

Abstract
Mutations in DJ-1 cause an autosomal recessive, early onset familial form of Parkinson disease (PD). However, little is presently known about the role of DJ-1 in the more common sporadic form of PD and in other age-related neurodegenerative diseases, such as Alzheimer disease (AD). Here we report that DJ-1 is oxidatively damaged in the brains of patients with idiopathic PD and AD. By using a combination of two-dimensional gel electrophoresis and mass spectrometry, we have identified 10 different DJ-1 isoforms, of which the acidic isoforms (pI 5.5 and 5.7) of DJ-1 monomer and the basic isoforms (pI 8.0 and 8.4) of SDS-resistant DJ-1 dimer are selectively accumulated in PD and AD frontal cortex tissues compared with age-matched controls. Quantitative Western blot analysis shows that the total level of DJ-1 protein is significantly increased in PD and AD brains. Mass spectrometry analyses reveal that DJ-1 is not only susceptible to cysteine oxidation but also to previously unsuspected methionine oxidation. Furthermore, we show that DJ-1 protein is irreversibly oxidized by carbonylation as well as by methionine oxidation to methionine sulfone in PD and AD. Our study provides new insights into the oxidative modifications of DJ-1 and indicates association of oxidative damage to DJ-1 with sporadic PD and AD.
Alzheimer disease (AD)2 and Parkinson disease (PD) are the two most common neurodegenerative disorders characterized by the selective loss of neurons in specific brain regions and the deposition of misfolded proteins into aggregates or inclusions, such as neurofibrillary tangles and amyloid plaques in AD and Lewy bodies in PD (1). The majority of AD and PD cases are sporadic with hereditary familial cases accounting for less than 10% (2, 3). The genetic defects underlying several monogenic familial forms of AD and PD have recently been identified (3). However, the causes of other AD and PD cases, particularly sporadic cases, remain unclear.
Increasing evidence indicates that oxidative stress plays a crucial role in the pathogenesis of idiopathic AD and PD (4-7). For example, both AD and PD have been associated with increased production of reactive oxygen species (ROS), which could result from a combination of aging, genetic predisposition, and environmental factors (6). Epidemiological studies suggest that exposure to pesticides, herbicides, and other environmental toxins that inhibit mitochondrial complex I can lead to excess production of ROS and increased incidence of sporadic PD (8). Furthermore, post-mortem analyses reveal that the overall levels of oxidative damage to proteins, lipids, and DNA are elevated in AD and PD brains (4, 9).
The most widely used marker for oxidative damage to proteins is the presence of carbonyl groups, which can be introduced into proteins by direct oxidation of Pro, Arg, Lys, or Thr side chains or by Michael addition reactions of Cys, His, or Lys residues with products of lipid peroxidation or glycooxidation (5, 10, 11). Elevation in the total level of protein carbonyls has been documented in both AD and PD (4, 9). Although it was initially thought that targets of oxidative damage by reactive oxygen species were random and indiscriminate, it has become increasingly clear that the susceptibility of proteins to oxidative damage is highly dependent on specific properties of individual proteins, such as unique sequence motifs, surface accessibility, protein folding, and subcellular localization (10-12). The identities of the oxidatively damaged proteins in AD and PD that have been modified by carbonylation or other types of oxidation remain largely unknown.
Mutations in DJ-1 has recently been linked to an autosomal recessive, early onset familial form of PD (13). DJ-1 is a ubiquitously expressed protein of the DJ-1/ThiJ/PfpI superfamily (14, 15). The precise biochemical function of DJ-1 is unknown, although DJ-1 has been proposed to act as a protease, chaperone, or antioxidant (15-19). The identification of a homozygous DJ-1 deletion that prevents the expression of DJ-1 protein in recessively transmitted PD (13) strongly suggests that loss of DJ-1 function leads to neurodegeneration. A loss-of-function pathogenic mechanism is also supported by analysis of PD-linked DJ-1 L166P mutation (17, 20-22) and DJ-1 knock-out studies in mice (23-25). Despite increasing genetic evidence indicating the importance of DJ-1 mutations in causing early onset familial PD (13, 26-28), the role of DJ-1 in sporadic PD is unknown. Interestingly, previous studies have shown that DJ-1 is rarely present in Lewy bodies in PD (29, 30). However, DJ-1 has been found to colocalize with β-synuclein-immunoreactive glial inclusions in multiple system atrophy and with a subset of pathological tau inclusions in a number of neurodegenerative tauopathies, including AD (29, 30). Here, we show that DJ-1 is extensively and irreversibly oxidized in brains of patients with idiopathic PD and AD. The observed oxidative damage to DJ-1 may have important implications for understanding the pathogenesis of sporadic PD and AD.
MATERIALS AND METHODS
Human Brain Samples—Brain tissues were obtained from the Emory Center for Neurodegenerative Disease Brain Bank. For biochemical studies, frontal cortex tissues from five PD cases, five AD cases, and five healthy non-demented control subjects were used (Table 1). The neuropathological diagnosis of PD was based on the presence of nigral degeneration and Lewy bodies. The neuropathological diagnosis of AD was made using Consortium to Establish a Registry for Alzheimer Disease criteria (31).
TABLE 1.
Demographic data of cases included in this study
| Case no. | Diagnosis | Age | Sex | PMIa |
|---|---|---|---|---|
| years | h | |||
| 1 | PD | 79 | Male | 2 |
| 2 | PD | 69 | Male | 3 |
| 3 | PD | 74 | Male | < 5 |
| 4 | PD | 74 | Male | 4.5 |
| 5 | PD | 66 | Male | 12 |
| 6 | AD | 76 | Female | 15 |
| 7 | AD | 91 | Female | 2.5 |
| 8 | AD | 70 | Male | 11 |
| 9 | AD | 85 | Female | 9.5 |
| 10 | AD | 50 | Female | 17 |
| 11 | Control | 74 | Female | 3 |
| 12 | Control | 68 | Female | 11 |
| 13 | Control | 65 | Female | 6 |
| 14 | Control | 75 | Female | 6 |
| 15 | Control | 87 | Male | 20.5 |
PMI, post-mortem interval.
2,4-Dinitrophenyl (DNP) derivatization, Two-dimensional Gel Electrophoresis, and Immunoblot Analysis—Protein extracts were prepared from human brain tissues as described (32). Protein samples (350 μg) were resolved by isoelectric focusing on 17-cm immobilized pH gradient strips (pH 3-10) followed by in-strip DNP derivatization (reacting with protein carbonyls) as described (32, 33). Second-dimensional separation was performed by electrophoresis on SDS-polyacrylamide gradient gels (10-20% porosity polyacrylamide) using the Ettan-DALT slab gel SDS-PAGE system (Amersham Biosciences). After electrophoresis, the gels were analyzed by Sypro Ruby staining and by immunoblotting as described (32) with anti-DNP antibody (1:16,000, Molecular Probes) or anti-DJ-1 antibody (P7F, 1:5000) (17). The protein level and the carbonyl level of each DJ-1 isoform in PD, AD, and controls were quantified using the two-dimensional gel analysis program PD Quest (Bio-Rad). Statistical comparison of the data obtained from five PD, five AD, and five control individuals were performed using analysis of variance. Significant difference was accepted at p < 0.05.
