

Abstract
Ovarian cancer is characterized by the rapid growth of solid intraperitoneal tumors and large volumes of ascitic fluid. Vascular endothelial growth factor (VEGF) augments tumor growth by inducing neovascularization and may stimulate ascites formation by increasing vascular permeability. We examined the role of VEGF in ovarian carcinoma using in vivo models in which intraperitoneal or subcutaneous tumors were induced in immunodeficient mice using the human ovarian carcinoma cell line SKOV-3. After tumor engraftment (7 to 10 days), some mice were treated with a function-blocking VEGF antibody (A4.6.1) specific for human VEGF. A4.6.1 significantly (P < 0.05) inhibited subcutaneous SKOV-3 tumor growth compared with controls. However, tumor growth resumed when A4.6.1 treatment was discontinued. In mice bearing intraperitoneal tumors (IP mice), ascites production and intraperitoneal carcinomatosis were detected 3 to 7 weeks after SKOV-3 inoculation. Importantly, A4.6.1 completely inhibited ascites production in IP mice, although it only partially inhibited intraperitoneal tumor growth. Tumor burden was variable in A4.6.1-treated IP mice; some had minimal tumor, whereas in others tumor burden was similar to that of controls. When A4.6.1 treatment was stopped, IP mice rapidly (within 2 weeks) developed ascites and became cachectic. These data suggest that in ovarian cancer, tumor-derived VEGF is obligatory for ascites formation but not for intraperitoneal tumor growth. Neutralization of VEGF activity may have clinical application in inhibiting malignant ascites formation in ovarian cancer.
Angiogenesis, the development of new blood vessels from existing vasculature, is an essential component of solid tumor growth and metastasis. 1-5 It is now generally accepted that solid tumor growth must be accompanied by angiogenesis to provide the vascular support essential for the expanding tumor mass. Several angiogenic factors are expressed by many tumors, suggesting that tumors promote their own vascularization by activating the host endothelium. The importance of angiogenesis in tumor progression is indicated by studies showing that the angiogenic potential of tumors, assessed by tumor microvessel density, directly correlates with poor prognosis. 6-11 However, the mechanism of solid tumor angiogenesis at the molecular level is not well understood, and the relative importance of specific angiogenic factors in mediating vasculogenesis in specific malignancies is not well defined.
One angiogenic factor that is thought to play a key role in the vascularization of normal and neoplastic tissue is vascular endothelial growth factor (VEGF), also known as vascular permeability factor. VEGF is a potent and specific mitogen for endothelial cells, 12-17 stimulates the full cascade of events required for angiogenesis in vitro and in vivo, 17,18 and markedly augments the permeability of existing microvasculatature. 19-21 VEGF is expressed in many animal and human malignancies and by most transformed cells lines. 21-41 The effect of VEGF on vascular permeability is believed to be crucial for malignant ascites formation. 19,42,43 The actions of VEGF are mediated by at least two cell surface receptors, flt-1 and KDR. 44,45 The central role of VEGF in tumor growth has been demonstrated in studies using animal models in which tumor growth and vascularization in vivo were inhibited if VEGF activity was neutralized by function-blocking antibodies 46 or expression of antisense VEGF mRNA, 47 or if signaling was disrupted by dominant-negative mutation of the KDR receptor. 48
Ovarian cancer is characterized by widespread intraperitoneal carcinomatosis and the formation of large volumes of ascitic fluid. 49 VEGF may play a major role in the progression of ovarian cancer by influencing tumor growth through its promotion of tumor angiogenesis and ascites production through its stimulation of vascular permeability. Although VEGF has been detected in ovarian cancer, 26,37,50,51 so, too, have most other known angiogenic factors; 37,52-59 therefore, the role of VEGF as a regulator of angiogenesis in ovarian cancer growth is unclear. However, several studies have indicated that VEGF-regulated angiogenesis is an important component of ovarian cancer growth. Microvessel density and the level of VEGF expression in ovarian cancer directly correlate with poor prognosis, suggesting that angiogenesis, possibly mediated at least in part by VEGF, influences disease progression. 26,50,51 In a murine model of ovarian cancer, the drug FR118487, which inhibits angiogenesis by inhibiting basic fibroblast growth factor and VEGF activities, 60 suppressed the in vivo growth and metastasis of a murine ovarian cancer cell line. 61 In the present study, we directly assessed the role of VEGF in the growth and progression of ovarian cancer. To that end, we used the human ovarian carcinoma cell line SKOV-3 to develop an in vivo model of ovarian cancer in immunodeficient mice that recapitulated the intraperitoneal carcinomatosis and ascites production seen in women with this disease. We then used a function-blocking monoclonal antibody, which blocks access of VEGF to both the flt-1 and KDR receptors, to specifically inhibit tumor-derived VEGF activity and assessed the consequences on tumor growth, ascites formation, and disease progression.
Materials and Methods
Materials
A mouse monoclonal antibody (A4.6.1) directed against human VEGF was used to neutralize VEGF activity in vivo. Characterization of this antibody, including its high specificity toward human VEGF and its ability to inhibit VEGF activity in vitro and in vivo, as well as to block binding of VEGF to its receptors in vivo, has been described previously. 46,62 The antibody does not inhibit the activity of mouse VEGF (unpublished data). The human ovarian cystadenocarcinoma cell line, SKOV-3, was obtained from the American Type Culture Collection (Manassas, VA). One-month-old female immunodeficient mice (BALB/c nu/nu) were obtained from Simonsen Laboratories (Gilroy, CA), housed in isolated conditions, and fed autoclaved standard pellets and water. All protocols involving immunodeficient mice were approved by the Committee on Animal Care, University of California, San Francisco.
