

Abstract
We analyzed the frequency and regional distribution of cells with genetic abnormalities of chromosomes 1, 14, and 22 in meningiomas. This data was evaluated for correlation to the clinical outcome of the patients. Eight defined areas of each of 77 paraffin-embedded meningioma samples (59 grade I, 13 grade II, and 5 grade III) were analyzed by fluorescent in situ hybridization using bacterial artificial chromosome probes localized to chromosomes 1p36.32, 1q25.3, 14q13.3, 14q32.12, 22q11.2, and 22q12.1-3. Chromosome deletion was considered to be regionally heterogeneous if <7 regions showed cells with chromosome deletions. Deletion of 1p occurred in 35% of the grade I tumors. Distribution of cells with 1p deletion was regionally heterogeneous in 25% and homogeneous in 10% of grade I tumors. Distribution of cells with deletion of 1p was regionally heterogeneous in 23% and homogeneous in 69% of the grade II tumors. All grade III meningiomas had homogeneous distribution of cells with deletion of chromosome 1p. Distribution of cells with deletion of 14q was regionally heterogeneous in 27% and homogeneous in 2% of the grade I meningiomas, heterogeneous in 31% and homogeneous in 62% of the grade II tumors, and heterogeneous in 40% and homogeneous in 60% of the grade III meningiomas. Distribution of cells with deletion of 22q was regionally heterogeneous in 15% and homogeneous in 3% of the grade I tumors, heterogeneous in 15% and homogeneous in 31% of grade II tumors, and homogeneous in 20% of the grade III meningiomas. Distribution of cells with trisomy 22q was regionally heterogeneous in 10% of grade I tumors, heterogeneous in 23% of grade II, and homogeneous in 80% of grade III meningiomas. The proportion of patients with a deletion of 22q (either homogeneous or heterogeneous) who had recurrence was greater than the proportion of those without 22q deletion who had recurrence, and deletion of 22q was significantly associated with radiologically detected recurrence (P < 0.05). We conclude that the appearance of chromosomal aberrations in different areas of the tumor demonstrates the importance of regional heterogeneity in the biological behavior of meningiomas.
Meningiomas are typically regarded as benign tumors that arise from arachnoid cap cells of the dura mater and account for about 20% of all primary intracranial tumors (Kujas, 1993). Although the clear majority are benign and potentially surgically curable, total resection is not always possible because of potential injury to neural structures or eloquent brain areas. According to WHO histological grading criteria, 85% to 94.3% of these tumors are grade I, 5% to 11% are grade II, and 1% to 3% are grade III (Jaaskelainen et al., 1986; Louis et al., 1997). Although recently revised grading schemes correlate well with overall biological behavior, meningiomas are like other brain tumors in that they display individual variability within a given histologic grade (Perry et al., 1997, 1999). Furthermore, a significant subset of these tumors possesses aggressive behavior as evidenced by recurrence and significant morbidity and mortality. Patients with anaplastic meningiomas have a median survival from diagnosis of approximately 1.5 years and a 5-year mortality rate of 68% (Perry et al., 1999). Atypical meningiomas (WHO grade II) are associated with a 40% 5-year recurrence rate, even when gross total resection is achieved. In addition, even histologically benign meningiomas (WHO grade I) may display an aggressive clinical course and are associated with a 9.5% recurrence after Simpson grade I removal (removal of suspected tumor-involved dura), 18.4% after Simpson grade II removal (coagulation of tumor-involved or surrounding dura), and 20% after Simpson grade III removal (excised, coagulated dura with tumor known to be remaining in bone or sinus) and grade IV removal (partial removal of tumor) (Adegbite et al., 1983; Marks et al., 1986; Simpson, 1957). As described in previous studies, patients whose tumors were not completely removed had a 4.2-fold higher excess risk of death during the second to fifteenth postoperative years as compared with patients whose tumors were completely removed (Kallio et al., 1992). However, there are also reports describing no regrowth of a meningioma after subtotal resection over extensive follow-up (Jung et al., 2000; Kleinpeter and Bock, 1990; Puchner et al., 1998).
Different techniques to assess the aggressiveness of these neoplasms have been explored, including assessment of nucleolar organizer regions (Plate et al., 1990), flow cytometry (Perry et al., 1998), telomerase activity (De-Masters et al., 1997; Langford et al., 1997; Simon et al., 2000, 2001), and immunohistochemical staining for cell proliferation markers such as MIB-1 (Ki-67) (Hsu et al., 1998; Perry et al., 1998). Meningiomas also express a variety of hormone receptors closely linked with histologic grade, most notably the progesterone receptor. The presence of this receptor, even in a small number of tumor cells, is a favorable prognostic factor for meningiomas (Hsu et al., 1997). Genetic characterization offers another possibility for stratifying meningiomas. It is known that aberrations of chromosome 22 are the most frequently reported genetic abnormalities in meningiomas (Maillo et al., 2001; Sayagues et al., 2002). The next most frequently reported genetic abnormalities are deletions of 1p and 14q chromosomes, which are thought to be involved in tumor progression (Bello et al., 1994, 2000; Cai et al., 2001; Ishino et al., 1998; Leone et al., 1999; Lomas et al., 2001; Lopez-Gines et al., 2001; Muller et al., 1999; Schneider et al., 1996; Simon et al., 1995; Ueki et al., 1999; Yakut et al., 2002). The potential clinical utility of studies such as these does not require the identification of specific genes. For example, it has been shown that loss of heterozygosity of chromosomes 1p and 19q has prognostic significance for therapy response and time to recurrence in oligodendrogliomas, even though the genes responsible for this behavior have not been identified (Cairncross et al., 1998; Ino et al., 2000).
In this study we have used fluorescent in situ hybridization (FISH)3 to analyze genetic alterations in a retrospective set of 77 meningiomas. This technique allows us to use paraffin-embedded archival specimens for which survival and follow-up data is often available. Specific chromosomal regions can be studied through the use of bacterial artificial chromosome (BAC) probes mapped to specific chromosomal regions. In addition, the cellular and regional heterogeneity of genetic alterations can be mapped in the tumor tissue.
We sought to examine the homogeneous or heterogeneous regional distribution of cells with aberrations of chromosomes 1, 14, or 22 in each tumor to evaluate the correlation to the clinical outcome of patients, taking into account that regional heterogeneity is a novel approach to the examination of chromosomal abnormalities in meningiomas.
