

Abstract
The incidence of adenocarcinoma of the esophagus has been increasing in developing countries over the last three decades and probably reflects a genuine increase in the incidence of its recognized precursor lesion, Barrett’s metaplasia. Despite advances in multimodality therapy, the prognosis for invasive esophageal adenocarcinoma is poor. An improved understanding of the molecular biology of this disease may allow improved diagnosis, therapy, and prognosis. We focus on recent developments in the molecular and cell biology of Barrett’s metaplasia, a heterogeneous lesion affecting the transitional zone of the gastro-esophageal junction whose associated molecular alterations may vary both in nature and temporally. Early premalignant clones produce biological and genetic heterogeneity as seen by multiple p53 mutations, p16 mutations, aneuploidy, and abnormal methylation resulting in stepwise changes in differentiation, proliferation, and apoptosis, allowing disease progression under selective pressure. Abnormalities in expression of growth factors of the epidermal growth factor family and cell adhesion molecules, especially cadherin/catenin complexes, may occur early in invasion. Exploitation of these molecular events may lead to a more appropriate diagnosis and understanding of these lesions in the future.
Gastroesophageal reflux disease is arguably the most common medical condition in Western countries; 30% of adults complain of heartburn at least once per month. 1 Chronic esophagitis has been shown to limit physical and social activity, resulting in quality of life scores as poor as those provided by angina patients awaiting coronary bypass surgery. Forty percent of patients with esophagitis will improve spontaneously, 50% will have persistent esophagitis, and up to 10% will progress to Barrett’s esophagus (BE). 2-4 Evidence indicates that the prevalence of BE 3 and its sequelae are both increasing, especially in the sixth decade of life in males. 2 There is compelling etiological evidence that acid refluxate is the major factor in progression from benign esophagitis to BE. 1 The association between pathological acid exposure and esophagitis, especially in short segment Barrett’s metaplasia, is, however, only 60%, which suggests that in up to 40% of cases other factors like nocturnal bile, 4 nonsteroidal anti-inflammatory drugs, radiotherapy, chemotherapy, 1 caustic agents, nitrosamines, Helicobacter colonization, or familial predisposition may be causative. 5
The classic endoscopic feature of BE is the presence of salmon pink mucosa. Histologically, the presence of specialized intestinal metaplasia containing goblet cells is characteristic (Figure 1) ▶ . Short-segment Barrett’s esophagus (SSBE), ie, Barrett’s metaplasia less than 3 cm in length, is found in 8–20% of adult individuals, making it more prevalent than long-segment Barrett’s esophagus (LSBE) (1% adult prevalence) 6-10 (Table 1) ▶ . Despite this fact, only 35% of esophageal adenocarcinomas arise in SSBE; therefore, the true cancer risk in SSBE is presently unclear but probably lies between 0.03–1% 10 (Table 1) ▶ . SSBE and, less commonly, LSBE have also been reported in at least one study to be associated with the occurrence of esophago-gastric adenocarcinomas and specialized intestinal metaplasia of the gastric cardia. 10 Esophageal and gastric cardia adenocarcinomas also share many features including increasing incidence, 11,12 male gender bias, tumor histology, 13 and common antigens such as bile duct mucins and large intestinal antigens. 14 Conversely, the risk factors, incidence, histopathology, and molecular biology of esophageal adenocarcinoma differ dramatically from those of squamous cell carcinoma. In particular, squamous cell carcinoma is associated with a poor diet, 15 cigarette smoking, and low socioeconomic status, 16 whereas adenocarcinoma is associated with obesity 17 and white race and is more prevalent in Caucasians in the richer North American and European countries. 18
Figure 1.
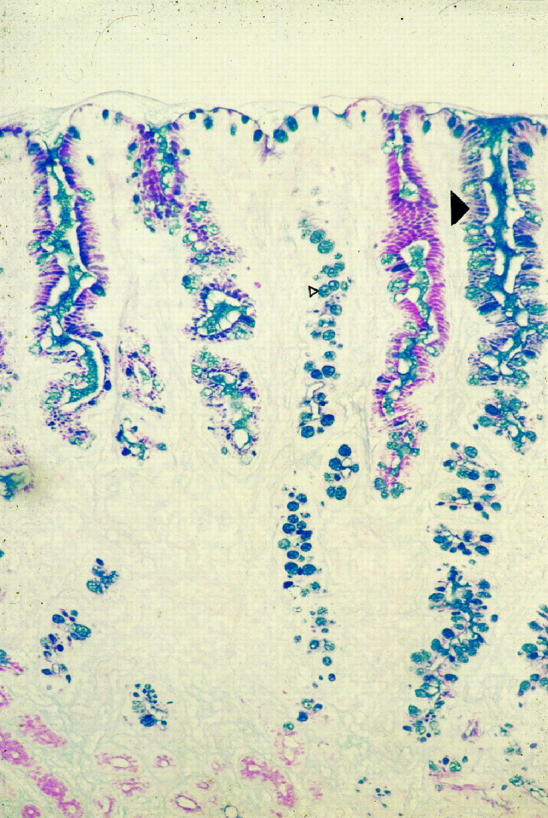
Photomicrograph of intestinal metaplasia in Barrett’s esophagus stained with Alcian blue/periodic acid-Schiff (mucins). Barrett’s esophagus is composed of columnar lined mucus-secreting cells and a proportion of the glands will be composed of goblet cells (small arrowhead). Alcian blue diastase periodic acid-Schiff staining indicates the heterogeneity of mucin phenotypes in esophageal cells: blue (basic), red (neutral), and purple (mixed) mucins (large arrowhead). Original magnification, ×250.
Table 1.
