

Abstract
Vascular endothelial growth factor (VEGF), a key regulator of vasculogenesis and embryonic angiogenesis, was recently found to be up-regulated in an animal model of stroke. Unlike VEGF, angiopoietin (Ang)-1 and -2, their receptor tie-2, and the associated receptor tie-1 exert their functions at later stages of vascular development, ie, during vascular remodeling and maturation. To assess the role of the angiopoietin/tie family in ischemia-triggered angiogenesis we analyzed their temporal and spatial expression pattern after middle cerebral artery occlusion (MCAO) using in situ hybridization and immunohistochemistry. Ang-1 mRNA was constitutively expressed in a subset of glial and neuronal cells with no apparent change in expression after MCAO. Ang-2 mRNA was up-regulated 6 hours after MCAO and was mainly observed in endothelial cell (EC) cord tips in the peri-infarct and infarct area. Up-regulation of both Ang-2 and VEGF coincided with EC proliferation. Interestingly, EC proliferation was preceded by a transient period of EC apoptosis, correlating with a change in VEGF/Ang-2 balance. Our observation of specific stages of vascular regression and growth after MCAO are in agreement with recent findings suggesting a dual role of Ang-2 in blood vessel formation, depending on the availability of VEGF.
Experimental models and studies on stroke patients have shown that hypoxia/ischemia induces vascular proliferation in the peri-infarct area. 1-4 Hypoxia/ischemia-induced angiogenesis is a tightly controlled multistep process by which new blood vessels are formed by sprouting from the pre-existing vasculature. 5 The induction of angiogenesis after cerebral ischemia could be interpreted as a natural defense mechanism helping to restore oxygen and nutrient supply in the respective tissue. Regulators of angiogenesis include at least two subfamilies of endothelial cell (EC)-specific transmembrane receptor tyrosine kinases and their ligands. Vascular endothelial growth factor (VEGF) and its receptors are central mediators of vasculogenesis and angiogenesis 6-9 and have been shown to be up-regulated after middle cerebral artery occlusion (MCAO) in rats. 3,4,10-13
Unlike VEGF, the angiopoietins (Ang)-1 and -2 and the receptor tyrosine kinases tie-1 and tie-2 exert their functions at later stages of vascular development, ie, during vascular remodeling and maturation. Tie-2 has at least four known ligands, Ang-1, Ang-2, and the yet less characterized Ang-3 and Ang-4. 14-19 Ligands for the tie-1 receptor have not been found up to date. Tie-1 and tie-2 mRNA are expressed in the vasculature of the developing brain but are down-regulated in the adult organism. 20-22 Knockout studies of the tie-1 or tie-2 gene in mice have shown that both receptors are essential for normal vascular development. Tie-1 and tie-2 deficient mice die at later times than VEGF receptor null mice, exhibiting phenotypical alterations consistent with a role in vascular remodeling. 21,23,24 Ang-1 and Ang-2 are secreted glycoproteins that share ∼60% amino acid identity and bind with similar affinity to tie-2. Although Ang-1 induces autophosphorylation of tie-2, Ang-2 is a naturally occurring antagonist that blocks Ang-1-induced tie-2 autophosphorylation. 16 Targeted gene inactivation of Ang-1 in mice reveals lethal embryonic defects similar to those seen in tie-2 knockout mice; namely a poorly organized subendothelial matrix, loosening of EC contacts to the basement membrane, and generalized lack of perivascular cells. 16 These findings suggest that Ang-1 is required for correct vascular assembly by recruiting and sustaining peri-endothelial cells. Consistent with its role as a tie-2 inhibitor, overexpression of Ang-2 results in lethal embryonic vascular defects reminiscent of those seen in Ang-1 or tie-2 knockout mice. Ang-2 expression occurs almost exclusively at sites of vascular remodeling processes and is highest at the leading edge of invading vascular sprouts. 16 It is hypothesized that Ang-2 blocks the stabilization or maturation function of Ang-1. Loosening of EC/pericyte contacts may thus allow vessels to convert into a plastic state. Acting in concert with VEGF, Ang-2 may initiate and stimulate angiogenesis in mature vessels. 16,25 However, other reports imply that in the absence of VEGF Ang-2 induces vessel regression. 26-28
The purpose of the present study was to investigate the participation of the angiopoietin/tie system in cerebral ischemia-induced angiogenesis, using the MCAO model. Ang-2, tie-1, and tie-2 were specifically up-regulated in the peri-ischemic vasculature. Surprisingly, before peri-infarct vessels started to proliferate a transient period of vessel regression occurred. Taken together these observations are highly suggestive of a VEGF-dependent dual role of Ang-2, namely promoting vessel regression or vessel proliferation.
Materials and Methods
MCAO Model
All experiments were performed in male Fisher 344 rats (Iffa Credo, L’Abresle, France) weighing 220 to 300 g. Animals were housed under standard conditions with free access to rat chow and tap water before and after surgery.
Irreversible occlusion of the right middle cerebral artery was performed as described previously. 3,29,30 Briefly, animals were anesthetized with 2% isoflurane in a 70/30 (by volume) nitrous/oxygen mixture and, using an operating microscope, the right middle cerebral artery was exposed by subtemporal craniectomy. The artery and its lenticulostriate branches were then occluded by bipolar electrocoagulation. Sham-operated control animals were prepared in similar manner, except that the exposed middle cerebral artery was not occluded. Afterward, retracted soft tissues were replaced, wounds were sutured, anesthesia was discontinued, and the rats were put back into their cages. Body temperature was maintained at 37°C by means of a rectal probe connected to a heating pad (CMA 150; Carnegie Medicine, Stockholm, Sweden) during surgery and until animals regained consciousness. Thereafter, rectal temperature was checked frequently (every 10 to 15 minutes) during the following 2 hours and, if necessary, it was corrected to 37°C by placing a heating pad under the cage. After MCAO the animals survived for 3 hours, 6 hours, 12 hours, 24 hours, 3 days, and 7 days (n = 4 to 6 per time). Thereafter the animals were decapitated under deep anesthesia. Brains were removed within 5 minutes after decapitation, frozen in OCT embedding medium (Sakura Finetec, Torrance, CA) on dry ice, and stored at −80°C until further processing. Sham-operated animals (at 3 hours and 24 hours) served as controls in all experiments mentioned.