Materials and Methods
Patient samples included 77 tumor samples from 73 patients (two patients were operated on twice for the same tumor, two patients had two separate meningiomas) who underwent surgical resection of their tumors between 1990 and 2000 at the Barrow Neurological Institute (Tables 1 and 2). Of the 73 patients analyzed, 71% were female and 29% were male. The mean overall age for the group was 55.57 years (SD = 14.91; range, 16–85 years). The mean age for grade I meningioma was 57.49 (SD = 11.99; range, 23–85 years), the mean age for grade II was 52.46 (SD = 23.79; range, 16–81 years), and the mean age for grade III was 41.0 (SD = 9.57; range, 28–52 years). There was a significant difference in mean age between all grades (P <0.05). The locations of the tumors are shown in Table 1. The diagnosis of meningioma was established in all cases according to standard histopathologic criteria (Perry et al., 1997). Following the WHO classification, 76.6% (n = 59) of the tumors were classified as grade I, 16.9% (n = 13) as grade II, and 6.5% (n = 5) as grade III. The histological subtypes are shown in Table 2. Six of the samples were from recurrent tumors: three grade I, two grade II, and one grade III.
Table 1.
Tumor location
| Grade | Location | Number | % |
|---|---|---|---|
| I | Skull base (including posterior fossa) | 33 | 42.9 |
| Cerebral convexity | 16 | 20.7 | |
| Falx and tentorial | 6 | 7.8 | |
| Spinal cord | 3 | 3.9 | |
| Ventricular | 1 | 1.3 | |
| II | Skull base (including posterior fossa) | 6 | 7.8 |
| Cerebral convexity | 5 | 6.5 | |
| Falx and tentorial | 2 | 2.6 | |
| III | Skull base (including posterior fossa) | 5 | 6.5 |
Table 2.
Tumor diagnosis
| Grade | Diagnosis | Number | % |
|---|---|---|---|
| I | transitional | 36 | 46.7 |
| meningothelial | 12 | 15.6 | |
| fibrous | 5 | 6.5 | |
| psammomatous | 3 | 3.9 | |
| microcystic | 2 | 2.6 | |
| angiomatous | 1 | 1.3 | |
| II | atypical | 11 | 14.3 |
| chordoid | 1 | 1.3 | |
| clear cell | 1 | 1.3 | |
| III | anaplastic | 3 | 3.9 |
| papillary | 2 | 2.6 |
We performed FISH analyses using tissue microarrays. Representative areas of tumor were identified on a hematoxylin & eosin–stained slide. We then removed four 0.6-mm cores from the corresponding areas on the paraffin block and placed them into a tissue microarray block using the Beecher Instruments Manual Tissue Arrayer I (Beecher Instruments, Sun Prairie, Wisc.).Within a case, there were no significant histological differences among the four areas sampled.
Probe Preparation
BACs mapped to chromosomes 22q11.2, 22q12.1-3, 1p36.32, 1q25.3, 14q13.3, and 14q32.12 were obtained from The Sanger Institute (Cambridge, U.K.) or Research Genetics (ResGen Invitrogen Corp., Carlsbad, Calif.) (Table 3). They were grown and purified according to standard protocols (Dunham et al., 1999a, b). Labeling with SpectrumGreen or SpectrumOrange was done with a Nick Translation kit (Vysis, Inc., Downers Grove, Ill.) and conditions specified by the manufacturer.
Table 3.
BAC information
| Chromosome Position | Clone | Size (bp) | bp Position (approx.) |
|---|---|---|---|
| 1p36.32 | RP11-1072A21 | 182,342 | 3145983-3328324 |
| CTD-3209F18 | 238,914 | 3313075-3551988 | |
| 1q25.3 | RP11-638L19 | 190,469 | 181801871-181992339 |
| CTD-2541K24 | 174,148 | 181909570-182083717 | |
| CTD-3204N6 | 171,016 | 182070927-182241942 | |
| 14q13.3 | RP11-381L10 | 209,869 | 35098955-35308823 |
| RP11-796F21 | 209,386 | 35567525-35776910 | |
| 14q32.12 | CTD-2017J9 | 196,909 | 89820634-90017542 |
| RP11-724F5 | 162,429 | 90032987-90195415 | |
| RP11-384J14 | 176,839 | 90195513-90372351 | |
| 22q11.2 | CTA-115F6 | 194,000 | 16153880-16340333 |
| CTA-154H4 | 245,000 | 16664796-16664940 | |
| CTA-433F6 | 145,374 | 19054092-19199465 | |
| 22q11.2-12.1 | CTA-526G4 | NAa | 27093082-NA |
| CTA-322B1 | 77,000 | 22624162-22716609 | |
| 22q11.23-12.1 | CTA-221G9 | 102,000 | 23874371-23976913 |
| CTA-9992D9 | 150,000 | 25736059-25891206 | |
| CTA-57G9 | 105,000 | 27837089-27950960 | |
| 22q12.1-12.3 | CTA-99F11 | 145,000 | 30021523-30021651 |
| CTA-415G2 | 190,000 | 31690492-31820448 | |
| CTA-221H1 | 49,000 | 32896025-32899047 | |
| CTA-212A2 | 230,000 | 34683353-34896105 |
NA, not available.
Pooled BAC probes contained 0.1 μg of each BAC DNA, 1 μg Cot-I DNA, 5 μg salmon sperm DNA, 4 μl sterile water, 1.2 μl 3M Na acetate (pH 5.6), and 30 μl 100% ethanol for each BAC DNA. The probes were stored at – 80°C for 10 min and centrifuged at 4°C at 14,000 rpm for 30 min. The pellets were resuspended in 3 μl sterile water and 7 μl LSI/WCP hybridization buffer (Vysis, Inc.), denatured at 73°C for 5 min, and then placed on the prepared slides.
Tissue Preparation and FISH Hybridization
Unstained sections of 5-μm thickness were cut from tissue microarrays. The tissues were deparaffinized in xylene, placed in Lugol’s iodine solution for 5 min, washed in 2.5% sodium thiocyanate until clear, and then dehydrated in ethanol. The slides were placed in 10 mM citrate acid pH 6 and microwaved on high for 5 min. The sections were then digested in a pepsin/0.9% NaCl solution, pH 1.5, for 60 min at 37°C, dehydrated in graded ethanols, and air dried. BAC probes were placed on the slides, sealed under a coverslip, denatured at 80°C for 3 min, and hybridized for 24 h at 37°C. Slides were then washed in 1.5 M urea/0.1× standard saline citrate (SSC) for 15 min at 45°C, rinsed briefly with 2× SSC, and then dried in darkness. Counterstaining was done using Vectashield counterstain with DAPI (Vector Laboratories, Burlingame, Calif.). After one set of probes was counted, the slides were washed in 2× SSC, dehydrated in graded ethanols, air dried, and rehybridized with a new set of probes.