Mass Spectrometry—Spots of interest were excised from the gels and digested in situ with trypsin (modified; Promega) or Lys-C (Sigma-Aldrich). The Lys-C digests were derivatized with ProteoMass Guanidination kit (Sigma-Aldrich) to increase the mass spectral signal intensities of C-terminal lysine-containing peptides (34). The digests were subjected to analysis by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF/MS), matrix-assisted laser desorption ionization time-of-flight tandem mass spectrometry (MALDI-TOF/TOF/MS/MS), and capillary HPLC-electrospray ionization tandem mass spectrometry (HPLC-ESI/MS/MS). MALDI-TOF/MS and MALDI-TOF-TOF/MS/MS mass spectra were acquired on an Applied Biosystems 4700 Proteomics discovery system. The peptide mass maps produced by MALDI-TOF/MS and MALDI-TOF/TOF/MS/MS were searched against the published databases by means of 4700 ExplorerTM software (Applied Biosystems). HPLC-ESI/MS/MS analysis was performed as described (32, 33). Assignment of the MS/MS fragments was verified by comparison with the predicted ions generated in silico by GPMAW (Lighthouse Data).
RESULTS
Accumulation of Acidic Isoforms of DJ-1 Monomer and Basic Isoforms of DJ-1 Dimer in PD and AD Brains—To investigate whether the expression and/or posttranslational modification(s) of DJ-1 is altered in sporadic PD and AD, we performed comparative high resolution, two-dimensional gel electrophoresis experiments on frontal cortex homogenates obtained from idiopathic PD and AD and age-matched controls. The frontal cortex was chosen because of tissue availability and its relevance to the pathology of both AD and PD. In AD the frontal cortex exhibits neuronal loss and is rich in neurofibrillary tangles and amyloid plaques. In PD, although neuronal loss mainly occurs in the substantia nigra, the frontal cortex contains Lewy bodies, the pathological hall-mark of PD. Analysis of DJ-1 expression and modifications in the substantia nigra or striatum might provide more PD-relevant information. Unfortunately, we had difficulty in obtaining sufficient amounts of these tissues for biochemical analyses.
Although DJ-1 exists as a homodimeric protein, DJ-1 migrates on SDS polyacrylamide gels as a 20-kDa monomer because of denaturation by SDS (17, 29). Immunoblot analysis of the two-dimensional SDS-PAGE gels with anti-DJ-1 antibody revealed the presence of at least six distinct isoforms of DJ-1 monomer that have the same apparent molecular mass of 20 kDa but different isoelectric points (5.5, 5.6, 5.7, 5.8, 6.1, and 6.4, respectively; Fig. 1, A and B). The predominant form of DJ-1 is the 20-kDa/pI 6.4 isoform, which is in good agreement with the predicted values of 19.9 kDa/pI 6.3 for DJ-1 monomer. Of the 6 identified DJ-1 monomer isoforms, the acidic isoforms (pI 5.5 and 5.7) of DJ-1 monomer were selectively accumulated in PD and AD compared with control brains (Fig. 1, B and C). The accumulation of these acidic DJ-1 isoforms is likely due to specific modifications of DJ-1 in PD and AD instead of nonspecific changes in DJ-1 occurring post-mortem because within each of the PD, AD, and control group, the relative levels of acidic DJ-1 isoforms (pI 5.5 and 5.7) were similar among samples with different post-mortem intervals (Fig. 1F).
FIGURE 1.
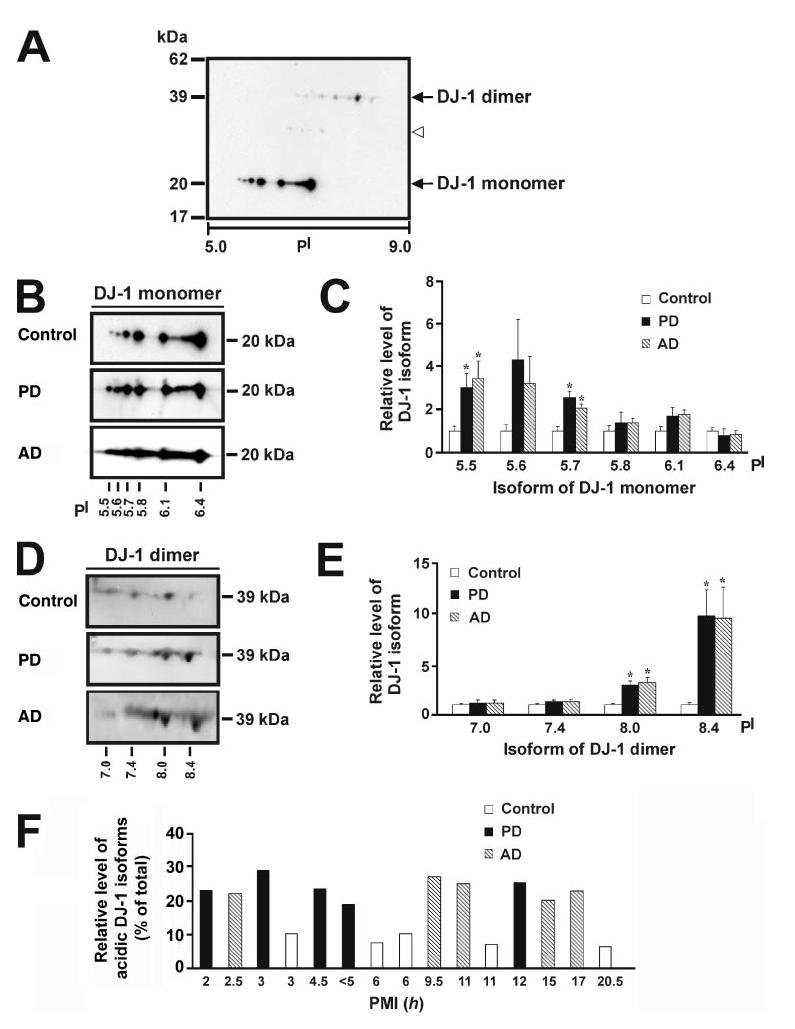
Altered expression of DJ-1 isoforms in PD and AD compared with control brains. A, two-dimensional immunoblot analysis with anti-DJ-1 antibody reveals the presence of multiple pI isoforms of DJ-1 monomer and dimer in human brain. The open arrowhead indicates the presence of weak DJ-1 immunoreactive spots at about 30 kDa (see “Results” for details). B and D, protein samples (350 μg) of control, PD, or AD brains were subjected to two-dimensional gel electrophoresis followed by immunoblotting with anti-DJ-1 antibody for detection of DJ-1 isoforms. C and E, the level of each DJ-1 isoform in PD or AD was quantified using the two-dimensional gel analysis program PD Quest and normalized to the level of the same isoform in the control brains. Values represent the mean ± S.E. for five PD, five AD, and five control individuals. The asterisk indicates a statistically significant ( p < 0.05) increase in the level of the indicated DJ-1 isoform in PD or AD versus the corresponding control. F, the relative level of acidic DJ-1 isoforms was measured by quantification of the intensity of the DJ-1 pI 5.5 and pI 5.7 isoforms and expressed as a percentage of the total level of all DJ-1 monomeric isoforms in each human sample. A histogram plot of relative level of acidic DJ-1 isoforms versus post-mortem interval (PMI) shows no correlation between accumulation of acidic DJ-1 isoforms and post-mortem interval.
