or protozoal pathogens4,5.
Host-cell recognition of β-glucan is mediated mainly by dectin-1, a myeloid-expressed type II transmembrane C-type lectin–like receptor that contains an immunoreceptor tyrosine-based activation motif in its cytoplasmic tail6,7. Dectin-1 binds to many fungal species, including saccharomyces8, candida, coccidioides9, pneumocystis10 and aspergillus11-13. In vitro, dectin-1 has been shown to mediate a variety of both TLR-dependent and TLR-independent antifungal cellular responses, including the respiratory burst14,15, phagocytosis15,16 and the production of many cytokines and chemokines8,10,14,17,18.
Here we have assessed the contribution of β-glucan recognition to the outcome of fungal infection in vivo by using dectin-1-deficient mice. We found that recognition of these carbohydrates had an essential function in antifungal immunity and host survival by promoting myeloid cell activation and regulating the subsequent inflammatory response. Our studies demonstrate a signaling non–Toll-like pattern-recognition receptor required for the induction of protective immune responses and provide new insights into the innate sensing of fungal pathogens.
RESULTS
Dectin-1-deficient mice show no gross abnormalities
To examine the function of β-glucan recognition in antifungal immunity, we generated mice deficient in dectin-1 (called ‘dectin-1-knockout mice’ here) using a conventional gene-targeting vector (Supplementary Fig. 1 online). We confirmed deletion of exons 1–3 of the gene encoding dectin-1 (Clec7a), corresponding to the cytoplasmic tail, transmembrane and stalk regions, by Southern blot with two external probes (Fig. 1a). Flow cytometry of peripheral leukocytes, done as described before19, confirmed that Clec7a expression was abrogated (Fig. 1b). Mice with heterozygous deficiency showed intermediate protein expression (Supplementary Fig. 1), suggesting a gene-dosage effect. The dectin-1-knockout mice were viable, had no gross abnormalities and had normal peripheral leukocyte counts (Table 1), and thioglycollate-elicited peritoneal macrophages from dectin-1-knockout and dectin-1-wild-type mice had a similar antigen phenotype and no abnormalities were evident other than the lack of expression of dectin-1 (Supplementary Fig. 1).
Figure 1.

Dectin-1-deficient mice. (a) Southern blot showing the wild-type 4-kilobase band in a nontargeted embryonic stem cell clone (+/+) and the presence of an additional 5.6-kilobase band in a heterozygous Clec7a-targeted embryonic stem cell clone (+/−). (b) Flow cytometry of mouse bone marrow, with gating on Gr-1hi7/4hi neutrophils (Neu) and monocytes (Mo; left), for analysis of dectin-1 expression by dectin-1-wild-type mice (dectin-1-WT; middle) and their dectin-1-knockout littermates (dectin-1-KO; right). Plots are representative of data obtained from three mice per group.
Table 1.
Differential splenocyte counts
| Cell type | Marker | WT cells (n) | KO cells (n) |
|---|---|---|---|
| B cells | B220+ | 41.25 ± 6.909 (4) | 41.58 ± 0.805 (4) |
| CD4+ T cells | CD3+CD4+ | 14.33 ± 3.135 (4) | 13.85 ± 2.123 (4) |
| CD4+CD25+ T cells | CD3+CD4+CD25+ | 1.928 ± 0.282 (4) | 2.219 ± 0.271 (4) |
| CD8+ T cells | CD3+CD8+ | 7.807 ± 1.119 (4) | 8.558 ± 0.640 (4) |
| CD8+CD25+ T cells | CD3+CD8+CD25+ | 0.328 ± 0.090 (4) | 0.352 ± 0.080 (4) |
| Natural killer cells | CD49b+CD3− | 1.203 ± 0.131 (4) | 1.394 ± 0.432 (4) |
| Dendritic cells | CD11chi | 1.201 ± 0.325 (10) | 1.269 ± 0.526 (10) |
| Red pulp macrophages | F4/80hi | 0.917 ± 0.131 (10) | 0.804 ± 0.128 (10) |
| Neutrophils | Gr-1hiCD11b+ | 1.311 ± 0.324 (10) | 1.09 ± 0.204 (10) |
| Eosinophils | Gr-1intCD11b+F4/80+ | 0.257 ± 0.033 (10) | 0.323 ± 0.040 (10) |
| ‘Resident’ monocytes | Gr-1−CD11b+F4/80+ | 0.485 ± 0.091 (10) | 0.581 ± 0.185 (10) |
| ‘Inflammatory’ monocytes | Gr-1+CD11b+F4/80+ | 0.444 ± 0.047 (10) | 0.330 ± 0.061 (10) |
Splenocytes were isolated from matched dectin-1-knockout mice (KO) and dectin-1-wild-type mice (WT) and cell types were identified with markers and flow cytometry. Red pulp macrophages were identified with autofluorescence and F4/80 staining; eosinophils and monocytes distinguished by forward- and side-scatter profiles. n = number of mice.
Data represent cell numbers × 106 (mean ± s.e.m.).
Impaired myeloid cell activation by fungal particles
We next assessed the ability of dectin-1-knockout macrophages to recognize and respond to zymosan, a β-glucan-rich particle derived from the cell wall of S. cerevisiae20. Consistent with dectin-1's being the main β-glucan receptor on macrophages21, thioglycollate-elicited macrophages from dectin-1-knockout mice had considerably impaired recognition of zymosan. The extent of recognition in dectin-1-knockout macrophages was similar to that obtained by treatment of dectin-1-wild-type cells with competing β-glucan (Fig. 2a,b). Prior opsonization with mouse serum restored the binding of these particles by promoting β-glucan-independent recognition through complement receptors21. However, the loss of dectin-1 also resulted in a failure to mount an inflammatory response to zymosan, assessed by release of tumor necrosis factor (TNF), which was not restored by serum opsonization (Fig. 2b). That defect was specific for fungal particles, as dectin-1-knockout macrophages showed no impairment in their response to other microbial stimuli, including lipopolysaccharide and the TLR2 agonist Pam3CSK4 (Fig. 2c). Thus, dectin-1 is required for inflammatory responses to both opsonized and unopsonized particles.
Figure 2.