Comparison of Long Segment BE, Short Segment BE, and Specialized Intestinal Metaplasia (SIM) at the Esophago-gastric Junction
| Long segment BE | Short segment BE | SIM at the gastro-esophageal junction | Reference | |
|---|---|---|---|---|
| Prevalence | 1.3% | 8–17% | 18–36% | 6, 7 |
| GERD | +++ | ++ | − | 6 |
| Helicobacter | + | ++ | +++ | 6, 7 |
| Racial bias | Caucasian | Caucasian | None | 18 |
| Age bias | +++ | +++ | +++ | 6, 18 |
| Gender bias | Male | Male | − | 8, 9, 10 |
| Esophageal cancer | ++ | + | +/− | 6–10 |
| Gastric cancer | +/− | + | − | 10 |
| Migration* | +++ | + | − | * |
| Glandular dysplasia | ++ | + | +/− | 10 |
SIMEGJ is very common but has little association with gastro-esophageal reflux disease malignant potential.
GERD, gastro-esophageal reflux disease; +++, very common (>50% of cases); ++, moderately common (>10% of cases); +, not common (<5% of cases); −, very uncommon (<1% of cases).
*Expanding dye fronts in Barrett’s tissue after 0.5 ml intraepithelial tattooing with India ink (J. Jankowski, MD thesis, Dundee University, Dundee, UK).
To date no treatments have been shown to reverse the progression of Barrett’s esophagus completely and convincingly or to alter its natural history once it has developed. 19 Moreover, developed photodynamic therapy has recently been associated with the subsequent occurrence of unusual neoplastic lesions lying deep in the submucosa. 19 Even after prolonged high-dose proton pump inhibition or successful antireflux surgery, fewer than 10% of Barrett’s cases regress and progression to cancer may occur over a short span of 3 years. 3,20 Cancers detected in endoscopic surveillance programs have a better prognosis, characterized by 5-year survival rates of 35–45% compared with 5–15% rates in cancers occurring outside surveillance populations, even when allowing for lead bias and earlier staging of detected lesions. 21,22
Genetic and Epigenetic Events Leading to Loss of Genomic Stability
Although the colorectal adenoma-carcinoma sequence (ACS) model has become the paradigm for researchers in molecular oncology, 23 a similar mechanistic representation is only now becoming accepted for the development of Barrett’s adenocarcinoma. Barrett’s is a heterogeneous metaplasia in which 2–5% of cases will have a lifetime risk of Barrett’s adenocarcinoma, the metaplasia-dysplasia-adenocarcinoma sequence (MCS). 3 MCS differs from ACS in several important regards. First, Barrett’s metaplasia, even when dysplastic, is rarely polypoid like colorectal adenoma. This has been attributed by some researchers to the high frequency of Ki-ras and adenomatous polyposis coli gene (APC) mutations in the colon and rectum, whereas these alterations are very uncommon in Barrett’s dysplasia. However, it seems that these latter genes, while permitting polypoid growth, may not be sufficient on their own, as many tumors expressing them are nonpolypoid. Second, colorectal adenomas arise in de novo epithelium, whereas in Barrett’s esophagus premalignant lesions arise in metaplastic tissue containing goblet cells. 24 Third, Barrett’s metaplasia arises in a background of reflux-induced chronic inflammation and ulceration, 25 whereas this does not occur in the ACS. In this regard, Barrett’s neoplastic progression does bear some similarity to that seen in idiopathic inflammatory bowel disease.
The progression of Barrett’s metaplasia to adenocarcinoma is associated with several changes in gene structure, gene expression, and protein structure. 26-34 The following sequence of events is not conclusive and is presented merely to reflect the potential interplay of multiple molecular pathways in the progression to adenocarcinoma. Perhaps one of the earliest molecular events is the selection and propagation of the metaplastic clones with specialized intestinal metaplasia (Figure 2) ▶ . Subsequently, loss of cell cycle check points and genomic instability may contribute to slow clonal expansion perhaps by increasing proliferation 32,33 (Figure 3) ▶ . Inhibition of apoptosis in BE occurs late, and then only in a select proportion of cells with high grade dysplasia. Invasive cancer may be preceded by alteration of cell adhesion, 34 whereas subsequent cumulative genetic errors may result in the generation of multiple clones of transformed cells, thereby expanding the population of altered cells with an angiogenic or metastatic potential.
Figure 2.



Schematic representation of adaptation during Barrett’s mucosa formation. The three compartments of the esophageal epithelium are represented on the diagrams. The bottom of each diagram shows basal compartment containing both stem cells (speckled nucleus) and proliferating cells; the middle, parabasal compartment containing proliferating cells; and the top, superficial compartment containing only mature differentiated cells. The uninflamed mucosa (right) is flat, whereas the inflamed mucosa (left) has invaginations of the basal layer, termed papillae. A: Damage to the esophageal differentiated cells in the superficial and parabasal compartments of the esophagus. B: Damage to a deeper compartment involving the squamous epithelial stem cells in the basal compartment of the papillae. C: Generation of clones with a mucin-secreting lineage resistant to acid/bile.
Figure 3.

Schematic representation of the key molecular events in Barrett’s dysplasia, the metaplasia-dysplasia-adenocarcinoma sequence (MCS). Acid and bile cause acute damage to the esophagus, which is rapidly healed by restitution or cellular replication (stages 1 and 2). In 10% of cases chronic damage to the epithelial stem cells allows rapid clonal replacement by lineages with a growth advantage containing p53 mutations (stage 2). The formation of each type of Barrett’s metaplasia is dependent on the stem cell from which it arises as well as the nature of the mucosal microenvironment. Appearance of dysplasia is associated in part with loss of heterozygosity of APC or alterations in the catenins (stage 3). In 1 in 100 cases, aneuploidy and errors in DNA repair represent final pathways which disrupt invasion suppressor genes (stages 4 and 5). The transition from high grade dysplasia to invasive cancer is rapid in all cases.
