

Abstract
Zellweger syndrome (cerebro-hepato-renal syndrome) is the most severe form of the peroxisomal biogenesis disorders leading to early death of the affected children. To study the pathogenetic mechanisms causing organ dysfunctions in Zellweger syndrome, we have recently developed a knockout-mouse model by disrupting the PEX5 gene, encoding the targeting receptor for most peroxisomal matrix proteins (M Baes, P Gressens, E Baumgart, P Carmeliet, M Casteels, M Fransen, P Evrard, D Fahimi, PE Declercq, D Collen, PP van Veldhoven, GP Mannaerts: A mouse model for Zellweger syndrome. Nat Genet 1997, 17:49–57 1 ). In this study, we present evidence that the absence of functional peroxisomes, causing a general defect in peroxisomal metabolism, leads to proliferation of pleomorphic mitochondria with severe alterations of the mitochondrial ultrastructure, changes in the expression and activities of mitochondrial respiratory chain complexes, and an increase in the heterogeneity of the mitochondrial compartment in various organs and specific cell types (eg, liver, proximal tubules of the kidney, adrenal cortex, heart, skeletal and smooth muscle cells, neutrophils). The changes of mitochondrial respiratory chainenzymes are accompanied by a marked increase ofmitochondrial manganese-superoxide dismutase, asrevealed by in situ hybridization and immunocytochemistry, suggesting increased production of reactive oxygen species in altered mitochondria. This increased oxidative stress induced probably by defective peroxisomal antioxidant mechanisms combined with accumulation of lipid intermediates of peroxisomal β-oxidation system could contribute significantly to the pathogenesis of multiple organ dysfunctions in Zellweger syndrome.
Peroxisomes are ubiquitous cell organelles, with diverse metabolic functions. The original discovery of H2O2-producing oxidases and catalase in them led de Duve and Baudhuin 2 to propose the term “peroxisome” for their designation. In the meantime other enzymes involved in generation and detoxification of reactive oxygen species (ROS) such as xanthine oxidase, superoxide dismutase, and glutathione peroxidase have been detected in peroxisomes of mammalian liver. 3 Moreover, oxidative stress as induced by hypoxia-reoxygenation, UV irradiation, or direct exposure to H2O2, has been shown to cause severe perturbation of the peroxisome compartment with evidence of peroxisome proliferation and induction of PEX genes, suggesting their involvement in cellular rescue from ROS. 3-5 The established functions of this organelle however lie in lipid metabolism. Peroxisomal β-oxidation protects living cells from the accumulation of highly insoluble (very long chain fatty acids), toxic (bile acid intermediates), and bioactive (leukotrienes and prostaglandins) lipid derivatives, 6 as well as from lipids involved in signal transduction pathways and apoptosis (such as arachidonic acid and polyunsaturated n-3 fatty acids). 7-9 In addition, peroxisomes are involved in the synthesis of cholesterol (all steps up to farnesyl-pyrophosphate) 10 and catalyze the first steps in ether-glycerolipid synthesis (plasmalogens). 11
The importance of peroxisomes for human health became evident by the identification of severe disorders of the Zellweger syndrome spectrum (Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum’s disease), in which functional peroxisomes are absent. 12-14 The patients with the cerebro-hepato-renal (Zellweger) syndrome, the most severe form of this disease spectrum, suffer from generalized hypotonia, exhibit hypomyelinization of neurons, and show neuronal migration defects associated with neonatal seizures. In addition, hepatic fibrosis/cirrhosis, adrenal insufficiency, and renal cysts occur in affected children who usually die during the first year of life. 15
The molecular defects in the diseases of the Zellweger syndrome spectrum are deletions or mutations in PEX genes, whose gene products—the peroxins (Pex-proteins)—are involved in the biogenesis of peroxisomes. 16,17 Malfunctions (or deficiencies) of some peroxins are associated with a plethora of metabolic disorders because of the absence of functional peroxisomes, such as accumulation of very long chain fatty acids and reduction of plasmalogens in plasma and various organs. 14,15 Because the pathogenesis of organ malformations and dysfunctions in patients with peroxisomal biogenesis disorders is not known and experimental studies in patients are hampered by medical ethical restrictions, we have generated a knockout-mouse model for Zellweger syndrome by disrupting the PEX5 gene. 1 This gene encodes Pex5p, the cytoplasmic shuttle receptor for the import of most peroxisomal matrix proteins. 16,17 PEX5−/− mice have a severe peroxisomal import defect, lack functioning peroxisomes, and exhibit all major signs, pathological defects, and biochemical abnormalities of Zellweger patients. 1 Therefore, this animal model provides a suitable tool for the study of the pathogenesis of organ dysfunctions in Zellweger syndrome and the investigation of potential therapeutic strategies. 18,19
Interestingly, in addition to the absence of peroxisomes, we found marked alterations of mitochondrial fine structure in the liver of PEX5−/− mice. 1 Indeed, mitochondrial alterations were also described in the original report by Goldfischer and colleagues 12 on the peroxisomal defects in Zellweger syndrome. Since then however, only a few reports have been published concerning ultrastructural changes and alterations of the mitochondrial respiratory chain enzymes in this disease. 20-24 Moreover, it is unclear whether those changes in the mitochondrial compartment are directly related to the pathogenesis of Zellweger syndrome.
Because the PEX5 knockout mouse provides an excellent model system for peroxisome biogenesis disorders and alterations of mitochondria are so prominent in the livers of those animals, a thorough analysis of the mitochondrial structure and of the function of the respiratory chain was conducted in different organs of PEX5−/− mice.
Materials and Methods
Materials for Morphological Investigations
All components for the preparation of Epon 812 were purchased from Fluka (Neu-Ulm, Germany). Paraffin (Paraplast plus) was from Sherwood Medical (St. Louis, MO). LR White, Unicryl, and osmium tetroxide were obtained from Polysciences (Heidelberg, Germany). Glutaraldehyde (25% stock solution), paraformaldehyde, PIPES, 3,3′-diaminobenzidine, cytochrome c, and di-sodium-cytidine-monophosphate were from Sigma (München, Germany). All other chemicals used for morphological experiments were obtained from Merck (Darmstadt, Germany).
Materials for Biochemical Investigations
Lactate dehydrogenase from hog muscle (complex V measurement), pyruvate kinase from rabbit muscle, 3-phosphoglycerate kinase from yeast, glyceraldehyde-3-phosphate dehydrogenase from rabbit skeletal muscle, β-hydroxybutyrate dehydrogenase grade II from Rhodobacter spheroides, lactate dehydrogenase from rabbit muscle (lactate and pyruvate measurements), glutamate-pyruvate transaminase from pig heart, NAD, and fat-free bovine serum albumin were purchased from Roche (Mannheim, Germany). Coenzyme Q1, decylubiquinone, cytochrome c from horse heart, NADH, n-dodecyl-β-d-maltoside, phosphoenolpyruvate, rotenone, antimycin A, oligomycin, glycerate-3-phosphate, lactate, pyruvate, and acetoacetate were from Sigma (Bornem, Belgium), ATP from Acros (Geel, Belgium), β-hydroxybutyrate from Serva (Heidelberg, Germany), PD-10 columns, Hybond-N+ nylon membranes and Protran nitrocellulose transfer membranes from Amersham Pharmacia Biotech Benelux (Roosendaal, The Netherlands), Dulbecco’s modified Eagle’s medium, fetal calf serum, l-glutamine, gentamicin, and Trizol reagent from Life Technologies (Merelbeke, Belgium), primers for polymerase chain reaction were obtained from Eurogentec (Seraing, Belgium).