FISH Analyses
FISH results were viewed on a Zeiss Pascal 5 laser scanning confocal microscope. For each hybridization, signals were counted for eight randomly chosen regions of 200 nonoverlapping nuclei. The x/y coordinates on the microscope for each region were recorded, which enabled us to compare the different chromosomal abnormalities on the same region of each tumor. Homogeneous regional distribution of chromosomal aberrations was defined as the occurrence of those abnormalities in seven or eight of the examined regions within one tumor. Heterogeneous regional distribution of aberrations was defined as the presence of the abnormalities in at least two but fewer than seven of the examined regions. At least two regions were required for a heterogeneous designation to allow for one false positive. Cutoffs for deletions and aberrations were based on the frequencies of signals for the same probes in leptomeningeal and non-neoplastic brain controls (median + 3 SD) and are 17% (chromosome 1), 18% (chromosome 14), 14% (monosomy 22), and 7% (trisomy 22). If the FISH signals were too weak to count, the hybridizations were considered noninformative.
Data Analysis and Statistical Methods
Summary statistics were completed for several variables for group comparison. Categorical variables summarized by frequencies and percentages and treatment groups were compared by using chi square or Fisher exact tests as appropriate. Continuous variables were computed by using analysis of variance. Logistic regression was utilized to examine radiological recurrence as a binary measure. We conducted all statistical tests employing a significance level of 0.05 (ClinMetrics, Inc., Scottsdale, Ariz.).
Results
Analysis of Individual Chromosomal Aberrations
The results of the analysis of chromosomal aberrations among the 77 cases studied for chromosomes 1, 14, and 22 are demonstrated in Fig. 1. In the group of grade I meningiomas (n = 59), heterogeneous distribution of cells with deletion of 1p was present in 25% of the tumors (n = 15), homogeneous distribution of cells with deletion of 1p was found in 10% (n = 6), and 65% of the tumors had no detectable abnormality of chromosome 1. Heterogeneous distribution of cells with deletion of 14q was found in 27% (n = 16) of the tumors, homogeneous distribution of cells with deletion of 14q was found in 2% (n = 1), and 71% had no detectable abnormality. Heterogeneous distribution of cells with deletion of 22 was present in 15% (n = 9), and homogeneous distribution of cells with deletion of 22 in 3% (n = 2) of the tumors. Heterogeneous distribution of cells with trisomy 22 was seen in 10% (n = 6) of the tumors, and no case had homogeneous distribution of cells with trisomy. Seventy-two percent of grade I meningiomas showed no detectable aberration of chromosome 22.
Fig. 1.

Analysis of individual chromosomal aberrations in grade I, II, and III meningiomas.
In the group of grade II meningiomas (n = 13), heterogeneous distribution of cells with deletion of 1p was present in 23% (n = 3) and homogeneous deletion of 1p in 69% (n = 9). One tumor (8%) had no detectable alterations of chromosome 1. Heterogeneous distribution of cells with deletion of 14q was seen in 31% (n = 4) of the tumors and homogeneous distribution of cells with deletion of 14q in 62% (n = 8). One tumor (8%) had no aberrations of chromosome 14. Heterogeneous distribution of cells with deletion of chromosome 22 was detected in 15% (n = 2) of the cases, homogeneous distribution of cells with deletion in 31% (n = 4), and heterogeneous distribution of cells with trisomy in 23% (n = 3), and no case had homogeneous distribution of cells with trisomy. Four tumors (31%) had no detectable alterations of chromosome 22.
All five grade III meningiomas had homogeneous distribution of cells with deletion of 1p. Two cases had heterogeneous and three had homogeneous distribution of cells with deletion of 14q. One meningioma had homogeneous distribution of cells with deletion of 22q, and the other four cases had homogeneous distribution of cells with trisomy 22q.
Analysis of Combined Chromosomal Aberrations
The combinations of chromosomal aberrations that we found are shown by meningioma grade in Table 4 and Fig. 2.
Table 4.
Percentages of single and combined chromosomal aberrations in grade I–grade III meningiomas
| Chromosomal Aberrations (homogeneous or heterogeneous)a | Grade I (n = 59)b | Grade II (n = 13)b | Grade III (n = 5)b |
|---|---|---|---|
| 1p deletion alone | 8.5 | 0 | 0 |
| 14q deletion alone | 6.8 | 0 | 0 |
| 22q trisomy alone | 8.5 | 0 | 0 |
| 1p deletion + 14q deletion | 6.8 | 23.1 | 0 |
| 1p deletion + 22q deletion | 5.1 | 0 | 0 |
| 1p deletion + 14q deletion + 22q deletion | 13.6 | 46.2 | 20 |
| 1p deletion + 14q deletion + 22q trisomy | 1.7 | 23.1 | 80 |
Deletion of 22q alone, deletion of 1p + trisomy of 22q, deletion of 14q + deletion of 22q, and deletion of 14q + trisomy of 22q were not found in any cases.
Chromosomal aberrations were found in 51% of grade I, 92.4% of grade II, and 100% of grade III meningiomas.
Fig. 2.