Pathophysiology of Chronic Esophagitis: Restitution and Replication
The development of esophagitis represents the failure of many mucosal defenses of the esophagus to counteract the refluxed acid or gastroduodenal contents. 35 The buffering activity of alkaline saliva and esophageal mucus and the esophageal peristaltic clearance are, we believe, usually sufficient to prevent mucosal damage from infrequent, transient lower esophageal sphincter relaxations. If, however, reflux is frequent or prolonged, episodes of gastro-esophageal reflux occur and tissue damage results, initially affecting the cells of the superficial compartment (Figure 2) ▶ . The regenerating inflamed epithelium contains immature squamous cells that are sensitive to acid or bile damage. 36-39
The proliferative hierarchy of the normal squamous cell-lined esophagus is relatively well understood. However, the mechanisms whereby focal areas of native squamous mucosa are replaced by metaplastic tissue are less certain; we present one favored current hypothesis. One of the early adaptive responses to increased cell loss in reflux esophagitis is an increase of the proliferative zone height to maintain or increase epithelial thickness by trophic stimulation of locally produced epidermal growth factor 40 (Figure 2A) ▶ . In addition, there is also an increased proliferative zone length as a result of folding of the basal epithelium (papillae formation). 3 The functional stem cells in the basal zone at the tip of the papillae remain in a relatively superficial position 30 in the epithelium, making them far more accessible and susceptible to refluxed or ingested chemical mutagens permeating through the thin upper layers than their counterparts in the flat basal layer deeper in the mucosa (Figure 2B) ▶ . Mucosal repair occurs more rapidly when reflux disease is treated, especially with the combined actions of epithelial migration and connective tissue contraction. 34 In 10% of cases, when treatment is insufficient the mucosal breach is more quickly and effectively replaced by de novo Barrett’s metaplasia (Figure 2C) ▶ .
Formation of Metaplasia: Replacement by Metaplastic Epithelium
Although the origin of BE is a matter of conjecture, one current theory holds that the stem cells of squamous mucosa or associated glandular ducts undergo altered differentiation, producing both microvilli and intercellular ridges, and express unique glandular phenotypes distinct from adjacent mucosal gastric stem cells. 41-44 This Barrett’s metaplastic lineage may give rise to Paneth cells and neuroendocrine cells in addition to gastric and intestinal cells and is therefore pluripotent. 42 Current theory indicates that these cells give rise to intestinal-type metaplasia. However, skeptics argue that gastric-type and fundic-type metaplasias are also discernible and that the three metaplastic types may more accurately be referred to as a mosaic, although a convincing paradigm is lacking (Figure 1) ▶ . The reason behind this heterogeneity of metaplastic phenotypes is unclear but the proportion of each has been attributed in part to the composition of the refluxate (environment). 40 The appearance of metaplasia during esophageal regeneration may also theoretically be selected for by several factors, including the degree of local stem cell enrichment, clonality, 34 number of DNA adducts accrued and alterations of xenobiotic metabolizing enzymes (mutagenesis), 45 and expression of homeobox genes such as members of the cdx family (differentiation pathways) (P. Traber, personal correspondence). Phenotypic heterogeneity may also be controlled genetically because clonal divergence in chromosomes 5, 8, 9, 12, 17, and 18 in nondysplastic Barrett’s cells can also be identified. 46,47
The location and composition of the proliferative compartment in the crypts of the metaplastic epithelium are not as well defined as in columnar lined epithelia of the stomach. 42 Interestingly, the degree to which differentiation occurs varies considerably. BE that appears in childhood differs from the adult variety in that intestinal mucins and cytokeratins are not present. 48 The adult variety also has an inflammatory cell infiltrate and may have Helicobacter-like organisms, both of which are less common in juvenile metaplasia. 49 Barrett’s intestinal phenotype has higher proliferative indices; this is associated with altered expression of multiple growth factors and inducible nitric oxide synthase (iNOS; NOS-2) and cyclooxygenase-2. 21,50
The replacement by LSBE has been reported in one study to be very rapid, with the maximal proximal colonization of the esophagus occurring within 3 years of initiation. Furthermore, once formed, the rate of surface area remains constant in most individuals. Only 5–10% of cases progress in surface area and 0–2% may partially regress in surface area. 10,35 More data are required about the natural history of benign Barrett’s metaplasia before these observations can be confirmed.
Dysplasia and Aneuploidy: Clonal Expansion by Increased Cell Cycle Abnormalities and Migration
Although the true prevalence of high grade dysplasia and aneuploidy are unknown because of referral bias, they have been reported in 2–24% of individuals with Barrett’s metaplasia and have a fourfold to eightfold greater risk of developing cancer 10 compared with the more common low grade dysplasia. 51-53 These high grade dysplastic lesions may already have irreversibly progressed; at least 50% have immediately adjacent adenocarcinoma and a variable proportion of the rest may remain static for at least 1 to 3 years regardless of the presence or absence of refluxed gastric or duodenal contents. 53,54
Dysplastic cells may have proliferative controls that are relaxed or uncoupled from the appropriate regulatory cues. In part this may be a result of altered expression of cytokines and growth factors, 40 although the acquisition of genomic alterations of cell cycle-associated genes also occurs. These cell cycle genes include increased cyclin D1 expression (chromosome 11q13), 55 hypermethylated or mutated p16 (chromosome 9p21), and mobilization of cells from G0 to G1 with subsequent accumulation in the G2 phase 33 (Figure 3) ▶ . Identification of increased telomerase RNA in early dysplastic lesions including Barrett’s metaplasia has been reported. 56 p53 mutations occur in only 1–5% of metaplastic diploid cell populations but are present in most aneuploid cells, suggesting they are usually not early events. 29,57-59 Furthermore, p53 is mutated increasingly in exons 5–8 during MCS: in 5–10% of cases with indeterminate dysplasia, in 65% of those with low grade dysplasia, in 75% of cases with high grade dysplasia, and in 50–90% of esophageal adenocarcinomas, suggesting that p53 mutations occur more often later in progression. 60-62 Epigenetic alterations in the expression of growth factors and their receptors, especially of the epidermal growth factor family, are also associated with these cell cycle changes in dysplastic Barrett’s mucosa. In particular, we believe increased expression of TGFα and its precursor, prepro TGFα (uncleaved TGFα, which is membrane-bound), may stimulate epidermal growth factor receptors in dysplastic cells by autocrine and paracrine mechanisms, respectively. 27