Besides DJ-1 monomeric forms, we observed at least four different isoforms of SDS-resistant DJ-1 dimer that migrated on the two-dimensional SDS-PAGE gels with an apparent molecular mass of 39 kDa and pI values of 7.0, 7.4, 8.0, and 8.4, respectively (Fig. 1, A and D). Interestingly, a previous study reported the presence of SDS-resistant DJ-1 dimers in the detergent-insoluble fraction of brains from patients with multiple system atrophy (30). To determine whether the accumulation of SDS-resistant dimeric DJ-1 is related to PD and AD, we compared the levels of DJ-1 dimeric forms in PD, AD, and age-matched control brains. The results showed that the levels of the basic isoforms (pI 8.0 and 8.4) of DJ-1 dimer were significantly increased in PD and AD brains compared with controls (Fig. 1, D and E).
In addition to the monomeric and dimeric forms of DJ-1, weak DJ-1-immunoreactive spots at about 30 kDa were occasionally observed in the control as well as in PD and AD brain samples (Fig. 1A). These 30-kDa DJ-1 spots might represent modified forms of DJ-1 that have undergone some types of post-translational modification, such as phosphorylation or mono-ubiquitination. Of note, the mono-ubiquitinated form of recombinant DJ-1 protein with similar apparent molecular mass (∼30 kDa) has been shown to exist in transfected SH-SY5Y cells (35). Furthermore, a 27-kDa DJ-1 species of unknown modification has been reported in the detergent-insoluble fraction of brains from patients with Pick's disease as well as of AD brains (30). Unfortunately, despite repeated attempts, we were unable to further characterize these 30-kDa DJ-1 species due to their extremely low abundance and the variability of their appearance from sample to sample.
The Total Level of DJ-1 Protein Is Increased in PD and AD Brains—Our two-dimensional immunoblotting data (Fig. 1) suggest that the total level of DJ-1 isoforms may be increased in PD and AD brains. To further investigate this possibility, we performed quantitative Western blot analysis using protein samples obtained from PD and AD brains and age-matched controls. The fact that all six DJ-1 monomer isoforms have the same molecular mass of 20 kDa and all four DJ-1 dimer isoforms are of 39 kDa (Fig. 1) allowed us to use one-dimensional gel electrophoresis for direct comparison of the total DJ-1 levels of various brain samples on the same gel. After electrophoresis the gels were subjected to immunoblotting with antibodies against DJ-1 and actin (Fig. 2A). Because the amount of protein extract (50 μg of protein/lane) loaded on one-dimensional gels were much less than the amount (350 μg of protein/gel) on two-dimensional gels, only a single DJ-1 band of 20 kDa was detected on the one-dimensional gels. The relative level of DJ-1 in each sample was measured by quantification of the intensity of 20-kDa DJ-1 band and normalized to the actin level in the corresponding sample. The results revealed a significant increase in the total level of DJ-1 in PD and AD brains compared with the controls (Fig. 2B).
FIGURE 2.
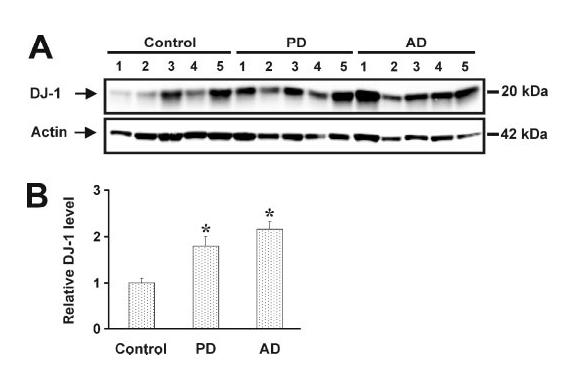
Quantitative Western blot analysis of the total level of DJ-1 protein in PD, AD, and control brains. A, protein extracts (50 μg of protein/lane) from PD, AD, or control brains were subjected to one-dimensional gel electrophoresis followed by immunoblotting with anti-DJ-1 antibody. Each lane represents a different individual from the PD, AD, or control group. B, the relative DJ-1 level was measured by quantification of the intensity of 20-kDa DJ-1 band and normalized to the actin level in the corresponding brain extract. The bar graph shows the results (mean ± S.E.) from five PD, five AD, and five control individuals. The asterisk indicates a statistically significant (p < 0.05) increase in the total level of DJ-1 protein in PD or AD versus control.
DJ-1 Protein Is Oxidatively Damaged in PD and AD Brains—Protein carbonylation is an irreversible oxidation that has been widely used as a biomarker of severe oxidative damage to proteins (5, 10, 11). Ample evidence indicates that the total level of protein carbonyls is increased in both AD and PD (4, 9). It has recently become clear that only a small subset of total brain proteins in PD and AD is carbonylated (32, 33, 36). However, the identities of these oxidatively damaged proteins are largely unknown. Furthermore, it remains to be determined whether the protein targets of oxidative damage are the same or are different in AD and PD.
To determine whether DJ-1 protein is oxidatively damaged in PD or AD brain, we used a well established assay to detect protein carbonyl groups by derivatization of the carbonyls with 2,4-dinitrophenylhydrazine and subsequent immuno-detection of the resulting hydrazones using anti-DNP antibodies (32, 33, 37, 38). Protein extracts from PD, AD, and control brains were resolved by isoelectric focusing on an immobilized pH-gradient strip followed by in-strip DNP derivatization of protein carbonyls and second-dimensional separation by SDS-PAGE and immunoblotting with anti-DJ-1 and anti-DNP antibodies (Fig. 3). We found that among the six identified monomeric forms of DJ-1, only the 20-kDa/pI 6.4 isoform was oxidatively modified, as evidenced by the significant increase in protein carbonyl content of this isoform in PD and AD brains (Fig. 3A). Of the four dimeric forms of DJ-1, three isoforms (39 kDa/pI 7.4, 39 kDa/8.0, and 39 kDa/pI 8.4) were found to exhibit elevated oxidative modification by carbonylation in PD and AD brains (Fig. 3B). No DNP immunoreactivity was observed in the controls where the primary anti-DNP antibody or the DNP derivatization procedure was omitted (data not shown), confirming the specificity of our observations. The lack of detection of carbonyls in the more acidic forms of DJ-1 monomers (pI 5.5-6.1) could mean that these isoforms represent un-carbonylated forms of DJ-1 or could be due to their lower abundance compared with the pI 6.4 DJ-1 isoform, making them undetectable because they might fall below the detection threshold required for anti-DNP immunoblotting. We favor the first interpretation, because the levels of these un-carbonylated DJ-1 monomeric forms were much higher than the dimeric forms of DJ-1 (pI 7.4-8.4) (Fig. 1A), which have been shown to be carbonylated by anti-DNP immunoblotting in the same experiments (Fig. 3B).
