

Abstract
Recently, vascular endothelial growth factor-C (VEGF-C or VEGF-2) was described as a specific ligand for the endothelial receptor tyrosine kinases VEGFR-2 and VEGFR-3. In vivo data, limited to constitutive overexpression in transgenic mice, have been interpreted as evidence that the growth-promoting effects of VEGF-C are restricted to development of the lymphatic vasculature. The current studies were designed to test the hypothesis that constitutive expression of VEGF-C in adult animals promotes angiogenesis. In vitro, VEGF-C exhibited a dose-dependent mitogenic and chemotactic effect on endothelial cells, particularly for microvascular endothelial cells (72% and 95% potency, respectively, compared with VEGF-A/VEGF-1). VEGF-C stimulated release of nitric oxide from endothelial cells and increased vascular permeability in the Miles assay; the latter effect was attenuated by pretreatment with the nitric oxide synthase inhibitor Nω-nitro-l-arginine methyl ester. Both VEGFR-2 and VEGFR-3 receptors were shown to be expressed in human saphenous vein and internal mammary artery. The potential for VEGF-C to promote angiogenesis in vivo was then tested in a rabbit ischemic hindlimb model. Ten days after ligation of the external iliac artery, VEGF-C was administered as naked plasmid DNA (pcVEGF-C; 500 μg) from the polymer coating of an angioplasty balloon (n = 8 each) or as recombinant human protein (rhVEGF-C; 500 μg) by direct intra-arterial infusion. Physiological and anatomical assessments of angiogenesis 30 days later showed evidence of therapeutic angiogenesis for both pcVEGF-C and rhVEGF-C. Hindlimb blood pressure ratio (ischemic/normal) after pcVEGF-C increased to 0.83 ± 0.03 after pcVEGF-C versus 0.59 ± 0.04 (P < 0.005) in pGSVLacZ controls and to 0.76 ± 0.04 after rhVEGF-C versus 0.58 ± 0.03 (P < 0.01) in control rabbits receiving rabbit serum albumin. Doppler-derived iliac flow reserve was 2.7 ± 0.1 versus 2.0 ± 0.2 (P < 0.05) for pcVEGF-C versus LacZ controls and 2.9 ± 0.3 versus 2.1 ± 0.2 (P < 0.05) for rhVEGF-C versus albumin controls. Neovascularity was documented by angiography in vivo (angiographic scores: 0.85 ± 0.05 versus 0.51 ± 0.02 (P < 0.001) for plasmid DNA and 0.74 ± 0.08 versus 0.53 ± 0.03 (P < 0.05) for protein), and capillary density (per mm2) was measured at necropsy (252 ± 12 versus 183 ± 10 (P < 0.005) for plasmid DNA and 229 ± 20 versus 164 ± 20 (P < 0.05) for protein). In contrast to the results of gene targeting experiments, constitutive expression of VEGF-C in adult animals promotes angiogenesis in the setting of limb ischemia. VEGF-C and its receptors thus constitute an apparently redundant pathway for postnatal angiogenesis and may represent an alternative to VEGF-A for strategies of therapeutic angiogenesis in patients with limb and/or myocardial ischemia.
Vascular endothelial growth factor (VEGF), 1-3 also known as vascular permeability factor, 4 is unique as an endothelial cell (EC) mitogen, because its high-affinity receptor tyrosine kinases VEGFR-1 (Flt-1) and VEGFR-2 (Flk-1/KDR) are expressed predominantly on ECs. VEGF has been established as critical for development 5-8 and maintenance 9-11 of the normal vasculature, as well as for therapeutic 12-15 and tumor-induced angiogenesis. 16-19
The recent purification and characterization of proteins with high homology to VEGF led to the designation of a vascular endothelial family of growth factors, consisting of four members to date. In addition to VEGF/vascular permeability factor, otherwise renamed VEGF-A or VEGF-1, two additional proteins, VEGF-C or VEGF-2 20 and VEGF-B or VEGF-3, 21 were isolated and shown to be abundantly expressed in highly vascularized tissues such as heart and skeletal muscle. In contrast to the widespread distribution of the VEGFs, the fourth member of the VEGF family, placenta growth factor (PlGF), 22 appears restricted in vivo to placenta and certain tumors. 23,24 All four proteins share structural homology among themselves as well as with the platelet-derived growth factor A and B polypeptides, including in particular a conserved motif consisting of eight cysteine residues in the putative receptor-binding domain. 25 Similarly to the A and B chains of platelet-derived growth factor, VEGF-A and PlGF can form heterodimers with demonstrable biological activity. 26-29 VEGF-B may also heterodimerize with VEGF-A in cells expressing both factors. 21 Moreover, it was recently shown that hypoxia, a principal stimulus for upregulation of VEGF-A, 18,30,31 may differentially regulate the induction of homo- and heterodimers of PlGF/VEGF-A. 28
Whereas VEGF-A is a high-affinity ligand for the receptors VEGFR-1 and VEGFR-2, VEGF-C was shown to bind another recently identified endothelium-specific receptor tyrosine kinase, VEGFR-3 (Flt-4); in addition, VEGF-C binds to VEGFR-2, but not to VEGFR-1. 20 Comparison of the amino acid sequence of an independently isolated specific activator of VEGFR-3, originally named vascular endothelial growth factor-related protein, 32 revealed its identity with VEGF-C. PlGF was shown to bind with high affinity to VEGFR-1, but not to VEGFR-2 26 or VEGFR-3. 33 The receptor(s) for VEGF-B 21 has not yet been characterized.
The role of VEGF-A in angiogenesis has been studied extensively in vivo and in vitro. 34 More recently, VEGF-A has been shown to be expressed in the media of human arteries and veins, 35 consistent with its proposed role in maintaining integrity of the vascular endothelium. The constitutive coexpression of all three VEGF types in many adult tissues 20,21,32 suggests an interactive or at least redundant capacity of the VEGF members to regulate angiogenesis and modulate EC function.
Data regarding the bioactivity of VEGF-C and VEGF-B are limited to date. 20,21,32,36 Accordingly, we sought to determine whether VEGF-C could promote angiogenesis when overexpressed in adult animals. Experiments in the present study confirm the mitogenic activity of VEGF-C and show its ability to stimulate cell migration for different EC types. VEGF-C is furthermore shown to stimulate release of nitric oxide (NO) from ECs, potentially an important mediator of VEGF-induced angiogenesis. 37-40 Indeed, when administered in vivo as plasmid DNA or recombinant protein, VEGF-C was shown to enhance vascular permeability and promote angiogenesis in a rabbit model of hindlimb ischemia. These findings thus indicate that VEGF-C may have utility as a primary or adjunctive agent for therapeutic angiogenesis.
Materials and Methods
Recombinant Protein
Heterodimeric recombinant human (rh) VEGF-A (VEGF-1) protein, purified from Escherichia coli, was a generous gift of Drs. N. Ferrara, B. Keyt, and S. Bunting at Genentech Inc. (South San Francisco, CA). E. coli-derived rhVEGF-A appeared as a single protein band on sodium dodecyl sulfate-polyacrylamide gel electrophoresis with a molecular weight of 39.8 kd under nonreducing conditions. rhVEGF-C (VEGF-2) was expressed in the BaculoGold Virus Expression Vector System (Pharmingen) and purified by ion exchange and hydrophobic interaction chromatography as described before. 41 Under nonreducing conditions, sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis yielded bands of 52 kd and 90 kd, representing multimeric, disulfide-linked rhVEGF-C complexes. 20 For all recombinant proteins, endotoxin levels were nondetectable using the limulus lysate assay.