Cell Culture
The SKOV-3 cells were cultured in Dulbecco’s modified Eagle’s medium H-21 containing 10% fetal calf serum, glucose (4.5 g/L), penicillin G (100 U/ml), streptomycin (2.5 μg/ml), glutamine (2 mmol/L), and fungizone (2.5 μg/ml). All cell culture reagents were obtained from the Cell Culture Facility, University of California, San Francisco. Before in vivo inoculation, SKOV-3 cells were grown to confluence, harvested by trypsinization, and resuspended in Ca2+/Mg2+-free phosphate buffered saline (PBS). In preliminary studies, we determined that SKOV-3 cells express VEGF in vivo and in vitro using reverse transcription-polymerase chain reaction and immunocytochemistry, respectively, and that A4.6.1 does not affect their proliferation in vitro (data not shown).
In Vivo Inoculation of SKOV3 Cells
The SKOV-3 cells were prepared for inoculation as described above and injected as a bolus either into the peritoneum (IP group; n = 31; 10 × 10 6 cells per mouse in 200 μl of PBS) or into the dorsal subcutaneous tissue (SC group; n = 8; 5 to 10 × 10 6 cells in 50 μl of PBS) of athymic mice. Some SC mice received two boluses of SKOV-3 cells, one in each flank. Seven to 10 days after SKOV-3 inoculation, some of the mice (IP group, n = 16; SC group, n = 5) were treated with A4.6.1 (100 μg in 0.1 ml of PBS, intraperitoneally, twice per week), and the rest were treated with the same volume of vehicle. A4.6.1 treatment was delayed to ensure that tumor engraftment was not inhibited. The size of subcutaneous tumors was measured twice weekly using calipers fitted with a Vernier scale. For each tumor, two perpendicular measurements were obtained from which an estimate of tumor radius was derived. Tumor volume was then calculated based on the assumption that tumors were spherical. At the end of the experimental period, mice were killed by anesthetic overdose. At autopsy, SKOV-3 tumors were excised, fixed in 4% paraformaldehyde/100 mmol/L PBS, pH 7.4, at 4°C for 24 hours, and embedded in paraffin. Paraffin sections (10 μm) were used for histochemical analysis.
Statistics
Data were analyzed using the unpaired Student’s t-test for statistical comparison between groups. Differences between groups were considered statistically significant at P < 0.05. Experiments were performed in triplicate.
Results
Experimental Model of Human Ovarian Cancer
All IP mice receiving PBS treatment (ie, control mice) developed a swollen abdomen, indicative of ascites formation and intraperitoneal carcinomatosis, within 3 to 6 weeks of SKOV-3 administration. Soon after (within 5 to 7 days) the appearance of abdominal swelling, PBS-treated IP mice became cachectic and as a consequence were euthanized in accordance with the animal care protocol. The intraperitoneal carcinomatosis in the IP mice closely resembled peritoneal metastases from poorly differentiated stage III papillary serous ovarian cancer. 63 The neoplasms were characterized by trabecular and solid patterns of growth with variable degrees of cytological atypia. Numerous mitotic figures were identified, and papillary growth was common on the luminal side. In most tumors, desmoplastic stroma typical of ovarian cancer metastases was seen. Tumors were found on the surfaces of the peritoneum, diaphragm, intestines (Figure 1) ▶ , uterus and associated fat, and stomach. Tumors were rarely found on the liver or spleen, and there was no evidence of visible metastasis to organs outside of the peritoneum. In general, the tumors did not invade the host tissue to which they were adherent; however, some focal invasion into muscle was seen. Extensive tumor growth was detected in the uterine fat, and foci of tumor were observed in the uterine lymphatics. The sites and extent of the peritoneal carcinomatosis and the production of ascitic fluid induced by intraperitoneal administration of SKOV-3 cells in immunodeficient mice were similar to those seen in women with ovarian epithelial cancer.
Figure 1.
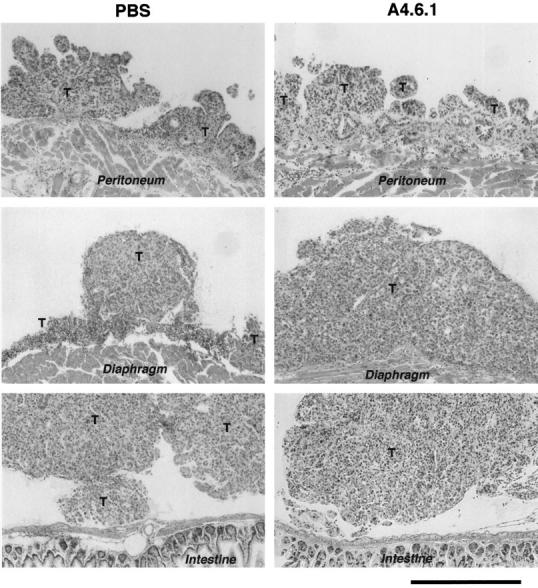
Histological appearance of intraperitoneal SKOV-3 tumors (T) derived from PBS-treated and antibody-treated (A4.6.1) mice approximately 5 weeks after intraperitoneal cell inoculation. H&E staining of 10-μm paraffin sections. Bar = 500 μm.