Primary Antibodies
The monospecificity of the polyclonal rabbit anti-rat liver catalase antibody was characterized previously. 25 Mouse monoclonal antibodies against the following subunits of the mitochondrial respiratory chain complexes were purchased from Molecular Probes (Leiden, The Netherlands): complex I, anti-bovine NADH-ubiquinol oxidoreductase 39-kd subunit (clone 20C11-B11-B11); complex II, anti-bovine succinate-ubiquinol oxidoreductase 30-kd subunit (clone 21A11-AE7); complex III, anti-bovine ubiquinol-cytochrome c oxidoreductase core 1 subunit (clone 13G12AF12-BB11); complex IV, anti-human cytochrome c oxidase subunit 1 (clone D6-E1-A8); complex V, anti-bovine mitochondrial ATP synthase α subunit (clone 7H10-BD4).
Polyclonal rabbit anti-rat manganese-superoxide dismutase (MnSOD) antibodies were purchased from Research Diagnostics Inc. (Flanders, NJ). In addition, for immunohistochemical studies of MnSOD, we used a polyclonal rabbit anti-rat MnSOD antibody, which was a generous gift of Prof. K. Kato (Aichi Human Service Center, Aichi, Japan). 26
PEX5 Knockout Mice
Homozygous PEX5−/− mice were obtained by breeding of the heterozygotes and identified by genotyping of tail DNA. They showed all characteristic metabolic changes and pathological alterations as described recently. 1 Pregnancy state of heterozygous mothers was calculated according to the appearance of the vaginal plug. Animals of embryonic day 18.5 (E18.5) or newborn mice (P0.5) were used for all investigations. The morning of the appearance of a vaginal plug was considered as E0.5. Pregnant mothers had access to food and water ad libitum and were exposed to a 12-hour light cycle.
Morphological Experiments
Perfusion Fixation of Animals
Newborn mice were anesthetized with ether and the tails cut for the determination of genotypes. After careful opening of the thorax, all animals of three families were perfused via the left ventricle. Before fixation by perfusion, blood cells were flushed out with physiological saline.
The fixative contained for light microscopical investigations, including immunohistochemistry and in situ hybridization, 4% depolymerized paraformaldehyde in phosphate-buffered saline (PBS), pH 7.4 (fixative a); or for electron microscopy, a mixture of 4% depolymerized paraformaldehyde and 0.05% glutaraldehyde with 2% sucrose in PBS, pH 7.4 (fixative b).
For perfusion of fetal (E18.5) mice, the fetuses of three families were delivered by cesarean section under ether anesthesia. To prevent anoxic changes in organs of fetuses, each fetus was perfusion-fixed before the removal of the next fetus from the uterus. After the perfusion, all animals were kept in the same fixatives (for electron microscopy in fixative b without glutaraldehyde) for several hours before further processing.
Light Microscopy
For light microscopic investigations, fixed animals were cut longitudinally into two halves and embedded in paraffin. After cutting of 1- to 3-μm sections with a sliding microtome (Leica, Bensheim, Germany), sections of complete fetuses or newborn animals were mounted on Superfrost Plus slides (Shandon, Frankfurt, Germany), dried overnight at 37°C, and processed either for immunohistochemistry or in situ hybridization.
Immunohistochemistry
Tissue pretreatment conditions and the immunohistochemical procedure for the localization of catalase and MnSOD proteins with polyclonal rabbit antibodies were modified from the method of Grabenbauer and colleagues, 27 developed in our laboratory for the localization of peroxisomal proteins and corresponding mRNAs on parallel sections of mouse fetuses. Briefly, before antibody incubation, antigens in tissue sections were either demasked by digestion with trypsin (5 minutes with 0.1% or 10 minutes with 0.01% trypsin) at 37°C in TNB buffer (0.1 mol/L Tris-HCl, pH 7.4, 0.15 mol/L NaCl; 0.5% blocking reagent; Roche Molecular Biochemicals, Mannheim, Germany) or by irradiation for 3 × 5 minutes in a microwave oven (850 W, in citrate buffer, pH 6). After blocking of endogenous peroxidase with 3% H2O2 and of endogenous biotin with an avidin/biotin blocking kit (NEN Life Science, Boston, MA), incubation with the primary antibody was performed overnight at 4°C in a moist chamber. Antigen-binding sites were detected with a peroxidase-coupled biotin/avidin system (rabbit extravidin kit; Sigma, München, Germany) and visualized by histochemical staining for peroxidase using Novared as substrate (Vector Laboratories).
A special mouse-on-mouse detection system (MOM kit, Vector Laboratories, Burlingame, CA) was used for the localization of respiratory chain enzymes with the monoclonal antibodies from mouse, because detection with the regular protocol and the mouse extravidin kit (Sigma, München, Germany) only resulted in nonspecific background staining. The pretreatment conditions for the localization of complexes III to V were the same as mentioned above. However, for the localization of complexes I and II, strong antigen retrieval by microwave irradiation (see above) and additional amplification with a tyramide signal amplification kit was necessary (TSA kit with biotinylated tyramines; NEN, Life Science, Boston, MA). 28
In Situ Hybridization
The cDNA for mouse mitochondrial MnSOD was amplified by reverse transcriptase-polymerase chain reaction on mouse liver RNA (for sequences of primers see below in Northern Blot section) and the generated 595-bp fragment cloned into pBluescript SK for in vitro transcription of riboprobes. The 1.6-kbp rat catalase cDNA was a generous gift of Prof. Takashi Hashimoto (Department of Pathology Northwestern University, Chicago, IL) and was subcloned in pGEM-7zf (−). 29
After preparation of digoxigenin-labeled cRNA probes by in vitro transcription (Roche Molecular Biochemicals, Mannheim, Germany), nonradioactive in situ hybridization for the localization of catalase or MnSOD mRNA was performed according to the protocols developed by Schad and colleagues 29 and recently modified for fetal mouse tissue by Grabenbauer and colleagues. 27 Very stringent hybridization (68°C) and washing (72°C) conditions were used to prevent nonspecific hybridization. Hybridization with digoxigenin-labeled sense probes was performed in parallel.
Electron Microscopy
For routine electron microscopy, microslicer sections of the liver (100 μm) and very thin razor blade sections of various other organs were prepared and were fixed for 15 minutes with 1% glutaraldehyde in 0.1 mol/L PIPES buffer, pH 7.4, washed briefly in 0.1 mol/L PIPES buffer, postfixed with reduced osmium, 30 and embedded in Epon 812.