Plasmids
Complementary DNA clones for rhVEGF-C, isolated from a human osteoclast cDNA library, were assembled into the pcDNAI/Amp mammalian expression vector that includes a cytomegalovirus promoter/enhancer to drive gene expression. This plasmid construct was named pcVEGF-C. The plasmid pGSVLacZ (courtesy of Dr. C. Bonnerot), containing a nuclear targeted β-galactosidase sequence coupled to the SV40 early promoter, was used for control transfection experiments. 42
Cell Culture
Human umbilical vein ECs (HUVECs) were isolated from umbilical cord vein by collagenase treatment as previously described 43 and grown in medium 199 (Life Technologies, Inc., Grand Island, NY) supplemented with 20% fetal bovine serum (FBS) (Life Technologies), 100 μg/ml EC growth supplement, and 50 U/ml heparin (Sigma Chemical Co., St. Louis, MO). HUVECs were used between passages 1 and 5.
Human microvascular (HM) ECs of dermal origin were purchased from Cascade Biologics, Inc. Immunostaining of representative plates with the antibody to PAL-E 44 was used to confirm the absence of lymphatic ECs. The cells were grown in EBM medium containing human epidermal growth factor (10 ng/ml), hydrocortisone (10 ng/ml), 5% FBS, and 0.4% bovine brain extract. HMECs were used between passages 4 and 6.
EC Proliferation Assay
Mitogenic activity was assayed using a previously validated 45 colorimetric MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay with the electron coupling reagent phenazine methosulfate (CellTiter 96 AQ; Promega, Madison, WI). Cells were seeded in a 96-well plate (5000 cells/well) in 0.1 ml serum-supplemented medium and allowed to attach overnight. VEGF (rhVEGF-A or rhVEGF-C) in 5% FBS with heparin (10 U/ml) was added to the wells for 48 hours. MTS/phenazine methosulfate mixture (20 μl; 1:0.05) was added per well and allowed to incubate for 1 hour at 37°C before absorbance was measured at 490 nm in an enzyme-linked immunosorbent assay plate reader. Background absorbance from control wells was subtracted, and seven wells were performed in parallel and averaged for each condition. Proliferation experiments were each repeated three times.
EC Migration Assay
EC migration assays were performed using a 48-well microchemotaxis chamber (Neuroprobe). 46 Polyvinylpyrrolidone-free polycarbonate filters with a pore size of 8 μm (Nuclepore) were coated with 0.1% gelatin for at least 6 hours at room temperature and dried under sterile air. Test substances were diluted to appropriate concentrations in medium 199 supplemented with 1% FBS, and 25 μl of the final dilution was placed in the lower chamber of a modified Boyden chamber. Subconfluent HUVEC or HMEC cultures were washed and trypsinized for the minimum time required to achieve cell detachment. After placing the filter between lower and upper chambers, 2.5 × 105 cells suspended in 50 μl medium 199 containing 1% FBS were seeded in the upper compartment. The apparatus was then incubated for 5 hours at 37°C in a humidified chamber with 5% CO2 to allow cell migration. After the incubation period, the filter was removed, and the upper side of the filter containing the nonmigrated cells was scraped with a rubber policeman. The filters were fixed with methanol and stained with Giemsa solution (Diff-Quick, Baxter Diagnostics, McGaw Park, IL). Migration was quantified by counting cells in three random high-power fields (×100) in each well. All groups were studied in quadruplicate.
NO Measurement
An NO-specific polarographic electrode connected to an NO meter (Iso-NO, World Precision Instruments, Sarasota, FL) 47 was used to measure NO released from a cultured HUVEC monolayer. Calibration of the NO electrode was performed according to the following equation:
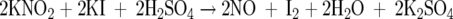 |
The standard calibration curve was obtained by adding graded concentrations of KNO2 (0, 5, 10, 25, 50, 100, 250, and 500 nmol/L) to the calibration solution containing KI and H2SO4. Specificity of the iso-NO electrode for NO was previously determined by measurement of NO from authentic NO gas. 48 The culture medium was removed, and HUVECs were washed twice with Dulbecco’s phosphate-buffered saline. Cells were then bathed in 5 ml of filtered Krebs-Henseleit solution in six-well plates, and the cell plates were placed on a slide warmer (Lab Line Instruments) to maintain the temperature at 37°C. The NO sensor probe was inserted vertically into the wells, with the tip of the electrode placed 2 mm under the surface of the solution before reagents were added. S-Nitroso-N-acetyl-penicillamine (Sigma Chemical Co.) was used as a positive control. The amount of released NO was expressed as pmol per 1 × 106 ECs. All values reported represent the mean of four to six measurements performed in each group (number of cell culture wells).
Miles Assay
Hairless male albino guinea pigs (200 to 600 g; Charles River Breeding Laboratories, Wilmington, MA) were lightly anesthetized with ether (Fisher Scientific, Fair Lawn, NJ). Evans blue dye (Sigma Chemical Co.) was filtered through 0.2-μm micropore filters (Corning Glass, Corning, NY) and then infused as a 0.5- to 1.0-ml bolus of 0.5% solution in saline via the left femoral vein. Evans blue dye binds to circulating plasma proteins and extravasates in response to certain reagents, rendering hyperpermeable dermal sites blue. 49 After the animals regained consciousness, reagents were injected intradermally in a volume of 0.1 ml. Intradermal injections were made into the trunk, posterior to the shoulder, 20 minutes after intravenous injection of Evans blue dye with a 30-gauge needle (Becton-Dickinson) causing a bleb of 9 to 11 mm in diameter. Increase in vascular permeability was assessed by leakage of blue dye into the bleb. 49 As originally described by Miles and Miles, 49 a small area of traumatic bluing 1 to 3 mm in diameter may be seen at the center of the bleb after intradermal injection of saline control. Intensity and area of the blue color changes within the blebs were identified and assessed, and the site of intradermal injection was photographed. The following reagents were injected intradermally: 1) saline alone (as vehicle control), 2) increasing concentrations of rhVEGF-A (4, 8, 16, 32, 64, and 128 ng), and 3) increasing concentrations of rhVEGF-C (4, 8, 16, 32, 64, and 128 ng).
To investigate the role of NO on VEGF-induced vascular permeability, Nω-nitro-l-arginine methyl ester (l-NAME; 20 mg/kg; Sigma Chemical Co.) was injected via the femoral vein immediately before administration of Evans blue dye. Twenty minutes later, the following were injected intradermally: 1) saline alone (negative control), 2) increasing concentrations of rhVEGF-A (8, 16, 32, 64, and 128 ng), and 3) increasing concentrations of rhVEGF-C (8, 16, 32, 64, and 128 ng).
Analysis of VEGF Receptor mRNA in Explanted Arterial and Venous Specimens
Total RNA from unused segments of human internal mammary artery and saphenous vein was isolated by phenol/chloroform extraction, 50 and RNA concentration was calculated from absorbance at 260 nm.
Reverse transcription 51 was performed by heating a 10-μl reaction mixture containing 1 μg total RNA and 20 μg/ml oligodeoxy-thymidine (Life Technologies) at 70°C for 10 minutes. After cooling, 60 units of human placenta ribonuclease inhibitor (Promega) and 200 U Moloney’s murine leukemia virus RNase H reverse transcriptase (Life Technologies) were added in a final 20-μl reaction mixture containing (mmol/L) deoxynucleotide triphosphate (Pharmacia) 1 each, dithiothreitol 10, Tris-HCl (pH 8.3) 25, KCl 75, and MgCl2 3; incubated for 1 hour at 42°C; heated 5 minutes at 95°C; and diluted to 50 μl with double-distilled water. For polymerase chain reaction (PCR) with a thermostable DNA polymerase, 52 a 50-μl reaction containing 5 μl of cDNA-RNA hybrids, 200 μmol/L deoxynucleotide triphosphate (Pharmacia), 20 mmol/L Tris-HCl (pH 8.55), 2.5 mmol/L MgCl2, 16 mmol/L (NH4)2SO4, 150 μg/ml bovine serum albumin, 1 μmol/L each oligonucleotide primer, and 1.25 U of Taq polymerase (Perkin-Elmer Corp., Norwalk, CT) was subjected to the following temperature cycles: 30 seconds of denaturing at 95°C, 5 seconds of annealing at 56°C, and 1 minute of extension at 72°C. At the end of the last cycle, a prolonged extension step was carried out for 10 minutes. PCR products were analyzed by electrophoresing 10 μl of each PCR reaction mixture in a 1.5% agarose gel, and bands were visualized by ethidium bromide staining. The results represent three independent amplifications from two separate studies.