Impaired β-glucan recognition by dectin-1-knockout macrophages. (a) ‘False-color’ photomicrographs of thioglycollate-elicited dectin-1-wild-type and dectin-1-knockout macrophages labeled with fluorescein isothiocyanate–tagged zymosan (green), after incubation for 30 min at 37 °C (25 zymosan particles per macrophage). Original magnification, ×20. (b) Fluorimetry for zymosan recognition (top) by dectin-1-wild-type macrophages (black bars) and dectin-1-knockout macrophages (gray bars), expressed as relative fluorescent units (RFU), and ELISA quantification of TNF production (bottom) after removal of zymosan and incubation for a further 3 h at 37 °C. Experiments were done in the presence (+) or absence (−) of competing β-glucans. Data represent mean (± s.e.m.) of four pooled, normalized experiments. (c) ELISA of TNF production by dectin-1-wild-type (filled circles) and dectin-1-knockout (open circles) thioglycollate-elicited macrophages after treatment with the soluble TLR2 agonist Pam3CSK4 or TLR4 agonist lipopolysaccharide. Data represent mean (± s.e.m.) of replicates from one representative experiment of two. (d) Respiratory burst of dectin-1-wild-type (filled circles) and dectin-1-knockout (open circles) thioglycollate-elicited macrophages in response to zymosan and complement-opsonized zymosan (Op-zymozan), as assessed by dihydrorhodamine 123. Dotted lines, basal H2O2 production in unstimulated cells. Data represent mean (± s.e.m.) of pooled normalized mean fluorescent intensity data (presented as cumulative H2O2) from three independent experiments. *, P < 0.05; **, P < 0.01; and ***, P < 0.001. (ANOVA with Bonferroni post-test (b); Student's t-test (d)).
As dectin-1 has been associated with the respiratory burst response to zymosan in macrophages14, we examined that response in peritoneal-elicited macrophages. Although low amounts of respiratory burst are induced in those cells22, there was no apparent defect in the respiratory burst after stimulation with serum-opsonized zymosan in the gene-targeted macrophages (Fig. 2d). Although we obtained a significant difference with unopsonized zymosan, that could be attributed to the lack of particle recognition of these cells (Fig. 2b). Thus dectin-1 seems to have a redundant function in the respiratory burst in macrophages.
Dendritic cells produce interleukin 10 (IL-10) and IL-12 in response to unopsonized yeast, through a mechanism dependent on the kinase Syk and the adaptor CARD9, which is thought to involve dectin-1 (refs. 14,17,23). However, we did not detect any substantial defect in the production of those cytokines in dectin-1-knockout bone marrow–derived dendritic cells cultured with zymosan (Supplementary Fig. 2 online). In contrast, the production of those cytokines was impaired substantially in elicited dectin-1-knockout macrophages (Supplementary Fig. 2). As dendritic cells are known to express other receptors involved in the recognition of yeast24, these data suggested that dectin-1 is dispensable for fungal recognition in those cells and that the phenotypes of cells with Syk or CARD9 deficiency may not be restricted to a specific blockade of the dectin-1 pathway.
We also examined the effect of dectin-1 deficiency on neutrophils, which are essential phagocytes in antifungal immunity1. In thioglycollate-elicited neutrophils, the absence of dectin-1 resulted in loss of the β-glucan-dependent recognition of unopsonized zymosan noted in dectin-1-wild-type cells (Fig. 3a). Although dectin-1-knockout neutrophils showed an attenuated respiratory burst when cultured together with unopsonized yeast, which may also be attributable to the lack of particle recognition, the response was not fully restored when we used opsonized zymosan particles (Fig. 3b). Thus, dectin-1 mediates recognition and cellular responses to fungal particles in neutrophils.
Figure 3.
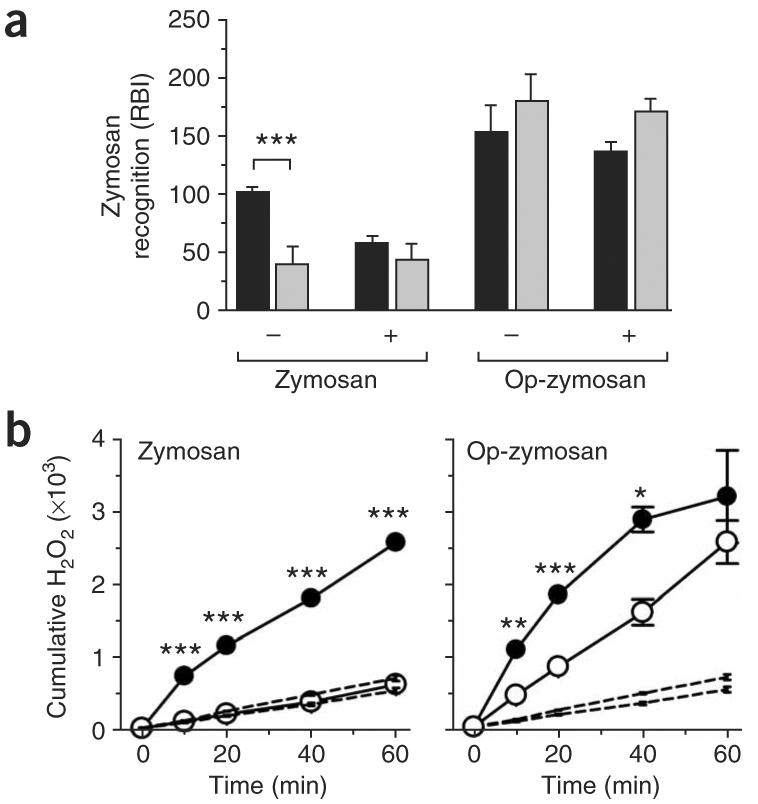
Impaired β-glucan recognition by dectin-1-knockout neutrophils. (a) Flourimetry of 129/Sv dectin-1-wild-type neutrophils (black bars) and dectin-1-knockout neutrophils (gray bars) incubated with fluorescein isothiocyanate–labeled zymosan or serum-opsonized zymosan (Op-zymosan), expressed as relative binding index (RBI), as described46. Experiments were done in the presence (+) or absence (−) of competing β-glucans. Data represent mean (± s.e.m.) of triplicates of one representative experiment from two. (b) Respiratory burst of dectin-1-knockout neutrophils (open circles) and dectin-1-wild-type neutrophils (filled circles) in response to opsonized zymosan (right) and unopsonized zymosan (left). Dotted lines, basal H2O2 production in unstimulated cells. Data represent mean (± s.e.m.) of triplicates of one representative experiment of two. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 (ANOVA with Bonferroni post-test (a); Student's t-test (b)).
Enhanced fungal dissemination in dectin-1-knockout mice
Because candida is one of the leading causes of nosocomial fungal infections and is well studied in animals1,25-27, we examined the function of dectin-1 in vivo by infecting mice intravenously with various doses of C. albicans SC5314 as a model of systemic candidiasis26. Dectin-1-knockout mice had much lower survival than wild-type mice after infection with a sublethal dose (1 × 104 colony-forming units (CFU)) or a lethal dose (1 × 105 CFU) of C. albicans (Fig. 4a). Dectin-1-knockout mice that succumbed to infection showed evidence of gastrointestinal involvement resulting macroscopically in considerable enlargement of the stomach (Fig. 4b and data not shown). Histological examination showed no substantial inflammation of the stomach and intestinal tissues, and the fungus seemed to be located mainly in the lumen (data not shown). However, the stomachs of infected dectin-1-knockout mice were full of food (data not shown), suggesting obstruction of the gastrointestinal tract.
Figure 4.