FIGURE 3.
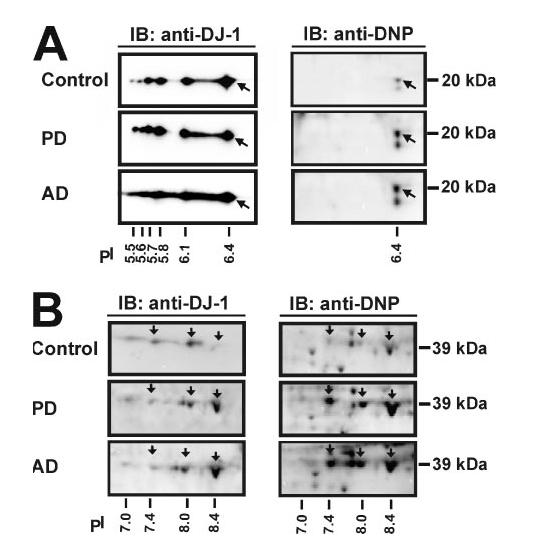
Increased oxidation of DJ-1 isoforms in PD and AD brains compared with controls. Protein samples (350 μg) of control, PD, or AD brains were subjected to two-dimensional gel electrophoresis followed by immunoblotting (IB) with anti-DJ-1 antibody for detection of DJ-1 isoforms or with anti-DNP antibody for detection of protein carbonyls. The arrow indicates the isoform of DJ-1 monomer (A) and of DJ-1 dimer (B) that exhibits elevated oxidation in PD or AD.
To quantitatively determine the differences in protein oxidation between the disease brains and the controls, we quantified the protein level and the carbonyl level of each DJ-1 isoform in five PD, five AD, and five control cases and obtained the specific oxidation index by normalization of the intensity of carbonyl level to the intensity of protein level for each DJ-1 isoform. Statistic analysis of the results showed a significant increase in the specific oxidation levels of the pI 6.4 monomeric and the dimeric forms (pI 7.4-8.4) of DJ-1 in PD as well as in AD compared with the age-matched control brains (Table 2).
TABLE 2.
Specific oxidation index of DJ-1 isoforms in PD, AD, and control brains. The specific oxidation index was obtained by normalization of the intensity of the carbonyl level to the intensity of protein level of the indicated DJ-1 isoform. Values represent the mean ± S.E. for five individuals of each of the PD, AD, or control group.
| DJ-1 isoform (Mr/pI) | Control | PD | AD |
|---|---|---|---|
| n = 5 | n = 5 | n = 5 | |
| 20/pI 6.4 | 0.30 ± 0.03 | 1.17 ± 0.23a | 1.07 ± 0.30a |
| 39/pI 7.4 | 0.43 ± 0.06 | 1.93 ± 0.50a | 2.06 ± 0.60a |
| 39/pI 8.0 | 0.73 ± 0.17 | 1.63 ± 0.26a | 1.67 ± 0.30a |
| 39/pI 8.4 | 0.97 ± 0.13 | 4.00 ± 0.40a | 3.66 ± 0.60a |
p < 0.05.
Identification of the Types and the Sites of DJ-1 Oxidative Modifications by Mass Spectrometry—In addition to carbonylation, proteins can undergo several types of oxidative modifications in response to oxidative stress (39). Thus, we used mass spectrometry to further characterize the oxidative modifications of DJ-1 in human brain samples. Mass spectrometry has emerged as a powerful tool for the analysis of post-translational modifications including oxidative modifications. However, despite recent advances in mass spectrometry technology, detection of modified peptides, and localization of modification sites of an endogenous protein remain a major challenge. Therefore, we focused our mass spectrometry study on PD and control samples. Individual DJ-1 protein spots were excised from the two-dimensional gels from five PD and five control brains, digested with a protease, and analyzed by mass spectrometry.
Identification of the types and the sites of DJ-1 oxidative modifications by mass spectrometry analysis is dependent on the ability to generate peptide fragments of appropriate size and adequate abundance that ideally should cover the entire length of DJ-1 amino acid sequence. To increase the sequence coverage, we tried digestion with different protease (trypsin or Lys-C protease), used a guanidination procedure for enhancing mass spectral signals of Lys-containing peptides (34), and employed a combination of different types of mass spectrometric analyses, including MALDI-TOF/MS, MALDI-TOF/TOF/MS/MS, and HPLC-ESI/MS/MS. Because of the low abundance of the SDS-resistant dimeric forms of DJ-1 in PD and control brains (Fig. 1), we were unable to detect oxidative modifications associated with the DJ-1 dimer isoforms. However, for each of the DJ-1 monomer isoforms in PD and control brains, we obtained sequence coverage of 50%, which matched to the primary sequence of DJ-1 (GenBankTM accession number NP_009193; Fig. 4 and additional data not shown).
FIGURE 4.
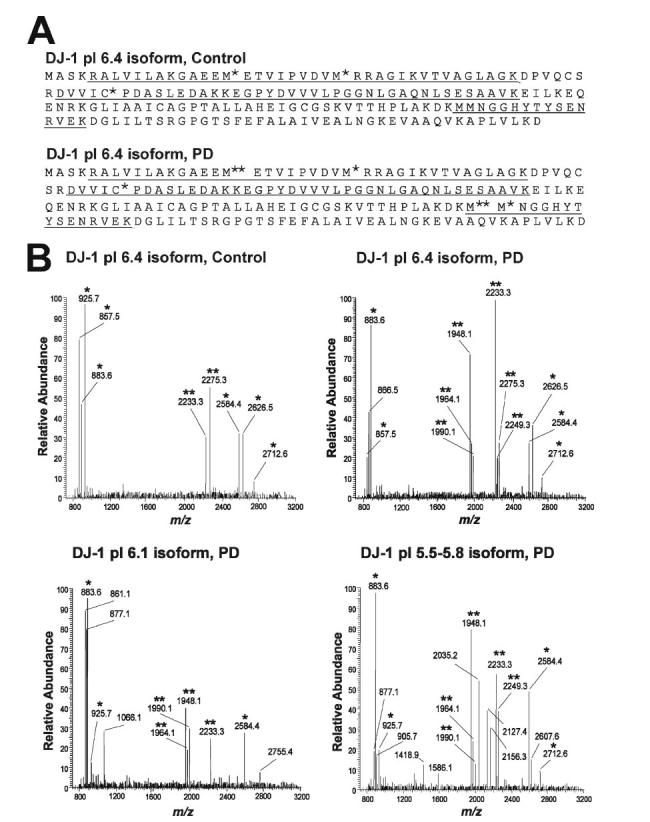
Characterization of DJ-1 oxidative modifications in PD and control brains by mass spectrometry. A, coverage map of DJ-1 pI 6.4 isoform from PD or control brains with underlined residues indicating peptides detected by the combination of MALDI-TOF/MS, HPLC-ESI/MS/MS, and MALDI-TOF/TOF/MS/MS. C*, cysteic acid; M*, methionine sulfoxide; M**, methionine sulfone. Similar coverage maps were obtained for all other 20-kDa DJ-1 pI isoforms in five PD and five control brains. B, MALDI-TOF/MS spectra of the Lys-C digest of DJ-1 isoforms from PD or control brains. *, peptides that match the theoretically predicted peptide masses in DJ-1. **, oxidatively modified peptides containing methionine sulfoxide and/or methionine sulfone. Note that the oxidized peptide fragments at m/z 1948.1, 1964.1, and 1990.1 and m/z 2249.1 in the spectrum of DJ-1 pI 6.4 isoform in PD are absent in the spectrum of DJ-1 pI 6.4 isoform in control brain. Similar results were obtained for five PD and five control brains.