The primers chosen for human VEGFR-3 (GenBank accession number X68203) were: for sense (5′ to 3′), CAG GAT GAA GAC ATT TGA, and for antisense (5′ to 3′), AAG AAA ATG CTG ACG TAT GC 53 (190 bp PCR-product); for human VEGFR-2 (GenBank accession number X61656), CAA CAA AGT CGG GAG AGG AG and ATG ACG ATG GAC AAG TAG CC (819 bp); for human VEGFR-1 (GenBank accession number X51602), CAG CGG CTT TTG TGG AAG ACT CAC and ACA TCT CGG TGT CAC TTC TTG GAC (735 bp); and for human glyceraldehyde-3-phosphate dehydrogenase, TGA AGG TCG GAG TCA ACG GAT TTG and CAT GTG GGC CAT GAG GTC CAC CAC (983 bp). The number of cycles was 40 for VEGFR-1, VEGFR-2, and VEGFR-3, and 30 for glyceraldehyde-3-phosphate dehydrogenase.
Animal Model
The potential for VEGF-C to promote angiogenesis in vivo was investigated in a previously described rabbit model of hindlimb ischemia. 54,55 All protocols were approved by the Institutional Animal Care and Use Committee of St. Elizabeth’s Medical Center. A total of 28 (seven per group) male New Zealand White rabbits (3.5 to 4.0 kg) (Pine Acre Rabbitry) were anesthetized with a mixture of ketamine (50 mg/kg) and acepromazine (0.8 mg/kg) after premedication with xylazine (2 mg/kg). The femoral artery of one hindlimb was completely excised from its proximal origin as a branch of the external iliac artery to the point distally where it bifurcates into the saphenous and popliteal arteries. Consequently, blood flow to the ischemic limb is dependent on collateral vessels originating from the internal iliac artery. 54,55 An interval of 10 days was allowed for postoperative recovery and development of endogenous collateral vessels. Beyond this time point, studies performed up to 90 days postoperatively 55 have demonstrated no significant collateral vessel augmentation.
Percutaneous Arterial Gene Transfer of pcVEGF-C
At 10 days postoperatively (day 0), after performing a baseline angiogram (see below), the internal iliac artery of the ischemic limb of eight animals was transfected with pcVEGF-C as naked DNA using a 2.0-mm hydrogel-coated balloon catheter (Slider with Hydroplus, Boston Scientific, Natick, MA). The angioplasty balloon was prepared ex vivo by first advancing the deflated balloon through a 5-F Teflon sheath (Boston Scientific), applying 500 μg of pcVEGF-C solution to the 20 μm-thick layer of hydrogel coating the external surface of the inflated balloon, and then retracting the inflated balloon back into the protective sheath. The sheath and angioplasty catheter were then introduced via the right carotid artery and advanced to the lower abdominal aorta using a 0.014-inch guide wire (Hi-Torque Floppy II, Advanced Cardiovascular Systems) under fluoroscopic guidance. The balloon catheter was then advanced into the internal iliac artery of the ischemic limb, inflated for 1 minute at 6 atmospheres, deflated, and withdrawn. An identical protocol was used to transfect the internal iliac artery of control animals (n = 8) with the promoter-matched plasmid pGSVLacZ containing a nuclear targeted β-galactosidase sequence.
On day 30, all measurements were repeated (see below), the animals were sacrificed, and tissue samples were retrieved for histological examination.
Intra-arterial Administration of rhVEGF-C Protein
Ten days postoperatively (day 0), baseline noninvasive and invasive hemodynamic parameters (see below) were recorded. A single intra-arterial bolus of rhVEGF-C (500 μg; n = 8) in 3 ml of phosphate-buffered saline or vehicle solution (3 ml of phosphate-buffered saline with 0.1% rabbit serum albumin (RSA), n = 8) was then administered over a period of 1 minute through a 3-F end-hole infusion catheter (Tracker-18; Target Therapeutics) positioned in the internal iliac artery of the ischemic limb. The catheter was then washed with 3 ml of phosphate-buffered saline containing 0.1% albumin. The dose of rhVEGF-C protein used in the present experiment (500 μg) has previously been shown for rhVEGF-1 to induce a maximal effect using a single bolus protocol. 12,56
Physiological Assessment
Calf Blood Pressure Ratio
Calf blood pressure was measured from the posterior tibial artery at day 0 and 30 in both hindlimbs using a Doppler Flowmeter (model 1050, Parks Medical Electronics) and cuff connected to a pressure manometer. 54,57 The calf blood pressure ratio was defined for each rabbit as the ratio of systolic pressure of the ischemic limb to that of the normal limb.
Intravascular Blood Flow Measurement
Blood flow was quantified in vivo before selective internal iliac angiography on day 30 with a 0.018-inch Doppler guide wire (Cardiometrics) introduced into the left common carotid artery as previously described. 58 The wire was advanced via a 3-F infusion catheter (Target Therapeutics) positioned at the origin of the common iliac artery to the proximal segment of the internal iliac artery supplying the ischemic limb. Average peak velocity (APV) was recorded at rest, and the maximum APV was evaluated after intra-arterial administration of nitroprusside (Sigma Chemical Co.) over 2 minutes via a constant infusion pump (1 ml/minute). The drug was administered at a dose of 1.5 μg/minutes/kg.
Angiographic luminal diameter of the internal iliac artery was determined, at rest and after drug infusion, using an automated edge-detection system (Quantum 2000I; QCS) as previously described. 54 The luminal diameter was measured at the site of the Doppler sample volume 5 mm distal to the wire tip. Cross-sectional area was calculated assuming a circular lumen.
Doppler-derived flow was calculated as QD = (πd2/4)(0.5 × APV), where QD is Doppler-derived time-averaged flow, d is vessel diameter, and APV is time average of the spectral peak velocity. 59 The mean velocity was estimated as 0.5 × APV by assuming a time-averaged parabolic velocity profile across the vessel. The Doppler-derived flow calculated in this fashion has been previously validated in vivo. 59 For the ischemic limb, in which the external iliac artery had been removed, flow through the internal iliac artery represented flow to the entire hindlimb. 54 Blood flow reserve was defined as the ratio of resting to maximum blood flow.
Anatomical Assessment
Selective Iliac Angiography
Angiographic analysis of collateral vessel development in the ischemic limb was performed using the 4-second angiograms recorded after injection of 0.25 mg of nitroglycerin and 5 ml of nonionic contrast medium (Isovue-370; Squibb Diagnostics, New Brunswick, NJ) into the internal iliac artery through a 3-F infusion catheter (Target Therapeutics). A grid overlay comprising 2.5 mm-diameter circles arranged in rows spaced 5 mm apart was placed over the angiogram at the level of the medial thigh. The number of contrast-opacified arteries crossing over circles and the total number of circles encompassing the medial thigh area were counted in single blinded fashion. An angiographic score was calculated as the ratio of circles crossed by opacified arteries divided by the total number of circles in the ischemic thigh. This angiographic score reflects vascular density in the medial thigh. 57
Capillary Density
Collateral vessel formation was further examined by measuring the number of capillaries in light microscopic sections taken from ischemic and nonischemic hindlimbs as previously described. 57 Tissue specimens were retrieved from each limb as transverse sections from the adductor and semimembranous muscles at the time of sacrifice (day 30). These two muscles were chosen because they are the two major muscles of the medial thigh, and each was originally perfused by the deep femoral artery, ligated at the time of the surgery. Samples were embedded in ornithine carbamoyltransferase compound (Miles Inc., Elkhart, IN) and snap-frozen in liquid nitrogen. Frozen sections (5 μm in thickness) were stained with alkaline phosphatase using an indoxyl-tetrazolium method to detect capillary ECs followed by eosin counterstaining. 60 Capillaries were counted under a ×20 objective to determine the capillary density (mean number of capillaries/mm2). A total of 20 different fields from the two muscles were randomly selected, and the number of capillaries was counted. To ensure that analysis of capillary density was not overestimated because of muscle atrophy, capillary density was also evaluated as a function of the number of muscle fibers in the histological section (capillary/myocyte ratio).