Role of VEGF in SKOV-3 Tumor Growth
To examine directly the role of tumor-derived VEGF in SKOV-3 tumor growth, we established an in vivo model in which SKOV-3 tumors were grown subcutaneously in immunodeficient mice. Well defined subcutaneous tumors developed within 7 days of SKOV-3 inoculation and were of sufficient size to permit accurate measurement. Tumor growth was rapid, and within 3 weeks the subcutaneous foci were 5 to 10 mm in diameter and began to exhibit vascular islands that eventually formed blood-filled cysts. By 6 weeks, the largest tumors were approximately 20 mm in diameter and contained numerous cysts that eventually ruptured. At this stage of tumor progression, the mice were killed.
To assess the role of VEGF in tumor growth, some SC mice (n = 5 mice; 11 tumors) were treated with A4.6.1. Control mice (n = 3 mice; 6 tumors) were treated with PBS. In preliminary studies, we found that treatment with a nonspecific antibody of the same IgG type had no effect on tumor growth and was essentially equivalent to vehicle alone (data not shown). A4.6.1 significantly inhibited the growth of subcutaneous SKOV-3 tumors within 2 weeks of treatment (Figure 2) ▶ ; tumor size did not progress beyond the size attained at the initiation of A4.6.1 treatment and the tumors did not form cysts. After cessation of A4.6.1 treatment (Figure 2B) ▶ , tumor growth resumed, and within 2 weeks the tumors developed blood-filled cysts and the mice had to be killed. In preliminary studies in which fewer cells were used for subcutaneous inoculation, after discontinuance of A4.6.1 treatment, tumor growth resumed and within 3 weeks the rate of growth paralleled that of controls. Tumor growth also was inhibited when A4.6.1 was administered late in tumor progression (30 days) (Figure 2B) ▶ . Interestingly, in these tumors A4.6.1 appeared to deplete the contents of the cysts; their volume was markedly reduced, and some cysts involuted without rupture. In addition, tumors lost their red coloration and became more skin toned in appearance.
Figure 2.
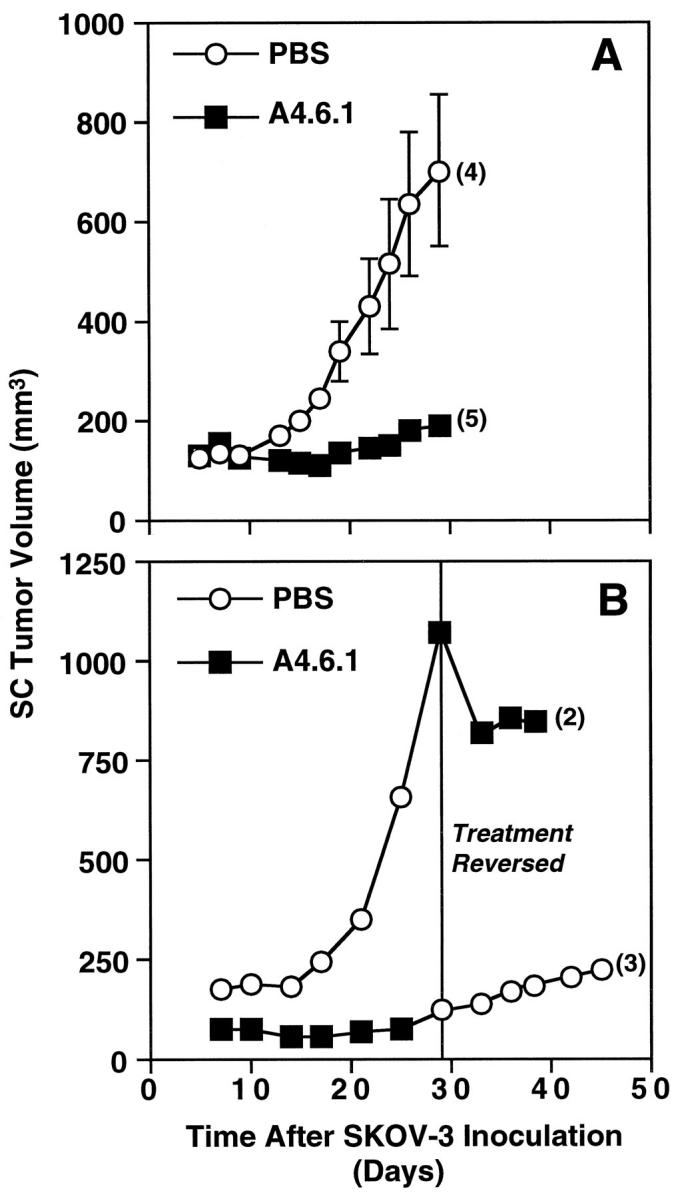
Effect of neutralization of tumor-derived VEGF activity on the growth of subcutaneous SKOV-3 tumors. Mice were treated with either PBS or antibody (A4.6.1, 100 μg) intraperitionally twice weekly for approximately 4 weeks. In A, all mice were killed at 30 days. In B, treatments were reversed at day 29. Numbers in parentheses represent the number of tumors analyzed in each group.
There was no evidence that A4.6.1 induced an antibody-dependent cellular cytotoxicity or a macrophage-mediated response that could have inhibited tumor growth. Histological examination of growth-inhibited subcutaneous tumors failed to demonstrate any significant inflammatory response (data not shown).
Role of VEGF in Intraperitoneal Tumor Growth
Studies of subcutaneous SKOV-3 tumor growth established the pivotal role of VEGF in tumor progression. However, as ovarian cancer is not a subcutaneous malignancy, we studied the effects of A4.6.1 treatment on the growth and progression of intraperitoneal SKOV-3 tumors. IP mice were treated with A4.6.1 (n = 16) or PBS (n = 15) in an identical fashion to those bearing subcutaneous tumors. However, intraperitoneal tumor growth could not be monitored directly and, because of its spread within the abdomen, could not be quantified accurately. Therefore, intraperitoneal tumor burden was assessed qualitatively at postmortem examination. In all animals, treatment was initiated 8 days after SKOV-3 inoculation and continued for various times.