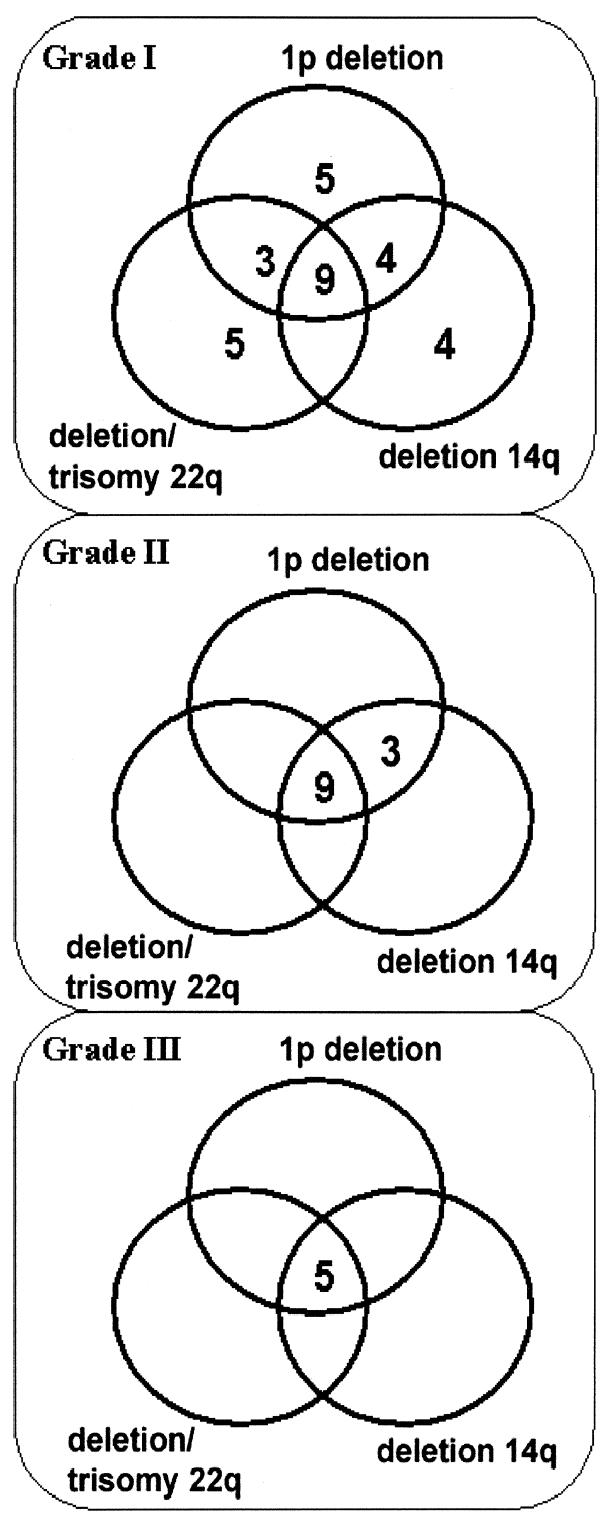
Numbers of patients with single or combined chromosomal aberrations in 59 grade I, 13 grade II, and 5 grade III meningiomas.
In the group of grade I meningiomas, one of six samples that had homogeneous regional distribution of cells with deletion of 1p and 4 of 15 samples that had heterogeneous distribution of cells with deletion of 1p had no additional chromosomal aberrations. In one case with homogeneous regional distribution of cells with deletion of 14q and in three cases with heterogeneous deletion of 14q, no additional chromosomal aberrations were found. No case of deletion of 22 was detected without homogeneous or heterogeneous regional distribution of cells with deletion of 1p. The combination of deletion of 1p and deletion of 14q was found in four cases, and deletion of 1p and deletion of 22q in three cases. The combination of deletion of 1p, 14q, and 22q was detected in eight cases. However, only one of six cases with heterogeneous distribution of cells with trisomy 22 was combined with deletion of 1p plus deletion of 14q. All three cases of recurrent grade I meningiomas had at least one chromosomal aberration. One case had heterogeneous regional distribution of trisomy 22, one case had heterogeneous distribution of cells with deletion of 1p, and one case had heterogeneous distribution of cells with deletion of 1p plus heterogeneous deletion of 14q plus homogeneous deletion of 22.
In contrast, there was no singular chromosomal aberration in grade II or grade III meningiomas. Three cases of grade II tumors had a homogeneous deletion 1p, two cases combined with homogenous deletion of 14q, and one case combined with heterogeneous deletion of 14q. Six cases demonstrated deletions of 1p, 14q, and 22, and three cases had deletions of 1p and 14q and trisomy 22. All five grade III meningiomas demonstrated deletion of 1p and 14q. One case had additional deletion of 22q, and four cases had additional trisomy 22.
Follow-Up of the Benign Meningiomas
In 11 of 55 patients with primary grade I meningiomas, the clinical follow-up was less than one year. No tumor regrowth was seen in these patients regardless of whether complete surgical resection was achieved. The other 44 patients had a mean clinical follow-up period of 6.7 years (range, 3–12 years). In the group of meningiomas without any chromosomal aberrations, there were 10 cases in which the patient received a gross total tumor resection and had no recurrent tumor. Out of the 15 patients who had subtotal tumor resection (four patients had an additional gamma knife treatment), three (without gamma knife treatment) demonstrated regrowth of the tumor (one case in two years and two cases in eight years).
In the group with chromosomal aberrations, 10 paients had a gross total tumor resection, and one of these patients had a recurrent tumor after five years. The remaining patients had a subtotal tumor removal, three of whom had an additional gamma knife treatment. Five of these patients (without gamma knife treatment) had tumor regrowth between one and eight years. Of the three patients with recurrent grade I meningiomas, two patients had gross total resection and showed no new recurrent tumor after three and nine years. One patient had subtotal resection, received additional gamma knife treatment, and remained stable for three years.
Follow-Up of Grade II and Grade III Meningiomas
Of the 13 grade II meningioma patients, five had a follow-up shorter than one year. Neither recurrent tumor nor tumor regrowth was detected within this time. The only patient without any chromosomal aberrations had no recurrent tumor after a gross total resection within a mean follow-up period of four years. The other patients demonstrated recurrent tumors following gross total resection after two and four years, or regrowths after subtotal resection within two years. Two patients succumbed to this disease: one after four years and one after seven years. Of five grade III tumors, two patients died three and eight years after surgery. The patient with grade III recurrent meningioma died 1.5 years after surgery. The other two patients had a gross total removal and showed no recurrent tumor after three and four years.
Statistical Analysis
Differences in chromosomal aberrations between different grades were calculated. Grades II and III were combined since there were only five cases in grade III. The proportion of aberrations was significantly different between grades (Fisher exact test, P < 0.05). We used binary logistic regression to examine the effect of chromosomal aberrations on clinical outcome, specifically, radiographic evidence of recurrence. Variables entered into the model were deletion of 1p, 14q, and 22q and trisomy 22q.
The only aberrations found to be significantly associated with radiographic evidence of recurrence were deletions of 22q (P < 0.05). The proportion of patients with a deletion of 22q (either homogeneous or heterogeneous) who had tumor recurrence was greater than the proportion of those without 22q deletion (35% compared to 13%) who had tumor recurrence.