Statistical Analysis
Results were expressed as mean ± standard error of the mean (SEM). Statistical significance was evaluated using unpaired Student’s t-test for comparisons between two means and analysis of variance followed by Scheffé’s procedure for more than two means. A value of P < 0.05 was interpreted to denote statistical significance.
Results
Mitogenic Effect of rhVEGF-C on ECs
The effects of rhVEGF-C on EC proliferation are shown in Figure 1 ▶ . At a concentration of 10 ng/ml, the mitogenic activity of VEGF-C protein on HUVECs was 35% of that seen with VEGF-A. This difference was statistically significant (P < 0.005) (Figure 1A) ▶ . The mitogenic effect of VEGF-C was more pronounced when applied to HMECs (Figure 1B) ▶ : at 10 ng/ml, the mitogenic response of VEGF-C was 72% as potent as VEGF-A (P < 0.01).
Figure 1.

Proliferative response of HUVECs (A) or HMECs (B) to rhVEGF-A/VEGF-1 or VEGF-C/VEGF-2 protein. Cells (5 × 103) were seeded per well, and increase in cell number was assessed 48 hours after addition of growth factors using the MTS-colorimetric assay. Data are means (bars, SEM) of parallel samples. *P < 0.05 VEGF-A/VEGF-1 versus VEGF-C/VEGF-2 protein.
Effect of rhVEGF-C on EC Migration
Both VEGF-A and VEGF-C protein exhibited a pronounced chemotactic effect on ECs. The dose-response curves showed a typical Gaussian distribution, beginning with an effect on migration that was evident at a minimal concentration of 0.1 ng/ml (Figure 2) ▶ . For HUVECs, at a concentration of 10 ng/ml, the chemotactic response of VEGF-C was 83% of that seen with VEGF-A (P < 0.05) (Figure 2A) ▶ ; for HMECs, the chemotactic response to VEGF-A and VEGF-C was indistinguishable (Figure 2B) ▶ .
Figure 2.

Migratory response of HUVECs (A) or HMECs (B) to rhVEGF-A/VEGF-1 or VEGF-C/VEGF-2 protein. Cells (2.5 × 104) were seeded in the upper wells of a 48-well microchemotaxis Boyden chamber and incubated for 4 hours at 37°C in medium 199 supplemented with 1% FBS. The lower wells contained different concentrations of growth factor. Cells migrating through a polycarbonate membrane with a pore size of 8 μm were quantified by staining the cells at the lower side of the membrane with Giemsa solution and counting three high-power fields (×100). Each condition was done in quadruplicate. Data are means (bars, SEM) of parallel samples. *P < 0.05 VEGF-A/VEGF-1 versus VEGF-C/VEGF-2 protein.
NO Release from Cultured HUVECs by VEGF-C
Treatment of HUVECs with VEGF-C (1, 10, and 100 ng/ml) resulted in a concentration-dependent increase in NO synthesis (Figure 3) ▶ . Peak NO synthesis was observed between 3 and 6 minutes after addition of VEGF-C. Treatment of HUVECs with VEGF-A (100 ng/ml) under the same experimental conditions induced NO synthesis as previously reported. 61 S-Nitroso-N-acetyl-penicillamine was used as a positive control, and 0.1 mmol/L resulted in a significant increase in NO concentration measured in the buffer medium. For comparison, peak NO measured in response to VEGF-C was 92.7% and 80.9% of that measured after the same dose of VEGF-A and 0.1 mmol/L S-nitroso-N-acetyl-penicillamine, respectively.
Figure 3.

Induction of NO release from HUVECs by VEGF-C/VEGF-2, measured with an NO-specific polarographic electrode connected to an NO meter. HUVECs in six-well plates were bathed with Krebs-Henseleit solution before addition of reagents. Baseline is defined as baseline fluctuation over a period of 5 minutes of NO production by cells not exposed to agonist. The NO donor S-nitroso-N-acetyl-penicillamine (0.1 mmol/L) and VEGF-A/VEGF-1 (100 ng/ml) were used as positive controls. Administration of the NO inhibitor l-NAME abrogated the effect of VEGF-C and served as a negative control. Data shown are means (bars, SEM) of four to six measurements in each group. *P < 0.05 versus 100 ng/ml VEGF-C.
Effect of VEGF-C on Vascular Permeability
Repeated intradermal injection of the vehicle control saline (0.1 ml) did not increase vascular permeability. Intradermal injection of either VEGF-A or VEGF-C (16, 32, 64, or 128 ng) significantly increased vascular permeability in a dose-dependent manner (Figure 4A) ▶ . Development of a positive (blue) response began 155 ± 6 seconds (n = 3) after intradermal injection. Repeated experiments (n = 3) did not show significant differences in the induction of vascular permeability between VEGF-A and VEGF-C. As a positive control, we used bradykinine and histamine (100 nmol/L), both of which increased vascular permeability at the injected sites (data not shown).
Figure 4.

Effect of VEGF-A and VEGF-C on vascular permeability (Miles assay). 49 A: VEGF-A as well as VEGF-C increased vascular permeability in a dose-dependent manner. Doses are indicated by the arrows. A small area of traumatic bluing was observed after injection of saline control. B: Effect of the NO synthase inhibitor l-NAME (20 mg/kg) on VEGF-A- and VEGF-C-induced vascular permeability. Systemic injection of l-NAME 20 minutes before administration of Evans blue dye attenuated VEGF-A- and VEGF-C-mediated vascular permeability. Photographs are representative of three experiments each.
To examine the role of NO in VEGF-C-mediated vascular permeability, the NO synthase inhibitor l-NAME (20 mg/kg) was injected systemically 20 minutes before the intradermal application of VEGF-A and VEGF-C. l-NAME markedly attenuated vascular permeability induced by both VEGF-A and VEGF-C (Figure 4B) ▶ . The inactive stereoisomer d-NAME (20 mg/kg), which does not inhibit endothelial NO synthesis, failed to inhibit the increase in vascular permeability observed with either VEGF-1 or VEGF-C (not shown). Thus, for both VEGF-A and VEGF-C, increased vascular permeability was dependent on local production of NO.
VEGF-C Receptor Expression
To identify potential target vessels for VEGF-C activity in adults, we studied VEGFR-3 and VEGFR-2 mRNA expression in explanted segments of human internal mammary artery and saphenous vein by reverse transcription-PCR. For these experiments we used human specimens, because the corresponding nucleotide sequences for rabbit species have not yet been published. Figure 5 ▶ demonstrates that the VEGF-C receptors VEGFR-2 and VEGFR-3, as well as VEGFR-1, are expressed in both venous and arterial tissues. As a positive control, we used HUVECs, for which coexpression of VEGFR-1, VEGFR-2, and VEGFR-3 has been previously described. 53 RNA from human fibroblasts, from which amplification never resulted in positive bands for these receptors, was used as a negative control.
Figure 5.

Reverse transcription-PCR demonstrates VEGFR-1, VEGFR-2, and VEGFR-3 mRNA expression in explanted segments of normal human saphenous vein (lane 1) and internal mammary artery (lane 2). HUVECs (lane 3), known to coexpress all three receptors, served as a positive control, whereas cultured human fibroblasts (lane 4) served as a negative control. M: DNA size marker. Sizes of the PCR products are indicated.