In experiment 1 (Figure 3A) ▶ , all animals were killed 21 days after SKOV-3 inoculation. At postmortem examination, two of the three PBS-treated animals exhibited abdominal swelling with a moderate level of ascites and had moderate and easily detectable intraperitoneal tumor burden. The remaining PBS-treated animal had no detectable abdominal swelling or ascites and only a mild tumor burden. None of the A4.6.1-treated IP animals showed signs of ascites formation or cachexia at the time of postmortem examination, and intraperitoneal SKOV-3 tumor burden was barely detectable. Interestingly, A4.6.1 inhibited the growth of small subcutaneous tumors at the site of SKOV-3 injection that developed in some IP animals.
Figure 3.

Effect of neutralization of tumor-derived VEGF on intraperitoneal SKOV-3 tumor development and ascites production in mice inoculated intraperitoneally with SKOV-3 cells. Shown are outcomes for individual animals in three separate experiments. Tumor burden was assessed qualitatively as follows: ++++, High, many clusters of large (5 to 10 mm in diameter) solid tumors easily visible in the peritoneal cavity; +++, Moderate, clusters of tumors (3 to 5 mm in diameter) readily visible but spread not as extensive as High; ++, Low, large tumors not apparent but small foci visible on peritoneum, omentum, uterine fat pads, and diaphragm; +, Scare, only small foci seen on peritoneum, omentum, and diaphragm. Extent of ascites production also was determined qualitatively as follows: +++, High, marked abdominal swelling with 5 to 10 ml of bloody ascites; ++, Moderate, distinct abdominal swelling with 3 to 5 ml of clear ascites; +, Low, no abdominal swelling, small volume of clear ascitic fluid detected at postmortem examination; −, No ascites detected.
In experiment 2 (Figure 3B) ▶ , the effects of prolonged A4.6.1 treatment (n = 6) were examined. All of the PBS-treated animals (n = 6) displayed signs of ascites formation and cachexia at various times after SKOV-3 inoculation; the first PBS-treated animal was killed at 4.5 weeks and the last at 6.5 weeks because of severe cachexia associated with the developing ascites. One A4.6.1-treated animal exhibited ascites and cachexia and therefore was killed at 5.5 weeks. This mouse had a high tumor burden. Four of the A4.6.1-treated animals were killed at 6.5 weeks. The remaining A4.6.1-treated IP mouse was killed the following day. Five of the six A4.6.1-treated IP animals appeared normal at the time of postmortem examination, and no ascites formation could be detected. In A4.6.1-treated mice, intraperitoneal tumor burden was variable: in three of the six animals it was high and comparable with tumor burden in those mice receiving PBS alone, whereas in the other IP animals tumor burden ranged from moderate to scarce. The reason for the apparent lack of response in the one A4.6.1-treated mouse is uncertain.
In experiment 3 (Figure 3C) ▶ , A4.6.1 treatment was stopped after the last PBS-treated animal was killed (approximately 7 weeks after SKOV-3 inoculation). At that time, three of the six A4.6.1-treated animals were killed. As with experiment 2, all of the PBS-treated animals exhibited abdominal swelling and became cachectic approximately 6 weeks after receiving SKOV-3 cells. All of these animals had severe ascites and a high intraperitoneal tumor burden. In contrast, the three A4.6.1-treated animals killed at 6.5 weeks had no detectable ascites and a variable, but clearly detectable, tumor burden. Importantly, within 2 to 3 weeks of cessation of A4.6.1 treatment, the remaining mice developed severe ascites, became cachectic, and had to be killed. The tumor burden in these animals varied from moderate to high.
Discussion
Ovarian cancer is a disease that begins in, and usually is limited to, the peritoneal cavity. The majority of women with ovarian cancer present with peritoneal carcinomatosis, the principal cause of morbidity and mortality. Ovarian cancer also is associated with malignant ascites formation; in most cases the first indication of ovarian cancer is swelling of the abdomen due to the accumulation of ascitic fluid. 63 VEGF is thought to play a major role in the progression of ovarian cancer by promoting the neovascularization and subsequent growth of solid intraperitoneal tumors and by inducing ascites formation by increasing the permeability of the tumor vasculature. Several studies have shown that VEGF is expressed by human ovarian carcinoma cells and that the level of expression directly correlates with poor prognosis. However, as ovarian carcinoma cells also express other angiogenic factors, the specific role of VEGF in the growth and progression of ovarian cancer is unclear. Therefore, the present study was conducted to determine the role of VEGF in the regulation of ovarian cancer growth and ascites formation.
We used the human cell line SKOV-3, derived from a human ovarian serous cystadenocarcinoma, which accounts for 40 to 50% of all ovarian epithelial cancers, to induce peritoneal carcinomatosis and ascites production in immunodeficient mice. This model closely mimicked human ovarian cancer in that: 1) carcinomatosis was confined to the peritoneum; 2) the progression of the disease involved ascites formation and cachexia (IP mice were asymptomatic until they began to develop ascites, which always was associated with a heavy intraperitoneal tumor burden); 3) the morphology of intraperitoneal SKOV-3 tumors closely resembled peritoneal metastases from poorly differentiated stage III ovarian cancer in women; and 4) in preliminary studies, we confirmed that SKOV-3 tumors, like human ovarian carcinomas, express VEGF.