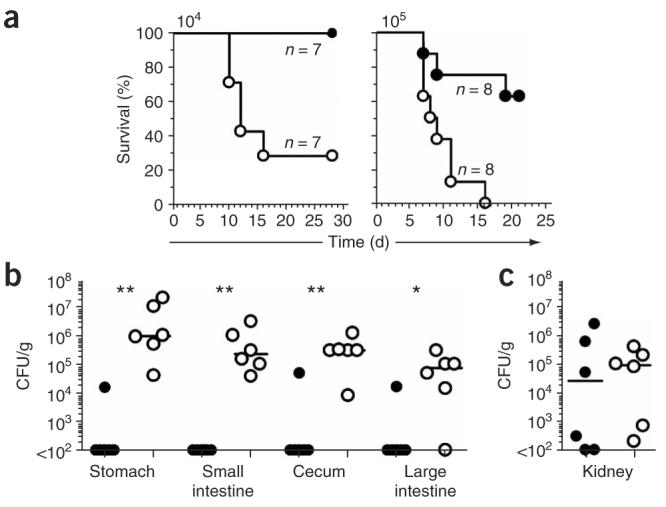
Dectin-1-knockout mice are more susceptible than dectin-1-wild-type mice to live C. albicans. (a) Survival curves of 129/Sv dectin-1-wild-type mice (filled circles) and dectin-1-knockout mice (open circles) infected intravenously with 1 × 104 CFU (left) or 1 × 105 CFU (right) C. albicans SC5314. P = 0.0066 (left) and P = 0.0035 (right; log-rank test). (b,c) Quantification of the live C. albicans fungal burden in the gastrointestinal tract (b) or kidneys (c) of dectin-1-wild-type mice (filled circles) and dectin-1-knockout mice (open circles) at 9 d after intravenous infection with 1 × 104 CFU. *, P < 0.05, and **, P < 0.01 (two-tailed Mann-Whitney test). Data are representative of two independent experiments.
To elucidate the development of disease, we also examined dectin-1-knockout mice before the onset of death. At day 9 after infection with 1 × 104 CFU C. albicans, dectin-1-knockout mice had significantly higher fungal burdens than those of dectin-1-wild-type mice throughout the gastrointestinal tract, although renal fungal loads were similar to those of wild-type mice at this time point (Fig. 4b,c). Enhanced systemic dissemination of C. albicans in the dectin-1-knockout mice was evident even within 24 h of infection (data not shown), consistent with a defect in the innate recognition and control of the fungal pathogen. However, we were unable to detect significant differences in cytokine production early in infection in systemically infected dectin-1-knockout and dectin-1-wild-type mice (data not shown). As gastrointestinal dissemination of C. albicans after intravenous challenge has not been widely reported (although it has been noted in other models; L. Romani, personal communication), we re-examined fungal distribution in dectin-1-wild-type mice infected with a lethal dose (2 × 105 CFU) of C. albicans. Those mice had gastrointestinal involvement similar to that of dectin-1-knockout mice, including enlargement of the stomach (Supplementary Fig. 3 online), which suggested that enlargement of the stomach was a normal pathological process in lethally infected mice.
Although the kidneys are the chief target organ of candida28, we never found a difference in renal fungal loads in dectin-1-knockout versus dectin-1-wild-type mice early in infection (Fig. 4c). However, the kidneys of dectin-1-knockout mice obtained at the time of death had enhanced colonization with the C. albicans, particularly in the pelvic region, with extension up the renal tubules. Such colonization was not evident in the kidneys of the dectin-1-wild-type mice, which had mostly cleared the infection by the end of the experiment (Fig. 5). Focally, the fungal hyphae extended through the tubular epithelium into the interstitium and were surrounded by acute neutrophilic inflammation, indicative of invasive candidiasis (Fig. 5 and data not shown). Thus, dectin-1 deficiency results in enhanced fungal dissemination and susceptibility to infection.
Figure 5.
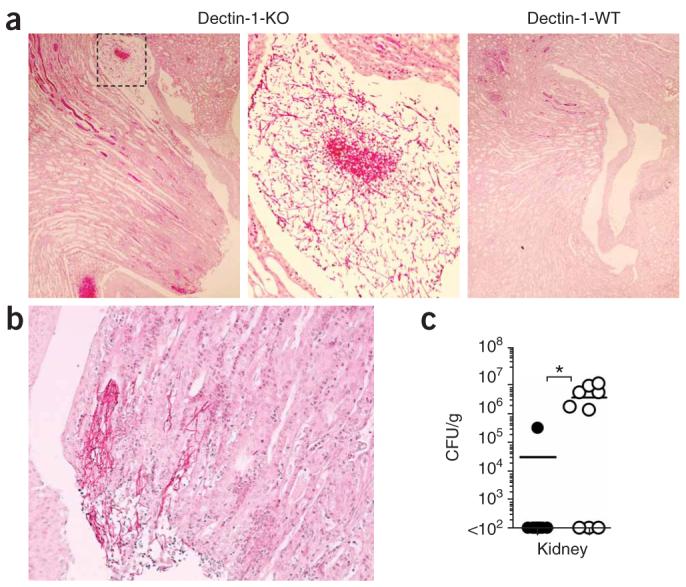
Kidney disease in dectin-1-knockout mice that succumb to infection. (a,b) Photomicrographs of fungal growth in periodic acid Schiff–stained kidneys of a representative dectin-1-knockout mouse (a, left) or dectin-1-wild-type mouse (a, right) obtained at death after intravenous challenge with 1 × 104 CFU C. albicans SC5314. Middle (a), enlargement of boxed area at left. b, hyphal tissue invasion in the kidney of a dectin-1-knockout mouse. Original magnification, ×40 (a, left, right) and ×100 (a, middle, and b). (c) Quantification of the fungal burden in dectin-1-wild-type kidneys (filled circles) and dectin-1-knockout kidneys (open circles) at the time of death or 28 d after intravenous infection with 1 × 104 CFU C. albicans. Mice with a fungal burden of less than 1 × 102 CFU had cleared the infection by the end of the experiment (day 28). Each symbol represents one mouse. *, P < 0.05 (Mann-Whitney nonparametric t-test). Data are representative of two independent experiments.
Leukocyte responses to fungal pathogens require dectin-1
To better understand the in vivo phenotype of dectin-1-deficient mice, we next examined the interaction of dectin-1-knockout leukocytes with C. albicans in vitro (Fig. 6). Although immune recognition of and response to C. albicans is thought to involve many pattern-recognition receptors25,27, we found that recognition of unopsonized yeast by dectin-1-knockout macrophages was much lower than that of dectin-1-wild-type macrophages (Fig. 6a). The lower recognition corresponded to significant impairment in the inflammatory response, as assessed by TNF release (Fig. 6a). Furthermore, the inflammatory response to the yeast was not restored after opsonization of the yeast with serum, despite its conferring normal fungal recognition to the dectin-1-deficient cells (Fig. 6b). Once recognized, dectin-1-knockout macrophages did not show any defect in their ability to kill serum-opsonized or unopsonized yeast (Fig. 6c and data not shown), which was consistent with our observation of a normal respiratory burst response to bound zymosan particles (Fig. 2d) and suggested that fungal recognition and killing occurred through different mechanism(s).