Apoptosis may also be inhibited late in a proportion of dysplastic cells that give rise to invasive or metastatic cells. 63 The bcl-2 gene is not overexpressed, as is recognized in colorectal adenomas, although p53 mutations may affect the proliferation/apoptosis ratio in the esophagus. 64 In addition, up-regulation of immunological death factors such as Fas ligand in the epithelium may not only protect Barrett’s dysplastic cells but also may selectively destroy cytotoxic T cells by crosslinking Fas. 65
In established Barrett’s mucosa, identical clonal cytogenetic abnormalities, aneuploidy, and gene amplification 66 are identifiable in diverse locations. If this is indeed the result of the lateral migration or clonal expansion of transformed clones and not of coincidental oligoclonal or field genetic changes, then catenin-regulated transcription may be partly responsible. Interestingly, recent evidence has shown that this process in Barrett’s metaplasia may partly involve down-regulation, 67 mutation, or phosphorylation of cadherin/catenin adhesion complexes, thereby increasing free cytosolic catenin. 7,68,69 In addition, the APC gene product (chromosome 5q), which has increasing loss of heterozygosity in the dysplastic progression of Barrett’s clones, 16,47 may lead to reduced β-catenin degradation. Increased β-catenin levels have been shown to subsequently aggregate with transcription factors in the nucleus, facilitating epithelio-mesenchymal transition and increased c-myc expression. 70-73
Development of Invasive Adenocarcinoma: Generation of Tumor Heterogeneity and Invasion
The identity of the cell from which esophageal adenocarcinoma originates is speculative because there are conflicting data as to whether invasive carcinomas arise from the interactions of multiple oligoclonal lesions (field cancerization) of malignant cells or from a single distinct clone of malignant cells. 74 In this regard, early neoplasia may be histologically distinct but can be multifocal or immediately juxtaposed with dysplastic tissue. Close but discontinuous dysplastic areas may, however, have different mutations of the p53 gene (chromosome 17p), whereas dysplastic regions contiguous with cancers usually express identical p53 mutations. 62,64 Although most research in Barrett’s esophagus has focused on mutations occurring in the mutation cluster region in exons 5–8 of the p53 gene, it is conceivable that mutations in other exons or in related proteins could also affect biological function. Interpretation is further complicated by the lack of data concerning the normal clonal colonization patterns of Barrett’s crypts such as have recently been noted in the colon and termed patch size. 75 Available data may suggest that one type of p53 mutation early in the disease is not sufficient to cause adenocarcinoma, 74 although it seems that p53 mutations accumulate in cancer cells because transformed cells select for specific p53 alterations according to their biological effects. Recently, Barrett’s tumors with synchronous high grade dysplasia and invasive cancer were analyzed and showed genetic alterations that were found to be conserved in the synchronous invasive cancers. 59 These data supported the paradigm of clonal derivation of the invasive cancer from the high grade dysplasia or early invasive cancer. Although these invasive tumors may or may not possess new mutations, a proportion of high grade lesions have genetic abnormalities that may develop but are not present in the synchronous invasive cancer. This heterogeneity indicates genetic divergence during the clonal evolution of cancer, particularly at the time when high grade dysplasia progresses to invasive cancer 76 (Figure 3) ▶ . Eight other tumor suppressor gene loci have loss of heterozygosity in Barrett’s adenocarcinoma, including VHL (chromosome 3p) in 64%, APC (chromosome 5q) in 45%, CDKN2 (chromosome 9p) in 52%, the retinoblastoma gene (Rb) (chromosome 13q) in 50%, the deleted in colorectal cancer gene (chromosome 18q) in 70%, 77 and the cgene in 20% of esophageal cancers. 78 Uncharacterized candidate oncosuppressor gene loci also include 9q (60%), 11p (61%), and 17q (46%). 76,79 Some gene loss-of-heterozygosity patterns are significantly associated, such as 5q and 9p. Interestingly, the Y chromosome is lost in 9% of Barrett’s metaplasia, in 38% of cases indefinite for dysplasia, and in 100% of high grade dysplasia cases, but the significance of this is uncertain because Y chromosomal loss increases with age and in highly proliferating cells. 80
In a proportion of esophageal adenocarcinomas (5–15%) the phenomenon of ubiquitous microsatellite instability occurs in both diploid and aneuploid Barrett’s cell populations, suggesting either that it is an early mutation 27 or that these lesions have accelerated the progression to invasive cancer. The genes involved in random error of replication tumors (microsatellite-positive) are similar to colorectal cancer MLH-1 and MSH-2. 81,82 In addition, these changes may also rarely be associated with transforming growth factor β type II receptor and insulin growth factor type II receptor mutations. 83,84
Unifying Molecular Framework and Outstanding Issues Requiring Further Research
In summary, it is postulated that esophageal squamous epithelium adapts to increased chemical damage and cell loss by acute and chronic responses. In the former, the esophagus increases the growth fraction (number of cells dividing) by hyperplasia and elongation of the proliferative compartment (Figure 2) ▶ . The chronic response occurs when the initial increase in cell proliferation fails to compensate for the cell loss. There is subsequently selection of specialized lineages of columnar mucosa brought about, in part, by changes in the genotype of the relatively exposed squamous or glandular stem cells. These novel lineages have specific functions including protection against acid, protection against bile (specialized intestinal metaplasia), and repair of ulceration (the ulcer-associated cell lineage). Subsequent mutagenesis and cell cycling abnormalities followed by epithelio-mesenchymal transition may allow invasive Barrett’s cancers to develop. The range of esophageal adaptive responses to environmental stimuli is diverse. 74,77
This analysis of the molecular biology of BE explains the long latency period of cancer development as multiple genetic events are required, some gene-environment interactions as well as gene-gene interactions, particularly during regeneration. Genetic differences with the ACS such as infrequent APC and Ki-ras mutations may explain the lack of exophytic growth.