Enzyme Cytochemistry
The catalase activity was localized with the alkaline 3,3′-diaminobenzidine (DAB) method 31 as modified subsequently by Angermüller and Fahimi. 32 Cytochrome c oxidase (mitochondrial complex IV) was localized by the DAB method 33 as modified by Angermüller and Fahimi. 32 For the localization of acid phosphatase activity the cerium technique 34 was used as described. 35 After the incubation, tissue sections were postfixed either with reduced osmium (catalase and acid phosphatase) or aqueous osmium (cytochrome c oxidase) and embedded in Epon 812.
Immunocytochemistry
After perfusion fixation, 100-μm microslices were directly dehydrated and embedded in LR White (soft grade) 36 or in Unicryl at low temperature. 37,38 Postembedding immunocytochemistry was performed according to the protocol of Baumgart. 39 After blocking nonspecific binding sites with TNB, LR White or Unicryl sections of liver tissue were incubated on drops of rabbit anti-rat MnSOD or anti-rat catalase, washed on a series of TBS drops and the antigen-antibody complexes visualized by incubation with protein A gold (15 nm). 39,40 In the case of the proteins of the mitochondrial respiratory chain enzymes, which were localized with the monoclonal mouse antibodies, the antigen-antibody complexes were detected with a secondary gold-labeled goat anti-mouse antibody (Polygold 15 nm; Polysciences, Heidelberg, Germany). All sections were contrasted with uranyl acetate and lead citrate before examination in a Philips 301 electron microscope.
Biochemical Experiments
Tissue Collection
Newborn and embryonic mice were decapitated and the livers and hearts were removed within seconds, snap-frozen in liquid nitrogen and stored at −80°C before use.
Measurement of Mitochondrial Respiratory Chain Complexes and Citrate Synthase Enzyme Activities in Liver and Heart
Individual livers (40 to 80 mg) and pooled hearts (30 to 40 mg) of PEX5−/− and control mice were homogenized in, respectively, 9 or 19 volumes of homogenization buffer (210 mmol/L mannitol, 70 mmol/L sucrose, 5 mmol/L HEPES, 1 mmol/L EGTA, pH 7.2) using a borosilicate glass hand homogenizer in ice. The homogenate was centrifuged at 600 × g, 4°C for 10 minutes to remove cell debris and nuclei. The supernatant with mitochondria was snap-frozen and thawed five times before measuring complex II, -III, -IV, and -V activities. For optimal measurements of complex I and citrate synthase activities, an additional sonication step (4 cycles of 10 seconds at amplitude 4, separated by a 20 second break) with a Soniprep 150 sonicator (MSE Scientific Instruments, Sussex, England) was required. The enzymatic reactions were performed at 30°C as described by Rahman and colleagues. 41 The change in absorbance was followed in a Uvikon 810 spectrophotometer (Kontron Instruments, Zurich, Switzerland). Complex III and IV activities are expressed as rate constants (min−1), whereas all other activities are expressed in units (μmol/min). All results were normalized to the activity of citrate synthase to compensate for variation in mitochondrial recovery.
Enzyme Measurements in Fibroblasts
Immortalized as well as primary fibroblasts of PEX5−/− and control mice were grown in Dulbecco’s modified Eagle’s medium with addition of 10% fetal calf serum, 2 mmol/L l-glutamine and 3 mg/L of gentamicin. The cells of eight (immortalized fibroblasts) or five (primary fibroblasts) in 10-cm culture dishes were harvested after trypsinization and washed three times with 10 ml of PBS, washed again with 1 ml homogenization buffer, and finally resuspended in 250 μl of the latter buffer. Before enzyme measurements, the cell suspension was frozen and thawed five times. Complex I and citrate synthase were measured after sonication as described above.
ATP Measurements
Pooled liver tissue (150 to 200 mg) was homogenized in 4 volumes of 0.9 mol/L perchloric acid. Homogenization was performed as follows: an appropriate volume of perchloric acid was frozen in a mortar precooled in liquid N2. The frozen tissue was added and ground together with the perchloric acid to a powder, which was transferred to a glass homogenizer for thawing and further homogenization at 0°C. After centrifugation (3000 × g at 4°C for 5 minutes) the pH of the supernatant was adjusted to ∼6 with K2CO3. Subsequently, a second centrifugation was performed (3000 × g at 4°C for 5 minutes) to remove the KClO4. ATP was determined in the supernatant by using the 3-phosphoglycerate kinase/glyceraldehyde-3-phosphate dehydrogenase method described by Jaworek and Welsch. 42
Lactate, Pyruvate, β-Hydroxybutyrate, and Acetoacetate Measurements
The quantification of these components was based on methods previously described 43-45 but slightly adjusted for fluorometric measurements except for lactate. The perchloric acid extracts prepared for ATP determination (see above) were used for the assays. All reactions were performed at room temperature and fluorescence was measured with a LS50 Luminescence Spectrometer (Perkin Elmer, Norwalk, CT) or absorbance with a Uvikon 810 spectrophotometer (lactate).
Northern Blot
Total RNA from mouse liver was extracted using the Trizol reagent. Thirty μg of RNA were loaded on a 1.2% (w/v) agarose gel containing 2.9% (w/v) formaldehyde. Gels were run in 200 mmol/L MOPS, pH 7.0, 50 mmol/L NaOAc, 10 mmol/L EDTA for 5 hours and RNA was transferred to a Hybond-N+ nylon membrane by capillary blotting. The 595-bp radioactive probe for detection of MnSOD mRNA was synthesized by performing a polymerase chain reaction on adult mouse liver cDNA using the primers 5′-TAG-TAG-GAA-TTC-GCA-CCA-CAG-CAA-GCA-CCA-TGC-GG-3′ and 5′-GAT-GAT-GGA-TCC-ATA-AAC-CAG-CCC-GGA-GCC-TGG-CC-3′. After stripping, the same blot was hybridized with a β-actin probe to check for equal loading of RNA.
Western Blot
Individual livers of newborn mice were homogenized in 4 volumes of 0.25 mol/L sucrose, 5 mmol/L MOPS, pH 7.2, 1 mmol/L ethylenediaminetetraacetic acid, 0.1% ethanol. Thirty or 15 μg of protein were separated on a 10% polyacrylamide gel and transferred to a Protran-nitrocellulose transfer membrane. Subunits of complexes I, II, III, IV, and V were detected using monoclonal mouse antibodies and MnSOD was visualized by a polyclonal rabbit antibody. Immunoreactive bands were stained with NBT/BCIP
Statistical Analysis
The unpaired t-test was used to compare the results between knockout and control mice.
Results
Mitochondria in Hepatocytes from PEX5−/− Mice Proliferate and Show Severe Ultrastructural Abnormalities
As previously described, 1 regular peroxisomes were easily identified by histochemical staining for catalase with 3,3′diaminobenzidine in hepatocytes of control mice at gestational day 18.5 (E18.5) (Figure 1A) ▶ , but were absent in the PEX5 knockouts of the same age (Figure 1B) ▶ . In ∼5% of hepatocytes of PEX5-deficient mice peroxisomal membrane-remnant ghosts consisting of double-membraned loops measuring ∼100 nm across were observed that were labeled with an antibody to 70-kd peroxisomal membrane protein. 1 Similar structures were also observed in the adrenal glands but not in the kidney tubules (data not shown).
Figure 1.