MALDI-TOF/MS analysis of the Lys-C digest of DJ-1 20-kDa/pI 6.4 isoform protein spot from PD brains suggested that six ions represented by m/z 1948.1, 1964.1, 1990.1, 2233.3, 2249.3, and 2275.3, respectively, could be peptides resulting from oxidative modification (Fig. 4). All six suspected oxidized DJ-1 peptides were also present in the MALDI-TOF mass spectra of DJ-1 20 kDa/pI 5.5, 20 kDa/pI 5.6, 20 kDa/pI 5.7, 20 kDa/pI 5.8 isoforms from PD brains, and all except the m/z 2249.3 fragment were found in the spectrum of DJ-1 20 kDa/pI 6.1 isoform from PD brains (Fig. 4). In contrast, only two (m/z 2233.3 and 2275.3) of the six suspected oxidized DJ-1 peptides were detected in the MALDI-TOF mass spectra of DJ-1 isoforms from control brains (Fig. 4).
All putative oxidized DJ-1 peptides were further analyzed by using MALDI-TOF-TOF/MS/MS to identify oxidatively modified amino acid residues. The results indicated that the three ions at m/z 1948.1, 1964.1, and 1990.1 are oxidatively modified peptides corresponding to amino acid residues 133-148 of DJ-1 (Fig. 5A and Table 3). The ion at m/z 1990.1 was generated from the m/z 1948.1 peptide by guanidination, a reaction that results in a 42-Da mass shift due to the conversion of C-terminal lysine residue to homoarginine (34). The MALDI-TOF-TOF tandem mass spectra of the m/z 1948.1 and 1990.1 peptides revealed the oxidative modification of both Met-133 and Met-134 residues to methionine sulfoxide (Fig. 5A and additional data not shown). In addition, we observed the oxidation of Met-133 to methionine sulfone and of Met-134 to methionine sulfoxide in the spectrum of the m/z 1964.1 peptide (data not shown). These results together with the MALDI-TOF/MS data showing the consistent presence of the m/z 1948.1, 1964.1, and 1990.1 peptides in 5 PD but not in 5 control brains (Fig. 4B; data not shown), suggest that the oxidation of DJ-1 Met-133 and Met-134 may be a phenomenon unique to PD.
FIGURE 5.
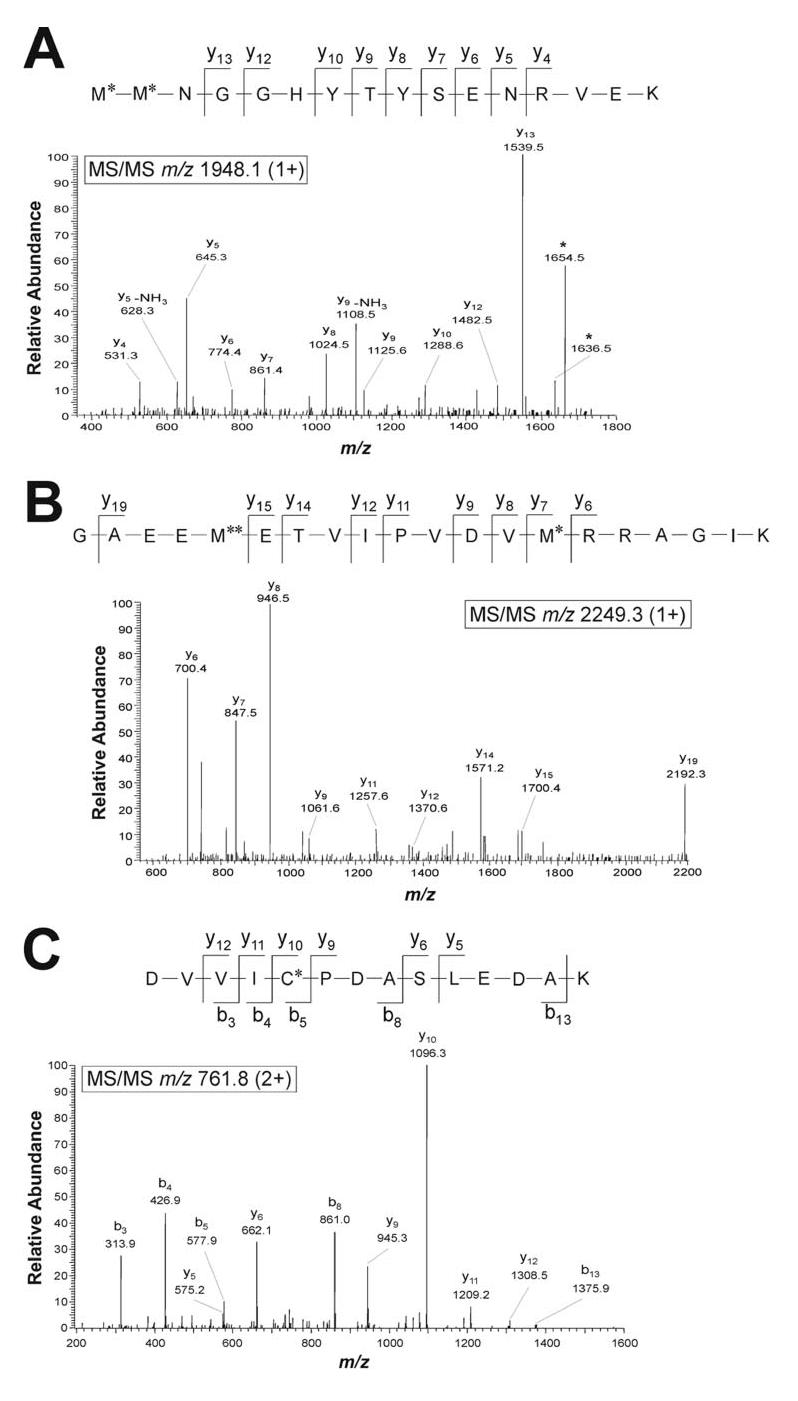
Identification of oxidation sites in DJ-1 by MALDI-TOF/TOF/MS/MS and HPLC-ESI/MS/MS analyses. A, MALDI-TOF/TOF/MS/MS spectrum of the ion at m/z 1948.1 (1+) from a Lys-C digest of DJ-1 pI 6.4 isoform in PD showing the identity as DJ-1 peptide 133-148 with oxidation of both Met-133 and Met-134 residues to methionine sulfoxide. The ion at m/z 1636.5 and 1654.4 are interpreted as b ion and b-H2O ion, respectively, corresponding to the internal fragment M*NGGHYTYSENRVE. B, MALDI-TOF/TOF/MS/MS spectrum of the ion at m/z 2249.3 (1+) from a Lys-C digest of DJ-1 pI 6.4 isoform in PD showing the identity as DJ-1 peptide 13-32 with oxidation of Met-17 to methionine sulfone and Met-26 to methionine sulfoxide. C, HPLC-ESI/MS/MS spectrum of the ion at m/z 761.8 (2+) from a tryptic digest of DJ-1 pI 6.4 isoform in control brain, showing the identity as DJ-1 peptide 49-62 with oxidation of Cys-53 to cysteic acid. Peptide fragments are indicated using the nomenclature of Roepstorff and Fohlman (52). C*, cysteic acid; M*, methionine sulfoxide; M**, methionine sulfone.