Effect of pcVEGF-C and rhVEGF-C on Blood Pressure and Flow
Calf blood pressure ratio was similar in all groups at day 0 (Figure 6A) ▶ . By day 30, blood pressure ratio in the pcVEGF-C and VEGF-C protein-treated groups had increased to 0.83 ± 0.03 and 0.76 ± 0.04, respectively. In both cases, this value exceeded that measured for the control groups (0.59 ± 0.04 and 0.58 ± 0.03; P = 0.002 and 0.009, respectively). Blood pressure ratio in rabbits undergoing gene transfer with phVEGF-C was not significantly different from that recorded in rabbits treated with VEGF-C recombinant protein.
Figure 6.

A: Ratio of hindlimb perfusion pressures at day 0 (immediately before treatment) and day 30. The blood pressure ratio was defined for each rabbit as the ratio of systolic pressure of the ischemic limb to systolic pressure of the normal limb (n = 8 each). At day 0, no differences were observed among the groups. At day 30, the blood pressure ratio is significantly greater in rabbits receiving rhVEGF-C/VEGF-2 protein versus RSA controls and in rabbits receiving pcVEGF-C/VEGF-2 plasmid versus pGSVLacZ controls. B: Angiographic score, derived by quantitative analysis of angiographically demonstrable vessels in the medial thigh of the ischemic hindlimb, at days 0 and 30. At day 0, there are no differences among the groups. At day 30, the number of vessels is significantly greater in the VEGF-C/VEGF-2 protein- and plasmid-treated groups compared with controls. Administration of plasmid DNA appeared to yield a more pronounced effect than use of recombinant protein (n = 8 each). C: Iliac blood flow reserve (ratio between blood flow at rest and maximal flow induced by nitroprusside) measured from the internal iliac artery of the ischemic limb at day 30, using intra-arterial Doppler wire. Both VEGF-C/VEGF-2 protein and plasmid significantly increased iliac flow reserve (n = 8 each). D: Capillary density evaluated at day 30 in histological sections harvested from the medial thigh muscles of the nonischemic and ischemic limbs. In the nonischemic limb, capillary density was not different among groups (not shown). In the ischemic limb, capillary density is increased significantly by VEGF-C/VEGF-2 protein and plasmid (n = 8 each). NS, not significant.
Blood flow was measured in the internal iliac artery of the ischemic limb at days 0 and 30. At day 0, there were no differences among groups in resting or maximum blood flow (not shown). For pcVEGF-C plasmid, 30-day maximum iliac blood flow was clearly increased compared with pGSVLacZ controls (53.7 ± 5.9 versus 31.6 ± 1.2 ml/min; P = 0.0009). At day 30, maximum flow after nitroprusside infusion (39.1 ± 6.7 ml/min) was significantly higher in the VEGF-C protein-treated group compared with RSA control (26.1 ± 5.4 ml/min; P = 0.03). Comparison between the plasmid and protein treatment groups disclosed no statistically significant difference (P = 0.17).
Iliac flow reserve, the ratio between blood flow at rest and after maximal pharmacological stimulation, was 2.90 ± 0.31 and 2.65 ± 0.14 for the pcVEGF-C- and rhVEGF-C-treated rabbits, respectively. In both cases, this represented a statistically significant increase compared with controls (pGSVLacZ = 2.10 ± 0.17 and RSA = 2.04 ± 0.23; P = 0.04 and P = 0.04, respectively) (Figure 6C) ▶ .
Effect of pcVEGF-C and rhVEGF-C on Anatomical Evidence of Collateral Development
Quantitative analysis of collateral vessel development in the medial thigh is summarized in Figure 6B ▶ . Before treatment (day 0), there were no significant differences among groups in angiographic score. By day 30, the angiographic score in both VEGF-C gene transfer (0.85 ± 0.05) and protein (0.74 ± 0.08) groups exceeded that measured in the control groups (pGSVLacZ = 0.51 ± 0.02 (P = 0.00003) and RSA = 0.53 ± 0.03 (P = 0.023), respectively). The angiographic score in the pcVEGF-C gene transfer group was significantly higher than that measured for the protein-treated group (P = 0.02). Figure 7 ▶ shows representative internal iliac angiograms recorded at day 30 from controls, VEGF-C plasmid, and VEGF-C recombinant protein-treated animals. In both treatment groups, collateral artery development was more marked compared with corresponding control groups.
Figure 7.

Selective internal iliac angiography at day 30 of animals receiving RSA control (A), rhVEGF-C/VEGF-2 protein (B), pGSVLacZ control plasmid (B), and pcVEGF-C plasmid DNA (D). In contrast to controls, treatment groups exhibited an increase in angiographically visible collateral blood vessels. (Ruler cm marking.)
Tissue sections from the medial thigh muscles of the ischemic and nonischemic limbs were examined by light microscopy at day 30. Histological evaluation after alkaline phosphatase staining revealed that the capillary density in the pcVEGF-C-transfected group (252 ± 12/mm2) and that in the VEGF-C protein-treated group (229 ± 20/mm2) were both significantly greater than values observed for the corresponding control groups (pGSVLacZ = 183 ± 10/mm2 (P = 0.001), and RSA = 164 ± 20/mm2 (P = 0.043), respectively) (Figure 6D) ▶ . Analysis of capillary/muscle fiber ratio yielded similar results (data not shown). Analysis of capillary density and capillary/muscle fiber ratio in the medial thigh muscle of the nonischemic limb showed no differences among groups (data not shown). Representative examples of histological sections stained for alkaline phosphatase in the different experimental groups are shown in Figure 8 ▶ .
Figure 8.

Histological sections retrieved at day 30 from ischemic hindlimb muscle (musculus adductor) and stained with alkaline phosphatase (counterstained with eosin). A: RSA control; B: rhVEGF-C/VEGF-2 protein; C: pGSVLacZ control plasmid; D: pcVEGF-C/VEGF-2 plasmid-treated rabbit. Administration of VEGF-C as recombinant protein or plasmid resulted in increased capillary density. Dots, which appear dark blue, indicate capillaries.
Discussion
The recent identification and purification of VEGF-C/VEGF-2 20 and VEGF-B/VEGF-3, 21 in addition to VEGF-A/VEGF-1 and PlGF, increased the number of known VEGF family members to four and correspondingly added another level of complexity to current concepts of angiogenesis and vascular remodeling. All four members are encoded by different genes, localized to different chromosomes, 23,62 but share considerable homology. In the case of VEGF-C, for example, 32% of the amino acid sequence is identical to VEGF121 and 27% to PlGF131.
The divergence in bioactivity, as shown for VEGF-A and PlGF, 26,28 is most likely due to different affinities of the four VEGF members to the three known endothelial-specific fms-like tyrosine kinases VEGFR-1, VEGFR-2, and VEGFR-3. VEGF-C was isolated as a ligand and specific activator of VEGFR-3 20,32 and, in addition, was shown to stimulate autophosphorylation of VEGFR-2. 20 Moreover, VEGFR-3 and VEGFR-2 mRNA expression was demonstrated in cultured HUVECs, 53 HMECs, and femoral vein ECs. 63 We therefore initiated our evaluation of VEGF-C bioactivity by performing proliferation and migration assays on different EC types in vitro. We observed clear mitogenic and chemotactic responses of ECs to VEGF-C, in agreement with previous reports. 20,32 Although the impact on cell migration was similar for VEGF-A and VEGF-C, the proliferative response to VEGF-C was less than that observed for VEGF-A, particularly in the case of HUVECs. Despite the latter, in vivo angiogenesis observed in response to VEGF-C was robust and indistinguishable from that observed previously in the same animal model using VEGF-A recombinant protein 12 or plasmid DNA. 64
The fact that the in vivo results of these experiments better parallel in vitro analyses of migration as opposed to proliferation is consistent with notions regarding the relative contribution of these activities to angiogenesis. In a classic experiment performed in the rat cornea, Sholley et al 65 showed that vascular sprouting could be induced and continue for more than 2 days, despite irradiation treatment sufficient to suppress DNA synthesis. Angiogenic activity in this model was thus interpreted to reflect the dominant impact of EC migration. Alternatively, it is possible that the somewhat one-dimensional in vitro assays commonly used to evaluate EC proliferation and migration do not accurately reflect the potential for angiogenesis to develop in vivo.