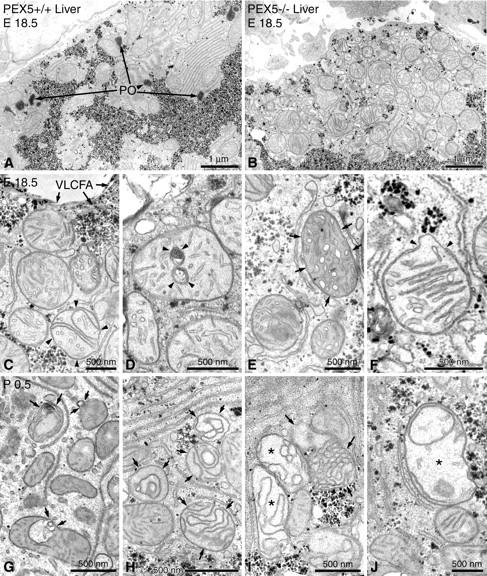
Electron micrographs of liver sections stained with alkaline DAB for the localization of peroxisomal catalase activity 31,32 and postfixed with reduced osmium. A: Low-magnification view of an hepatocyte from a control animal of gestational age E18.5. Peroxisomes (PO) are easily recognized by the DAB reaction product in their matrix and are often found in the vicinity of glycogen areas. B: Low-magnification view of a similar region of an hepatocyte in a liver section of a PEX5−/− mouse. Note the absence of peroxisomes near the glycogen areas and the strong proliferation of pleomorphic mostly round, large mitochondria, some of which exhibit stacks of parallel cristae. C–F: Higher magnification views of altered mitochondria in hepatocytes of PEX5−/− mice (E18.5). C: Curvilinear alterations of cristae are already observed in some hepatic mitochondria of PEX5−/− mice before birth (arrowheads). Because of the defect in peroxisomal β-oxidation, crystals of very long chain fatty acids (VLCFA) accumulate on the surface of regular lipid droplets (arrows). D: Myelin-like or ring-shaped alterations of mitochondrial cristae (arrowheads). E: Mitochondria with tubular cristae (small arrows). F: A mitochondrion with parallel-stacked cristae, exhibiting a focal disruption of the outer membrane (arrowheads) and protrusion of the inner membrane through the gap. G–J: Mitochondrial alterations in hepatocytes of newborn PEX5−/− animals (P0.5) that are more severe than in E18.5. G: Mitochondria with evaginations and invaginations of the outer membrane (small arrows). H: Severe curvilinear alterations of mitochondrial cristae (small arrows), with the formation of different types of mitochondrial ghosts (I, small arrows). I and J: Swollen mitochondria with less dense matrix and curvilinear cristae (I, asterisks) or rarefication of cristae (J, asterisk).
Strikingly, large aggregates of pleomorphic mitochondria were observed in many hepatocytes of PEX5-deficient mice, indicating focal proliferation of the mitochondrial compartment (Figure 1B) ▶ . Such mitochondrial aggregates were randomly distributed in the liver and were often found under the sinusoidal and the basolateral surface of hepatocytes (Figure 1B) ▶ . By electron microscopy different types of alterations of mitochondria were present, which were more pronounced in newborn (Figure 1; G to J ▶ ) as opposed to E18.5 PEX5−/− mice (Figure 1; C to F ▶ ). These alterations involved all subcompartments of the mitochondria (outer membrane, inner membrane, and the matrix space). Alterations of the outer membrane consisted of 1) membrane whorls on the surface of mitochondria (Figure 1E) ▶ , 2) small blebs with electronlucent contents (Figure 1G) ▶ , or 3) protrusions of the inner membrane through focal interruptions of the outer membrane (Figure 1F) ▶ . In mitochondrial cristae, more obvious morphological changes were observed that consisted of 1) rarefication of cristae (Figure 1, B and J) ▶ , 2) arrangements into parallel stacks (Figure 1, B and F) ▶ , 3) curvilinear and circular forms (Figure 1; C and H to J ▶ ), 4) onion-like concentric rings (Figure 1, D and G) ▶ , 5) dilation with the formation of tubular cristae similar to those seen in steroid hormone-producing cells (Figure 1E) ▶ . In hepatocytes of newborn PEX5 knockout mice, we also observed ghost-like mitochondrial rest structures (Figure 1, H and I ▶ ; small arrows), swollen mitochondria with strong rarefication of cristae, and reduced electron density of their matrix (Figure 1, I and J ▶ ; asterisks). Mitochondrial alterations were quite heterogeneous in different hepatocytes and even within the same cell severely altered mitochondria were observed adjacent to normal ones (Figure 1; B, C, and G to J ▶ ). The mitochondria in sinusoidal cells (endothelial cells and Kupffer cells), in contrast to the ones in hepatocytes, appeared normal as did the mitochondria in fibroblasts of the sparse connective tissue of the fetal or newborn liver.
Mitochondrial Alterations Are also Present in Other Tissues and in Blood Cells
In addition to changes of the mitochondrial compartment in hepatocytes, structural alterations of the mitochondrial population of several lipid-metabolizing organs and tissues, which are also pathologically altered in Zellweger syndrome, were observed. In the epithelial cells of the adrenal cortex of PEX5−/− mice, especially in the inner zona fasciculata, severely altered mitochondria were found (Figure 2, A and B) ▶ , showing either enlarged lamellated mitochondrial granules (Figure 2A) ▶ or accumulation of electron-dense material in the intermembrane space of tubular cristae (Figure 2B) ▶ . Focal proliferation of pleomorphic mitochondria with different types of lamellated granules were also seen in epithelial cells of proximal tubules of the kidney, known to be rich in peroxisomes (Figure 2, C and D) ▶ . In contrast, the mitochondrial compartment in other parts of the nephron was not altered. In cardiomyocytes of PEX5−/− mice, routine electron microscopy revealed an increase in size and number of mitochondrial granules and the presence of megamitochondria (Figure 2F) ▶ . Pathological alterations in skeletal muscle were dependent on the region of the body and the specific muscles investigated. Most severe alterations were observed in the diaphragm, consisting of blebs on the surface of mitochondria (not shown) as well as rarefication and curvilinear alterations of mitochondrial cristae. In some myocytes of leg muscles subsarcolemmal aggregates of mitochondria and megamitochondria with bizarre forms were present (not shown). On the other hand, mitochondria in smooth muscle cells of the intestine displayed only slight alterations, which consisted either of the enlargement of the intermembrane space of the cristae (Figure 2G) ▶ and blebs on the surface, or invaginations into their matrix (Figure 2H) ▶ . In light of the hematopoietic capacity of the liver in newborn mice, mitochondrial alterations were also evaluated in blood cells present in this organ. Whereas mitochondria were completely normal in precursors of red blood cells, such as normoblasts (not shown), they were abnormal in a small portion of developing neutrophils and lymphocytes (Figure 2I) ▶ . The specific alterations of blood cells were independent of the severity of mitochondrial changes in neighboring hepatocytes and were rather cell type-specific. Whereas in neutrophilic granulocytes, mitochondria with circular cristae were frequently observed (Figure 2I) ▶ , the alterations in lymphocytes were more discrete and resembled those of smooth muscle cells showing enlargement of the intercristae space with formation of tubular cristae (not shown).