Magnetic Resonance Imaging
To verify successful MCAO and to visualize the lesioned territory all brains were characterized by nuclear magnetic resonance imaging before brain removal as previously described. 3 Briefly, in animals surviving 24 hours or longer, infarct volume was determined by means of T2-weighted quantitative in vivo magnetic resonance imaging. 31 The rat was anesthetized with 1% to 1.5% isoflurane delivered via a face mask and positioned with its head in the resonator. Each animal was then subjected to one imaging cycle in which 13 contiguous T2-weighted coronal slices of the brain (1.2 mm thick) were taken using a RARE sequence (optimized parameters: repetition time, 3,000 ms; effective echo time, 66 ms; spatial resolution in plane, 156 μm2). The total measuring time was 5 minutes.
At early times (up to 12 hours) after MCAO, the ischemic lesion was visualized in diffusion-weighted magnetic resonance images. Eleven 1.2-mm-thick contiguous coronal slices were acquired in 5 minutes (TR = 1,500 ms; TE = 37.2 ms, two averages) using a multislice Stejskal-Tanner-like spin echo sequence. 32 The diffusion gradient was applied along the left-right axis of the animal, choosing a nominal b value of 1,500 s/mm2. The in-plane spatial resolution was 312 μm2.
In Situ Hybridization
Ten-μm-thick frozen sections were melted on silanized (3-aminopropyltriethoxysilane; Fluka, Buchs, Switzerland) glass slides, dried at 50°C, and fixed for 15 minutes in 4% paraformaldehyde/phosphate-buffered saline (PBS) followed by dehydration through ethanol (30%, 60%, 80%, 95%, and 100% ethanol, 5 minutes each). Slides were incubated in 0.2 mol/L HCl for 10 minutes at room temperature followed by digestion with Proteinase K (10 μg/ml) (Sigma Chemical Co., Deisenhofen, Germany) for 10 minutes at room temperature and acetylation with 0.1 mol/L triethanolamine (Sigma Chemical Co.) mixed with 0.25% acetic anhydride (Fluka) for 10 minutes at room temperature. Sections were then prehybridized in 4× standard saline citrate (SSC), 0.02% sodium dodecyl sulfate, 5× Denhardt’s solution, 50% ultrapure formamide (Life Technologies, Inc., Karlsruhe, Germany), 5% dextran sulfate (Sigma Chemical Co.), and 0.5 mg/ml yeast tRNA (Sigma Chemical Co.) for 5 hours at room temperature. Hybridization was performed with a digoxigenin-labeled (Boehringer Mannheim, Mannheim, Germany) cRNA generated by in vitro transcription using the following cDNA templates: a 560-bp NotI-EcoRI Ang-1 cDNA fragment, a 640-bp EcoRI-XhoI Ang-2 cDNA fragment, a 560-bp SacII-NotI tie-2 cDNA fragment encoding part of the murine tie-2 extracellular domain, and a 600-bp XbaI-XhoI tie-1 cDNA encoding part of the murine tie-1 extracellular domain. Labeled cRNA probes were used at a concentration of 0.5 ng RNA/μl. Hybridization with sense probe served as control. Tissue sections were incubated in a humidified chamber under glass coverslips at 70°C (hybridization oven, Biometra, Göttingen, Germany) for 16 hours to 18 hours. Posthybridization stringency washes included 0.2×SSC for 30 minutes at 70°C, 2×SSC for 2 minutes at room temperature, 0.2×SSC for 15 minutes at 70°C, and 2×SSC for 5 minutes at room temperature. Each wash was performed twice. Hybridized probes were detected by an anti-digoxigenin antibody conjugated to alkaline phosphatase (diluted 1:500, 1 hour at room temperature; Boehringer Mannheim) using nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate solution as substrate (Boehringer Mannheim). Color reaction time ranged from 2 hours to 2 days, after which slides were rinsed in PBS, counterstained with hematoxylin or methyl green, finally rinsed in aqua dest, and mounted in elvanol.
Immunohistochemistry
Immunohistochemistry was performed as described previously. 3 The following antibodies were used: rabbit polyclonal anti-human VEGF-A20 (0.5 μg/ml; Santa Cruz, Heidelberg, Germany), mouse monoclonal anti-rat CD11b (20 μg/ml; Serotec, Oxford, UK), mouse monoclonal anti-rat ED1 (Serotec), polyclonal anti-Ibal antibody (14.6 μg/ml, gift of Dr. Yoshinori Imai, Tokyo, Japan), monoclonal anti-glial fibrillary acid protein (GFAP) (5 μg/ml; Boehringer), rabbit polyclonal anti-human von Willebrand Factor (29 μg/ml; DAKO, Glostrup, Denmark), mouse monoclonal anti-human tie-2 (10 μg/ml; gift of Dr Kevin Peters, Durham, NC), mouse monoclonal anti-human tie-1 (15 μg/ml; gift of Dr. Kari Alitalo, University of Helsinki, Helsinki, Finland), rabbit polyclonal anti-mouse Ki67 (Dianova, Hamburg, Germany) and mouse monoclonal anti-rat CD31 (6 μg/ml; Serotec). In all immunohistochemical experiments omission of the primary antibody served as control. According to the primary antibody the following secondary antibodies were used: biotinylated rat anti-mouse, rabbit anti-mouse, or goat anti-rabbit antibodies (diluted 1:300 in 20% goat serum/PBS/0.1% Triton; Dianova), respectively. Sections stained for tie-1 were additionally incubated with biotinylated goat anti-rabbit sandwich antibody for 1 hour. Thereafter, slides were washed and incubated with peroxidase-conjugated streptavidin (Vectastain Kit ABC; Vector Laboratories, Burlingame, CA) for 1 hour at room temperature and then rinsed in PBS/0.1%Triton. The immunoperoxidase reaction was visualized with 3,3′-diaminobenzidine-HCl buffer tablets (Sigma Chemical Co.) or 3-amino-9-ethylcarbazole (Vector Laboratories) and 0.006% H2O2. The slides were briefly counterstained with hematoxylin or methyl green, finally rinsed in aqua dest, and mounted in elvanol.