Figure 6.
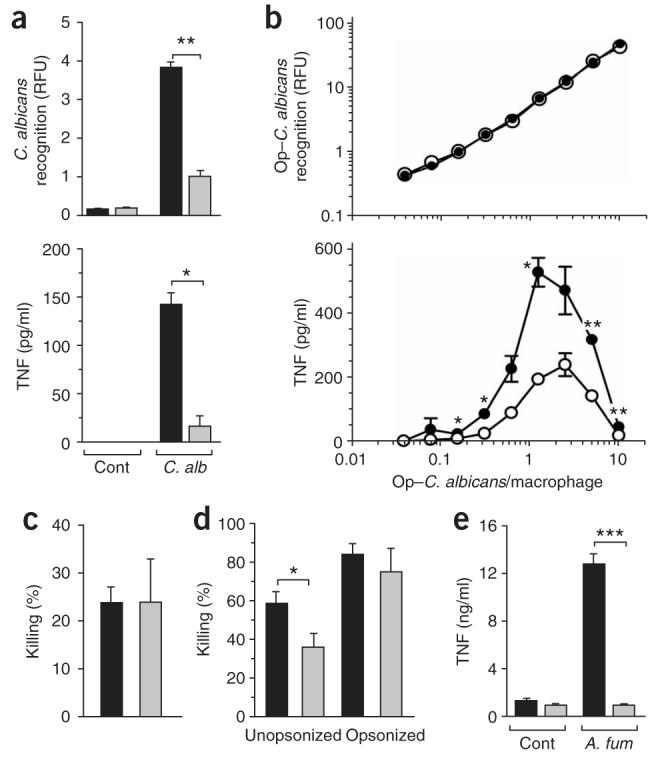
The function of dectin-1 in the recognition and killing of live fungal particles. (a) Fluorimetry (top) of dectin-1-wild-type macrophages (black bars) and dectin-1-knockout macrophages (gray bars) incubated for 30 min at 37 °C with live rhodamine green-X–labeled C. albicans (C. alb; 1.25:1, candida/macrophage ratio), expressed as relative fluorescent units (RFU), and ELISA of TNF production (bottom) after removal of unbound fungi, determined during the next 3 h at 37 °C. Cont, control (medium alone). Data represent mean (± s.e.m.) of replicates from one representative assay of two. (b) Fluorimetry (top) of dectin-1-wild-type macrophages (filled circles) and dectin-1-knockout macrophages (open circles) incubated for 30 min at 37 °C with serum-opsonized C. albicans (Op–C. albicans; yeast/ cell ratio, horizontal axis), and ELISA of TNF production (bottom). Data represent mean (± s.e.m.) of replicates from one representative assay of two. (c,d) Killing assay of dectin-1-wild-type (black bars) and dectin-1-knockout (gray bars) macrophages (c) and neutrophils (d) incubated with opsonized C. albicans (c) or either unopsonized or opsonized C. albicans (d). Data represent mean (± s.e.m.) of replicates from one representative assay of two (c) or from four independent paired data sets (d). (e) ELISA of TNF production by dectin-1-wild-type macrophages (black bars) and dectin-1-knockout macrophages (gray bars) in response to live A. fumigatus (A. fum). Data represent mean (± s.e.m.) from one representative experiment of two. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 (Student's t-test).
Dectin-1-deficient neutrophils, however, had significantly less ability than dectin-1-wild-type neutrophils to kill unopsonized C. albicans in coculture, although killing was restored when the yeast were opsonized with serum (Fig. 6d). In addition to studying the response to C. albicans, we also examined the inflammatory response of elicited macrophages to unopsonized live Aspergillus fumigatus and found that TNF production was also significantly lower in the dectin-1-knockout cells than in dectin-1-wild-type cells (Fig. 6e). Thus, dectin-1 is required for normal leukocyte responses to live fungal particles.
Dectin-1 required for inflammation in candida infection
As our data from systemic infection with C. albicans suggested that the susceptibility of the dectin-1-knockout mice was due to a defect in the early innate response, we further explored how the loss of β-glucan recognition by leukocytes affected inflammation in response to fungi in vivo using a peritoneal infection model. We injected mice intraperitoneally with 1 × 105 C. albicans yeast and assessed inflammatory leukocytes in the peritoneum after 4 h by flow cytometry, as described29. Flow cytometry with the granulocyte and monocyte markers Gr-1 and 7/4 and the macrophage marker F4/80 showed that dectin-1-knockout mice had many fewer recruited cells than did dectin-1-wild-type mice, including Gr-1hi7/4hi neutrophils, Gr-1+7/4hiF4/80+ inflammatory monocytes and Gr-1int7/4loF4/80+ side-scatter-high eosinophils, and also had a reproducible trend of less resident macrophage emigration (Fig. 7a,b). The lower inflammatory cell recruitment was also associated with many defects in the production of specific cytokines and growth factors in the inflammatory lesion, with considerable reproducible defects evident in the production of IL-6, the chemokines CCL2 and CCL3, and granulocyte and granulocyte-monocyte colony-stimulating factors (Fig. 7c). A similar impairment in inflammation was also evident after intraperitoneal injection of mice with zymosan particles but not after injection of thioglycollate broth (Supplementary Fig. 4 online). Thus, dectin-1-knockout mice have an intrinsic defect in their inflammatory response to fungal particles in vivo.
Figure 7.