Figure 2.
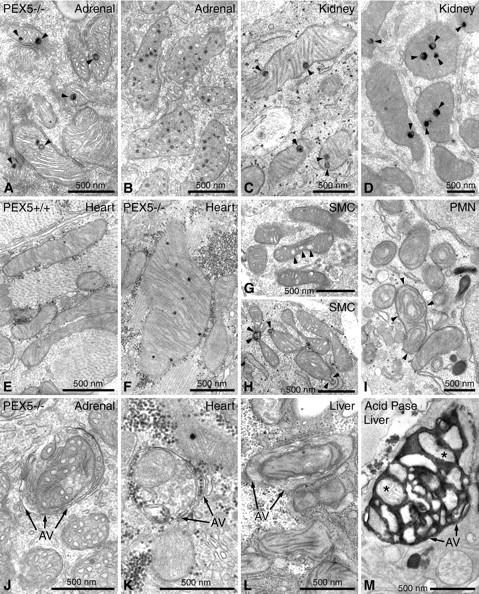
Mitochondrial alterations of extrahepatic cells of PEX5−/− mice of gestational stage E18.5, stained for the localization of peroxisomal catalase activity 31,32 with the alkaline DAB method (A–L) or for the localization of lysosomal acid-phosphatase activity 34 with cerium chloride (M). A and B: Mitochondria in epithelial cells of the zona fasciculata of the adrenal gland with large lamellated granules (arrowheads) (A) and accumulation of electron-dense material in the intermembrane space of cristae (B). C and D: Kidney, large inclusions in mitochondrial matrix of proximal tubules (arrowheads). E and F: Cardiomyocytes of normal (E) and PEX5−/−(F) mice. Megamitochondria with an increase in the number of dense granules are shown (F) as compared to the low number of granules and regular size of mitochondria (E) in a control animal of the same gestational stage (E18.5). G and H: Smooth muscle cells (SMCs) of the intestinal wall of PEX5−/− mice showing enlargement of the intermembrane space in cristae (arrowheads, G) and in- or evaginations of the mitochondrial outer membrane (arrowheads, H). I: A neutrophil (PMN) with several abnormal mitochondria with circular cristae (arrowheads), in a focus of extramedullary hematopoiesis of a PEX5−/− mouse liver. J–L: Autophagic vacuoles (AVs) containing mitochondria in an epithelial cell of the zona fasciculata of the adrenal gland (J), a cardiomyocyte (K), or a hepatocyte (L). M: By using the cytochemical staining technique for acid phosphatase activity with cerium chloride, large autophagic vacuoles containing many mitochondria (two of them marked with asterisks) are identified in a hepatocyte of a PEX5−/− mouse.
Finally, electron microscopic analysis of the neocortex of PEX5 knockout mice revealed no major ultrastructural changes in the mitochondria of neurons (not shown), despite the accumulation of different classes of lipids in those cells, as shown previously. 1
Damaged Mitochondria Are Removed from the Cytoplasm by Increased Autophagocytosis
In addition to ultrastructural changes of the mitochondria, we observed other signs suggestive of increased cellular toxicity and damage to mitochondria. Thus, many mitochondria were found in autophagic vacuoles of adrenal cortical cells, cardiomyocytes, and hepatocytes of PEX5−/− mice (Figure 2, J to L) ▶ . Indeed, with cytochemical staining for acid phosphatase activity, large autophagic vacuoles were seen containing many mitochondria (Figure 2M) ▶ .
Reduction of Complex I Activity Is the Most Consistent Biochemical Finding in Mitochondria of Liver and Heart
Because the five multimeric complexes (complexes I to V) of the electron transport chain reside in the mitochondrial inner membrane and severe ultrastructural changes of this compartment were found in PEX5−/− mice, we investigated the activities and the protein contents of the respiratory chain enzymes. Liver, displaying the strongest morphological alterations of mitochondria, and heart, with more subtle changes, were analyzed. In Table 1 ▶ the activities of mitochondrial respiratory chain enzymes in liver and heart of newborn mice, expressed relative to the activity of citrate synthase are summarized. In comparison to control levels, the activity in liver of newborn PEX5−/− mice was reduced to 45% for complex I and to 65% for complex V, whereas complex IV activity was increased up to 180%. In contrast, no change was observed in the activities of complexes II and III. These differences in respiratory chain complex activities were already present at day 18.5 of embryonic development, except for complex V, which was only reduced by 25%, a statistically insignificant change (not shown).
Table 1.
Activities of the Respiratory Chain Complexes in Newborn Mice
| Complex | Liver | Heart | ||
|---|---|---|---|---|
| Control mice | PEX5−/− | Control mice | PEX5−/− | |
| I | 0.39 ± 0.08 (7) | 0.17 ± 0.01 (7)† | 0.27 ± 0.04 (5) | 0.18 ± 0.02 (4)* |
| II | 0.66 ± 0.11 (7) | 0.61 ± 0.11 (7) | 0.41 ± 0.03 (5) | 0.42 ± 0.03 (4) |
| III | 72.6 ± 20.6 (7) | 64.1 ± 11.6 (5) | 57.0 ± 12.7 (5) | 54.7 ± 8.3 (4) |
| IV | 55.5 ± 15.6 (7) | 97.6 ± 19.2 (7)† | 34.3 ± 3.5 (5) | 41.5 ± 6.9 (4) |
| V | 1.66 ± 0.33 (6) | 1.13 ± 0.20 (7)* | 1.03 ± 0.08 (5) | 1.05 ± 0.06 (4) |
Activities of the complexes are expressed relative to the activity of citrate synthase (CS), a mitochondrial marker enzyme, in order to correct for the recovery of mitochondria. The activities were expressed in units for the complexes I, II, and V and as a rate constant (min−1) for the complexes III and IV, before dividing by the CS activity (expressed in units). Mean ± SD (N). The unpaired t-test was used to compare both groups of mice (*, P < 0.005; †, P < 0.001).
In heart tissue (Table 1) ▶ , which showed only slight ultrastructural alterations, the differences in activities of the mitochondrial respiratory chain enzymes between control and PEX5−/− mice were also less severe than in liver. Complex I activity of newborn PEX5−/− mice was reduced to 65% of the control value, but there was no significant difference in E18.5 animals (data not shown). On the other hand, complex IV activity was 20% higher in heart of newborn PEX5−/− mice, but this difference was not statistically significant.
Corresponding to the normal appearance of mitochondria in fibroblasts in primary cultures of PEX5 knockout mice (not shown) or in situ in various organs, complex I activity was not altered in fibroblast cultures of these mice (either immortalized fibroblasts or primary cultures of passage 3 or 4) (data not shown).
Western blot analysis of different subunits of respiratory chain complexes from liver mitochondria (Figure 3) ▶ revealed an estimated 50% reduction of the 39-kd subunit of complex I, a less-pronounced reduction of the core I subunit of complex III and subunit I of complex IV. In contrast the analyzed subunits of complexes II and V were not significantly altered (Figure 3) ▶ .
Figure 3.