TABLE 3.
Oxidized peptides of DJ-1 detected by mass spectrometric analysis. Oxidized fragments at m/z 1990.1 and 2275.3 were generated by guanidation (Gu) of lysine at the C terminus of 1948.1 and 2233.3 peptides, respectively. C*, cysteic acid; M*, methionine sulfoxide; M**, methionine sulfone.
| Group | m/z | Oxidized peptide sequence |
|---|---|---|
| Control | 761.8 (2+) | DVVIC*PDASLEDAK |
| 2233.3 (1+) | GAEEM*ETVIPVDVM*RRAGIK | |
| 2275.3 (1+) | GAEEM*ETVIPVDVM*RRAGIK(Gu) | |
| PD | 761.8 (2+) | DVVIC*PDASLEDAK |
| 1948.1 (1+) | M*M*NGGHYTYSENRVEK | |
| 1964.1 (1+) | M**M*NGGHYTYSENRVEK | |
| 1990.1 (1+) | M*M*NGGHYTYSENRVEK(Gu) | |
| 2233.3 (1+) | GAEEM*ETVIPVDVM*RRAGIK | |
| 2249.3 (1+) | GAEEM**ETVIPVDVM*RRAGIK | |
| 2275.3 (1+) | GAEEM*ETVIPVDVM*RRAGIK(Gu) |
MALDI-TOF-TOF/MS/MS analysis also showed that the three ions at m/z 2233.3, 2249.3, and 2275.3 are oxidatively modified peptides corresponding to amino acid residues 13-32 of DJ-1 (Fig. 5B and Table 3). The spectra of the m/z 2233.3 peptide and its guanidinated product at m/z 2275.3 revealed the oxidative modification of both Met-17 and Met-26 residues to methionine sulfoxide (data not shown). These oxidation products were found in all 20-kDa DJ-1 pI isoforms from both PD and control brains (Fig. 4 and Table 3). However, the m/z 2249.3 fragment, a more severely oxidized form of DJ-1 peptide 13-32 generated from the conversion of Met-17 into methionine sulfone and Met-26 into methionine sulfoxide (Fig. 5B), was only found in the 20-kDa DJ-1 pI 5.5-5.8 and pI 6.4 isoforms from 5 PD brains but not in the DJ-1 isoforms from the 5 controls (Fig. 4 and Table 3). In addition, our MALDI-TOF/MS and HPLC-ESI-MS/MS analyses of trypsin-digested DJ-1 revealed that Cys-53 residue was oxidized to cysteic acid (also known as cysteine sulfonic acid) in all 20-kDa DJ-1 pI isoforms from both PD and control brains (Fig. 5C and Table 3), suggesting the Cys-53 oxidation may occur under relatively mild oxidative stress conditions related to aging.
DISCUSSION
Our study demonstrates for the first time the oxidative damage to DJ-1 in idiopathic PD and AD brains. We showed that human DJ-1 exists in 10 different isoforms; that is, 6 monomeric forms and 4 SDS-resistant dimeric forms. Although DJ-1 adopts a homodimeric structure in solution as well as in the crystal (14, 15, 17), DJ-1 usually appears as a 20-kDa monomer on the SDS-PAGE gel (17, 19, 29, 40, 41). Consistent with a recent report (42), we found that the acidic pI isoforms (pI 5.5 and 5.7) of DJ-1 monomer are selectively accumulated in PD brains. Because the 20-kDa DJ-1 has been shown to undergo acidic pI shift upon exposure of cells to oxidative stress (19, 40, 41, 43, 44), the observed accumulation of the acidic forms of DJ-1 monomer in PD provides a first clue that the oxidative modifications of DJ-1 may be elevated in PD brains. A similar accumulation of the acidic forms of DJ-1 monomer was also found in AD brains, suggesting that increased oxidation of DJ-1 may occur in AD as well.
In addition to the monomeric forms of DJ-1, we found four different isoforms of SDS-resistant DJ-1 dimer, of which the basic isoforms (pI 8.0 and 8.4) are selectively accumulated in PD and AD brains. Although these dimeric DJ-1 isoforms have never been reported, SDS-resistant DJ-1 dimer has been seen in the detergent-insoluble fraction of multiple system atrophy brains (30). The nature of chemical modifications that render these DJ-1 dimers to become SDS-resistant is unknown. The observation that three of the four isoforms of SDS-resistant DJ-1 dimer are irreversibly oxidized by carbonylation in PD and AD brains suggests that oxidative modifications may be involved in conferring the SDS resistance.
Consistent with the accumulation of several DJ-1 isoforms, our quantitative one-dimensional immunoblot analysis has revealed a significant increase in the total level of DJ-1 in PD and AD brains compared with age-matched controls. Previous studies have reported mixed results regarding the DJ-1 levels in PD and AD brains (29, 35, 42). Bandopadhyay et al. (42) failed to find any obvious difference in the total level of DJ-1 between PD and controls (42), whereas Moore et al. (35) found a >5-fold increase in the DJ-1 level in the detergent-insoluble fraction of PD brains compared with control and AD brains. In contrast, Rizzu et al. (29) observed a dramatic increase in the DJ-1 level in the detergent-insoluble fraction of AD brains. The reason(s) for these discrepancies is unclear but could be due to differences in anti-DJ-1 antibodies, patient samples, and extraction conditions used in the immunoblot analyses.
Previous studies have shown that, in cultured mammalian cells, Cys-53 and Cys-106 residues of DJ-1 can undergo reversible oxidation in response to oxidative stress induced by H2O2 and PD-linked environmental toxins, such as paraquat and MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) (19, 40, 41). The reversible oxidation of these residues has been proposed to regulate the molecular chaperone function (19, 45) or mitochondrial localization of DJ-1 (40). Our mass spectrometry analysis has shown that Cys-53 of DJ-1 is oxidized to cysteine sulfonic acid in PD brains as well as in the age-matched controls. As pointed out earlier, despite our repeated efforts we could only achieve 50% sequence coverage. Because we did not recover any peptide containing Cys-106, we could not determine the oxidation status of this residue in human brain samples. Future studies to increase sequence coverage are needed to identify all oxidative modifications of endogenous DJ-1 in PD and AD.