Double-Labeling Experiments
Combined In Situ Hybridization/Immunohistochemistry
In situ hybridization was performed as described above, using the murine Ang-2 cRNA probe. Color reaction time ranged from 6 hours to 16 hours, after which slides were rinsed in PBS and overlaid for 30 minutes with PBS containing 5% bovine serum (Fraction 5; Sigma Chemical Co.) and then for 1 hour with PBS containing 20% normal goat serum to block nonspecific binding. Immunohistochemistry was performed essentially as described above, using the following antibodies: polyclonal anti-Ibal antibody, monoclonal anti-CD-11b, monoclonal anti-GFAP antibody, polyclonal anti-von Willebrand Factor (29 μg/ml; DAKO), and biotin-labeled lectin from lycopersicon esculentum (10 μg/ml; Sigma Chemical Co.). After six washes in PBS/0.1% Triton, slides were incubated for 1 hour at room temperature with biotinylated rat anti-mouse or goat anti-rabbit (dilution 1:200 and 1:500 in PBS containing 10% normal goat serum, respectively; Dianova) secondary antibody. Slides were rinsed three times in PBS/0.1% Triton and then incubated for 1 hour at room temperature with peroxidase-conjugated streptavidin (Vectastain KIT ABC; Vector Laboratories). Thereafter, slides were rinsed four times with PBS/0.1%Triton and then incubated with 3-amino-9-ethylcarbazole (Vector Laboratories) and 0.006% H2O2. Color developed within 20 minutes. Sections were rinsed in aqua dest, counterstained with hematoxylin, and mounted in elvanol.
Double-Immunofluorescence Labeling
To show proliferation of blood vessels and capillaries around the infarct zone double-immunofluorescence labeling was performed with rabbit polyclonal anti-mouse Ki-67 (Dianova) and mouse monoclonal anti-rat CD31 (6 μg/ml; Serotec).
Briefly, cryosections were washed thoroughly in PBS before and after each incubation. Nonspecific binding sites were blocked by incubation in 20% normal goat serum and 5% bovine serum albumin/PBS. Goat anti-rabbit rhodamine red-X-conjugated IgG (7.5 μg/ml; Jackson Immunoresearch Laboratories, West Grove, PA) and sheep anti-mouse Cy2-conjugated IgG (7.5 μg/ml; Jackson Immunoresearch Laboratories) were used as secondary antibodies. Evaluation of double staining was performed using a fluorescence microscope.
EC Apoptosis
After performing the terminal dUTP nick-end labeling (TUNEL) assay (Intergen, Purchase, NY) on cryosections according to the manufactures instructions, ECs were labeled by incubation with biotin-labeled lectin from lycopersicon esculentum (10 μg/ml; Sigma Chemical Co.). After three rinses in PBS/0.1% Triton and a 1 hour incubation with peroxidase-conjugated streptavidin (Vectastain KIT ABC; Vector Laboratories) at room temperature, binding was visualized by staining with 3-amino-9-ethylcarbazole (Vector Laboratories) and 0.006% H2O2. Sections were rinsed in aqua dest, counterstained with hematoxylin, and mounted in elvanol.
Semiquantitative Analysis (Table 1) ▶
Table 1.
Semiquantitative Analysis at Different Times after MCAO
| 3 hours | 6 hours | 12 hours | 24 hours | 3 days | 7 days | 24 hours sham | |
|---|---|---|---|---|---|---|---|
| Ang-2 mRNA | − | + | +++ | ++ | ++ | + | − |
| Ang-2 mRNA contralateral hemisphere | − | − | + | + | − | − | − |
| Tie-1 mRNA | − | − | − | − | ++ | − | − |
| Tie-1 protein | − | − | − | − | +++ | − | − |
| Tie-2 mRNA | − | − | − | − | ++ | − | − |
| Tie-2 protein | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| Apoptosis in ECs | − | − | ++ | − | − | − | − |
| Apoptosis in ECs contralateral hemisphere | − | − | + | − | − | − | − |
| Proliferating ECs | − | − | − | + | ++ | + | − |
| Ang-1 mRNA | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
−, no staining; +, 1 to 3 cells; ++, 4 to 9 cells; +++, >9 cells.
For semiquantitative analysis two to four randomly chosen high magnifications fields (×200) in the peri-infarct and the corresponding area in the contralateral hemisphere at different times after MCAO were assessed. Areas analyzed are shown in Figure 1 ▶ .
Figure 1.

H&E-stained rat brain 24 hours after MCAO. I, infarct area; P, peri-infarct area. For analysis, corresponding areas were taken from the ipsilateral and contralateral hemispheres relative to the side of MCAO. Scale bar, 2 mm.
Ang-2 mRNA, tie-2 mRNA, and protein-expressing blood vessels, as well as proliferating and apoptotic vessels were counted. In contrast, for tie-1 mRNA and protein analysis only vessels with increased staining levels compared to vessels in the contralateral hemisphere were taken for analysis. For Table 1 ▶ stained blood vessels were classified as follows: −, no staining; +, 1 to 3 blood vessels; ++, 4 to 9 blood vessels; +++, >9 blood vessels.
Ang-1 mRNA expression in astrocytes and neurons was assessed in a similar manner using the following classification: −, no staining, +, 1 to 3 cells; ++, 4 to 9 cells; +++, >9 cells.
Results
Expression of Ang-1 mRNA and Ang-2 mRNA
Ang-1 mRNA was constitutively expressed in astrocytes and in cerebellar Purkinje cells, whereas other neurons showed little expression. Blood vessels did not express Ang-1. No alteration of Ang-1 mRNA expression was observed after occlusion of the middle cerebral artery (Table 1) ▶ .
Ang-2 mRNA signal was first detected in single cells in the infarct area 6 hours after MCAO and reached a maximum 12 to 24 hours after MCAO. Expression was confined to single cells localized in the infarct area, in the peri-infarct zone and, although in fewer cells, in the corresponding areas of the contralateral hemisphere (Figure 2 ▶ , expressing cells marked by arrows). Both the ipsilateral and contralateral hippocampal formation showed induction of Ang-2 mRNA as well. The majority of Ang-2 mRNA-expressing cells seemed to be ECs. No Ang-2 transcripts were found in sham-operated animals.