To examine the role of VEGF in ovarian cancer, we specifically ablated tumor-derived VEGF activity in IP mice using the function-blocking antibody, A4.6.1. This neutralizing antibody, which blocks access of VEGF to both VEGF receptors, inhibits the activity of human, but not mouse, VEGF and therefore specifically blocks the activity of tumor-derived VEGF. The effects of A4.6.1 on SKOV-3 tumor development were first examined using the subcutaneous model in which tumor growth could be monitored directly. We found that A4.6.1 was tumoristatic for SKOV-3 tumors grown subcutaneously; tumors failed to grow beyond the size attained at the beginning of the A4.6.1 treatment. However, when A4.6.1 treatment was stopped, the growth of subcutaneous tumors resumed. This observation is consistent with the concept that VEGF regulates, and is essential for, tumor neovascularization. Tumors could not grow further, because they could not stimulate the necessary vascularization to support a greater tumor mass. However, the existing vasculature likely was not affected by A4.6.1 treatment and therefore remained sufficient to maintain tumor size. Interestingly, when A4.6.1 was administered to SC mice bearing advanced tumors (10 to 15 mm in diameter with blood-filled cysts), tumor growth was inhibited and cysts regressed and involuted without rupturing. The regression of blood-filled cysts in response to VEGF inhibition suggests that tumor cyst formation is influenced by VEGF. This observation is consistent with the increase of vascular permeability caused by VEGF and suggests that VEGF may induce cyst formation by augmenting microvessel permeability. These data clearly demonstrated that tumor-derived VEGF is a necessary component of subcutaneous SKOV-3 tumor growth. However, ovarian cancer is not a subcutaneous disease but instead usually is limited to the peritoneal cavity. Therefore, we performed similar experiments using the intraperitoneal model.
In IP mice, inhibition of tumor-derived VEGF activity by A4.6.1 prolonged life and completely inhibited ascites formation. However, unlike its consistent tumoristatic action in SC mice, A4.6.1 only partially inhibited SKOV-3 tumor growth in IP mice. In some A4.6.1-treated IP mice, the extent of intraperitoneal tumor burden was similar to that of PBS-treated animals, whereas in others it was minimal. The reason that tumor-derived VEGF was obligatory for subcutaneous, but not intraperitoneal, SKOV-3 tumor growth is unclear. The peritoneal cavity offers a markedly different environment for tumor growth and spread than does the subcutaneous space. Within the peritoneum, SKOV-3 cells are not confined, as they are when administered as a subcutaneous bolus. Consequently, subcutaneous tumors grew only as a spherical mass under the skin, whereas in the peritoneum, tumors grew as thin sheets over a relatively large surface area with the occasional formation of solid tumor foci extending into the peritoneal cavity. With this mode of tumor growth and spread, it is likely that dependency on angiogenesis would be minimal, as the thin layers of tumor and some of the tumor buds would be small enough to survive by passive diffusion of nutrients from the underlying host vasculature and the surrounding peritoneal fluid. However, neovascularization clearly occurred in some intraperitoneal tumors, particularly the large solid tumors that formed on the pelvic organs. Thus, intraperitoneal carcinomatosis appears to have angiogenesis-independent and -dependent components; ie, formation and growth of the thin layers of tumor and some of the smaller solid buds would be independent of angiogenesis and be maintained by the pre-existing vasculature, whereas the larger solid intraperitoneal tumors would require neovascularization for continued growth. If this concept is correct, then inhibition of angiogenesis would only inhibit the development and growth of the larger intraperitoneal tumors. Consistent with this notion, we found that A4.6.1 did not inhibit the formation of tumor sheets on peritoneal surfaces or the formation of small solid tumor foci. However, large and presumably angiogenesis-dependent SKOV-3 tumors were detected in some A4.6.1-treated mice. It is unlikely that the dose of A4.6.1 was insufficient to inhibit SKOV-3 activity by intraperitoneal tumors, as the same dose was sufficient to act systemically and completely inhibit subcutaneous SKOV-3 tumor growth in SC mice. Furthermore, in some IP mice it was sufficient to inhibit the growth of a subcutaneous tumor that developed at the site of SKOV-3 inoculation. This indicates that VEGF-regulated angiogenesis may not be an essential factor in the growth of intraperitoneal carcinomatosis. Indeed, it is possible that other angiogenic factors, many of which have been detected in ovarian carcinoma cells, may have compensated for the lack of VEGF or may play a more prominent role in the control of intraperitoneal tumor angiogenesis. However, it is difficult to reconcile this possibility with an explanation of why such factors would not have supported angiogenesis in the subcutaneous model. It appears that the role of VEGF in tumor growth may be influenced by the site of tumor engraftment.
A4.6.1 completely inhibited ascites formation in IP mice, even though some animals had a tumor burden that was similar to that of controls that developed ascites and cachexia. When A4.6.1 treatment was stopped, IP mice rapidly developed ascites and became cachectic. VEGF is a potent stimulator of vascular permeability and is thought to play a major role in the development of malignant ascites. 43 Our data support the hypothesis that ascites formation in ovarian cancer is regulated by tumor-derived VEGF via the augmentation of tumor microvessel permeability. Ascites formation, as indicated by abdominal swelling, only became apparent relatively late in disease progression when tumor burden was high. It is possible that ascites was produced earlier; however, its rate of production was likely less than its rate of clearance from the peritoneal cavity. As tumor burden increased, the rate of ascites production likely became greater than the capacity for clearance and resulted in ascites accumulation and abdominal swelling with associated cachexia. Interestingly, in the SC mice, A4.6.1 reduced the volume of existing tumor cysts, suggesting that A4.6.1 could possibly reverse ascites accumulation if administered to IP mice exhibiting abdominal swelling. This possibility has important clinical implications, as it suggests that inhibition of VEGF activity may reverse the accumulation of ascitic fluid in women with ovarian cancer, which could significantly contribute to treatment of the disease. In summary, these data suggest that tumor-derived VEGF is not an essential regulator of peritoneal ovarian cancer growth but plays a pivotal role in malignant ascites formation likely by increasing vascular permeability.