NO is released from ECs and arterial segments in cell and organ culture, respectively, 58,61 and has been shown to mediate VEGF-induced neovascularization in a rabbit cornea pocket assay. 66 More recently, experiments that we have performed in mice lacking endothelial NO synthase 67 have established that endogenous NO sulfate is essential for endogenous angiogenesis that develops in response to limb ischemia, and that the absence of endogenous NO sulfate precludes a favorable response to rhVEGF165. 67 Our finding that VEGF-C promotes NO release in vitro that is comparable to VEGF-A is consistent with the demonstration that VEGF-C promotes angiogenesis in vivo.
In accordance with this finding, VEGF-C enhanced permeability in the Miles assay, an effect that was almost completely inhibited by prior administration of an inhibitor of NO synthase. We have recently reported similar results for VEGF-A. 68 Given the extent to which angiogenesis and permeability appear to be linked, 69 this finding provides additional evidence for involvement of NO in mediating VEGF (A and C)-mediated angiogenesis. To our knowledge, the results obtained using the Miles assay establish VEGF-C as the only growth factor other than VEGF-A to augment vascular permeability.
Subsequent to initial descriptions of VEGF-C and VEGF-B20, 62 serious questions have been raised regarding the bioactivity of these two gene products. In vitro, both factors were found to exhibit mitogenic activity, 20,21,32 and VEGF-C stimulated bovine EC outgrowth in a collagen gel assay. 20 Overexpression of VEGF-C in the skin of transgenic mice, however, was observed by Jeltsch et al 36 to result in lymphatic vessel hyperplasia; because no evidence of angiogenesis was observed, these findings were interpreted to indicate that VEGF-C does not promote angiogenesis but is instead a specific ligand for lymphatic endothelium. 36 As the authors acknowledged, this outcome was “unexpectedly specific,” because VEGF-C has been shown to transduce the VEGFR-2 receptor, the principal receptor responsible for VEGF-A-induced angiogenesis.
Our findings contradict the assertion of Jeltsch et al. 36 Using a previously established rabbit ischemic hindlimb model, 54,55 we demonstrated that VEGF-C clearly promotes angiogenesis in vivo. Anatomical and physiological evidence of augmented angiogenesis observed in this animal model have been previously shown to result from marked enhancement of EC proliferative activity in response to an endothelial mitogen 70 and thus implies the development of vascular sprouts. Preclinical studies in this animal model using naked DNA encoding for VEGF-A have been duplicated in human subjects. 15,71 Similar to results reported previously for VEGF-A, 12,64 VEGF-C—administered as naked DNA or recombinant protein—increased flow reserve, hindlimb perfusion pressure, angiographically visible collateral vessels, and capillary density. In those experiments in which VEGF-A was transferred as plasmid DNA, reverse transcription-PCR followed by Southern blot analysis detected expression of the transferred gene up to 21 days posttransfection in the arterial wall. 72 Such continuous expression of the introduced gene constitutes one potential explanation for the more pronounced effect on physiological and anatomical parameters after gene transfer versus single bolus administration of recombinant VEGF-C protein. Alternatively, VEGF-C, once transcribed and translated, may form intracellular heterodimers with other VEGF family members, as shown for VEGF-A 27-29 and VEGF-C, 21 thereby compounding effects attributable to one factor alone.
There are at least two potential explanations for the differing outcomes observed previously in transgenic mice and those observed currently in the rabbit model. First, physiological function of any ligand is dependent on the temporal and spatial expression of its specific receptors. It remains to be determined which of the two receptors, VEGFR-2 or VEGFR-3, or both, transduce the effects of VEGF-C, given that autophosphorylation of both receptors has been shown to occur after stimulation with VEGF-C, 20,32 and both are shown here to be expressed postnatally in segments of arteries and veins. (Similar results have been previously reported in cultured HMECs and femoral and umbilical vein ECs. 53,63 ) Even if VEGFR-3 function in adults is limited to maintenance of lymphatic vessels, as demonstrated recently for embryonic development, 36,73 postnatal angiogenesis may be exclusively mediated by the VEGFR-2 receptor. 36 In addition, the possible formation of VEGFR-2/VEGFR-3 heterodimers could lead to more cell type and/or tissue-specific consequences. 36
Furthermore, previous studies from our laboratory 74 and those of others 75 have shown that ischemia leads to regional upregulation of VEGFR-2, the principal receptor mediating VEGF-A-induced angiogenesis. In the absence of such ischemia-induced upregulation, we have never observed angiogenesis in response to transient overexpression of VEGF-A. 12,76 Consequently, in the absence of appropriate receptor upregulation, even transient overexpression of VEGF might have been interpreted not to promote angiogenesis in postnatal animal models.
A second potential explanation involves the construct used by Jeltsch et al 36 to generate VEGF-C transgenic mice. Because it utilizes the human keratin 14 (K14) promoter, gene expression is targeted to the basal cells of stratified squamous epithelium. It may thus not be valid to extrapolate these results to gene expression in other tissues or organs.
In summary, the current study demonstrates that despite evidence of specificity for lymphatic endothelium in a developmental model, VEGF-C, when administered to adult animals in the setting of tissue ischemia, promotes angiogenesis and augments flow to ischemic tissues. These findings underscore the principle that constitutive overexpression of a given protein during embryogenesis is not necessarily equivalent to transient overexpression of the same protein postnatally; postnatal receptor modulation associated with specific pathological processes, in this case tissue ischemia, may in part explain the differing outcomes observed in the embryonic versus adult organism. Thus, VEGF-C may indeed have utility as a primary or adjunctive agent for therapeutic angiogenesis.
Footnotes
Address reprint requests to Dr. Jeffrey M. Isner, St. Elizabeth’s Medical Center of Boston, 736 Cambridge Street, Boston, MA 02135. E-mail: jisner@opal.tufts.edu.