In addition to cysteine oxidation, our mass spectrometry analysis has revealed that DJ-1 is susceptible to previously unsuspected methionine oxidation. We identified four methionine residues (Met-17, Met-26, Met-133, and Met-134) as the sites of DJ-1 oxidation. We found that all four methionine residues were oxidized to methionine sulfoxide in PD, whereas only two of them (Met-17 and Met-26) were oxidized to methionine sulfoxide in age-matched controls. A variety of reactive oxygen species (e.g. O2.,H2O2, ·OH, or peroxynitrite) can oxidize methionine residues to methionine sulfoxide (46), and this oxidation can be reversed by the enzyme peptide methionine sulfoxide reductase (MsrA) in a thioredoxin-dependent manner (47). The reversible methionine oxidation/reduction has been suggested to act as a signaling device analogous to phosphorylation/dephosphorylation for regulating protein function and cellular processes (46). It is, thus, tempting to speculate that the methionine oxidation of DJ-1 identified here represents a specific, reversible mechanism for regulation of DJ-1 activity by intracellular redox status. In addition, the reversible oxidation of methionine residues has been proposed to serve as an important defense mechanism for scavenging ROS (48). Such a mechanism would support an antioxidant role of DJ-1 in protecting cells from oxidative damage (19, 24). Interestingly, Met-26, a methionine residue of DJ-1 found to be oxidized in this study, is mutated to isoleucine (M26I) in a rare, autosomal recessive form of familial PD (26). Thus, the reversible oxidation of Met-26 as well as of other identified methionine oxidation sites (Met-17, Met-133, and Met-134) may have a role in regulation of DJ-1 function and/or localization. Future studies are needed to test these hypotheses and determine the functional consequences of DJ-1 methionine oxidation.
In addition to the reversible methionine oxidation, we found that Met-17 and Met-133 were irreversibly oxidized to methionine sulfone in PD but not in controls. Unlike the reversible oxidation of methionine residues to methionine sulfoxide, which occurs under physiological conditions, the irreversible oxidation to methionine sulfone is rare and only takes place in the presence of strong oxidants (46). This irreversible oxidation is often associated with pathophysiological conditions and results in functional impairment of the oxidized proteins (46, 49).
Another important finding of the present study is the oxidative damage to DJ-1 in PD and AD by the irreversible carbonylation. Protein carbonylation occurs under stronger oxidative stress than that causing cysteine and methionine oxidation and has been associated with functional impairment in a variety of structural proteins and enzymes (10, 11). For example, actin carbonylation occurs after its Cys and Met residues have already been oxidized, and the carbonylation leads to the disruption of actin filaments and the inhibition of F-actin formation (50). The lipid peroxidation product 4-hydroxynonenal induces carbonylation of ubiquitin C-terminal hydrolase L1 (UCH-L1), resulting in a dramatic reduction in the hydrolase activity of UCH-L1 (51). Thus, the observed DJ-1 carbonylation together with irreversible methionine oxidation may cause functional impairment of DJ-1 and contribute to the pathophysiology and progression of sporadic PD and AD. Further investigation of the relationship between oxidative modifications of DJ-1 as well as of other proteins and neuronal dysfunction should generate new insights into the mechanisms of oxidative damage in the pathogenesis of PD and AD and provide new opportunities for developing therapeutic strategies for treating these devastating diseases.
Acknowledgments
Acknowledgments—We thank Christopher A. Carroll of the Institutional Mass Spectrometry Laboratory at the University of Texas Health Science Center at San Antonio (supported in part by National Institutes of Health Grant CA54174) for mass spectrometric analyses.
The abbreviations used are:
- AD
Alzheimer disease
- PD
Parkinson disease
- DNP
2,4-dinitrophenyl
- ROS
reactive oxygen species
- MALDI-TOF/MS
matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- MALDI-TOF/TOF/MS/MS
MALDI-TOF tandem mass spectrometry
- HPLC
high performance liquid chromatography
- ESI/MS/MS
electrospray ionization tandem mass spectrometry.
Footnotes
This work was supported by National Institutes of Health Grants NS047199, NS047575, NS050650, AG021489, and AG025688. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Ross CA, Poirier MA. Nat. Med. 2004;10(suppl):10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 2.Cordato DJ, Chan DK. J. Clin. Neurosci. 2004;11:119–123. doi: 10.1016/j.jocn.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Pardo LM, van Duijn CM. Mutat. Res. 2005;592:89–101. doi: 10.1016/j.mrfmmm.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Jenner P. Ann. Neurol. 2003;53:Suppl–3. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- 5.Levine RL, Stadtman ER. Exp. Gerontol. 2001;36:1495–1502. doi: 10.1016/s0531-5565(01)00135-8. [DOI] [PubMed] [Google Scholar]
- 6.Beal MF. Free Radic. Biol. Med. 2002;32:797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 7.Ischiropoulos H, Beckman JS. J. Clin. Investig. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenner P. Trends Neurosci. 2001;24:245–247. doi: 10.1016/s0166-2236(00)01789-6. [DOI] [PubMed] [Google Scholar]
- 9.Giasson BI, Ischiropoulos H, Lee VM, Trojanowski JQ. Free Radic. Biol. Med. 2002;32:1264–1275. doi: 10.1016/s0891-5849(02)00804-3. [DOI] [PubMed] [Google Scholar]
- 10.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Trends Mol. Med. 2003;9:169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 11.Nystrom T. EMBO J. 2005;24:1311–1317. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gracy RW, Talent JM, Kong Y, Conrad CC. Mutat. Res. 1999;428:17–22. doi: 10.1016/s1383-5742(99)00027-7. [DOI] [PubMed] [Google Scholar]
- 13.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 14.Huai Q, Sun Y, Wang H, Chin LS, Li L, Robinson H, Ke H. FEBS Lett. 2003;549:171–175. doi: 10.1016/s0014-5793(03)00764-6. [DOI] [PubMed] [Google Scholar]