Acknowledgments
We thank Janet Lee, Paul Goldsmith, Evelyn Garrett, Sharon Lojun, and Charles Zaloudek for their invaluable assistance.
Footnotes
Address reprint requests to Dr. Robert B. Jaffe, Reproductive Endocrinology Center, University of California, San Francisco, Box 0556, 505 Parnassus Ave., San Francisco, CA 94143-0556. E-mail: robert_jaffe@quickmail.ucsf.edu.
Presented in part at the 43rd Annual Meeting of the Society for Gynecological Investigation, San Diego, CA, 1997.
Supported in part by National Institutes of Health grant PO1 CA64602.
SM’s present address is Maternal Health Research Centre, Endocrine Unit, John Hunter Hospital, Newcastle, New South Wales, Australia.
References
- 1.Folkman J, Watson K, Ingber D, Hanahan D: Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339:58-61 [DOI] [PubMed] [Google Scholar]
- 2.Liotta LA, Kleinerman J, Saidel GM: Quantitative relationships of intravascular tumor cells, tumor vessels, and pulmonary metastases following tumor implantation. Cancer Res 1974, 4:997-1004 [PubMed] [Google Scholar]
- 3.Weidner N, Semple JP, Welch WR, Folkman J: Tumor angiogenesis and metastasis: correlation in invasive breast carcinoma. N Engl J Med 1991, 324:1-8 [DOI] [PubMed] [Google Scholar]
- 4.Folkman J, Klagsbrun M: Vascular physiology: a family of angiogenic peptides. Nature 1987, 329:671-672 [DOI] [PubMed] [Google Scholar]
- 5.Folkman J: What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1990, 82:4-6 [DOI] [PubMed] [Google Scholar]
- 6.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J: Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol 1993, 143:401-409 [PMC free article] [PubMed] [Google Scholar]
- 7.Olivarez D, Ulbright T, DeRiese W, Foster R, Reister T, Einhorn L, Sledge G: Neovascularization in clinical stage A testicular germ cell tumor: prediction of metastatic disease. Cancer Res 1994, 54:2800-2082 [PubMed] [Google Scholar]
- 8.Maeda K, Chung YS, Takatsuka S, Ogawa Y, Sawada T, Yamashita Y, Onoda N, Kato Y, Nitta A, Arimoto Y, Kondo Y, Sawa M: Tumor angiogenesis as a predictor of recurrence in gastric carcinoma. J Clin Oncol 1995, 13:477-481 [DOI] [PubMed] [Google Scholar]
- 9.Macchiarini P, Fontaini G, Hardin MJ, Sqartini F, Angeletti CA: Relation of neovascularization to metastasis in non-small cell lung cancer. Lancet 1992, 340:145-146 [DOI] [PubMed] [Google Scholar]
- 10.Hollingsworth HC, Kohn EC, Steinberg SM, Rothenberg ML, Merino MJ: Tumor angiogenesis in advanced stage ovarian carcinoma. Am J Pathol 1995, 147:33-41 [PMC free article] [PubMed] [Google Scholar]
- 11.van Diest PJ, Zevering JP, Zevering LC, Baak JP: Prognostic value of microvessel quantitation in cisplatin treated FIGO 3 and 4 ovarian cancer patients. Pathol Res Pract 1995, 191:25-30 [DOI] [PubMed] [Google Scholar]
- 12.Conn G, Bayne ML, Soderman DD, Kwok PW, Sullivan KA, Palisi TM, Hope DA, Thomas KA: Amino acid and cDNA sequence of a vascular endothelial cell mitogen that is homologous to platelet-derived growth factor. Proc Natl Acad Sci USA 1990, 87:2628-2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J: Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest 1989, 84:1470-1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrara N, Henzel WJ: Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 1989, 161:851-858 [DOI] [PubMed] [Google Scholar]
- 15.Gospodarowicz D, Abraham JA, Schilling J: Isolation and characterization of a vascular endothelial cell mitogen produced by pituitary-derived folliculostellate cells. Proc Natl Acad Sci USA 1989, 86:7311-7315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT: Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989, 246:1309-1312 [DOI] [PubMed] [Google Scholar]
- 17.Leung DW, Cachiane G, Kuang WJ, Goeddel DV, Ferrara N: Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246:1306-1309 [DOI] [PubMed] [Google Scholar]
- 18.Ferrara N, Jakeman L, Houck K, Leung DW: Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev 1992, 13:18-32 [DOI] [PubMed] [Google Scholar]
- 19.Senger DR, Van de Water L, Brown LF, Nagy JA, Yeo KT, Yeo TK, Berse B, Jackman RW, Dvorak AM, Dvorak HF: Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev 1993, 12:303-324 [DOI] [PubMed] [Google Scholar]
- 20.Dvorak HF, Sioussat TM, Brown LF, Berse B, Nagy JA, Sotrel A, Manseau EJ, Van de Water L, Senger DR: Distribution of vascular permeability factor (vascular endothelial growth factor) in tumors: concentration in tumor blood vessels. J Exp Med 1991, 174:1275-1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dvorak HF, Brown LF, Detmar M, Dvorak AM: Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995, 146:1029-1039 [PMC free article] [PubMed] [Google Scholar]