Discussion
Tumor heterogeneity may be defined as variation in the tumor’s genotype and/or phenotype (Coons and Johnson, 1993a). Heterogeneity in brain tumors has been identified by using histological description (Burger and Kleihues, 1989; Paulus and Peiffer, 1989), cytogenetics (Bigner, 1981; Shapiro and Shapiro, 1985), variable expression of growth factors and their receptors (Scheck et al., 1991; Strommer et al., 1990), proliferation markers (Coons and Johnson, 1993b; Deckert et al., 1989), flow cytometry (Coons et al., 1995), and oncogene expression (Sidransky et al., 1992). Two patterns of heterogeneity have been recognized. The first one is a distribution of different cell types, exemplified by the admixture of different cytological subtypes (e.g., meningotheliomatous and fibrillary cell types within meningiomas). The second pattern of heterogeneity is the regional distribution of cellular features. Individual cells tend to be similar to their neighbors, while more distant cells may have markedly different characteristics. The importance of regional heterogeneity in gliomas has been examined in numerous studies (Coons and Johnson, 1992, 1993a, b; Coons et al., 1995). Scheck et al. described the case of a glioblastoma multiforme recurring as a histologically characterized low-grade astrocytoma. However, molecular genetic analyses of the recurrent tumor showed signs of a higher grade tumor (Scheck et al., 1996). As this case exhibits, molecular changes may precede histopathological changes. The heterogeneity that results from this has clinical implications in that removal of the more malignant areas of a heterogeneous tumor can have significant impact on patient survival (Scheck et al., 1996).
To our knowledge no studies have addressed genetic heterogeneity in meningiomas. When examination of multiple regions of a tumor leads to discovery of aberrations in only selected regions of that tumor and this data is used to predict outcome, the tumor can be mistakenly described as more benign if only one region is examined and no aberrations are found. Previous studies that have used FISH on paraffin-embedded tissue have either neglected the contribution of regional heterogeneity or considered samples to be abnormal even if the alteration is only focally detectable. In this current study we used the FISH method to regionally analyze each tumor using defined regions, comparing possible chromosomal aberrations of one chromosome in each region with possible aberrations of another chromosome in the same region (Fig. 3). For this reason, we were able to examine not only homogeneous chromosomal aberrations (defined as aberrations in at least seven of the examined eight regions) but also heterogeneous chromosomal aberrations (defined as aberrations in at least two but less than seven examined regions). Because we examined different regions of each tumor, we detected chromosomal aberrations that would not have been detected if we had only examined one region. This method might explain the higher percentage of chromosome deletions in our study as compared to other studies (Cai et al., 2001; Maillo et al., 2001). Additionally, we looked at correlations of homogeneous or heterogeneous aberrations between chromosomes. Of our grade I meningiomas, 51% demonstrated chromosomal aberrations, showing at least heterogeneous deletion of 1p, 14q, or 22q or trisomy 22. Of these grade I meningiomas, 47% showed aberrations in one chromosome, 23% showed aberrations in two chromosomes, and 30% showed aberrations in three chromosomes.
Fig. 3.

FISH analyses of chromosomes 1, 14, and 22 in the same region of a benign meningioma. A. Chromosome 1: p36 (green), q25 (orange). No deletion of 1p36 (long arrows). B. Chromosome 14: q13 (green), q32 (orange). No deletion of 14q in both areas (arrowheads). C. Chromosome 22: q11 (orange), q12 (green). No deletion of 22q (arrowheads), trisomy 22q (long arrows).
The proportion of patients with deletion of 22q who had tumor recurrence was greater than the proportion of those without deletion of 22q who had recurrence. This was detected for patients with homogeneous as well as with heterogeneous aberrations (P < 0.05). In 15 grade I meningioma cases without chromosomal aberrations where subtotal tumor resection was performed, only three patients showed regrowth of the tumor during mean clinical follow-up. However, five of the nine patients in the group of subtotally resected grade I meningiomas with chromosomal aberrations presented with tumor regrowth within their mean clinical follow-up. Analysis of the different chromosomal aberrations of these five patients revealed that three patients with a regrowth in seven and eight years had only heterogeneous regional distribution of chromosomal aberrations of one chromosome. The other two patients, showing regrowth within one and 1.5 years, had heterogeneous regional distribution of aberrations in chromosomes 1p, 14q, and 22q. The mean time to regrowth was shorter in the group with aberrations (4.8 years) than in the group with no chromosomal aberrations (six years). Four patients without chromosomal aberrations who had subtotal tumor removal, as well as three patients with chromosomal aberrations, had additional gamma knife treatment of the residual tumor. Both groups showed no regrowth of the residual tumor within their mean follow-up (6.7 years). Gamma knife was also used in treatment after regrowth in one tumor without chromosomal aberrations and in two tumors with chromosomal aberrations. In all three gamma knife–treated cases, the tumors remained stable after a mean follow-up of 2.6 years.
Chromosomal aberrations were present in all three cases of recurrent grade I meningiomas. Gross total resection was done in two cases. One case with single heterogeneous regional distribution of trisomy 22 recurred after resection 14 years earlier, and another case with a single deletion of 1p showed recurrence within three years. Subtotal resections plus gamma knife treatment were done on the third case. The meningioma remained stable for three years.
Our findings confirm that trisomy 22 predicts worse outcome (Maillo et al., 2001). In our study, this finding was consistent when homogeneous or heterogeneous regional distribution of cells with trisomy was detected. Analysis of the grade II meningiomas showed that tumors that recurred or regrew within four years demonstrated homogeneous regional distribution of cells with deletion of 1p, 14q, and 22q. Tumors that recurred or regrew earlier showed heterogeneous regional distribution of trisomy. The only patient with a grade II tumor who had no chromosomal aberrations had no recurrent tumor after a gross total resection within a follow-up period of four years.
We found homogeneous regional distribution of cells with trisomy 22 in four of five anaplastic meningiomas. One patient who had a homogeneous deletion of 22 presented no recurrent tumor after four years and initially seemed better than the other patients with grade III meningiomas. Deletion of 22 is discussed controversially in the literature. Maillo et al. (2001) did not find significantly higher rates of deletion of 22 in higher grade II or III meningiomas than in grade I meningiomas. We confirmed a higher percentage of deletion of 22q in grade II meningiomas, which was observed by Zattara-Cannoni et al. (1998).