Figure 2.
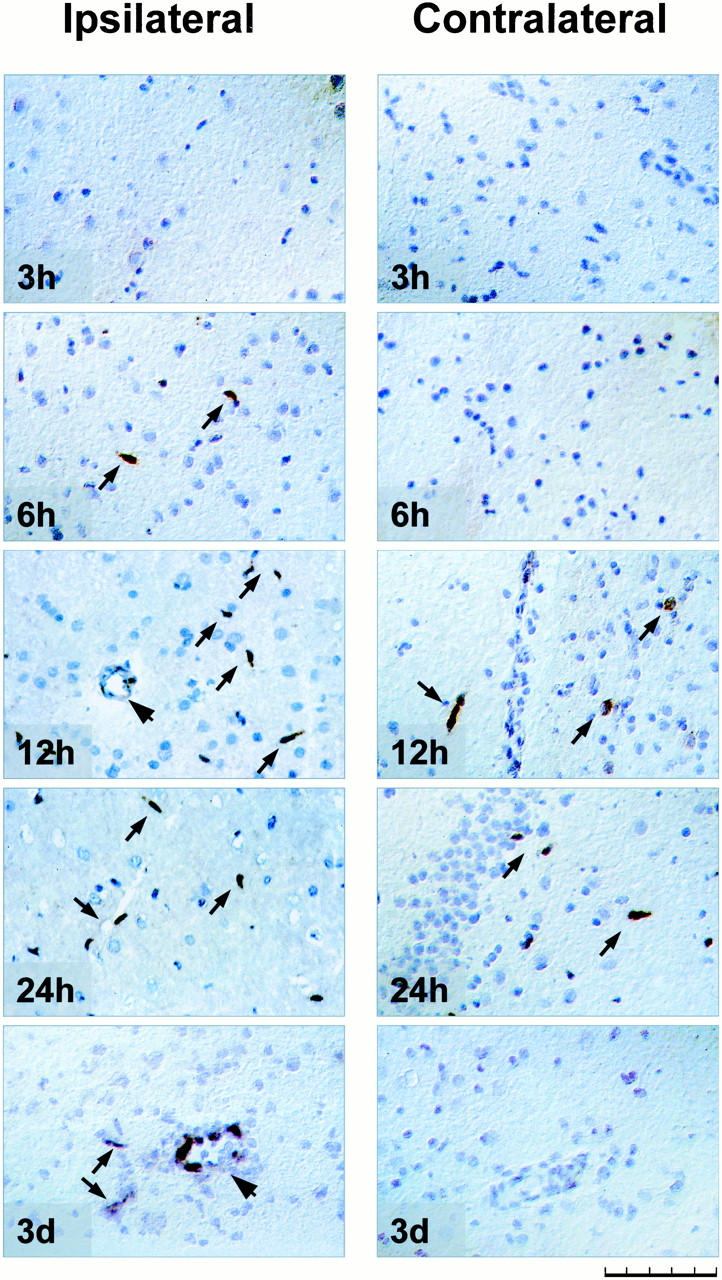
In situ hybridization for Ang-2 mRNA in rat brain subjected to MCAO. Figure ▶ shows ipsilateral (left) peri-infarct cortex and corresponding area in the contralateral (right) hemisphere relative to the side of MCAO, except for 24-hour contralateral, where the hippocampus is shown. Time indicates hours after permanent MCAO. Note up-regulation of Ang-2 mRNA in the contralateral hemisphere 12 and 24 hours after MCAO in single cells (arrows), low-expressing large vessel (arrowhead) at 12 hours, and marked Ang-2 induction at 3 days in a large vessel (arrowhead) as well as smaller vessels (arrows). Scale bar, 100 μm.
Three days after MCAO the Ang-2 mRNA expression pattern considerably changed. Ang-2 mRNA was expressed at high levels in small and larger vessels around the infarct area (marked by arrows and arrowhead, respectively, in Figure 2 ▶ ; compare low-expressing large vessel at 12 hours MCAO marked by arrowhead). However, in contrast to Ang-2 mRNA induction in single cells during the first 24 hours, transcripts were now detected in all ECs of an expressing vessel. Yet, Ang-2 mRNA in the contralateral hemisphere had totally disappeared. Seven days after MCAO Ang-2 mRNA expression had almost returned to baseline levels. Results are summarized in Table 1 ▶ and are illustrated in Figure 2 ▶ .
Characterization of Ang-2 mRNA-Expressing Cell Types
To characterize the cell types expressing Ang-2 mRNA, double-labeling experiments combining in situ hybridization and immunohistochemistry were performed. Anti-GFAP, anti-αIbaI, and anti-vWF antibodies were used for detection of astrocytes, macrophages/microglial cells, and ECs, respectively.
Most Ang-2 mRNA-expressing cells also expressed vWF and were thus identified as ECs (Figure 3a) ▶ . Ang-2 mRNA-expressing cells were mainly located at the tip of an EC cord, although after 3 days of MCAO larger vessels around the infarct area also expressed high levels of Ang-2 mRNA. In addition, GFAP immunohistochemistry detected Ang-2 mRNA up-regulation in astrocytes in the peri-infarct area 24 hours after MCAO (Figure 3b) ▶ . No co-localization of Ang-2 mRNA and IbaI protein was found at any time of investigation (not shown), suggesting that microglial cells/macrophages did not express Ang-2 mRNA.
Figure 3.

Characterization of Ang-2 mRNA-expressing cell types. Double labeling for Ang-2 mRNA (shown in black) and vWF (a) or GFAP immunostaining (b) in the peri-infarct area 24 hours after MCAO. Immunoreactivity for the proteins is indicated by brown color. a: Ang-2 mRNA-expressing blood vessel. b: Ang-2 mRNA-expressing astrocyte. Scale bar, 20 μm.