- 22.Conn G, Soderman DD, Schaeffer MT, Wile M, Hatcher VB, Thomas KA: Purification of a glycoprotein vascular endothelial cell mitogen from rat glioma-derived cell line. Proc Natl Acad Sci USA 1990, 87:1323-1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weindel K, Marme D, Weich H: AIDS-associated Kaposi’s sarcoma cells in culture express vascular endothelial growth factor. Biochem Biophys Res Commun 1992, 183:1167-1174 [DOI] [PubMed] [Google Scholar]
- 24.Weindel K, Moringlane J, Marme D, Weich H: Detection and quantification of vascular endothelial growth factor/vascular permeability factor in brain tumor tissue and cyst fluid: the key to angiogenesis? Neurosurgery 1994, 35:439-448 [DOI] [PubMed] [Google Scholar]
- 25.Berse B, Brown LF, Van de Water L, Dvorak HF, Senger DR: Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol Biol Cell 1992, 3:211-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boocock CA, Charnock-Jones S, Sharkey AM, McLaren J, Baker PJ, Wright KA, Twentyman PR, Smith SK: Expression of vascular endothelial growth factor and its receptors flt and KDR in ovarian carcinoma. J Natl Cancer Inst 1995, 87:506-516 [DOI] [PubMed] [Google Scholar]
- 27.Brown LF, Berse B, Jackman RW, Tognazzi K, Guidi AJ, Dvorak HF, Senger DR, Connolly JL, Schnitt SJ: Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in breast cancer. Hum Pathol 1995, 26:86-91 [DOI] [PubMed] [Google Scholar]
- 28.Brown LF, Berse B, Jackman RW, Tognazzi K, Manseau EJ, Senger DR, Dvorak HF: Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in adenocarcinomas of the gastrointestinal tract. Cancer Res 1993, 53:4727-4735 [PubMed] [Google Scholar]
- 29.Brown LF, Berse B, Jackman RW, Tognazzi K, Manseau EJ, Dvorak HF, Senger DR: Increased expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in kidney and bladder carcinomas. Am J Pathol 1993, 143:1255-1262 [PMC free article] [PubMed] [Google Scholar]
- 30.Detmar M, Brown LF, Claffey KP, Yeo KT, Kocher O, Jackman RW, Berse B, Dvorak HF: Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J Exp Med 1994, 180:1141-1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guidi AJ, Abu-Jawdeh G, Berse B, Jackman RW, Tognazzi K, Dvorak HF, Brown LF: Angiogenesis and vascular permeability factor (vascular endothelial growth factor) expression in cervical neoplasia. J Natl Cancer Inst 1995, 87:1237-1245 [DOI] [PubMed] [Google Scholar]
- 32.Guidi AJ, Abu-Jawdeh G, Tognazzi K, Dvorak HF, Brown LF: Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in endometrial carcinoma. Cancer 1996, 78:454-460 [DOI] [PubMed] [Google Scholar]
- 33.Plate KH, Breier G, Weich HA, Risau W: Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359:845-848 [DOI] [PubMed] [Google Scholar]
- 34.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF: Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 279:983-985 [DOI] [PubMed] [Google Scholar]
- 35.Senger DR, Perruzzi CA, Feder J, Dvorak HF: A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res 1986, 46:5629-5632 [PubMed] [Google Scholar]
- 36.Yeo K-T, Wang HH, Nagy JA, Sioussat TM, Ledbetter SR, Hoogewerf AJ, Zhou Y, Masse EM, Senger DR, Dvorak HF, Yeo T-K: Vascular permeability factor (vascular endothelial growth factor) in guinea pig and human tumor and inflammatory effusions. Cancer Res 1993, 53:2912-2918 [PubMed] [Google Scholar]
- 37.Olson TA, Mohanraj D, Carson LF, Ramakrishnan S: Vascular permeability factor gene expression in normal and neoplastic human ovaries. Cancer Res 1994, 54:276-280 [PubMed] [Google Scholar]
- 38.Takeshita S, Zheng LP, Brogi E, Kearney M, Pu LQ, Bunting S, Ferrara N, Symes JF, Isner JM: Therapeutic angiogenesis: a single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. J Clin Invest 1994, 93:662-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alvarez JA, Baird A, Tatum A, Daucher J, Chorsky R, Gonzalez AM, Stopa EG: Localization of basic fibroblast growth factor and vascular endothelial growth factor in human glial neoplasms. Mod Pathol 1992, 5:303-307 [PubMed] [Google Scholar]
- 40.Doldi N, Origoni M, Bassan M, Ferrari D, Rossi M, Ferrari A: Vascular endothelial growth factor: expression in human vulvar neoplastic and nonneoplastic tissues. J Reprod Med 1996, 41:844-848 [PubMed] [Google Scholar]
- 41.Doldi N, Bassan M, Gulisano M, Broccoli V, Boncinelli E, Ferrari A: Vascular endothelial growth factor messenger ribonucleic acid expression in human ovarian and endometrial cancer. Gynecol Endocrinol 1996, 10:375-382 [DOI] [PubMed] [Google Scholar]
- 42.Sioussat TM, Dvorak HF, Brock TA, Senger DR: Inhibition of vascular permeability factor (vascular endothelial growth factor) with antipeptide antibodies. Arch Biochem Biophys 1993, 301:15-20 [DOI] [PubMed] [Google Scholar]