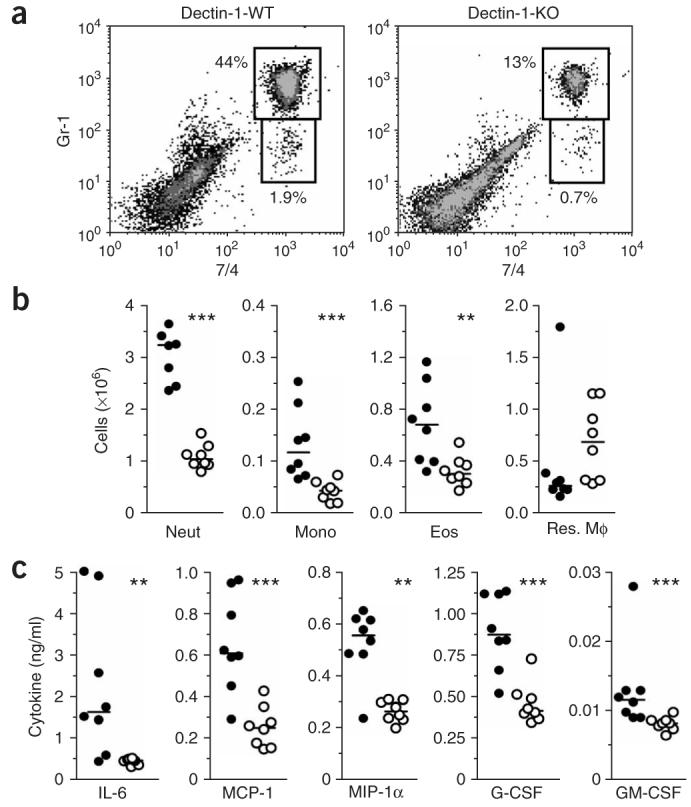
Abnormal antifungal inflammatory response in vivo in the absence of dectin-1. (a) Flow cytometry for Gr-1hi7/4hi neutrophils and Gr-1+7/4hi inflammatory monocytes in dectin-1-wild-type mice (left) and dectin-1-knockout mice (right) after 4 h of intraperitoneal infection with 1 × 105 live C. albicans. Numbers adjacent to outlined areas indicate percent neutrophils (top) and monocytes (bottom). Data are representative of three experiments with seven to nine mice in each. (b) Scatter plots of myeloid cell subsets in the peritoneal cavities of dectin-1-wild-type mice (filled circles ) and dectin-1-knockout mice (open circles) after 4 h of intraperitoneal infection with 1 × 105 live C. albicans. Each symbol represents an individual mouse; horizontal bars, medians. Neut, neutrophil; Mono, monocyte; Eos, eosinophil; Res. Mφ, resident macrophage. Data represent one of three experiments. (c) Bio-Plex assays for cytokines, chemokines and growth factors in lavage fluid from the inflamed peritoneal cavities of dectin-1-wild-type mice (filled circles ) and dectin-1-knockout mice (open circles) after 4 h of intraperitoneal infection with 1 × 105 live C. albicans. Horizontal bars, medians. Only inflammatory mediators with reproducible differences in separate experiments are presented. MCP-1, chemokine CCL2; MIP-1a, chemokine CCL3; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-monocyte colony-stimulating factor. Data are representative of three independent experiments. **, P < 0.01, and ***, P < 0.001 (Mann-Whitney nonparametric t-test).
DISCUSSION
Here we have shown that the recognition of β-glucan by dectin-1 is an essential component of the innate immune response to fungal pathogens. Recognition of those carbohydrates by dectin-1, along with ‘collaborative’ TLR signaling8,14, induces a proinflammatory response that results in the rapid recruitment of inflammatory leukocytes, including neutrophils, and the containment and killing of the fungus. In the absence of dectin-1, defective activation of tissue resident macrophages impeded the subsequent inflammatory response, leading to impaired recruitment of myeloid cells, which is key in controlling the pathogen. That occurred in the context of a deficiency in inflammatory mediators and growth factors, which would normally enhance neutrophil activation and neutrophil-mediated killing30. Ultimately, the absence of dectin-1 and of a normal inflammatory response resulted in considerably enhanced dissemination of the pathogen and increased host susceptibility.
Our study has also emphasized the importance of the non–Toll-like receptors in immune responses. Pattern-recognition receptors such as CD14 (ref. 31) and CD36 (ref. 32) are important in the recognition of other pathogens and their components, although the main function of those receptors seems to be the capture of PAMPs, rendering them accessible to the TLRs. As TLRs seem to recognize fungal PAMPs other than β-glucan, at least on zymosan14, dectin-1 may also serve a similar function in PAMP presentation. However, the inability to restore TLR-mediated inflammatory responses after cellular targeting of fungal particles by serum opsonization indicated that the signaling pathways induced by dectin-1 (refs. 18,23) are essential for those responses. Furthermore, many of the dectin-1-mediated activities, including phagocytosis16, the respiratory burst14,15 and the production of certain cytokines17, are TLR independent. Thus, our data have demonstrated the requirement of a cell surface, signaling, non–Toll-like pattern-recognition receptor for the induction of protective immune responses.
Given the importance of dectin-1 in the recognition of β-glucan, it is relevant to consider the function of CR3, lactosylceramide and scavenger receptors, all of which have been proposed to be β-glucan receptors involved in fungal recognition18. CR3 has also been linked to the β-glucan-mediated enhancement of antitumor monoclonal immunotherapy33 and was initially proposed to be the main β-glucan receptor on myeloid cells. As our data have demonstrated that dectin-1 has a nonredundant function in β-glucan recognition, the function of those other receptors is unclear. It is possible that they are involved in promoting intracellular signaling33,34, and they may also contribute to β-glucan recognition in cell types lacking dectin-1 (ref. 35).
As recognition of β-glucans is central to antifungal immunity, it is not unexpected that fungi may have evolved to avoid immune recognition by hiding these carbohydrates. Although yeast forms of candida and saccharomyces can induce inflammatory responses by means of exposed β-glucans, most of those polysaccharides are concealed by a mannoprotein layer that is thought to prevent excessive immune activation36. In addition, both A. fumigatus11-13 and C. albicans3 seem to completely mask their β-glucans after transition from the yeast to the hyphal form, a morphogenic trait proposed to be linked to virulence37,38. β-Glucans may also be masked by encapsulation, as occurs in Cryptococcus neoformans39, or may be lower in abundance in the cell wall, as occurs with Paracoccidioides brasiliensis after infection of the lung40. Avoidance of β-glucan recognition may therefore be a common immune evasion strategy in fungi, supporting the hypothesis that targeting the unmasking of cell-wall β-glucans may lead to new antimycotic agents36.
In addition to recognizing β-glucans, dectin-1 can recognize an unidentified ligand on lymphocytes and can modulate T cell function6,41,42. Dectin-1 is expressed by macrophages and dendritic cells in the T cell areas of the spleen and lymph nodes43, suggesting involvement in the interactions between antigen-presenting cells and T cells. However, after examining the antibody response to ovalbumin, we found that the dectin-1-deficient mice had normal T cell–dependent immune responses, with no evident T helper type 1–type 2 bias or effect on isotype switching (data not shown). Thus, the function of dectin-1 in the adaptive response, if any, remains to be clarified.
In summary, our studies of dectin-1-deficient mice have emphasized the fundamental function of β-glucan recognition in the development of an antifungal inflammatory response and the restriction of fungal dissemination in vivo. Our studies, therefore, have demonstrated a previously unknown mechanism underlying the recognition and initiation of the protective responses to these pathogens and distinguish dectin-1 as a cell surface non-TLR necessary for antifungal immunity. In addition, given the similarity between the mouse and human systems44, our studies suggest that modulation of dectin-1 (ref. 45) may also represent an alternative approach for the treatment of fungal infections.
METHODS
Generation of dectin-1-deficient mice
The generation of the dectin-1-knockout mice is described in the Supplementary Methods online. Mice were maintained in accordance with institutional guidelines at the University of Oxford, London (Imperial College) and the University of Cape Town. Clec7a+/+ and Clec7a−/− mice were on a mixed 129/Sv × C57BL/6 genetic background unless stated otherwise.