Consideration of heterogeneity may help to explain typically disparate analyses of chromosomal aberrations. For example, Cai et al. (2001) cited deletion of 1p in 23% of benign, 56% of atypical, and 75% of anaplastic meningiomas, while Ishino et al. (1998) cited deletion of 1p in 11.8% of benign, 60% of atypical, and 85.7% of anaplastic meningiomas. Cai et al. (2001) cited deletion of 14q in 31% of benign, 57% of atypical, and 67% of anaplastic meningiomas, while Zattara-Cannoni et al. (1998) cited deletion of 14q only in anaplastic meningiomas (without a mentioned percentage). Maillo et al. (2001) found deletion of chromosome 22 in 49% and chromosome 22 trisomy/tetrasomy in 9% of tumors without specifying grade; Zattara-Cannoni et al. (1998) found deletion of 22 in 19% of grade I and in 47% of grade II meningiomas; and Sayagues et al. (2002) reported deletion of chromosome 22 in 53% and chromosome 22 trisomy/tetrasomy in 9% of meningiomas. It has been reported in previous studies that trisomy/tetrasomy is significantly correlated with a shorter disease-free survival (Maillo et al., 2001). Also, a trend has been reported for patients with anaplastic meningiomas with deletion of 14q to have poorer overall survival, as well as for patients with atypical meningiomas with combined deletion of 1p and deletion of 14q (Cai et al., 2001). These authors used FISH for their analyses; however, they did not report on the presence or absence of regional heterogeneity in their samples.
Our study demonstrates that molecular genetics may be an important adjunct to standard pathology. The appearance of multiple chromosomal aberrations in different regions demonstrates the importance of regional heterogeneity in the biological behavior of meningiomas. When detecting chromosomal aberrations in subtotally resected meningiomas, it is reasonable to assume that residual portions likely possess chromosomal aberrations. Thus, it would seem helpful for genetic studies of a subtotally resected meningioma to be done using tissue obtained as close as possible to the margin, remaining tumor mass, or areas of suspected tumor. Because of the high incidence of regional heterogeneity in the distribution of genetic aberrations in low-grade meningiomas, well-designed FISH studies of benign tumors such as meningiomas will need to incorporate studies of regional heterogeneity.
Acknowledgments
The authors gratefully acknowledge the generous gift of chromosome 22 BACs from Dr. I. Dunham at The Sanger Institute, Cambridge, U.K. We thank Kathy Goehring for the preparation of the paraffin slides and Aaron Coons for the preparation of the tissue microarrays. We also thank the neurosurgeons and operating room staff of the Barrow Neurological Institute for providing the patient samples.
Footnotes
This work was supported by NIH CA25956 and The Barrow Neurological Foundation.
Abbreviations used are as follows: BAC, bacterial artificial chromosome; FISH, fluorescent in situ hybridization; SSC, standard saline citrate.
References
- Adegbite AB, Khan MI, Paine KW, Tan LK. The recurrence of intracranial meningiomas after surgical treatment. J Neurosurg. 1983;58:51–56. doi: 10.3171/jns.1983.58.1.0051. [DOI] [PubMed] [Google Scholar]
- Bello MJ, de Campos JM, Kusak ME, Vaquero J, Sarasa JL, Pestana A, Rey JA. Allelic loss at 1p is associated with tumor progression of meningiomas. Genes Chromosomes Cancer. 1994;9:296–298. doi: 10.1002/gcc.2870090411. [DOI] [PubMed] [Google Scholar]
- Bello MJ, de Campos JM, Vaquero J, Kusak ME, Sarasa JL, Rey JA. High-resolution analysis of chromosome arm 1p alterations in meningioma. Cancer Genetics Cytogenet. 2000;120:30–36. doi: 10.1016/s0165-4608(99)00249-6. [DOI] [PubMed] [Google Scholar]
- Bigner DD. Biology of gliomas: Potential clinical implications of glioma cellular heterogeneity. Neurosurgery. 1981;9:320–326. [PubMed] [Google Scholar]
- Burger PC, Kleihues P. Cytologic composition of the untreated glioblastoma with implications for evaluation of needle biopsies. Cancer. 1989;63:2014–2023. doi: 10.1002/1097-0142(19890515)63:10<2014::aid-cncr2820631025>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Cai DX, Banerjee R, Scheithauer BW, Lohse CM, Kleinschmidt-DeMasters BK, Perry A. Chromosome 1p and 14q FISH analysis in clinicopathologic subsets of meningioma: Diagnostic and prognostic implications. J Neuropathol Exp Neurol. 2001;60:628–636. doi: 10.1093/jnen/60.6.628. [DOI] [PubMed] [Google Scholar]
- Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Findekstein DM, Hammond RR, Silver JS, Stark PC, Macdonald DR, Ino Y, Ramsay DA, Louis DN. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473–1479. doi: 10.1093/jnci/90.19.1473. [DOI] [PubMed] [Google Scholar]
- Coons SW, Johnson PC. Regional heterogeneity in the Ki67 labeling indexes of gliomas. J Neuropathol Exp Neurol. 1992;51:331. doi: 10.1097/00005072-199311000-00008. [DOI] [PubMed] [Google Scholar]
- Coons SW, Johnson PC. Regional heterogeneity in the DNA content of human gliomas. Cancer. 1993a;72:3052–3060. doi: 10.1002/1097-0142(19931115)72:10<3052::aid-cncr2820721030>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Coons SW, Johnson PC. Regional heterogeneity in the proliferative activity of human gliomas as measured by the Ki-67 labeling index. J Neuropathol Exp Neurol. 1993b;52:609–618. doi: 10.1097/00005072-199311000-00008. [DOI] [PubMed] [Google Scholar]
- Coons SW, Johnson PC, Shapiro JR. Cytogenetic and flow cytometry DNA analysis of regional heterogeneity in a low grade glioma. Cancer Res. 1995;55:1569–1577. [PubMed] [Google Scholar]
- Deckert M, Reifenberger G, Wechsler W. Determination of the proliferative potential of human brain tumors using the monoclonal antibody Ki-67. J Cancer Res Clin Oncol. 1989;115:179–188. doi: 10.1007/BF00397921. [DOI] [PubMed] [Google Scholar]
- DeMasters BK, Markham N, Lillehei KO, Shroyer KR. Differential telomerase expression in human primary intracranial tumors. Am J Clin Pathol. 1997;107:548–554. doi: 10.1093/ajcp/107.5.548. [DOI] [PubMed] [Google Scholar]
- Dunham, I., Dewar, K., Kim, U.-J., and Ross, M.T. (1999a) Bacterial Cloning Systems. In: Birren, B., Green, E.D., Klapholz, S., Myers, R.M., Riethman, H., and Roskams, J. (Eds.), Cloning Systems (Genome Analysis: A Laboratory Manual Series, Vol. 3). Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press, pp. 1–87.
- Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, Bruskiewich R, Beare DM, Clamp M, Smink LJ, Ainscough R, Almeida JP, Babbage A, Bagguley C, Bailey J, Barlow K, Bates KN, Beasley O, Bird CP, Blakey S, Bridgeman AM, Buck D, Burgess J, Burrill WD, O’Brien KP, et al. The DNA sequence of chromosome 22. Nature. 1999b;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- Hsu DW, Efird JT, Hedley-Whyte ET. Progesterone and estrogen receptors in meningiomas: Prognostic considerations. J Neurosurg. 1997;86:113–120. doi: 10.3171/jns.1997.86.1.0113. [DOI] [PubMed] [Google Scholar]
- Hsu DW, Efird JT, Hedley-Whyte ET. MIB-1 (Ki-67) index and transforming growth factor-alpha (TGF alpha) immunoreactivity are significant prognostic predictors for meningiomas. Neuropathol Appl Neurobiol. 1998;24:441–452. doi: 10.1046/j.1365-2990.1998.00150.x. [DOI] [PubMed] [Google Scholar]
- Ino Y, Zlatescu MC, Sasaki H, Macdonald DR, Stemmer-Rachamimov AO, Jhung S, Ramsay DA, von Deimling A, Louis DN, Cairn-cross JG. Long survival and therapeutic responses in patients with histologically disparate high-grade gliomas demonstrating chromosome 1p loss. J Neurosurg. 2000;92:983–990. doi: 10.3171/jns.2000.92.6.0983. [DOI] [PubMed] [Google Scholar]
- Ishino S, Hashimoto N, Fushiki S, Date K, Mori T, Fujimoto M, Nakagawa Y, Ueda S, Abe T, Inazawa J. Loss of material from chromosome arm 1p during malignant progression of meningioma revealed by fluorescent in situ hybridization. Cancer. 1998;83:360–366. [PubMed] [Google Scholar]
- Jaaskelainen J, Haltia M, Servo A. Atypical and anaplastic meningiomas: Radiology, surgery, radiotherapy, and outcome. Surg Neurol. 1986;25:233–242. doi: 10.1016/0090-3019(86)90233-8. [DOI] [PubMed] [Google Scholar]
- Jung HW, Yoo H, Paek SH, Choi KS. Long-term outcome and growth rate of subtotally resected petroclival meningiomas: Experience with 38 cases. Neurosurgery. 2000;46:567–574. doi: 10.1097/00006123-200003000-00008. [DOI] [PubMed] [Google Scholar]
- Kallio M, Sankila R, Hakulinen T, Jaaskelainen J. Factors affecting operative and excess long-term mortality in 935 patients with intracranial meningioma. Neurosurgery. 1992;31:2–12. doi: 10.1227/00006123-199207000-00002. [DOI] [PubMed] [Google Scholar]
- Kleinpeter G, Bock F. Invasion of the cavernous sinus by medial sphenoid meningioma—”radical” surgery and recurrence. Acta Neurochir. 1990;103:87–91. doi: 10.1007/BF01407511. [DOI] [PubMed] [Google Scholar]
- Kujas M. Meningioma. Curr Opin Neurol. 1993;6:882–887. doi: 10.1097/00019052-199312000-00009. [DOI] [PubMed] [Google Scholar]
- Langford LA, Piatyszek MA, Xu R, Schold SC, Jr, Wright WE, Shay JW. Telomerase activity in ordinary meningiomas predicts poor outcome. Hum Pathol. 1997;28:416–420. doi: 10.1016/s0046-8177(97)90029-0. [DOI] [PubMed] [Google Scholar]
- Leone PE, Bello MJ, de Campos JM, Vaquero J, Sarasa JL, Pestana A, Rey JA. NF2 gene mutations and allelic status of 1p, 14q and 22q in sporadic meningiomas. Oncogene. 1999;18:2231–2239. doi: 10.1038/sj.onc.1202531. [DOI] [PubMed] [Google Scholar]
- Lomas J, Bello MJ, Arjona D, Gonzalez-Gomez P, Alonso ME, de Campos JM, Vaquero J, Ruiz-Barnes P, Sarasa JL, Casartelli C, Rey JA. Analysis of p73 gene in meningiomas with deletion at 1p. Cancer Genetics Cytogenet. 2001;129:88–91. doi: 10.1016/s0165-4608(01)00430-7. [DOI] [PubMed] [Google Scholar]
- Lopez-Gines C, Cerda-Nicolas M, Gil-Benso R, Barcia-Salorio JL, Llombart-Bosch A. Loss of 1p in recurrent meningiomas. A comparative study in successive recurrences by cytogenetics and fluorescence in situ hybridization. Cancer Genetics Cytogenet. 2001;125:119–124. doi: 10.1016/s0165-4608(00)00365-4. [DOI] [PubMed] [Google Scholar]
- Louis, D.N., Budka, H., and von Deimling, A. (1997) Meningiomas. In: Kleihues, P., and Cavenee, W.K. (Eds.), Pathology and Genetics of Tumours of the Nervous System. Lyon: International Agency for Research on Cancer, pp. 133–148.