- 43.Nagy JA, Masse EM, Herzberg KT, Meyers MS, Yeo KT, Yeo TK, Sioussat TM, Dvorak HF: Pathogenesis of ascites tumor growth: vascular permeability factor, vascular hyperpermeability, and ascites fluid accumulation. Cancer Res 1995, 55:360-368 [PubMed] [Google Scholar]
- 44.Terman BI, Carrion ME, Kovacs E, Rasmussen BA, Eddy RL, Shows TB: Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene 1991, 6:1677-1683 [PubMed] [Google Scholar]
- 45.de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT: The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 255:989-991 [DOI] [PubMed] [Google Scholar]
- 46.Kim KJ, Li B, Winer J, Armanini M, Gillet N, Phillips HS, Ferrara N: Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362:841-844 [DOI] [PubMed] [Google Scholar]
- 47.Claffey KP, Brown LF, del Aguila LF, Tognazzi K, Yeo KT, Manseau EJ, Dvorak HF: Expression of vascular permeability factor/vascular endothelial growth factor by melanoma cells increases tumor growth, angiogenesis, and experimental metastasis. Cancer Res 1996, 56:172-181 [PubMed] [Google Scholar]
- 48.Millauer B, Shawver LK, Plate KH, Risau W, Ullrich A: Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature 1994, 367:576-579 [DOI] [PubMed] [Google Scholar]
- 49.Perez RP, Godwin AK, Hamilton TC, Ozols RF: Ovarian cancer biology. Semin Oncol 1991, 18:186-204 [PubMed] [Google Scholar]
- 50.Abu-Jawdeh GM, Faix JD, Niloff J, Tognazzi K, Manseau E, Dvorak HF, Brown LF: Strong expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in ovarian borderline and malignant neoplasms. Lab Invest 1996, 74:1105-1115 [PubMed] [Google Scholar]
- 51.Paley PJ, Staskus KA, Gebhard K, Mohanraj D, Twiggs LB, Carson LF, Ramakrishnan S: Vascular endothelial growth factor expression in early stage ovarian carcinoma. Cancer 1997, 80:98-106 [DOI] [PubMed] [Google Scholar]
- 52.Kutteh WH, Kutteh CC: Quantitation of tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-6 in the effusions of ovarian epithelial neoplasms. Am J Obstet Gynecol 1992, 167:1864-1869 [DOI] [PubMed] [Google Scholar]
- 53.Stromberg K, Johnson GR, O’Connor DM, Sorensen CM, Gullick WJ, Kannan B: Frequent immunohistochemical detection of EGF supergene family members in ovarian carcinogenesis. Int J Gynecol Pathol 1994, 13:342-347 [DOI] [PubMed] [Google Scholar]
- 54.Kurachi H, Morishige K, Amemiya K, Adachi H, Hirota K, Miyake A, Tanizawa O: Importance of transforming growth factor α/epidermal growth factor receptor autocrine growth mechanism in an ovarian cancer cell line in vivo. Cancer Res 1991, 51:5956-5959 [PubMed] [Google Scholar]
- 55.Reynolds K, Farzaneh F, Collins WP, Campbell S, Bourne TH, Lawton F, Moghaddam A, Harris AL, Bicknell R: Association of ovarian malignancy with expression of platelet-derived endothelial cell growth factor. J Natl Cancer Inst 1994, 86:1234-1238 [DOI] [PubMed] [Google Scholar]
- 56.Henriksen R, Funa K, Wilander E, Backstrom T, Ridderheim M, Oberg K: Expression and prognostic significance of platelet-derived growth factor and its receptors in epithelial ovarian neoplasms. Cancer Res 1993, 53:4550-4554 [PubMed] [Google Scholar]
- 57.Henriksen R, Gobl A, Wilander E, Oberg K, Miyazono K, Funa K: Expression and prognostic significance of TGF-beta isotypes, latent TGF-beta 1 binding protein, TGF-beta type I and type II receptors, and endoglin in normal ovary and ovarian neoplasms. Lab Invest 1995, 73:213-220 [PubMed] [Google Scholar]
- 58.Di Blasio AM, Cremonesi L, Vigano P, Ferrare M, Gospodarowicz D, Vignali M, Jaffe RB: Basic fibroblast growth factor and its receptor messenger ribonucleic acids are expressed in human ovarian epithelial neoplasms. Am J Obstet Gynecol 1993, 169:1517-1523 [DOI] [PubMed] [Google Scholar]
- 59.Crickard K, Gross JL, Crickard U, Yoonessi M, Lele S, Herblin WF, Eidsvoog K: Basic fibroblast growth factor and receptor expression in human ovarian cancer. Gynecol Oncol 1994, 55:277-284 [DOI] [PubMed] [Google Scholar]
- 60.Otsuka T, Ohkawa T, Shibata T, Oku T, Okuhara M, Terano H, Kohsaka M, Imanaka H: A new potent angiogenesis inhibitor, FR-118487. J Microbiol Biotech 1991, 1:163-168 [Google Scholar]
- 61.Mu J, Abe Y, Tsutsui T, Yamamoto N, Tai XG, Niwa O, Tsujimura T, Sato B, Terano H, Fujiwara H, Hamaoka T: Inhibition of growth and metastasis of ovarian carcinoma by administering a drug capable of interfering with vascular endothelial growth factor activity. Jpn J Cancer Res 1996, 87:963-971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim KJ, Li B, Houck K, Winer J, Ferrara N: The vascular endothelial growth factor proteins: identification of biologically relevant regions by neutralizing monoclonal antibodies. Growth Factors 1992, 7:53-64 [DOI] [PubMed] [Google Scholar]
- 63.Czernobilsky B: Common epithelial tumors of the ovary. Blaustein’s Pathology of the Female Genital Tract. Edited by RJ Kurman. New York, Springer-Verlag, 1987