Western blots of subunits of the respiratory chain complexes in liver homogenates of newborn (P0.5) PEX5-deficient and control mice. Thirty μg of total protein was loaded onto an SDS-PAGE gel under reducing conditions, except for complex I (15 μg). The blots were incubated with antibodies against the 39-kd subunit of complex I, the 30-kd subunit of complex II, the subunit core 1 of complex III, subunit 1 of complex IV, and the α subunit of complex V. Note the reductions of complexes I, III, and IV.
Proliferated Mitochondria Form Large Aggregates in Hepatocytes of PEX5−/− Mice
In addition to electron microscopy, we used an extravidin/peroxidase kit to detect mitochondria by light microscopy, based on their high content of biotinylated proteins (Figure 4; A to D ▶ ). 46 The staining pattern corresponded to the intracellular distribution of mitochondria in electron microscopical preparations. In contrast to the granular particulate appearance of mitochondria, some of which were localized around glycogen deposits, in hepatocytes of control mice (Figure 4, A and C) ▶ , aggregates of proliferated mitochondria in subplasmalemmal regions were observed in E18.5 PEX5 knockout mice (Figure 4B) ▶ . The abundance and proliferation of the mitochondrial aggregates that filled up some hepatocytes was even more pronounced in newborn PEX5−/− mice (Figure 4D) ▶ , which in addition exhibited reduced glycogen deposits.
Figure 4.
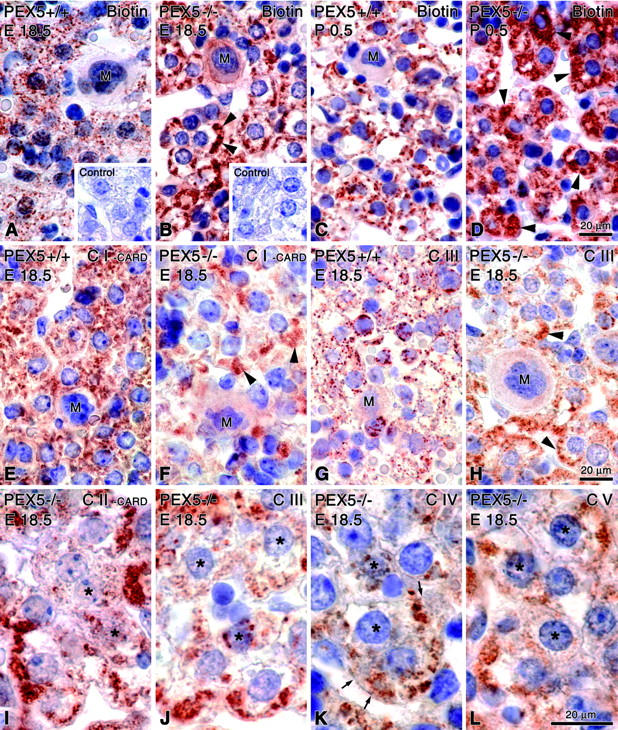
Light microscopical visualization of mitochondria with different techniques in paraffin sections of the liver of PEX5+/+ (E18.5: A, E, G; P0.5: C) or PEX5−/− (E18.5: B, F, H, I–L; P0.5: D) mice. A–D: Localization of biotinylated proteins in mitochondria with extravidin-peroxidase. Insets in A and B depict appropriate negative controls after blocking of endogenous biotin groups. Note the granular appearance of mitochondria in control mice (A and C) in contrast to the large subsinusoidal aggregates (arrowheads) in PEX5-deficient E18.5 mice (B). In P0.5 animals the mitochondrial aggregates fill up the cytoplasm of most PEX5−/− hepatocytes (D, arrowheads). E–L: Indirect immunohistochemical localization of distinct proteins of different mitochondrial respiratory chain complexes (CI to CV) with appropriate antibodies. For optimal visualization, the signals for complexes I and II had to be amplified with catalyzed reporter deposition (E, F, I). Hepatocytes in the liver of PEX5−/− mice with strong proliferation of mitochondria are indicated by arrowheads (F and H). Asterisks in I–L indicate hepatocytes with heterogeneous staining of their mitochondrial populations (marked in detail with small arrows in K). M, megakaryocytes. Note the large aggregates of proliferated mitochondria in hepatocytes of PEX5−/− mice. Bars on the right indicate magnifications for all pictures in the corresponding row.
Proteins of Respiratory Chain Complexes Are Heterogeneously Distributed and Focally Reduced in Hepatocyte Mitochondria of PEX5−/− Mice
To provide further insight into mitochondrial alterations, the localization of different mitochondrial respiratory chain complexes was investigated by immunohistochemistry on paraffin sections (Figure 4; E to L ▶ ). Optimal light microscopical localization of proteins of respiratory chain enzymes in hepatocytes was observed only after antigen retrieval by 3 × 5-minute microwave irradiation of paraffin sections. The localization of complexes III to V was possible thereafter using our standard procedure. 27 However, localization of complexes I and II was only achieved by means of additional catalyzed reporter deposition with biotinylated tyramine (TSA, tyramide signal amplification). 28 By applying these methods, a marked intercellular heterogeneity in the distribution of the mitochondrial respiratory chain enzymes was observed in hepatocytes of E18.5 and P0.5 PEX5 knockout mice (shown only for E18.5 fetuses in Figure 4; F, and H to L ▶ ). The strongest reduction of overall staining intensity was seen in preparations for complex I (Figure 4, E ▶ versus F), but a reduction, albeit to a lesser extent, was also present in preparations for other respiratory chain complexes. Despite aggregation of proliferated mitochondria in most hepatocytes, the stain intensity was patchy showing heterogeneity in neighboring cells of PEX5−/− mice (Figure 4; H to L ▶ ).
The intracellular heterogeneity of mitochondrial respiratory chain enzymes was further substantiated by electron microscopical localization of cytochrome c oxidase enzyme activity in the liver of PEX5−/− mice (both in E18.5 and P0.5; Figure 5, B and C ▶ ). In contrast, mitochondria in hepatocytes of control animals exhibited a uniform distribution of the cytochrome c oxidase activity (Figure 5A) ▶ . Interestingly, the majority of ghost-like or severely altered mitochondria were either negative or exhibited low-cytochrome c oxidase activity (Figure 5B) ▶ . On the other hand, the mitochondria with parallel-stacked cristae in hepatocytes of PEX5−/− mice showed very strong staining of their longitudinal cristae, indicating increased cytochrome c oxidase activity (Figure 5C) ▶ . Interestingly, despite the relatively subtle changes of mitochondrial morphology in cardiomyocytes of PEX5 knockout mice, strong heterogeneity of cytochrome c oxidase staining with negative organelles next to heavily stained ones was also found in the heart (Figure 5F) ▶ . On the other hand, a uniform cytochrome c oxidase-staining pattern was present in the diaphragm, despite the more severe alterations of the mitochondrial ultrastructure (Figure 5, G and H) ▶ .
Figure 5.