Expression of Tie-1 mRNA and Immunolocalization of Tie-1 Protein
During the first 24 hours after MCAO tie-1 mRNA and protein were expressed at low levels (not shown). A marked up-regulation of both tie-1 mRNA and protein was observed 3 days after MCAO. Labeling was confined to small and larger vessels in the peri-infarct zone. Seven days after MCAO tie-1 mRNA and protein expression returned to baseline levels. Results are summarized in Table 1 ▶ and illustrated in Figure 4 ▶ .
Figure 4.

Tie-1 immunostaining at 3 days MCAO of the ipsilateral peri-infarct (left) area and the corresponding area in the contralateral cortex (right); note the up-regulation of tie-1 protein in peri-infarct vessels. Scale bar, 100 μm.
Expression of Tie-2 mRNA and Immunolocalization of Tie-2 Protein
Tie-2 mRNA was first detected 3 days after MCAO in vessels immediately adjacent to the infarct area. At 7 days after MCAO tie-2 mRNA levels had declined and staining was no longer seen. In contrast, tie-2 protein was broadly expressed in the endothelium of the quiescent vasculature of the rat brain. Interestingly, no ischemia-induced up-regulation could be observed at any time. Results are summarized in Table 1 ▶ and are illustrated in Figure 5 ▶ .
Figure 5.
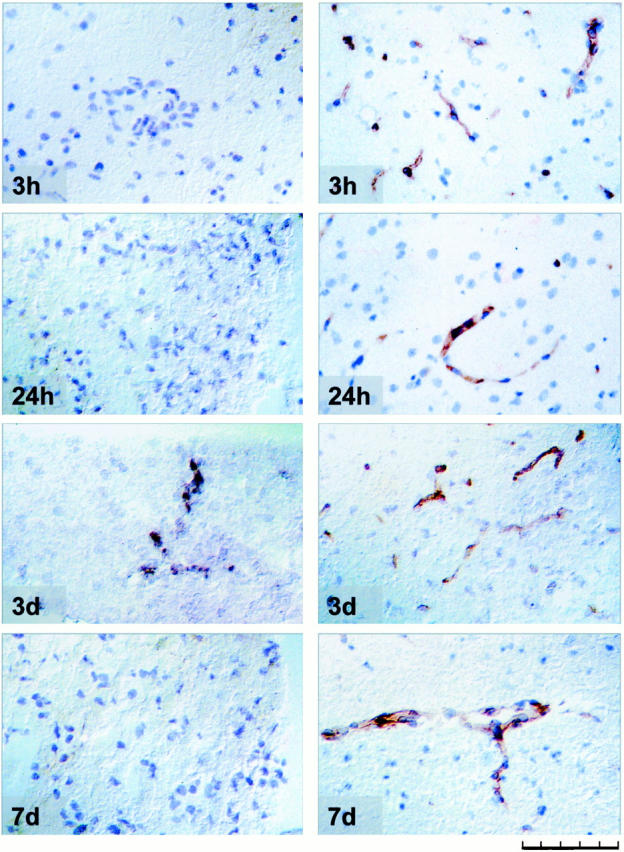
Tie-2 in situ hybridization and immunolocalization following different times after MCAO. Left: Tie-2 mRNA expression. Right: Immunolocalization of tie-2 protein in vessels in the peri-infarct cortical area. Scale bar, 100 μm.
Vascular Remodeling, EC Apoptosis, and Proliferation
It is suggested that Ang-2 has a dual role in vascular remodeling. It may mediate vessel regression or vascular proliferation depending on the absence or presence of VEGF. Our interest was to determine whether the spatial and temporal expression pattern of Ang-2 and VEGF would suggest a similar mechanism in MCAO-induced angiogenesis. Apoptotic (regressing) ECs were detected by combining the TUNEL assay with lectin staining. In sham-operated control animals and in 3- and 6-hour MCAO animals no apoptotic ECs were detectable. Twelve hours after MCAO the first apoptotic ECs appeared in the ischemic area and in the peri-ischemic zone (Figure 6a) ▶ . Notably, single apoptotic cells were also located in the contralateral hemisphere (insert in 6a). All cells undergoing apoptosis in the contralateral hemisphere were invariably identified as ECs. By 24 hours the number of apoptotic cells including ECs in the center of the ischemic lesion had considerably increased, however no apoptotic ECs in the peri-infarct area or the contralateral hemisphere were detectable at that time (Figure 6b) ▶ . Three and 7 days after MCAO no apoptotic ECs could be identified in the peri-infarct zone or in the contralateral hemisphere.
Figure 6.

Apoptosis versus proliferation in ECs in the peri-infarct area. TUNEL staining combined with lectin staining 12 hours (a) and 24 hours (b) after MCAO. Dotted line in a and b demarcates the infarct area. Arrow in a indicates an apoptotic vessel in the peri-infarct area, insert in a shows a high magnification view of an apoptotic vessel in the contralateral hemisphere at 12 hours. Double immunofluorescence for CD31 (green color) and Ki67 (red color) at 24 hours (c) and 3 days MCAO (d). Arrows in c and d indicate proliferating vessels. Note large proliferating blood vessel in d, insert in d depicts small proliferating vessels. Scale bar, 100 μm (a and b), 50 μm (c and d, insert in a and d).
To identify proliferating vessels, we performed double immunohistochemistry using anti-CD31 and anti-Ki67 an tibodies. Proliferating ECs were only detectable in the peri-infarct area. Co-expression started 24 hours after MCAO, reached a maximum at 3 days MCAO, and decreased thereafter (Figure 6, c and d) ▶ . At 24 hours proliferation was confined to small seized vessels, at 3 days also larger vessels showed proliferating ECs. Results are summarized in Table 1 ▶ .
Discussion
We and others have recently described the up-regulation of VEGF, a key regulator of angiogenesis, in an animal model of stroke. 3,4,10-13 VEGF expression was accompanied by an increase in vessel density in the peri-infarct area. 3 Several reports suggest a crucial role of the angiopoietin/tie system in governing the late stages of vascularization in different angiogenic settings, eg, in embryonic development, 21,33 tumor growth, 28,34 ovarian follicle growth, 26 and wound healing. 35 This prompted us to investigate whether ischemic triggered angiogenesis is guided by the same mechanisms. In this report we analyzed the temporal and spatial expression pattern of the angiopoietin/tie family after MCAO in the rat.