Fungus propagation, labeling and opsonization
C. albicans SC5314 was streaked onto yeast peptone dextrose (YPD) or Sabouraud agar plates for isolation of individual colonies. Colonies were cultured in a shaking incubator for 16 h at 37 °C in 50 ml of YPD broth, for in vitro assays, or at 30 °C in Sabouraud broth for in vivo infections. A. fumigatus isolate 13073 was cultured and used as described11. For labeling with Rhodamine Green-X (Invitrogen), live C. albicans was washed extensively in PBS before being resuspended at a density of 3.2 × 108 yeast per ml. Rhodamine Green-X was added to a final concentration of 200 μg/ml and cells were rotated gently for 30 min at 25 °C. Then, C. albicans was washed extensively with PBS to remove free Rhodamine Green-X before use. For opsonization of yeast particles, mouse blood was collected by cardiac puncture and was immediately placed at 4 °C. Blood cells were removed by centrifugation 10,000g for 5 min at 4 °C and serum was stored in aliquots at −80 °C as soon as possible after blood was collected, to preserve complement activity. Aliquots were defrosted before use and were discarded after use. Yeast particles, typically up to 1 × 108 zymosan or C. albicans, were resuspended in 200 μl RPMI medium with 10% (volume/volume) heat-inactivated FCS and then were mixed for 15–30 min at 37 °C with 200 μl freshly defrosted mouse serum with frequent agitation. Yeast particles were then washed at least four times to remove residual complement and serum proteins before subsequent use.
In vitro fungus-recognition assays
These assays were done essentially as described7,8,21,46-48, with some modifications. For in vitro recognition of zymosan and live fungal particles by macrophages, peritoneal exudate cells were isolated by lavage with ice-cold 5 mM EDTA in PBS from mice that had been treated intraperitoneally 4 d before with thioglycollate broth (Difco). The thioglycollate broth used does not contain yeast extract. Macrophages were plated in 24-well plates at a density of 2.5 × 105 cells per well (for zymosan-recognition assays) or 1 × 106 cells per well (for live C. albicans–recognition assays) in RPMI medium with 10% (volume/volume) heat-inactivated FCS. Cells were washed three times with medium before the addition of yeast particles. Fluorescein isothiocyanate–labeled zymosan (Invitrogen) or Rhodamine Green-X–labeled live C. albicans SC5314 were used in recognition assays at macrophage/particle ratios of 25:1 for zymosan and 10:1 to 0.04:1 for live C. albicans. After incubation for 30 min at 37 °C to allow efficient recognition of the fungal particles by both isoforms of dectin-1 (ref. 47), unbound yeast cells were washed away by four washes with RPMI medium plus 10% (volume/volume) heat-inactivated FCS. The medium was replaced and cells were cultured for 3 h at 37 °C for analysis of production of proinflammatory cytokines or for 20 h at 37 °C for measurement of IL-12p40 and IL-10. After incubation, supernatants were stored at −80 °C until use, cells were lysed in 3% (volume/volume) Triton X-100, pH 7.5, and fluorescence was measured with a Titer-Tek Fluoroskan II (Labsystems). Inflammatory responses to A. fumigatus (multiplicity of infection, 25:1) were measured after 20 h of fungus–macrophage coculture, as described11.
For in vitro recognition of zymosan by neutrophils, peritoneal inflammatory cells were collected with 5 mM EDTA in PBS at 16–18 h after thioglycollate administration. Cells were pretreated for 30 min on ice with 100 μg/ml of β-glucan. Inflammatory cells (5 × 105) were mixed with fluorescein isothiocyanate–labeled zymosan particles (1 × 106 particles; 2:1 zymosan/inflammatory cell ratio) in 100 μl of RPMI medium plus 10% (volume/volume) heat-inactivated FCS in 5-ml polystyrene tubes and were centrifuged at 350g for 5 min at 4 °C. Inflammatory cells and zymosan were then incubated for 1 h at 37 °C. Cellular recognition of zymosan was determined by flow cytometry. Where required, zymosan was opsonized with complement as described above.
For in vitro recognition of zymosan by bone marrow–derived dendritic cells, the cells were incubated with various concentrations of unlabeled zymosan (Sigma) for 20 h at 37 °C, then supernatants were collected and were stored at −80 °C until use for cytokine analysis.
Cytokine production and respiratory burst
Cytokine, chemokine and growth factor concentrations were measured with the Bio-Plex Protein Array System (Bio-Rad) as directed by the manufacturer. TNF, IL-12p40, and IL-10 were also measured by commercial enzyme-linked immunosorbent assay (ELISA; OptEIA cytokine ELISA sets; BD Pharmingen). For analysis of hydrogen peroxide (H2O2) generation, inflammatory cells were ‘loaded’ with dihydrorhodamine 123 at a final concentration of 1 μM. After incubation with zymosan, cells were detached and the conversion of dihydrorhodamine 123 was assessed by flow cytometry and is expressed as mean fluorescent intensity or arbitrary units. Cells ‘loaded’ with dihydrorhodamine 123 but not treated with zymosan were used to assess background H2O2 production.
In vitro C. albicans killing assay
This assay was based on published assays49 with some modifications. Where required, complement opsonisation was done as described above. Thioglycollate-elicited peritoneal macrophages, pooled from three to five mice per group, were plated at a density of 1 × 106 macrophages per well of a 24-well plate on the day before the assay. Cells were washed three times with medium before the addition of live C. albicans at a ratio of 0.4 yeast per macrophage. Cells were allowed to interact for 30 min at 37 °C with C. albicans, then unbound particles were removed by four washes with medium and then cells were returned to the incubator for 4 h to allow fungal killing. Control plates were kept at 4 °C to provide a measure of live fungi in the wells. After incubation, medium was removed and cells were lysed, with scraping, by incubation for 5 min at 25 °C with water at a pH of 11, as described50. Lysis buffer was neutralized with excess PBS and CFU C. albicans was determined by plating on YPD agar and incubation overnight at 37 °C.
Thioglycollate-elicited peritoneal exudate cells were collected from at least two mice per group by peritoneal lavage with ice-cold RPMI medium plus 10% (volume/volume) heat-inactivated FCS at 14 h after the administration of thioglycollate and were pooled. Those cells were used as a source of inflammatory granulocytes (mainly neutrophils) and monocytes. Inflammatory leukocytes (0.6 × 106) were mixed with live C. albicans (0.3 × 104 to 1 × 104) in the wells of a 48-well plate and were kept for 60 min at 4 °C to allow the cells to ‘settle’, before being transferred to an incubator at 37 °C for a further 60 min. Control plates were kept in parallel at 4 °C during that incubation. After incubation, cells were mixed, with scraping, and were plated on YPD agar for counting of viable C. albicans after incubation overnight at 37 °C.
In vivo model of systemic candidiasis
Cultures of C. albicans SC5314 grown for 24 h in Sabouraud broth were washed twice in PBS and were resuspended at the required concentration. Female mice (12–15 weeks old; 129/Sv genetic background) were anaesthetized intraperitoneally (12% (volume/volume) ketamine and 8% (volume/volume) xylazine in PBS) and were weighed before infection. Live C. albicans was administered intravenously in a final volume of 100 μl in PBS. Mice were weighed and monitored daily with an animal welfare scoring sheet. Mice were killed by CO2 asphyxiation after a certain score was achieved, as determined by the animal welfare scoring sheet, or when they had lost 20% of their body weight. Experiments continued for a maximum of 28 d, when all surviving mice were killed and analyzed.