Several issues are incompletely elucidated at present. First, the origin of stem cells that give rise to Barrett’s metaplasia are unknown. Second, it is not known whether acid or indeed bile reflux is frequently necessary to initiate metaplastic formation. Third, the clonality of metaplastic glands and the tissue patch size (mucosal surface area of contiguous cells arising from the same clone) are unclear. Fourth, the nature of the mechanism governing the expansion of metaplastic glands into the proximal esophagus is ambiguous. Fifth, the natural history of dysplastic glands, especially the more common low grade dysplasia, is a matter of contention. Sixth, we do not know which biological processes are essential determinants of early invasion.
In conclusion, there is a need for improved understanding of the molecular biology of BE, particularly because the premalignant areas are often not visible endoscopically and may occur over a wider surface area compared with colorectal adenomatous polyps. Although no common and simple molecular pathway of progression is evident, we can correlate the pathophysiology with specific molecular alterations (Figure 3) ▶ . The corollary is that molecular genetics, when applied to histological material, will also increase accurately our knowledge of the natural history of specific lesions found in the metaplasia-dysplasia-adenocarcinoma sequence.
Acknowledgments
We thank everybody whose comments or ideas helped to focus important issues, in particular Dr. Fiona Bedford of the Molecular Cell Biology Institute of University College (London) and Dr. Neil Shepherd of the Department of Pathology of the Gloucester Royal Hospital (Gloucester, UK).
Footnotes
Address reprint requests to Janusz A Jankowski, M.D., Ph.D., Epithelial Laboratory, Institute for Cancer Studies, University Hospital, Birmingham, B15 2TJ, United Kingdom. E-mail: j.jankowski@bham.ac.uk.
Supported by grants from the U.S. Department of Veterans Affairs, the National Institutes of Health, the Imperial Cancer Research Fund (UK), and the Cancer Research Campaign (UK).
On behalf of the Molecular Biology representatives of the International Society for Diseases of the Esophagus (ISDE), the International Organisation for Statistical Studies on Diseases of the Esophagus (OESO) and the Oesophageal Section of the British Society of Gastroenterology (BSG).
References
- 1.Spechler SJ: Barrett’s esophagus. Sem Oncol 1994, 21:431-437 [PubMed] [Google Scholar]
- 2.Winter C, Jr, Spurling TJ, Chobabian SJ, Curtis DJ, Esposito RL, Hacker JF, III, Johnston DA, Cruess DF, Cotelingam JD, Gurney MS, Cattau EL, Jr: Barrett’s esophagus: a prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987, 92:118-124 [PubMed] [Google Scholar]
- 3.Prach AT, MacDonald TA, Hopwood DA, Johnston DA: Increasing incidence of Barrett’s oesophagus: education, enthusiasm or epidemiology. Lancet 1997, 350:933. [DOI] [PubMed] [Google Scholar]
- 4.Beynon J, Pye JK, Howell P, Nehra D: Assessment of combined bile acid and pH profiles using an automated sampling device in gastro-oesophageal reflux disease. Br J Surg 1998, 85:134-137 [DOI] [PubMed] [Google Scholar]
- 5.Romero Y, Cameron AJ, Locke GR, Schaid DJ, Melton LJ, Slexak J: Familial gastroesophageal reflux: association with Barrett’s esophagus and adenocarcinoma. Gastroenterology 1997, 110:456. [DOI] [PubMed] [Google Scholar]
- 6.Spechler SJ, Zeroogian JM, Antonioli DA, Wang HH, Goyal RK: Prevalence of metaplasia at the gastro-oesophageal junction. Lancet 1994, 344:1533-1536 [DOI] [PubMed] [Google Scholar]
- 7.Johnston MH, Hammond AS, Laskin W, Jones DM: The prevalence and clinical characteristics of short segments of specialised intestinal metaplasia in the distal esophagus on routine endoscopy. Am J Gastroenterol 1996, 91:1507-1511 [PubMed] [Google Scholar]
- 8.Cameron AJ, Lomboy CT: Barrett’s esophagus: age, prevalence, and extent of columnar epithelium. Gastroenterology 1992, 103:1241-1245 [DOI] [PubMed] [Google Scholar]
- 9.Chalasani N, Wo JM, Hunter JG, Waring JP: Significance of intestinal metaplasia in different areas of esophagus including the esophagogastric junction. Dig Dis Sci 1997, 42:603-607 [DOI] [PubMed] [Google Scholar]
- 10.Morales TG, Bhattacharyya A, Johnson C, Sampliner RE: Is Barrett’s esophagus associated with intestinal metaplasia of the gastric cardia? Am J Gastroenterol 1997, 92:1818-1822 [PubMed] [Google Scholar]
- 11.Kim R, Weissfeld JL, Reynolds JC, Keller LH: Etiology of Barrett’s metaplasia and esophageal adenocarcinoma. Cancer Epidem Biomarkers Prevent 1997, 6:369-377 [PubMed] [Google Scholar]
- 12.Powell J, McConkey CE: The rising trend in oesophageal adenocarcinoma and gastric cardia cancer. Eur J Cancer Prev 1992, 1:265-269 [DOI] [PubMed] [Google Scholar]
- 13.Paraf F, Flejou JF, Pignon JP, Fekete F, Potet F: Surgical pathology of arising in Barrett’s-esophagus: analysis of 67 cases. Am J Surg Pathol 1995, 19:183-191 [DOI] [PubMed] [Google Scholar]
- 14.Das KM, Prasad I, Garla S, Amenta PS: Detection of a shared colon epithelial epitope on Barrett’s epithelium by a novel monoclonal antibody. Ann Int Med 1994, 120:753-756 [DOI] [PubMed] [Google Scholar]