Expression of Angiopoietins
Ang-1 is hypothesized to be required for the stabilization of peri-endothelial contacts with surrounding smooth muscle cells in mature vessels. 36 In our study, Ang-1 mRNA was constitutively expressed in a subset of glial and neuronal cells throughout the cortex and cerebellum as previously described. 34 Corresponding with reports showing that hypoxia fails to up-regulate Ang-1 mRNA 37-39 we observed no change in its expression after MCAO.
Ang-2 is thought to play a role at sites of vascular remodeling in the adult by blocking the constitutive expression of Ang-1 and allowing the vessel to revert to a more plastic and unstable state. 36 Our finding of an early Ang-2 mRNA up-regulation in the tips of EC cords is reminiscent of the Ang-2 induction at the invading front of vascular sprouts in the developing corpus luteum. 16
Ang-2 mRNA up-regulation may be because of various mechanisms. Hypoxia and VEGF have been reported to increase Ang-2 mRNA in ECs in vitro as early as 2 hours with VEGF acting in a time- and concentration-dependent manner. 39 After MCAO, VEGF mRNA is up-regulated as early as 3 hours with a peak at 12 hours after MCAO. VEGF protein is first detected by 6 hours reaching a maximum in protein expression at 24 hours. 3 Ang-2 induction by VEGF would be expected to occur later than 6 hours, because it would require diffusion to EC, binding to VEGF receptor, and cell signaling. Thus, it seems likely that during the first hours of ischemia induction of Ang-2 is mainly hypoxia-induced, whereas VEGF-induced up-regulation might be involved at later times. The early induction of Ang-2 by hypoxia would render ECs more accessible to angiogenic inducers such as VEGF by loosening the EC/pericyte cell contact. The observed Ang-2 expression in astrocytes, a component of the blood-brain barrier, may contribute to this process. This would lead to further Ang-2 up-regulation, successive vessel outgrowth, and vascular proliferation.
An unexpected observation was the increase of Ang-2 transcripts in ECs in both hippocampal formations. Interestingly, the hippocampal formation is known to be vulnerable to hypoxic and ischemic events. 40 Neither VEGF up-regulation nor reductions in cerebral blood flow 41 have been described in this area. It is tempting to speculate that the secretion of a yet unidentified factor, likely transmitted by neuronal signaling, contributes directly or indirectly to Ang-2 mRNA up-regulation in ECs. The presence of such a factor might also help to explain Ang-2 up-regulation as well as the reported VEGF induction and microglia activation in the contralateral hemisphere. 3,12
Potential Role of Ang-2 in Vascular Remodeling after MCAO
It is suggested that vessel destabilization by Ang-2 in the presence of high VEGF levels primes the vessel to mount a robust angiogenic response state. 36 In our model, double immunofluorescence with the EC marker CD31 and proliferation marker Ki67 was used to identify proliferating ECs. Vessel growth started at 24 hours after MCAO with a maximum induction at 3 days MCAO. Interestingly, Ang-2 expression pattern and vessel proliferation seemed to correlate, eg, both were seen in small vessels at 24 hours and in small and large blood vessels at 3 days after MCAO. These results suggest that Ang-2 up-regulation is an important component and prerequisite for the induction of vascular proliferation. Neither in the contralateral hemisphere nor in the hippocampal formation was vessel proliferation or an increase in vessel density detected. Possibly, VEGF expression in these areas is not sufficient to induce vascular proliferation.
Vessel destabilization mediated by Ang-2 in the absence of VEGF is proposed to lead to frank vessel regression as seen in atretic follicles 26 or in tumor formation. 27,28 We supposed that Ang-2 mRNA-expressing EC cord tips might undergo regression at early times in the peri-infarct area or in the contralateral hemisphere, when VEGF protein expression has not yet reached its maximum. To investigate apoptotic EC death after MCAO we combined the TUNEL assay with a vessel-specific lectin labeling. The first apoptotic cells were seen in the peri-ischemic zone in 6-hour MCAO animals. Apoptotic ECs could first be detected 12 hours after MCAO, mainly located in the ischemic area and in the peri-ischemic zone. Notably, single apoptotic ECs were also located in the contralateral hemisphere. At 24 hours MCAO, the time showing maximal VEGF expression, no apoptotic vessels around the infarct zone or in the contralateral hemisphere were detectable any longer. In contrast, total apoptotic cell number in the infarct area was increased.
For technical reasons, we could not investigate induction of Ang-2 and apoptosis within the same sections. However, Ang-2 mRNA-expressing ECs and apoptotic ECs were localized in the same regions, namely the peri-infarct area and the contralateral hemisphere. In addition, ECs undergoing apoptosis and expressing Ang-2 were both situated predominantly in the tips of EC cords. Taken together, these findings are highly suggestive of an Ang-2-mediated induction of EC cell death at 12 hours after MCAO. Although VEGF protein is present in these brain regions starting at 6 hours, the very concentration at the EC might not be sufficient to rescue the cells from apoptosis. Ang-2-induced susceptibility of ECs for apoptosis would thus be a transient process, lasting less than 12 hours. Increasing VEGF concentrations at later times probably counteracts this signal. Our observations support the hypothesis that Ang-2 induction leads to vessel proliferation or regression depending on the presence or absence of VEGF. 42
Expression of the Tie-Receptor Family
The receptor tie-2 has been implicated in vascular maintenance as well as angiogenesis. 22,34 It has been shown that both hypoxia and VEGF only slightly modulate tie-2 expression in vitro. 38,39 In our MCAO model we observed differing regulation patterns of tie-2 mRNA and protein levels. Tie-2-protein was widely expressed in the quiescent adult vasculature of the rat brains consistent with previous reports, 22 whereas tie-2 mRNA transcripts were barely detectable. At 3 days after MCAO a marked induction of tie-2 mRNA was seen in vessels in the peri-infarct area, which, surprisingly, did not lead to a concomitant increase in tie-2 protein. A similar discrepancy between tie-2 mRNA and protein regulation was observed in postnatal brain vasculature of the rat (our own unpublished observations). However, as minor alterations in protein levels might not be detected by immunohistochemistry, small increases of tie-2 protein cannot be excluded. Tie-2 mRNA levels gradually decreased with brain maturation, yet tie-2 protein remained constitutively expressed at high levels.