In vivo inflammatory response models
Mice were injected intraperitoneally with 1 × 106 zymosan or 1 × 105 live C. albicans and were killed after 4 h. The inflammatory infiltrate was collected by lavage with ice-cold 5 mM EDTA in PBS. Inflammatory cell populations were counted and then were analyzed by flow cytometry to determine the leukocyte composition as described19,29. Cytokines in peritoneal lavage fluid were determined as described above.
In vivo immune response study
For analysis of antibody responses, blood was obtained from mice on day 0 for preimmune serum, and then mice were immunized intraperitoneally with 50 μg ovalbumin in alum on days 1 and 14. On day 21, blood was again obtained from mice for immune serum. Ovalbumin-specific antibodies in sera from days 0 and 21 were measured by ELISA with isotype-specific antibodies (Southern Biotechnology).
Antibodies
The following antibodies, along with the appropriate isotype controls and streptavidin complexes, were used: 2A11 (antibody to dectin-1 (anti-dectin-1)19,21); fluorescein isothiocyanate–conjugated 7/4 (antibody to neutrophils and monocytes), phycoerythrin-conjugated anti-CD19 (clone 6D5) and biotin-conjugated anti-F4/80 (Serotec); CyChrome-conjugated anti–CD3 molecular complex (clone 17A2), peridinine chlorophyll protein– and cyanin 5.5–conjugated anti-CD11b (clone M1/70), phycoerythrin-conjugated anti-CD11c (clone HL3), fluorescein isothiocyanate–conjugated anti-CD21,CD35 (clone 7G6), phycoerythrin-conjugated anti-CD23 (clone B3B4), biotin-conjugated anti-CD35 (clone 8C12), peridinine chlorophyll protein– and cyanin 5.5–conjugated anti-B220 (clone RA3-6B2), phycoerythrin-conjugated anti-CD49b (clone DX5), phycoerythrin-conjugated anti-Gr-1 (anti-Ly6G/C; clone RB6-8C5) and streptavidin conjugated to fluorescein isothiocyanate, phycoerythrin or allophycocyanin (BD Pharmingen); fluorescein isothiocyanate–conjugated anti-CD3 (clone HM3401), phycoerythrin-conjugated anti-CD4 (clone RM4-5), fluorescein isothiocyanate–conjugated anti-CD8a (clone 5H10) and biotin-conjugated anti-CD25 (clone PC61 5.3; Caltag Laboratories); and streptavidin–AlexaFluor 488 (Invitrogen).
Statistical analysis
One-way analysis of variance (ANOVA) with Bonferroni post-tests was used when multiple groups were analyzed and the two-tailed Student's t-test was used for analysis of two groups, with paired analysis when appropriate. For the analysis of nonparametrically distributed data, the two-tailed Mann-Whitney test was used. Survival data were analyzed with the log-rank test. Results were considered statistically significant with P values of less than 0.05.
ACKNOWLEDGMENTS
We thank the animal facility staff of for care of the animals; and A. Bygrave, P. Norsworthy, L. Fick, M. Tyler, D. Williams, C. Huysamen, L. Graham and C. Maske for help, reagents and technical assistance with the generation and study of the gene-targeted mice and histology. We dedicate this paper to the memory of Albert Beyers. S.G. and G.D.B. share senior authorship. Supported by the Wellcome Trust (055109, 070579, 071467 and 072420; Research Career Development Fellowship, P.R.T.; Senior Fellowship in Biomedical Science in South Africa, G.D.B.), the Cancer Association of South Africa, the Medical Research Council South Africa, the National Institutes of Health (1RO1HL080317) and the Biotechnology and Biological Sciences Research Council (02/B1/P/08210, 60/P17835 and BBS/B/10331).
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Romani L. Immunity to fungal infections. Nat. Rev. Immunol. 2004;4:1–23. doi: 10.1038/nri1255. [DOI] [PubMed] [Google Scholar]
- 2.Calderone RA, Braun PC. Adherence and receptor relationships of Candida albicans. Microbiol. Rev. 1991;55:1–20. doi: 10.1128/mr.55.1.1-20.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005;24:1277–1286. doi: 10.1038/sj.emboj.7600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross GD, Vetvicka V, Yan J, Xia Y, Vetvickova J. Therapeutic intervention with complement and β-glucan in cancer. Immunopharmacology. 1999;42:61–74. doi: 10.1016/s0162-3109(99)00013-2. [DOI] [PubMed] [Google Scholar]
- 5.Tzianabos AO. Polysaccharide immunomodulators as therapeutic agents: structural aspects and biologic function. Clin. Microbiol. Rev. 2000;13:523–533. doi: 10.1128/cmr.13.4.523-533.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ariizumi K, et al. Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J. Biol. Chem. 2000;275:20157–20167. doi: 10.1074/jbc.M909512199. [DOI] [PubMed] [Google Scholar]
- 7.Brown GD, Gordon S. Immune recognition. A new receptor for β-glucans. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 8.Brown GD, et al. Dectin-1 mediates the biological effects of β -glucans. J. Exp. Med. 2003;197:1119–1124. doi: 10.1084/jem.20021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viriyakosol S, Fierer J, Brown GD, Kirkland TN. Innate immunity to the pathogenic fungus Coccidioides posadasii is dependent on Toll-like receptor 2 and Dectin-1. Infect. Immun. 2005;73:1553–1560. doi: 10.1128/IAI.73.3.1553-1560.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steele C, et al. Alveolar macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the Dectin-1 β-glucan receptor. J. Exp. Med. 2003;198:1677–1688. doi: 10.1084/jem.20030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steele C, et al. The β-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. 2005;1:e42. doi: 10.1371/journal.ppat.0010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hohl TM, et al. Aspergillus fumigatus triggers inflammatory responses by stage-specific β-glucan display. PLoS Pathog. 2005;1:e30. doi: 10.1371/journal.ppat.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gersuk GM, Underhill DM, Zhu L, Marr KA. Dectin-1 and TLRs permit macrophages to distinguish between different Aspergillus fumigatus cellular states. J. Immunol. 2006;176:3717–3724. doi: 10.4049/jimmunol.176.6.3717. [DOI] [PubMed] [Google Scholar]
- 14.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood. 2005;106:2543–2550. doi: 10.1182/blood-2005-03-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herre J, et al. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood. 2004;104:4038–4045. doi: 10.1182/blood-2004-03-1140. [DOI] [PubMed] [Google Scholar]
- 17.Rogers NC, et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 2005;22:507–517. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Brown GD. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 2006;6:33–43. doi: 10.1038/nri1745. [DOI] [PubMed] [Google Scholar]