- 15.Tuyns AJ: Oesophageal cancer in France and Switzerland: recent time trends. Eur J Cancer Prev 1992, 1:275-278 [DOI] [PubMed] [Google Scholar]
- 16.Day NE, Munoz N, Ghadirian P: Epidemiology of esophageal cancer: a review. Correa P Haenszel W eds. Epidemiology of Cancer of the Digestive Tract. 1982, :pp 21-57 The Hague, Martinus Nijhoff [Google Scholar]
- 17.Brown LM, Swanson CA, Gridley G, Swanson GM, Schoenberg JB, Greenberg RS, Silverman DT, Pottern LM, Hayes RB, Schwartz AG, Liff JM, Fraumeni JF, Hoover RN: Adenocarcinoma of the esophagus: role of obesity and diet. J Natl Cancer Inst 1995, 87:1104-1109 [DOI] [PubMed] [Google Scholar]
- 18.Blot WJ, Devesa SS, Kneller RW, Fraumeni JF: Rising incidence of adenocacinoma of the esophagus and gastric cardia. J Am Med Asso 1993, 265:1287-1289 [PubMed] [Google Scholar]
- 19.Barr H, Shepherd NA, Dix A, Roberts DJH, Tan WC, Krasner N: Eradication of high grade dysplasia in columnar-lined esophagus by photodynamic therapy with endogenously generated photoporphyrin-IX. Lancet 1996, 348:584-585 [DOI] [PubMed] [Google Scholar]
- 20.Sagar PM, Ackroyd R, Hosie KB, Patterson JE, Stoddard CJ, Kingsnorth AN: Regression and progression of Barrett’s oesophagus after antireflux surgery. Br J Surg 1995, 82:806-810 [DOI] [PubMed] [Google Scholar]
- 21.Provenzale D, Kemp JA, Arora S, Wong JB: A guide for surveillance of patients with Barrett’s esophagus. Am J Gastroenterol 1994, 89:670-680 [PubMed] [Google Scholar]
- 22.Streitz JM, Andrews CW, Ellis FH: Endoscopic surveillance of Barrett’s esophagus. Does it help? J Thorac Cardiovasc Surg 1993, 105:383-387 [PubMed] [Google Scholar]
- 23.Fearon E, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990, 61:759-767 [DOI] [PubMed] [Google Scholar]
- 24.Weinstein WM, Ippoliti AF: The diagnosis of Barrett’s esophagus: goblets, goblets, goblets. Gastrointest Endosc 1996, 44:91-94 [DOI] [PubMed] [Google Scholar]
- 25.Mason RJ, Bremner CG: Gastritis in Barrett’s esophagus. World J Surg 1995, 19:96-101 [DOI] [PubMed] [Google Scholar]
- 26.Jankowski J, Coghill G, Hopwood D, Wormsley K: Oncogenes and anti-oncogenes in adenocarcinoma of the oesophagus. Gut 1992, 33:1033-1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brito M, Filipe MI, Linehan J, Jankowski J: Transforming growth factor α expression in gastro-oesophageal tumorigenesis may reflect altered processing of the precusor peptide. Int J Cancer 1995, 60:27-327814148 [Google Scholar]
- 28.Blount PL, Galipeau PC, Sanchez CA, Neshat K, Levine DS, Yin J, Suzuki H, Abraham JM, Meltzer SJ: Analyses in diploid cells of patients with Barrett’s esophagus who develop aneuploidy. Cancer Res 1994, 54:2292-2295 [PubMed] [Google Scholar]
- 29.Blount PL, Meltzer SJ, Yin J, Huang Y, Krasna MJ, Reid BJ, Blount PL, Meltzer SJ, Yin J, Huang Y, Krasna MJ, Reid BJ: Clonal ordering of 17p and 5q allelic losses in Barrett’s dysplasia and adenocarcinoma. Proc Natl Acad Sci USA 1993, 90:3221-3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jankowski J, Austin W, Howat K, Coghill G, Dover R, Hopwood D, Wormsley K: Proliferating cell nuclear antigen labelling in the oesophagus; correlation with autoradiography. Eur J Gastroenterol Hepatol 1992, 4:579-584 [Google Scholar]
- 31.Boynton RF, Blount PL, Yin J, Brown VL, Huang Y, Tong Y, McDaniel T, Newkirk C, Resau JH, Raskind WH, Haggitt RC, Reid BJ, Meltzer SJ: Loss of heterozygosity involving the APC and MCC genetic loci occurs in the majority of human esophageal cancers. Proc Natl Acad Sci USA 1992, 89:3385-3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, Boynton RF, Blount PL, Silverstein RJ, Yin J, Tong Y, McDaniel TK, Newkirk C, Resau JH, Sridhara R, Reid BJ, Meltzer SJ: Loss of heterozygosity involves multiple tumour suppressor genes in human esophageal cancers. Cancer Res 1992, 52:6525-6530 [PubMed] [Google Scholar]
- 33.Reid BJ, Sanchez CA, Blount PL, Levine DS: Barrett’s esophagus: cell cycle abnormalities in advancing stages of neoplastic progression. Gastroenterology 1993, 105:119-129 [DOI] [PubMed] [Google Scholar]
- 34.Bailey T, Biddleston L, Shepherd N, Barr H, Warner P, Jankowski J: Altered cadherin/catenin complexes in the dysplasia-adenocarcinoma sequence: correlation with disease progression and dedifferentiation. Am J Pathol 1998, 152:135-144 [PMC free article] [PubMed] [Google Scholar]
- 35.Spechler SJ, Goyal RK: The columnar-lined esophagus, intestinal metaplasia and Norman Barrett. Gastroenterology 1996, 110:6114-6621 [DOI] [PubMed] [Google Scholar]
- 36.Hopwood D, Milne G, Bouchier I: Effect of bile acids and hydrogen ions on the fine ultrastructure of the oesophageal epithelium. Gut 1981, 22:305-311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tobey NA, Orlando RC: Mechanisms of acid injury to rabbit esophageal epithelium. Gastroenterology 1991, 101:1220-1228 [DOI] [PubMed] [Google Scholar]