Tie-1 is also known to be implicated in angiogenesis. 35 In most adult organs except lung and heart only little tie-1 message can be detected. 43 Hypoxia and VEGF have been shown to up-regulate tie-1 protein in vitro in a time-dependent manner as early as 1 hour on stimulus. 44 Further, tie-1-mRNA expression has recently been described to be increased in microvessels residing in the peri-infarct area in an embolic MCAO rat model. 45 In our model tie-1 mRNA and protein levels were up-regulated only at 3 days after MCAO, which interestingly coincides with high expressions levels of the Ang-2/tie-2 system. Tie-1 was subsequently down-regulated to baseline levels at 7 days.
The failure of angiogenesis and insufficient growth of collateral vessels is a major problem in vascular diseases, such as stroke. It is of considerable importance for therapeutic purposes to elucidate the exact mechanisms by which the growth factor signaling cascade can be recruited for an angiogenic response in adults. Our study might provide a new experimental basis for treating cerebro-ischemic diseases by rescuing ECs from undergoing apoptosis and stimulating vessel proliferation, which could probably reduce infarct size.
Acknowledgments
We thank Dr. George D. Yancopoulos (Tarrytown, NY) for providing Ang-1 and Ang-2 cDNA clones; Dr. Tom Sato (Dallas, TX) for providing tie-1 and tie-2 cDNA clones; Dr. Kevin Peters (Durham, NC) for the gift of the monoclonal tie-2 antibody; Dr. Kari Alitalo (Helsinki, Finland) for tie-1 antibody; Drs. Yoshinori Imai and Shinichi Kohsaka (Tokyo, Japan) for kindly supplying the Iba1 antibody; and Francesco D’Amato and Elife Iyen for their technical assistance.
Footnotes
Address reprint requests to Dr. Heike Beck, Abteilung Neuropathologie, Friedrich-Alexander-Universität Erlangen-Nürnberg, Krankenhausstrasse 8-10, D-91054 Erlangen, Germany. E-mail: heikebeck@gmx.de.
Supported by Bundesministerium für Bildung und Forschung (grant 01KV/9922/6) and Deutsche Krebshilfe (Grant 10-1302-Ri3) to K. H. P.
H. B. and T. A. contributed equally to the article.
References
- 1.Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM: Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25:1794-1798 [DOI] [PubMed] [Google Scholar]
- 2.Marchal G, Serrati C, Rioux P, Petit-Taboue MC, Viader F, de IS V, Le Doze F, Lochon P, Derlon JM, Orgogozo JM: PET imaging of cerebral perfusion and oxygen consumption in acute ischaemic stroke: relation to outcome. Lancet 1993, 341:925-927 [DOI] [PubMed] [Google Scholar]
- 3.Plate KH, Beck H, Danner S, Allegrini PR, Wiessner C: Cell type specific upregulation of vascular endothelial growth factor in an MCA-occlusion model of cerebral infarct. J Neuropathol Exp Neurol 1999, 58:654-666 [DOI] [PubMed] [Google Scholar]
- 4.Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, Risau W: Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol 2000, 156:965-976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Risau W: Mechanisms of angiogenesis. Nature 1997, 386:671-674 [DOI] [PubMed] [Google Scholar]
- 6.Plate KH: Mechanisms of angiogenesis in the brain. J Neuropathol Exp Neurol 1999, 58:313-320 [DOI] [PubMed] [Google Scholar]
- 7.Breier G, Albrecht U, Sterrer S, Risau W: Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development 1992, 114:521-532 [DOI] [PubMed] [Google Scholar]
- 8.Breier G, Clauss M, Risau W: Coordinate expression of vascular endothelial growth factor receptor-1 (flt-1) and its ligand suggests a paracrine regulation of murine vascular development. Dev Dyn 1995, 204:228-239 [DOI] [PubMed] [Google Scholar]
- 9.Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NP, Risau W, Ullrich A: High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993, 72:835-846 [DOI] [PubMed] [Google Scholar]
- 10.Cobbs CS, Chen J, Greenberg DA, Graham SH: Vascular endothelial growth factor expression in transient focal cerebral ischemia in the rat. Neurosci Lett 1998, 249:79-82 [DOI] [PubMed] [Google Scholar]
- 11.Kovacs Z, Ikezaki K, Samoto K, Inamura T, Fukui M: VEGF and flt. Expression time kinetics in rat brain infarct. Stroke 1996, 27:1865-1872 [DOI] [PubMed] [Google Scholar]
- 12.Lennmyr F, Ata KA, Funa K, Olsson Y, Terent A: Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol 1998, 57:874-882 [DOI] [PubMed] [Google Scholar]
- 13.Hayashi T, Abe K, Suzuki H, Itoyama Y: Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke 1997, 28:2039-2044 [DOI] [PubMed] [Google Scholar]
- 14.Davis S, Yancopoulos GD: The angiopoietins: Yin and Yang in angiogenesis. Curr Top Microbiol Immunol 1999, 237:173-185 [DOI] [PubMed] [Google Scholar]
- 15.Grosios K, Leek JP, Markham AF, Yancopoulos GD, Jones PF: Assignment of ANGPT4, ANGPT1, and ANGPT2 encoding angiopoietins 4, 1 and 2 to human chromosome bands 20p13, 8q22.3–>q23 and 8p23.1, respectively, by in situ hybridization and radiation hybrid mapping. Cytogenet Cell Genet 1999, 84:118-120 [DOI] [PubMed] [Google Scholar]
- 16.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD: Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277:55-60 [DOI] [PubMed] [Google Scholar]
- 17.Nishimura M, Miki T, Yashima R, Yokoi N, Yano H, Sato Y, Seino S: Angiopoietin-3, a novel member of the angiopoietin family. FEBS Lett 1999, 448:254-256 [DOI] [PubMed] [Google Scholar]
- 18.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD: Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87:1171-1180 [DOI] [PubMed] [Google Scholar]