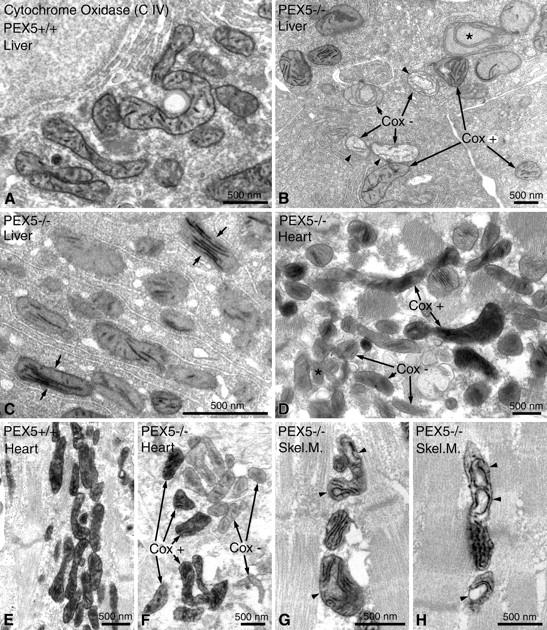
Enzyme cytochemical staining for cytochrome c oxidase (Cox) activity in mitochondria of hepatocytes (A–C), of cardiomyocytes (D–F), and of myocytes of the diaphragm (Skel. M., skeletal muscle) (G, H) in control (A, E) and in PEX5−/− mice (B–D, F–H) of gestational age E18.5. Note the marked intracellular heterogeneity of Cox activity in hepatocytes (B, C) and cardiomyocytes (D, F) of PEX5−/− mice. B: A swollen mitochondrion with circular cristae and a negative Cox reaction is marked with an asterisk; mitochondria with a fluffy matrix and negative Cox reaction (Cox−) are marked with arrowheads. C: Note the strong Cox activity in mitochondria with parallel-stacked cristae in hepatocytes of PEX5−/− mice (small arrows). G, H: Mitochondria in myocytes of the diaphragm with altered, curvilinear cristae and positive Cox activity are marked with arrowheads.
The ultrastructural localization of the respiratory chain enzyme proteins in hepatocytes was only possible with the antibody against complex III, which also gave the best results in light microscopical preparations. The labeling of complex III (not shown) revealed a strong reduction (more than five times) in the mitochondria of PEX5−/− mice, when the ratio of immunogold labeling was determined by morphometry in 500 mitochondria. Thus, in contrast to 20.8% of mitochondria in controls only 4.2% of them from PEX5−/− mice were labeled with gold particles representing antigenic sites for complex III. In addition, a strong heterogeneity of labeling of mitochondria in neighboring hepatocytes of PEX5 knockout mice was observed. Similar to the cytochemical results for cytochrome oxidase, a clearly stronger labeling for complex III was observed in normal-appearing mitochondria or in mitochondria with stacks of parallel cristae whereas mitochondria with abnormal cristae or mitochondrial ghosts contained less gold particles.
ATP Levels and Redox State in Hepatocytes of PEX5 Knockout Mice
The question whether changes in the overall activities of complex I and complex V would lead to reduced ATP levels in hepatocytes of PEX5 knockout mice, was investigated by measuring the ATP content in the liver (Table 2) ▶ . The mean value obtained for PEX5−/− mice (2.69 ± 0.02 μmol/g liver) was slightly higher than the control value (2.14 ± 0.18 μmol/g liver), indicating that there is no deficiency in the steady-state level of hepatic ATP in peroxisome-deficient mice.
Table 2.
ATP, Lactate, Pyruvate, β-Hydroxybutyrate, and Acetoacetate in Liver
| Control mice | PEX5−/− | |
|---|---|---|
| ATP | 2.14 ± 0.18 (3) | 2.69 ± 0.02 (2) |
| Lactate | 1.16 ± 0.25 (4) | 0.29 ± 0.04 (3) |
| Pyruvate | 0.045 ± 0.009 (4) | 0.032 ± 0.005 (3) |
| L/P | 26.48 ± 7.25 (4) | 9.13 ± 0.49 (3) |
| β-Hydroxybutyrate | 0.281 ± 0.071 (4) | 0.022 ± 0.005 (3) |
| Acetoacetate | 0.132 ± 0.028 (4) | 0.038 ± 0.010 (3) |
| HB/AA | 2.13 ± 0.31 (4) | 0.59 ± 0.12 (3) |
Results are expressed as μmol/g liver, except for the lactate/pyruvate (L/P) and β-hydroxybutyrate/acetoacetate ratios, which are dimensionless. Mean ± SD (N). Three or four livers were pooled for each measurement.
Because lactate/pyruvate and β-hydroxybutyrate/acetoacetate ratios reflect the redox state in the cytoplasm and in the mitochondria of cells, respectively, the four compounds were measured in livers of newborn mice (see Table 2 ▶ ). They were all reduced in PEX5 knockout livers in comparison to controls (percentage of control levels: lactate, 25%; pyruvate, 71%; β-hydroxybutyrate, 8%; and acetoacetate, 29%). These low values are probably because of starvation of the peroxisome-deficient mice as a consequence of the severe hypotonia and may therefore invalidate the ratios of lactate/pyruvate and β-hydroxybutyrate/acetoacetate as indicators for respiratory chain function. Contrary to what one would expect in case of a deficiently functioning respiratory chain, both ratios were strongly reduced in PEX5−/− mice (34 and 27% of control values).
Induction of MnSOD in Different Organs of PEX5 Knockout Mice Suggests an Increase of ROS
Because some of the features of mitochondria in PEX5−/− hepatocytes were reminiscent of conditions associated with oxidative stress and because complex I deficiency is known to be accompanied by the production of ROS, 47 we looked for further indications of oxidative stress in the liver of peroxisome-deficient mice. Because in several experimental conditions associated with increased oxidative stress mitochondria express increased amounts of MnSOD protein, 47 in situ hybridization and immunocytochemical procedures were optimized to localize the mRNA and protein of this enzyme. A strong and reproducible immunohistochemical signal for MnSOD was obtained with a polyclonal anti-MnSOD antibody received through the courtesy of Professor Kato. 26 When compared to wild-type littermates, PEX5−/− mice at E18.5 and older, showed strong increases of MnSOD mRNA and protein expression in their livers (Figure 6; A to F ▶ ). In addition to the alterations in liver, heterogeneous induction of the MnSOD mRNA and protein was also found in cardiomyocytes (Figure 6; I to L ▶ ).
Figure 6.