- 38.Kaver WKH, Peters JH, Demeester TR, Ireland AP, Bremner CG, Hagen JA: Mixed reflux of gastric and duodenal juices is more harmful to the esophagus than gastric juice alone: the need for surgical therapy re-emphasised. Ann Surg 1995, 222:525-533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaezi MF, Richter JE: Role of acid and duodenogastric reflux in esophageal mucosal injury: a review of animal and human studies. Gastroenterology 1995, 108:1897-1907 [DOI] [PubMed] [Google Scholar]
- 40.Jankowski J: Gene expression in Barrett’s mucosa: modulation of acute and chronic adaptive respones. Gut 1993, 34:1012-1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanby A, Jankowski J, Elia G, Poulsom R, Wright NA: Expression of the trefoil peptides pS2 and human spasmolytic polypeptide (hSP) in Barrett’s metaplasia and the native oesophageal epithelium differentiates secretory lineages. J Pathol 1994, 168:210-216 [DOI] [PubMed] [Google Scholar]
- 42.Jankowski J, Wright NA: Epithelial stem cells in the gastrointestinal tract: structure, function and adaptation. Sem Cell Biol 1992, 3:445-456 [DOI] [PubMed] [Google Scholar]
- 43.Shields HM, Sawhney RA, Zwas F, Boch JA, Kim S, Goran D, Antonioli DA: Scanning electron microscopy of the human esophagus: application to Barrett’s esophagus, a precancerous lesion. Microsc Res Tech 1995, 31:248-256 [DOI] [PubMed] [Google Scholar]
- 44.Boch JA, Shields HM, Antonioli DA, Zwas F, Sawhney RA, Trier JS: Distribution of cytokeratin markers in Barrett’s specialised columnar epithelium. Gastroenterology 1997, 112:760-765 [DOI] [PubMed] [Google Scholar]
- 45.Patel HR, Hewer A, Phillips DH, Hayes JD, Wolf CR, Campbell FC: Metabolic competence and susceptibility of intestinal epithelium to genotoxic injury during regeneration. Carcinogenesis 1997, 18:2171-2177 [DOI] [PubMed] [Google Scholar]
- 46.MacKay CK, Stuart RC, Going J, Baxter JN, Keith WN: Interphase cytogenetics of non-dysplastic Barrett’s esophagus. Gastroenterology 1997, 112:A607 [Google Scholar]
- 47.Wu TT, Watanabe T, Heitmiller R, Zahurak M, Forastiere AA, Hamilton SR: Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction region. Am J Pathol 1998, 153:287-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu GD, Beer DG, Moore JH, Orringer MB, Appelamn HD, Traber PG: Sucrase-isomaltase gene expression in Barrett’s esophagus and adenocarcinoma. Gastroenterology 1993, 105:837-844 [DOI] [PubMed] [Google Scholar]
- 49.Weston AP, Cherian R, Horum RT, Lawrinenko V, Dixon A, McGregor D: Mucosa associated lymphoid tissue (MALT) in Barrett’s esophagus: prospective evaluation and association with gastric MALT, MALT lymphoma and HLO’s. Am J Gastroenterol 1997, 92:800-804 [PubMed] [Google Scholar]
- 50.Wilson KT, Fu S, Ramanujam KS, Meltzer SJ: Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in Barrett’s esophagus and associated adenocarcinomas. Cancer Res 1998, 58:2929-2934 [PubMed] [Google Scholar]
- 51.Wright TA: High grade dysplasia in Barrett’s metaplasia. Br J Surg 1997, 84:760-766 [PubMed] [Google Scholar]
- 52.Altorki NK, Sunagawa M, Little AG, Skinner DB: High grade dysplasia in the columnar-lined esophagus. Am J Surg 1991, 161:97-99 [DOI] [PubMed] [Google Scholar]
- 53.Levine DS, Haggitt RC, Blount PL, Weinstein WM, Reid BR: An endoscopic biopsy protocol can differentiate high grade dysplasia from early adenocarcinoma in Barrett’s esophagus. Gastroenterology 1995, 105:40-50 [DOI] [PubMed] [Google Scholar]
- 54.Reid BJ, Blount PL, Rubin CE, Levine DS, Haggitt RC, Rabinovitch PS: Flow-cytometric and histological progression to malignancy in Barrett’s esophagus: prospective endoscopic surveillance of a cohort. Gastroenterology 1992, 102:1212-1219 [PubMed] [Google Scholar]
- 55.Coppola D, Falcone R, Livingston S, Karl R, Nicosia S, Cacho CM: Cyclin D1 expression correlates with degrees of dysplasia in Barrett’s esophagus. Lab Invest 1997, 76:298-302 [Google Scholar]
- 56.Morales CP, Lee EL, Shay JW: In situ hybridisation for the detection of telomerase RNA in the progression from Barrett’s esophagus to esophageal adenocarcinoma. Cancer 1998, 83:652-659 [PubMed] [Google Scholar]
- 57.Wong DJ, Barrett MT, Stoger R, Emond MJ, Reid BJ: p16 (ink4) promoter is hypermethylated at a high frequency in esophageal adenocarcinoma. Cancer Res 1997, 57:2619-2622 [PubMed] [Google Scholar]
- 58.Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, Levine DS, Rabinovitch PS, Reid BJ: 17p (p53) allelic losses, 4N (G(2)/tetraploid) populations, and progression to aneuploidy in BE. Proc Natl Acad Sci USA 1996, 93:7081-7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neshat K, Sanchez CA, Galipeau PC, Blount PL, Levine DS, Joslyn G, Reid BJ: p53 mutations in Barrett’s adenocarcinoma and high grade dysplasia. Gastroenterology 1994, 106:1589-1595 [DOI] [PubMed] [Google Scholar]
- 60.Casson AG, Mukhopadhay T, Cleary KR, Ro JY, Levin B, Roth JA: p53 gene mutations in Barrett’s epithelium and esophageal cancer. Cancer Res 1991, 51:4495-4499 [PubMed] [Google Scholar]
- 61.Younes M, Lebovitz RM, Lechago LV, Lechago J: p53 protein accumulation in Barrett’s metaplasia, dysplasia and carcinoma: a follow up study. Gastroenterology 1993, 105:1637-1642 [DOI] [PubMed] [Google Scholar]