- 19.Valenzuela DM, Griffiths JA, Rojas J, Aldrich TH, Jones PF, Zhou H, McClain J, Copeland NG, Gilbert DJ, Jenkins NA, Huang T, Papadopoulos N, Maisonpierre PC, Davis S, Yancopoulos GD: Angiopoietins 3 and 4: diverging gene counterparts in mice and humans. Proc Natl Acad Sci USA 1999, 96:1904-1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koblizek TI, Runting AS, Stacker SA, Wilks AF, Risau W, Deutsch U: Tie2 receptor expression and phosphorylation in cultured cells and mouse tissues. Eur J Biochem 1997, 244:774-779 [DOI] [PubMed] [Google Scholar]
- 21.Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y: Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 1995, 376:70-74 [DOI] [PubMed] [Google Scholar]
- 22.Wong AL, Haroon ZA, Werner S, Dewhirst MW, Greenberg CS, Peters KG: Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res 1997, 81:567-574 [DOI] [PubMed] [Google Scholar]
- 23.Puri MC, Rossant J, Alitalo K, Bernstein A, Partanen J: The receptor tyrosine kinase TIE is required for integrity and survival of vascular endothelial cells. EMBO J 1995, 14:5884-5891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumont DJ, Gradwohl G, Fong GH, Puri MC, Gertsenstein M, Auerbach A, Breitman ML: Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev 1994, 8:1897-1909 [DOI] [PubMed] [Google Scholar]
- 25.Hanahan D: Signaling vascular morphogenesis and maintenance. Science 1997, 277:48-50 [DOI] [PubMed] [Google Scholar]
- 26.Goede V, Schmidt T, Kimmina S, Kozian D, Augustin HG: Analysis of blood vessel maturation processes during cyclic ovarian angiogenesis. Lab Invest 1998, 78:1385-1394 [PubMed] [Google Scholar]
- 27.Holash J, Wiegand SJ, Yancopoulos GD: New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene 1999, 18:5356-5362 [DOI] [PubMed] [Google Scholar]
- 28.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ: Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284:1994-1998 [DOI] [PubMed] [Google Scholar]
- 29.Tamura A, Graham DI, McCulloch J, Teasdale GM: Focal cerebral ischaemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metab 1981, 1:53-60 [DOI] [PubMed] [Google Scholar]
- 30.Sauer D, Allegrini PR, Cosenti A, Pataki A, Amacker H, Fagg GE: Characterization of the cerebroprotective efficacy of the competitive NMDA receptor antagonist CGP40116 in a rat model of focal cerebral ischemia: an in vivo magnetic resonance imaging study. J Cereb Blood Flow Metab 1993, 13:595-602 [DOI] [PubMed] [Google Scholar]
- 31.Allegrini PR, Sauer D: Application of magnetic resonance imaging to the measurement of neurodegeneration in rat brain: MRI data correlate strongly with histology and enzymatic analysis. Magn Reson Imaging 1992, 10:773-778 [DOI] [PubMed] [Google Scholar]
- 32.Stejskal EO, Tanner JE: Spin diffusion measurements: spin echoes in the presence of a time-dependent field gradient. J Chem Phys 1965, 42:282-292 [Google Scholar]
- 33.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD: Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87:1171-1180 [DOI] [PubMed] [Google Scholar]
- 34.Stratmann A, Risau W, Plate KH: Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol 1998, 153:1459-1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korhonen J, Partanen J, Armstrong E, Vaahtokari A, Elenius K, Jalkanen M, Alitalo K: Enhanced expression of the tie receptor tyrosine kinase in endothelial cells during neovascularization. Blood 1992, 80:2548-2555 [PubMed] [Google Scholar]
- 36.Lauren J, Gunji Y, Alitalo K: Is angiopoietin-2 necessary for the initiation of tumor angiogenesis? Am J Pathol 1998, 153:1333-1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Enholm B, Paavonen K, Ristimaki A, Kumar V, Gunji Y, Klefstrom J, Kivinen L, Laiho M, Olofsson B, Joukov V, Eriksson U, Alitalo K: Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 1997, 14:2475-2483 [DOI] [PubMed] [Google Scholar]
- 38.Mandriota SJ, Pepper MS: Regulation of angiopoietin-2 mRNA levels in bovine microvascular endothelial cells by cytokines and hypoxia. Circ Res 1998, 83:852-859 [DOI] [PubMed] [Google Scholar]
- 39.Oh H, Takagi H, Suzuma K, Otani A, Matsumura M, Honda Y: Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine microvascular endothelial cells. J Biol Chem 1999, 274:15732-15739 [DOI] [PubMed] [Google Scholar]
- 40.Auer RN, Benveniste H: Hypoxia and related conditions. Graham DI Lantos PL eds. Greenfield’s Neuropathology. 1997, :pp 263-314 Oxford University Press, New York [Google Scholar]
- 41.Tamura A, Asano T, Sano K: Correlation between rCBF and histological changes following temporary middle cerebral artery occlusion. Stroke 1980, 11:487-493 [DOI] [PubMed] [Google Scholar]
- 42.Cheung AH, Stewart RJ, Marsden PA: Endothelial Tie2/Tek ligands angiopoietin-1 (ANGPT1) and angiopoietin-2 (ANGPT2): regional localization of the human genes to 8q22.3-q23 and 8p23. Genomics 1998, 48:389-391 [DOI] [PubMed] [Google Scholar]
- 43.Korhonen J, Polvi A, Partanen J, Alitalo K: The mouse tie receptor tyrosine kinase gene: expression during embryonic angiogenesis. Oncogene 1994, 9:395-403 [PubMed] [Google Scholar]
- 44.McCarthy MJ, Crowther M, Bell PR, Brindle NP: The endothelial receptor tyrosine kinase tie-1 is upregulated by hypoxia and vascular endothelial growth factor. FEBS Lett 1998, 423:334-338 [DOI] [PubMed] [Google Scholar]
- 45.Lin TN, Wang CK, Cheung WM, Hsu CY: Induction of angiopoietin and Tie receptor mRNA expression after cerebral ischemia-reperfusion. J Cereb Blood Flow Metab 2000, 20:387-395 [DOI] [PubMed] [Google Scholar]