- 15.Tao X, Tong L. J. Biol. Chem. 2003;278:31372–31379. doi: 10.1074/jbc.M304221200. [DOI] [PubMed] [Google Scholar]
- 16.Lee SJ, Kim SJ, Kim IK, Ko J, Jeong CS, Kim GH, Park C, Kang SO, Suh PG, Lee HS, Cha SS. J. Biol. Chem. 2003;278:44552–44559. doi: 10.1074/jbc.M304517200. [DOI] [PubMed] [Google Scholar]
- 17.Olzmann JA, Brown K, Wilkinson KD, Rees HD, Huai Q, Ke H, Levey AI, Li L, Chin LS. J. Biol. Chem. 2004;279:8506–8515. doi: 10.1074/jbc.M311017200. [DOI] [PubMed] [Google Scholar]
- 18.Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A. PLoS Biol. 2004;2:1764–1773. doi: 10.1371/journal.pbio.0020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller DW, Ahmad R, Hague S, Baptista MJ, Canet-Aviles R, McLendon C, Carter DM, Zhu PP, Stadler J, Chandran J, Klinefelter GR, Blackstone C, Cookson MR. J. Biol. Chem. 2003;278:36588–36595. doi: 10.1074/jbc.M304272200. [DOI] [PubMed] [Google Scholar]
- 21.Macedo MG, Anar B, Bronner IF, Cannella M, Squitieri F, Bonifati V, Hoogeveen A, Heutink P, Rizzu P. Hum. Mol. Genet. 2003;12:2807–2816. doi: 10.1093/hmg/ddg304. [DOI] [PubMed] [Google Scholar]
- 22.Moore DJ, Zhang L, Dawson TM, Dawson VL. J. Neurochem. 2003;87:1558–1567. doi: 10.1111/j.1471-4159.2003.02265.x. [DOI] [PubMed] [Google Scholar]
- 23.Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 24.Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Proc. Natl. Acad. Sci. U. S. A. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Cagniard B, Mathews T, Jones S, Koh HC, Ding Y, Carvey PM, Ling Z, Kang UJ, Zhuang X. J. Biol. Chem. 2005;280:21418–21426. doi: 10.1074/jbc.M413955200. [DOI] [PubMed] [Google Scholar]
- 26.Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW. Ann. Neurol. 2003;54:283–286. doi: 10.1002/ana.10675. [DOI] [PubMed] [Google Scholar]
- 27.Hedrich K, Schafer N, Hering R, Hagenah J, Lanthaler AJ, Schwinger E, Kramer PL, Ozelius LJ, Bressman SB, Abbruzzese G, Martinelli P, Kostic V, Pramstaller PP, Vieregge P, Riess O, Klein C. Ann. Neurol. 2004;55:145. doi: 10.1002/ana.10816. [DOI] [PubMed] [Google Scholar]
- 28.Gorner K, Holtorf E, Odoy S, Nuscher B, Yamamoto A, Regula JT, Beyer K, Haass C, Kahle PJ. J. Biol. Chem. 2004;279:6943–6951. doi: 10.1074/jbc.M309204200. [DOI] [PubMed] [Google Scholar]
- 29.Rizzu P, Hinkle DA, Zhukareva V, Bonifati V, Severijnen LA, Martinez D, Ravid R, Kamphorst W, Eberwine JH, Lee VM, Trojanowski JQ, Heutink P. Ann. Neurol. 2004;55:113–118. doi: 10.1002/ana.10782. [DOI] [PubMed] [Google Scholar]
- 30.Neumann M, Muller V, Gorner K, Kretzschmar HA, Haass C, Kahle PJ. Acta Neuropathol. (Berl) 2004;107:489–496. doi: 10.1007/s00401-004-0834-2. [DOI] [PubMed] [Google Scholar]
- 31.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 32.Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. J. Biol. Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 33.Choi J, Rees HD, Weintraub ST, Levey AI, Chin LS, Li L. J. Biol. Chem. 2005;280:11648–11655. doi: 10.1074/jbc.M414327200. [DOI] [PubMed] [Google Scholar]
- 34.Beardsley RL, Reilly JP. Anal. Chem. 2002;74:1884–1890. doi: 10.1021/ac015613o. [DOI] [PubMed] [Google Scholar]
- 35.Moore DJ, Zhang L, Troncoso J, Lee MK, Hattori N, Mizuno Y, Dawson TM, Dawson VL. Hum. Mol. Genet. 2005;14:71–84. doi: 10.1093/hmg/ddi007. [DOI] [PubMed] [Google Scholar]
- 36.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Free Radic. Biol. Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 37.Levine RL. Free Radic. Biol. Med. 2002;32:790–796. doi: 10.1016/s0891-5849(02)00765-7. [DOI] [PubMed] [Google Scholar]
- 38.Butterfield DA, Castegna A. Amino Acids. 2003;25:419–425. doi: 10.1007/s00726-003-0027-7. [DOI] [PubMed] [Google Scholar]
- 39.Stadtman ER. Ann. N. Y. Acad. Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x. [DOI] [PubMed] [Google Scholar]
- 40.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. Proc. Natl. Acad. Sci. U. S. A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kinumi T, Kimata J, Taira T, Ariga H, Niki E. Biochem. Biophys. Res. Commun. 2004;317:722–728. doi: 10.1016/j.bbrc.2004.03.110. [DOI] [PubMed] [Google Scholar]
- 42.Bandopadhyay R, Kingsbury AE, Cookson MR, Reid AR, Evans IM, Hope AD, Pittman AM, Lashley T, Canet-Aviles R, Miller DW, McLendon C, Strand C, Leonard AJ, Abou-Sleiman PM, Healy DG, Ariga H, Wood NW, De Silva R, Revesz T, Hardy JA, Lees AJ. Brain. 2004;127:1–11. doi: 10.1093/brain/awh054. [DOI] [PubMed] [Google Scholar]
- 43.Mitsumoto A, Nakagawa Y. Free Radic. Res. 2001;35:885–893. doi: 10.1080/10715760100301381. [DOI] [PubMed] [Google Scholar]
- 44.Mitsumoto A, Nakagawa Y, Takeuchi A, Okawa K, Iwamatsu A, Takanezawa Y. Free Radic. Res. 2001;35:301–310. doi: 10.1080/10715760100300831. [DOI] [PubMed] [Google Scholar]
- 45.Martinat C, Shendelman S, Jonason A, Leete T, Beal MF, Yang L, Floss T, Abeliovich A. PLoS Biol. 2004;2:1754–1763. doi: 10.1371/journal.pbio.0020327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoshi T, Heinemann S. J. Physiol. (Lond.) 2001;531:1–11. doi: 10.1111/j.1469-7793.2001.0001j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weissbach H, Etienne F, Hoshi T, Heinemann SH, Lowther WT, Matthews B, St John G, Nathan C, Brot N. Arch. Biochem. Biophys. 2002;397:172–178. doi: 10.1006/abbi.2001.2664. [DOI] [PubMed] [Google Scholar]
- 48.Levine RL, Mosoni L, Berlett BS, Stadtman ER. Proc. Natl. Acad. Sci. U. S. A. 1996;93:15036–15040. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stadtman ER, Van Remmen H, Richardson A, Wehr NB, Levine RL. Biochim. Biophys. Acta. 2005;1703:135–140. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 50.Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Di Simplicio P, Colombo R, Milzani A. Free Radic. Biol. Med. 2002;32:927–937. doi: 10.1016/s0891-5849(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 51.Nishikawa K, Li H, Kawamura R, Osaka H, Wang YL, Hara Y, Hirokawa T, Manago Y, Amano T, Noda M, Aoki S, Wada K. Biochem. Biophys. Res. Commun. 2003;304:176–183. doi: 10.1016/s0006-291x(03)00555-2. [DOI] [PubMed] [Google Scholar]
- 52.Roepstorff P, Fohlman J. Biomed. Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]