References
- 1.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N: Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246:1306-1309 [DOI] [PubMed] [Google Scholar]
- 2.Connolly DT, Hewelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel RN, Leimgruber RS, Feder J: Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest 1989, 84:1470-1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plouet J, Schilling J, Gospodarowicz D: Isolation and characterization of a newly identified endothelial cell mitogen produced by AtT-20 cells. EMBO J 1989, 8:3801-3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF: Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219:983-985 [DOI] [PubMed] [Google Scholar]
- 5.Breier G, Albrecht U, Sterrer S, Risau W: Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development 1992, 114:521-532 [DOI] [PubMed] [Google Scholar]
- 6.Drake CJ, Little CD: Exogenous vascular endothelial growth factor induces malformed and hyperfused vessels during embryonic neovascularization. Proc Natl Acad Sci USA 1995, 92:7657-7661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Kendraprasad H, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A: Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380:435-439 [DOI] [PubMed] [Google Scholar]
- 8.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hilan KJ, Moore MW: Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380:439-442 [DOI] [PubMed] [Google Scholar]
- 9.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E: Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995, 1:1024-1028 [DOI] [PubMed] [Google Scholar]
- 10.Peters KG, deVries C, Williams LT: Vascular endothelial growth factor receptor expression during embryogenesis and tissue repair suggests a role in endothelial differentiation and blood vessel growth. Proc Natl Acad Sci USA 1993, 90:8915-8919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsurumi Y, Murohara T, Krasinski K, Dongfen C, Witzenbichler B, Kearney M, Couffinhal T, Isner JM: Reciprocal relationship between VEGF and NO in the regulation of endothelial integrity. Nat Med 1997, 3:879-886 [DOI] [PubMed] [Google Scholar]
- 12.Takeshita S, Zheng LP, Brogi E, Kearney M, Pu LQ, Bunting S, Ferrara N, Symes JF, Isner JM: Therapeutic angiogenesis: a single intra-arterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hindlimb model. J Clin Invest 1994, 93:662-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearlman JD, Hibberd MG, Chuang ML, Harada K, Lopez JJ, Gladston SR, Friedman M, Sellke FW, Simons M: Magnetic resonance mapping demonstrates benefits of VEGF-induced myocardial angiogenesis. Nat Med 1995, 1:1085-1089 [DOI] [PubMed] [Google Scholar]
- 14.Banai S, Jaklitsch MT, Shou M, Lazarous DF, Scheinowitz M, Biro S, Epstein SE, Unger EF: Angiogenic-induced enhancement of collateral blood flow to ischemic myocardium by vascular endothelial growth factor in dogs. Circulation 1994, 89:2183-2189 [DOI] [PubMed] [Google Scholar]
- 15.Isner JM, Pieczek A, Schainfeld R, Blair R, Haley L, Asahara T, Rosenfield K, Razvi S, Walsh K, Symes J: Clinical evidence of angiogenesis following arterial gene transfer of phVEGF165. Lancet 1996, 348:370-374 [DOI] [PubMed] [Google Scholar]
- 16.Folkman J: Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995, 1:27-30 [DOI] [PubMed] [Google Scholar]
- 17.Plate KH, Breier G, Millauer B, Ullrich A, Risau W: Up-regulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res 1993, 53:5822-5827 [PubMed] [Google Scholar]
- 18.Plate KH, Breier G, Weich HA, Risau W: Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359:845-848 [DOI] [PubMed] [Google Scholar]
- 19.Kim KJ, Li B, Winer J, Armanini M, Billett N, Phillips HS, Ferrara N: Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362:841-844 [DOI] [PubMed] [Google Scholar]
- 20.Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, Saksela O, Kalkkinen N, Alitalo K: A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J 1996, 15:290-298 [PMC free article] [PubMed] [Google Scholar]
- 21.Olofsson B, Pajusola K, Kaipainen A, vonEuler G, Joukov V, Saksela O, Orpana A, Pettersson RF, Alitalo K, Eriksson U: Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc Natl Acad Sci USA 1996, 93:2576-2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG: Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci USA 1991, 88:9267-9271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maglione D, Guerriero V, Viglietto G, Ferraro MG, Aprelikova O, Alitalo K, Del Vecchio S, Lei KJ, Chou JY, Persico MG: Two alternative mRNAs coding for the angiogenic factor, placenta growth factor (PIGF), are transcribed from a single gene of chromosome 14. Oncogene 1993, 8:925-931 [PubMed] [Google Scholar]
- 24.Hauser S, Weich HA: A heparin-binding form of placenta growth factor (PIGF-2) is expressed in human umbilical vein endothelial cells, and in placenta. Growth Factors 1993, 9:259-268 [DOI] [PubMed] [Google Scholar]
- 25.Park JE, Keller G-A, Ferrara N: The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial ECM and bioactivity of ECM-bound VEGF. Mol Biol Cell 1993, 4:1317-1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park JE, Chen HH, Winer J, Houck KA, Ferrara N: Placenta growth factor: potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem 1994, 269:25646-25654 [PubMed] [Google Scholar]
- 27.DiSalvo J, Bayne ML, Conn G, Kwok PW, Trivedi PG, Soderman DD, Palisi TM, Sullivan KA, Thomas KA: Purification and characterization of a naturally occurring vascular endothelial growth factor: placenta growth factor heterodimer. J Biol Chem 1995, 270:7717-7723 [DOI] [PubMed] [Google Scholar]
- 28.Cao Y, Linden P, Shima D, Browne F, Folkman J: In vivo angiogenic activity and hypoxia induction of heterodimer of placenta growth factor/vascular endothelial growth factor. J Clin Invest 1996, 98:2507-2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao Y, Chen H, Zhou L, Chiang MK, Anand-Apte B, Weatherbee JA, Wang Y, Fang F, Flanagan JG, Tsang MLS: Heterodimers of placenta growth factor/vascular endothelial growth factor: endothelial activity, tumor cell expression and high affinity binding to Flk-1/KDR. J Biol Chem 1996, 271:3154-3162 [DOI] [PubMed] [Google Scholar]
- 30.Shweiki D, Itin A, Soffer D, Keshet E: Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359:843-845 [DOI] [PubMed] [Google Scholar]
- 31.Brogi E, Wu T, Namiki A, Isner JM: Indirect angiogenic cytokines upregulate VEGF and bFGF gene expression in vascular smooth muscle cells, while hypoxia upregulates VEGF expression only. Circulation 1994, 90:649-652 [DOI] [PubMed] [Google Scholar]
- 32.Lee J, Gray A, Yuan J, Luoh S, Avraham H, Wood WI: Vascular endothelial growth factor-related protein: a ligand and specific activator of the tyrosine kinase receptor Flt4. Proc Natl Acad Sci USA 1996, 93:1988-1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pajusola K, Aprelikova O, Pelicci G, Weich H, Claesson Welsh L, Alitalo K: Signaling properties of FLT4, a proteolytically processed receptor tyrosine kinase related to two VEGF receptors. Oncogene 1994, 9:3545-3555 [PubMed] [Google Scholar]
- 34.Ferrara N, Davis-Smyth: The biology of vascular endothelial growth factor. Endocr Rev 1997, 18:4-25 [DOI] [PubMed] [Google Scholar]
- 35.Couffinhal T, Kearney M, Witzenbichler B, Chen D, Murohara T, Losordo DW, Symes JF, Isner JM: Vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) in normal and atherosclerotic human arteries. Am J Pathol 1997, 150:1673-1685 [PMC free article] [PubMed] [Google Scholar]
- 36.Jeltsch M, Kaipainen A, Joukov V, Meng X, Lakso M, Rauvala H, Swartz M, Fukumura D, Jain RK, Alitalo K: Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science 1997, 276:1423-1425 [DOI] [PubMed] [Google Scholar]
- 37.Leibovich SJ, Polverini PJ, Fong TW, Harlow LA, Koch AE: Production of angiogenic activity by human monocytes requires an l-arginine/nitric oxide-synthase-dependent effector mechanism. Proc Natl Acad Sci USA 1994, 91:4190-4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asahara T, Bauters C, Wu T, Zheng LP, Chen D, Symes J, Isner JM: Nitric oxide precursor augments angiogenesis and attenuates endothelial dysfunction in collateral circulation in rabbit ischemic hindlimb in vivo (Abstract). FASEB J 1996, 10:A545 [Google Scholar]
- 39.Morbidelli L, Chang C-H, Douglas JG, Granger HJ, Ledda F, Ziche M: Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am J Physiol 1995, 270:H411-H415 [DOI] [PubMed] [Google Scholar]