- 62.Hamelin R, Flejou JF, Muzeau F, Potet F, Laurent-Purg P, Fekete F, Thomas G: TP53 mutations, and p53 protein immunoreactivity in malignant and premalignant Barrett’s oesophagus. Gastroenterology 1994, 107:1012-1018 [DOI] [PubMed] [Google Scholar]
- 63.Katada N, Hinder RA, Smyrk TC, Hirabayashi N, Perdikis G, Lund RJ, Woodward T, Klinger PJ: Apoptosis is inhibited ealy in the dysplasia-carcinoma sequence of Barrett’s esophagus. Arch Surg 1997, 132:728-732 [DOI] [PubMed] [Google Scholar]
- 64.Goldblum JR, Rice TW: Bcl-2 protein expression in Barrett’s metaplasia, dysplasia carcinoma sequence. Mod Pathol 1995, 8:866-869 [PubMed] [Google Scholar]
- 65.Fan XJ, Crowe SE, Bamford K, Van Houten N, Reyes VE, Ernst PB: Fas-mediated apoptosis of gastric epithelial cells. Gastroenterology 1997, 110:A13 [Google Scholar]
- 66.Raskind WH, Norwood T, Levine DS, Haggitt RC, Rabinovitch PS, Reid BJ: Persistent clonal areas and clonal expansion in Barrett’s esophagus. Cancer Res 1994, 52:2946-2950 [PubMed] [Google Scholar]
- 67.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW: Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275:1787-1790 [DOI] [PubMed] [Google Scholar]
- 68.Zhuang Z, Vortmeyer AD, Mark RJ, Odze R, Emmert-Buch MR, Merino MJ, Moen H, Liotta CA, Durage OH: Barrett’s esophagus metaplastic cells with loss of heterozygosity at the APC gene loci are clonal precursors to invasive adenocarcinoma. Cancer Res 1996, 56:1961-1964 [PubMed] [Google Scholar]
- 69.Powell S, Papadopoulos N, Kinzler KW, Smolinski K, Meltzer SJ: Truncating APC gene mutations in the mutation cluster region are rare in esophageal cancers. Gastroenterology 1994, 107:1759-1763 [DOI] [PubMed] [Google Scholar]
- 70.Jankowski J, Bruton R, Shepherd N, Sanders S: Catenin regulated transcription provides a global mechanism for cancer progression. Mol Pathol 1997, 50:1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H: Depletion of epithelial stem cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 1998, 19:379-383 [DOI] [PubMed] [Google Scholar]
- 72.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW: Identification of c-MYC as a target of the APC pathway. Science 1998, 281:1509-1512 [DOI] [PubMed] [Google Scholar]
- 73.Hsu SC, Galceran J, Grosschedl R: Modulation of transcriptional regulation by LEF-1 in response to Wnt-1 signaling and association with β-catenin. Mol Cell Biol 1998, 8:4807-4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jankowski J: Clonality of esophageal carcinomas: genetic and epigenetic events leading to loss of genomic stability. Dis Esophagus 1997, 10:143-144 [DOI] [PubMed] [Google Scholar]
- 75.Novelli MR, Williamson JA, Tomlinson IP, Elai G, Hodgson SV, Talbot IC, Bodmer WF, Wright NA: Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science 1996, 272:1187-1190 [DOI] [PubMed] [Google Scholar]
- 76.Montesano R, Hollstein M, Hainaut P: Genetic alterations in esophageal cancer and their relevance to etiology and pathogenesis: a review. Int J Cancer 1996, 69:225-235 [DOI] [PubMed] [Google Scholar]
- 77.Reid BJ, Barrett MT, Galipeau PC, Sanchez CA, Neskat K, Cowan DS, Levine DS: Barrett’s esophagus: ordering the events that lead to cancer. Eur J Cancer Prevent 1996, 52:57-65 [DOI] [PubMed] [Google Scholar]
- 78.Dolan K, Garde J, Gosney J, Sissons M, Wright T, Kingsnorth AN, Walker SJ, Sutton R, Meltzer SJ, Field JK: Alleotype analysis of oesophageal adenocarcinoma: loss of heterozygosity occurs at multiple sites. Br J Cancer 1998, 78:950-957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aoki T, Mori T, Du X, Nishihira T, Marsbra T, Nakamura Y: Alleotype study of oesophageal cancer. Genes Chromosomes Cancer 1994, 10:177-182 [DOI] [PubMed] [Google Scholar]
- 80.Barrett MT, Galipeau PC, Sanchez CA, Emond MJ, Reid BJ: Determination of the frequency of loss of heterozygosity in esophageal adenocarcinoma by cell sorting, whole genome amplification and microsatellite polymorphisms. Oncogene 1996, 12:1873-1878 [PubMed] [Google Scholar]
- 81.Meltzer SJ, Yin J, Manin B, Rhyu MG, Cottrell J, Hudson E, Redd JL, Krasna MJ, Abraham JM, Reid BJ: Microsatellite instability occurs frequently and in both diploid and aneuploid cell populations of Barrett’s associated esophageal adenocarcinomas. Cancer Res 1994, 54:3379-3382 [PubMed] [Google Scholar]
- 82.Gleeson CM, Sloan JM, McGuigan JA, Ritchie AJ, Weber JL, Russell SH: Ubiquitous somatic alterations at microsatellite alleles occur infrequently in Barrett’s associated esophageal adnocarcinoma. Cancer Res 1996, 56:259-26 [PubMed] [Google Scholar]
- 83.Garrigue-Antar L, Souza RF, Vellucci VF, Meltzer SJ, Reiss M: Loss of transforming growth factor β type II receptor gene expression in human esophageal cancer. Lab Invest 1996, 75:263-272 [PubMed] [Google Scholar]
- 84.Wang S, Souza RF, Kong D, Yin J, Smolinski KN, Zou TT, Frank T, Young J, Flanders KC, Sugimura H, Abraham JM, Meltzer SJ: Deficient transforming growth factor-β1 activation and excessive insulin-like growth factor II (IGFII) expression in IGFII receptor-mutant tumors. Cancer Res 1997, 57:2543-2546 [PubMed] [Google Scholar]