- 19.Taylor PR, et al. The β-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J. Immunol. 2002;169:3876–3882. doi: 10.4049/jimmunol.169.7.3876. [DOI] [PubMed] [Google Scholar]
- 20.Di Carlo FJ, Fiore JV. On the composition of zymosan. Science. 1958;127:756–757. doi: 10.1126/science.127.3301.756-a. [DOI] [PubMed] [Google Scholar]
- 21.Brown GD, et al. Dectin-1 is a major β-glucan receptor on macrophages. J. Exp. Med. 2002;196:407–412. doi: 10.1084/jem.20020470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nathan CF, Root RK. Hydrogen peroxide release from mouse peritoneal macrophages: dependence on sequential activation and triggering. J. Exp. Med. 1977;146:1648–1662. doi: 10.1084/jem.146.6.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gross O, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006;442:651–656. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- 24.Cambi A, et al. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur. J. Immunol. 2003;33:532–538. doi: 10.1002/immu.200310029. [DOI] [PubMed] [Google Scholar]
- 25.Bellocchio S, et al. The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J. Immunol. 2004;172:3059–3069. doi: 10.4049/jimmunol.172.5.3059. [DOI] [PubMed] [Google Scholar]
- 26.MacCallum DM, Odds FC. Temporal events in the intravenous challenge model for experimental Candida albicans infections in female mice. Mycoses. 2005;48:151–161. doi: 10.1111/j.1439-0507.2005.01121.x. [DOI] [PubMed] [Google Scholar]
- 27.Netea MG, et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Invest. 2006;116:1642–1650. doi: 10.1172/JCI27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brieland J, et al. Comparison of pathogenesis and host immune responses to Candida glabrata and Candida albicans in systemically infected immunocompetent mice. Infect. Immun. 2001;69:5046–5055. doi: 10.1128/IAI.69.8.5046-5055.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor PR, Brown GD, Geldhof AB, Martinez-Pomares L, Gordon S. Pattern recognition receptors and differentiation antigens define murine myeloid cell heterogeneity ex vivo. Eur. J. Immunol. 2003;33:2090–2097. doi: 10.1002/eji.200324003. [DOI] [PubMed] [Google Scholar]
- 30.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 31.Beutler B. Tlr4: central component of the sole mammalian LPS sensor. Curr. Opin. Immunol. 2000;12:20–26. doi: 10.1016/s0952-7915(99)00046-1. [DOI] [PubMed] [Google Scholar]
- 32.Hoebe K, et al. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 33.Hong F, et al. Mechanism by which orally administered β-1,3-glucans enhance the tumoricidal activity of antitumor monoclonal antibodies in murine tumor models. J. Immunol. 2004;173:797–806. doi: 10.4049/jimmunol.173.2.797. [DOI] [PubMed] [Google Scholar]
- 34.Rice PJ, et al. Oral delivery and gastrointestinal absorption of soluble glucans stimulate increased resistance to infectious challenge. J. Pharmacol. Exp. Ther. 2005;314:1079–1086. doi: 10.1124/jpet.105.085415. [DOI] [PubMed] [Google Scholar]
- 35.Evans SE, et al. Pneumocystis cell wall β-glucans stimulate alveolar epithelial cell chemokine generation through nuclear factor-κB-dependent mechanisms. Am. J. Respir. Cell Mol. Biol. 2005;32:490–497. doi: 10.1165/rcmb.2004-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wheeler RT, Fink GR. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog. 2006;2:e35. doi: 10.1371/journal.ppat.0020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo HJ, et al. Nonfilamentous C. albicans mutants are avirulent. Cell. 1997;90:939–949. doi: 10.1016/s0092-8674(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 38.Gale CA, et al. Linkage of adhesion, filamentous growth, and virulence in Candida albicans to a single gene, INT1. Science. 1998;279:1355–1358. doi: 10.1126/science.279.5355.1355. [DOI] [PubMed] [Google Scholar]
- 39.Cross CE, Bancroft GJ. Ingestion of acapsular Cryptococcus neoformans occurs via mannose and β-glucan receptors, resulting in cytokine production and increased phagocytosis of the encapsulated form. Infect. Immun. 1995;63:2604–2611. doi: 10.1128/iai.63.7.2604-2611.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borges-Walmsley MI, Chen D, Shu X, Walmsley AR. The pathobiology of Paracoccidioides brasiliensis. Trends Microbiol. 2002;10:80–87. doi: 10.1016/s0966-842x(01)02292-2. [DOI] [PubMed] [Google Scholar]
- 41.Grunebach F, Weck MM, Reichert J, Brossart P. Molecular and functional characterization of human Dectin-1. Exp. Hematol. 2002;30:1309–1315. doi: 10.1016/s0301-472x(02)00928-1. [DOI] [PubMed] [Google Scholar]
- 42.Willment JA, Gordon S, Brown GD. Characterization of the human β-glucan receptor and its alternatively spliced isoforms. J. Biol. Chem. 2001;276:43818–43823. doi: 10.1074/jbc.M107715200. [DOI] [PubMed] [Google Scholar]
- 43.Reid DM, et al. Expression of the β-glucan receptor, Dectin-1, on murine leukocytes in situ correlates with its function in pathogen recognition and reveals potential roles in leukocyte interactions. J. Leukoc. Biol. 2004;76:86–94. doi: 10.1189/jlb.0104031. [DOI] [PubMed] [Google Scholar]
- 44.Willment JA, et al. The human β-glucan receptor is widely expressed and functionally equivalent to murine Dectin-1 on primary cells. Eur. J. Immunol. 2005;35:1539–1547. doi: 10.1002/eji.200425725. [DOI] [PubMed] [Google Scholar]
- 45.Willment JA, et al. Dectin-1 expression and function are enhanced on alternatively activated and GM-CSF-treated macrophages and are negatively regulated by IL-10, dexamethasone, and lipopolysaccharide. J. Immunol. 2003;171:4569–4573. doi: 10.4049/jimmunol.171.9.4569. [DOI] [PubMed] [Google Scholar]
- 46.Taylor PR, et al. The role of SIGNR1 and the β-glucan receptor (dectin-1) in the nonopsonic recognition of yeast by specific macrophages. J. Immunol. 2004;172:1157–1162. doi: 10.4049/jimmunol.172.2.1157. [DOI] [PubMed] [Google Scholar]
- 47.Heinsbroek SEM, et al. Expression of functionally different dectin-1 isoforms by murine macrophages. J. Immunol. 2006;176:5513–5518. doi: 10.4049/jimmunol.176.9.5513. [DOI] [PubMed] [Google Scholar]
- 48.Lutz MB, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 49.Vonk AG, Wieland CW, Netea MG, Kullberg BJ. Phagocytosis and intracellular killing of Candida albicans blastoconidia by neutrophils and macrophages: a comparison of different microbiological test systems. J. Microbiol. Methods. 2002;49:55–62. doi: 10.1016/s0167-7012(01)00348-7. [DOI] [PubMed] [Google Scholar]
- 50.Decleva E, Menegazzi R, Busetto S, Patriarca P, Dri P. Common methodology is inadequate for studies on the microbicidal activity of neutrophils. J. Leukoc. Biol. 2006;79:87–94. doi: 10.1189/jlb.0605338. [DOI] [PubMed] [Google Scholar]