- 40.Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, Geppetti P, Ledda F: Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest 1994, 94:2036-2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu J-S, Hastings GA, Cherry S, Gentz R, Ruben S, Coleman TA: A novel regulatory function of proteolytically cleaved VEGF-2 for vascular endothelial and smooth muscle cells. FASEB J 1997, 11:498-504 [DOI] [PubMed] [Google Scholar]
- 42.Bonnerot C, Rocancourt D, Briand P, Grimber G: A β-galactosidase hybrid protein targeted to nuclei as a marker for developmental studies. Proc Natl Acad Sci USA 1987, 84:6795-6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaffe EA, Nachman RL, Becker CG, Minick CR: Culture of human endothelial cells derived from umbilical veins. J Clin Invest 1973, 52:2745-2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlingemann RO, Domgjan GM, Emeis JJ, Blok J, Warnaar SO, Ruiter DJ: Monoclonal antibody PAL-E specific for endothelium. Lab Invest 1985, 52:71-76 [PubMed] [Google Scholar]
- 45.Buttke TM, McCubrey JA, Owen TC: Use of an aqueous soluble tetrazolium/formazan assay to measure viability and proliferation of lymphokine-dependent cell lines. J Immunol Methods 1993, 157:233-240 [DOI] [PubMed] [Google Scholar]
- 46.Falk W, Goodwin RH, Leonard EJ: A 48 well microchemotaxis assembly for rapid and accurate measurement of leukocyte migration. J Immunol Methods 1980, 33:239-247 [DOI] [PubMed] [Google Scholar]
- 47.Shibuki K, Okada D: Endogenous nitric oxide release required for long-term synaptic depression in the cerebellum. Nature 1991, 358:676-678 [DOI] [PubMed] [Google Scholar]
- 48.Weyrich AS, Ma X-l, Buerke M, Murohara T, Armstead VE, Lefer AM, Nicolas JM, Thomas AP, Lefer DJ, Vinten-Johansen J: Physiological concentrations of nitric oxide do not elicit an acute negative inotropic effect in unstimulated cardiac muscle. Circ Res 1994, 75:692-700 [DOI] [PubMed] [Google Scholar]
- 49.Miles AA, Miles EM: Vascular reactions to histamine, histamine liberators or leukotoxins in the skin of the guinea pig. J Physiol 1952, 118:228-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 51.Sambrook J, Fritsch EF, Maniatis T: ed 2 Molecular Cloning: A Laboratory Manual, 1989, :pp 8.60-8.63 Cold Spring Harbor Laboratory Press, New York [Google Scholar]
- 52.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KG, Ehrlich HA: Primer-directed enzymatic amplification of DNA with thermostable DNA polymerase. Science 1988, 239:487-491 [DOI] [PubMed] [Google Scholar]
- 53.Hewett PW, Murray JC: Coexpression of flt-1, flt-4 and KDR in freshly isolated and cultured human endothelial cells. Biochem Biophys Res Commun 1996, 221:697-702 [DOI] [PubMed] [Google Scholar]
- 54.Bauters C, Asahara T, Zheng LP, Takeshita S, Bunting S, Ferrara N, Symes JF, Isner JM: Physiologic assessment of augmented vascularity induced by VEGF in ischemic rabbit hindlimb. Am J Physiol 1994, 267:H1263-H1271 [DOI] [PubMed] [Google Scholar]
- 55.Pu LQ, Jackson S, Lachapelle KJ, Arekat Z, Graham AM, Lisbona R, Brassard R, Carpenter S, Symes JF: A persistent hindlimb ischemia model in the rabbit. J Invest Surg 1994, 7:49-60 [DOI] [PubMed] [Google Scholar]
- 56.Takeshita S, Pu L-Q, Zheng L, Ferrara N, Stein LA, Sniderman AD, Isner JM, Symes JF: Vascular endothelial growth factor induces dose-dependent revascularization in a rabbit model of persistent limb ischemia. Circulation 1994, 90:II-228-II-234 [PubMed] [Google Scholar]
- 57.Walter DH, Hink U, Asahara T, Van Belle E, Horowitz J, Tsurumi Y, Vandlen R, Heinsohn H, Keyt B, Ferrara N, Symes JF, Isner JM: The in vivo bioactivity of vascular endothelial growth factor/vascular permeability factor is independent of N-linked glycosylation. Lab Invest 1996, 74:546-556 [PubMed] [Google Scholar]
- 58.Ku DD, Zaleski JK, Liu S, Brock TA: Vascular endothelial growth factor induces EDRF-dependent relaxation in coronary arteries. Am J Physiol 1993, 265:H586-H592 [DOI] [PubMed] [Google Scholar]
- 59.Doucette JW, Corl PD, Payne HM, Flynn AE, Goto M, Nassi M, Segal J: Validation of a Doppler wire for intravascular measurements of coronary artery flow velocity. Circulation 1992, 85:1899-1911 [DOI] [PubMed] [Google Scholar]
- 60.Ziada AM, Hudlicka O, Tyler KR, Wright AJ: The effect of long-term vasodilation on capillary growth and performance in rabbit heart and skeletal muscle. Cardiovasc Res 1984, 18:724-732 [DOI] [PubMed] [Google Scholar]
- 61.van der Zee R, Murohara T, Luo Z, Zollmann F, Passeri J, Lekutat C, Isner JM: Vascular endothelial growth factor (VEGF)/vascular permeability factor (VPF) augments nitric oxide release from quiescent rabbit and human vascular endothelium. Circulation 1997, 95:1030-1037 [DOI] [PubMed] [Google Scholar]
- 62.Paavonen K, Horeli-Kuitunen N, Chilov D, Kukk E, Pennanen S, Kallioniemi O, Pajusola K, Olofsson B, Eriksson U, Joukov V, Palotie A, Alitalo K: Novel human vascular endothelial growth factor genes VEGF-B and VEGF-C localize to chromosomes 11q13 and 4q34, respectively. Circulation 1996, 93:1079-1082 [DOI] [PubMed] [Google Scholar]
- 63.Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VWM, Fang T-H, Dumont D, Breitman M, Alitalo K: Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci USA 1995, 92:3566-3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takeshita S, Tsurumi Y, Couffinhal T, Asahara T, Bauters C, Symes JF, Ferrara N, Isner JM: Gene transfer of naked DNA encoding for three isoforms of vascular endothelial growth factor stimulates collateral development in vivo. Lab Invest 1996, 75:487-502 [PubMed] [Google Scholar]
- 65.Sholley MM, Ferguson GP, Seibel HR, Montour JL, Wilson JD: Mechanisms of neovascularization: vascular sprouting can occur without proliferation of endothelial cells. Lab Invest 1984, 51:624-634 [PubMed] [Google Scholar]
- 66.Ziche M, Morbidelli L, Choudhuri R, Zhang H-T, Donnini S, Granger HJ, Bicknell R: Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not fibroblast growth factor-induced angiogenesis. J Clin Invest 1997, 99:2625-2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM: Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest 1998, 101:2567-2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murohara T, Horowitz J, Silver M, Tsurumi Y, Sullivan A, Isner JM: Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation 1998, 97:99-107 [DOI] [PubMed] [Google Scholar]
- 69.Dvorak HF, Brown LF, Detmar M, Dvorak AM: Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995, 146:1029-1039 [PMC free article] [PubMed] [Google Scholar]
- 70.Takeshita S, Rossow ST, Kearney M, Zheng LP, Bauters C, Bunting S, Ferrara N, Symes JF, Isner JM: Time course of increased cellular proliferation in collateral arteries following administration of vascular endothelial growth factor in a rabbit model of lower limb vascular insufficiency. Am J Pathol 1995, 147:1649-1660 [PMC free article] [PubMed] [Google Scholar]
- 71.Baumgartner I, Pieczek A, Manor O, Blair R, Kearney M, Walsh K, Isner JM: Constitutive expression of phVEGF165 following intramuscular gene transfer promotes collateral vessel development in patients with critical limb ischemia. Circulation 1998, 97:1114-1123 [DOI] [PubMed] [Google Scholar]
- 72.Takeshita S, Weir L, Chen D, Zheng LP, Riessen R, Bauters C, Symes JF, Ferrara N, Isner JM: Therapeutic angiogenesis following arterial gene transfer of vascular endothelial growth factor in a rabbit model of hindlimb ischemia. Biochem Biophys Res Commun 1996, 227:628-635 [DOI] [PubMed] [Google Scholar]
- 73.Kukk E, Lymboussaki A, Taira S, Kaipainen A, Jeltsch M, Joukov V, Alitalo K: VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development 1996, 122:3829-3837 [DOI] [PubMed] [Google Scholar]
- 74.Brogi E, Schatteman G, Wu T, Kim EA, Varticovski L, Keyt B, Isner JM: Hypoxia-induced paracrine regulation of VEGF receptor expression. J Clin Invest 1996, 97:469-476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Waltenberger J, Mayr U, Pentz S, Hombach V: Functional upregulation of the vascular endothelial growth factor receptor KDR by hypoxia. Circulation 1996, 94:1647-1654 [DOI] [PubMed] [Google Scholar]
- 76.Isner JM, Walsh K, Symes J, Pieczek A, Takeshita S, Lowry J, Rosenfield K, Weir L, Brogi E, Jurayj D: Arterial gene transfer for therapeutic angiogenesis in patients with peripheral artery disease. Hum Gene Ther 1996, 7:959-988 [DOI] [PubMed] [Google Scholar]