- Maillo A, Diaz P, Sayagues JM, Blanco A, Tabernero MD, Ciudad J, Lopez A, Goncalves JM, Orfao A. Gains of chromosome 22 by fluorescence in situ hybridization in the context of an hyperdiploid karyotype are associated with aggressive clinical features in meningioma patients. Cancer. 2001;92:377–385. doi: 10.1002/1097-0142(20010715)92:2<377::aid-cncr1333>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Marks SM, Whitwell HL, Lye RH. Recurrence of meningiomas after operation. Surg Neurol. 1986;25:436–440. doi: 10.1016/0090-3019(86)90081-9. [DOI] [PubMed] [Google Scholar]
- Muller P, Henn W, Niedermayer I, Ketter R, Feiden W, Steudel W-I, Zang KD, Steilen-Gimbel H. Deletion of chromosome 1p and loss of expression of alkaline phosphatase indicate progression of meningiomas. Clin Cancer Res. 1999;5:3569–3577. [PubMed] [Google Scholar]
- Paulus W, Peiffer J. Intratumoral histologic heterogeneity of gliomas. A quantitative study. Cancer. 1989;64:442–447. doi: 10.1002/1097-0142(19890715)64:2<442::aid-cncr2820640217>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Perry A, Stafford SL, Scheithauer BW, Suman VJ, Lohse CM. Meningioma grading: An analysis of histologic parameters. Am J Surg Pathol. 1997;21:1455–1465. doi: 10.1097/00000478-199712000-00008. [DOI] [PubMed] [Google Scholar]
- Perry A, Stafford SL, Scheithauer BW, Suman VJ, Lohse CM. The prognostic significance of MIB-1, p53, and DNA flow cytometry in completely resected primary meningiomas. Cancer. 1998;82:2262–2269. [PubMed] [Google Scholar]
- Perry A, Scheithauer BW, Stafford SL, Lohse CM, Wollan PC. “Malignancy” in meningiomas: A clinicopathologic study of 116 patients, with grading implications”. Cancer. 1999;85:2046–2056. doi: 10.1002/(sici)1097-0142(19990501)85:9<2046::aid-cncr23>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Plate KH, Ruschoff J, Mennel HD. Nucleolar organizer regions in meningiomas. Correlation with histopathologic malignancy grading, DNA cytometry and clinical outcome. Anal Quant Cytol Histol. 1990;12:429–438. [PubMed] [Google Scholar]
- Puchner MJ, Fischer-Lampsatis RC, Herrmann HD, Freckmann N. Suprasellar meningiomas—neurological and visual outcome at long-term follow-up in a homogeneous series of patients treated micro-surgically. Acta Neurochir. 1998;140:1231–1238. doi: 10.1007/s007010050243. [DOI] [PubMed] [Google Scholar]
- Sayagues JM, Tabernero MD, Maillo A, Diaz P, Rasillo A, Bortoluci A, Gomez-Moreta J, Santos-Briz A, Morales F, Orfao A. Incidence of numerical chromosome aberrations in meningioma tumors as revealed by fluorescence in situ hybridization using 10 chromosome-specific probes. Cytometry. 2002;50:153–159. doi: 10.1002/cyto.10075. [DOI] [PubMed] [Google Scholar]
- Scheck AC, Beikman MK, Korn MC, Shapiro JR. Regional analysis of genes potentially involved in resistance to BCNU in human malignant gliomas. Proc Am Assoc Cancer Res. 1991;32:358. abstract 2125. [Google Scholar]
- Scheck AC, Shapiro JR, Coons SW, Norman SA, Johnson PC. Biological and molecular analysis of a low grade recurrence of a glioblastoma multiforme. Clin Cancer Res. 1996;2:187–199. [PubMed] [Google Scholar]
- Schneider BF, Shashi V, von Kap-Herr C, Golden WL. Loss of chromosomes 22 and 14 in the malignant progression of meningiomas. A comparative study of fluorescence in situ hybridization (FISH) and standard cytogenetic analysis. Cancer Genetics Cytogenet. 1996;85:101–104. doi: 10.1016/0165-4608(95)00154-9. [DOI] [PubMed] [Google Scholar]
- Shapiro JR, Shapiro WR. The subpopulations and isolated cell types of freshly resected high grade human gliomas: Their influence on the tumor’s evolution in vivo and behavior and therapy in vitro. Cancer Metastasis Rev. 1985;4:107–124. doi: 10.1007/BF00050691. [DOI] [PubMed] [Google Scholar]
- Sidransky D, Mikkelsen T, Schwechheimer K, Rosenblum ML, Cavenee W, Vogelstein B. Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature. 1992;355:846–847. doi: 10.1038/355846a0. [DOI] [PubMed] [Google Scholar]
- Simon M, von Deimling A, Larson JL, Wellenreuther R, Kaskel P, Waha A, Warnick RE, Tew JM, Jr, Menon AG. Allelic losses on chromosomes 14, 10, and 1 in atypical and malignant meningiomas: A genetic model of meningioma progression. Cancer Res. 1995;55:4696–4701. [PubMed] [Google Scholar]
- Simon M, Park TW, Leuenroth S, Hans VH, Loning T, Schramm J. Telomerase activity and expression of the telomerase catalytic subunit, hTERT, in meningioma progression. J Neurosurg. 2000;92:832–840. doi: 10.3171/jns.2000.92.5.0832. [DOI] [PubMed] [Google Scholar]
- Simon M, Park TW, Koster G, Mahlberg R, Hackenbroch M, Bostrom J, Loning T, Schramm J. Alterations of INK4a(p16-p14ARF)/INK4b(p15) expression and telomerase activation in meningioma progression. J Neurooncol. 2001;55:149–158. doi: 10.1023/a:1013863630293. [DOI] [PubMed] [Google Scholar]
- Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatr. 1957;20:22–39. doi: 10.1136/jnnp.20.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strommer K, Hamou MF, Diggelmann H, de Tribolet N. Cellular and tumoural heterogeneity of EGFR gene amplification in human malignant gliomas. Acta Neurochir. 1990;107:82–87. doi: 10.1007/BF01405784. [DOI] [PubMed] [Google Scholar]
- Ueki K, Wen-Bin C, Narita Y, Asai A, Kirino T. Tight association of loss of merlin expression with loss of heterozygosity at chromosome 22q in sporadic meningiomas. Cancer Res. 1999;59:5995–5998. [PubMed] [Google Scholar]
- Yakut T, Bekar A, Doygun M, Acar H, Egeli U, Ogul E. Evaluation of relationship between chromosome 22 and p53 gene alterations and the subtype of meningiomas by the interphase-FISH technique. Teratogenesis Carcinog Mutagen. 2002;22:217–225. doi: 10.1002/tcm.10013. [DOI] [PubMed] [Google Scholar]
- Zattara-Cannoni H, Gambarelli D, Dufour H, Figarella D, Vollot F, Grisoli F, Vagner-Capodano AM. Contribution of cytogenetics and FISH in the diagnosis of meningiomas. A study of 189 tumors. Ann Genet. 1998;41:164–175. [PubMed] [Google Scholar]