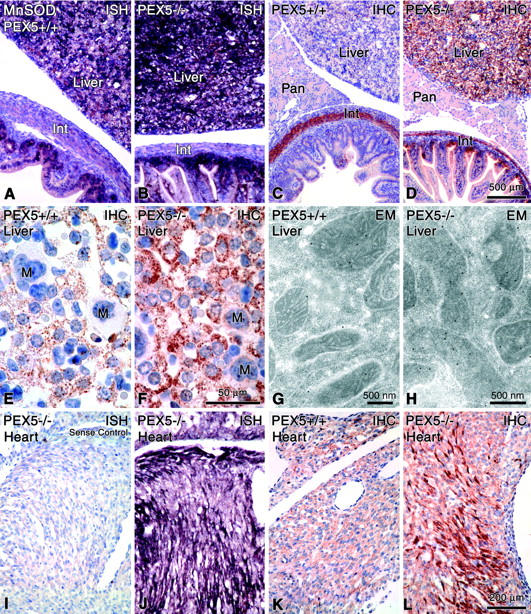
Detection of MnSOD mRNA (A, B, J; negative control = I) by nonradioactive in situ hybridization, MnSOD protein (C-F, K, L) by immunohistochemistry (IHC), and G, H by immunoelectron microscopy (EM) in different organs of control (A, C, E, G, K) and PEX5−/− mice (B, D, F, H, I, J, L). Note the strong increases of MnSOD mRNA and protein in the liver (B, D, F) and the heterogeneous induction of mRNA and proteins in cardiomyocytes (J, L). Proliferated mitochondria in hepatocytes of PEX5−/− mice are stronger stained for MnSOD protein (F) than the mitochondria of control animals (E) and by electron microscopy the immunolabeling is significantly higher in PEX5-deficient mice (H) than in controls (G). Int, intestine; Pan, pancreas; M, megakaryocytes. Bars on the right indicate magnifications for all figures in the corresponding row (except for electron micrographs).
Electron microscopical localization of MnSOD in liver sections embedded in Unicryl, revealed intra- and intercellular heterogeneous expression of this protein in mitochondria of hepatocytes (Figure 6, G and H) ▶ . Analysis of 200 mitochondria by morphometry revealed that the immunolabeling for MnSOD was increased in PEX5−/− mice. Thus, in contrast to 45% labeling in control mice, 59% of mitochondria were labeled in PEX5 knockouts. Interestingly, in hepatocytes of PEX5−/− mice, mostly the normal-appearing, or ring-shaped mitochondria were labeled with the MnSOD antibody, whereas severely altered mitochondria were unlabeled.
Despite clear evidence by in situ hybridization and immunocytochemistry (Figure 6) ▶ the analysis of MnSOD mRNA and protein by Northern and Western blotting revealed no significant alterations in liver tissue of newborn peroxisome-deficient mice as compared to wild-type controls (data not shown). However, a twofold increase in MnSOD transcripts was found in two PEX5-deficient mice that exceptionally survived for 2 days (not shown).
Discussion
In this study, we report on the pathological alterations of the mitochondrial compartment in PEX5−/− mice, a model of peroxisome biogenesis disorders.
Severe Mitochondrial Alterations Are Found in PEX5−/− Mice, Corroborating the Fine Structural Changes Reported in Patients with Zellweger Syndrome
In PEX5−/− mice the most prominent mitochondrial alterations were found in hepatocytes, exhibiting heterogeneous, focal proliferation of pleomorphic mitochondria in some cells and involving all mitochondrial subcompartments (outer membrane, mitochondrial cristae, and matrix space). These alterations are already present in E18.5 PEX5−/− fetuses but become more pronounced after birth (P0.5), occasionally leading to severe and degenerative mitochondrial ghost forms (Figure 1; H to J ▶ ). Alterations of the mitochondrial fine structure in Zellweger patients were already described in the original report on the absence of peroxisomes by Goldfischer and colleagues. 12 Those structural changes were, however, less severe in nature and consisted of irregular and twisted cristae, dilatation of the intracristae space, as well as occurrence of attenuated organelles, corresponding to the subtle changes in the livers of our E18.5 PEX5−/− mice. In several subsequent ultrastructural studies, 20-22,24,48 mitochondrial alterations of varying types and severity were reported, although in a few cases there also were no changes noted. 49,50
In addition to the liver, changes of mitochondria in Zellweger patients have been reported in skeletal muscle. 51,52 Consistent with the finding of Sarnat and colleagues, 51 the diaphragm of the PEX5−/− mice was more affected than muscles of the limbs, which is interesting, because respiratory failure could be partly responsible for the death of some patients and possibly also of our mice. Furthermore, PEX5−/− mice showed abnormalities of mitochondria with different severity in other tissues known to be altered in Zellweger syndrome, such as epithelial cells of the inner zona fasciculata of the adrenal cortex and the renal proximal tubules, smooth muscle cells of the duodenum, cardiomyocytes, and some blood cells (developing lymphocytes and neutrophils). Although Zellweger patient fibroblasts have been extensively studied, mitochondrial alterations in those cells have not been observed, which is consistent with our observations in PEX5−/− fibroblasts. Finally, despite the depositions of different lipid derivatives in brain, 1 no obviously altered mitochondria were found in neurons of the cortical plate of PEX5 knockout mice. Mitochondrial changes were also not observed in neurons of Zellweger patients 53-55 but were seen in astrocytes, ranging from severely altered, degenerative forms 12 to mitochondrial proliferation. 50 Because pathological malformations of the brain occur focally, additional studies of the brain in PEX5 knockout mice are necessary before definitive conclusions on mitochondrial alterations in this organ can be drawn.
Ultrastructural Alterations of Mitochondria Are Associated with Changes in Respiratory Chain Complexes
As shown by a combination of activity measurements and Western blot analysis of liver homogenates, immunocytochemical localization of the enzyme proteins, as well as enzyme cytochemical staining for cytochrome c oxidase, there are profound alterations in the activity and content of different respiratory chain enzymes in mitochondria of PEX5−/− mice. The most consistent finding was a twofold reduction of NADH-ubiquinone oxidoreductase (complex I) activity that correlated with the reduced levels of a complex I subunit in liver samples of newborn PEX5−/− mice (Table 1) ▶ . These results are in line with the studies of Goldfischer and colleagues, 12 who noted a 70% decrease in O2 consumption in mitochondrial fractions of liver of Zellweger patients when the fractions were incubated with complex I-linked substrates. In contrast to Zellweger patients who also displayed a reduced activity of complex II, this activity was in the normal range in liver or heart homogenates of PEX5−/− mice. For complex III, there was a marked heterogeneity in immunocytochemical staining despite unchanged levels of total enzyme activity in homogenates. Importantly, severely altered mitochondria seemed to contain much less complex III than less affected mitochondria. Enzyme histochemical staining for cytochrome c oxidase (complex IV) also revealed a heterogeneous staining of the mitochondrial cristae. In individual hepatocytes, ultrastructurally normal mitochondria and mitochondria with parallel stacks of cristae exhibited a strong cytochrome c oxidase activity, whereas mitochondria with altered cristae showed a significantly weaker staining with mitochondrial ghosts exhibiting only minimal staining. The elevation of complex IV activity observed in liver homogenates is not in contradiction with the reduction observed for other respiratory chain enzymes, because this protein is differently regulated. 56 Indeed, in some patients with a reduced activity of complex I, an elevation of the activity of complex IV has been reported. 57
On the other hand, a significant 35% reduction of complex V was found in liver homogenates of newborn PEX5 knockout mice, and this was also reflected in reduced and heterogeneous staining of complex V in light microscopic preparations. No evidence of uncoupling of the mitochondria in liver and heart of PEX5−/− mice was obtained by enzyme histochemical staining for ATPase activity (M. Borgers, data not shown). This is in agreement with an earlier report on liver mitochondria of Zellweger patients, 48 although in skeletal muscle a slight uncoupling of mitochondria has been described. 52
Taken together, the ultrastructural alterations of liver mitochondria are accompanied by an altered activity and distribution of the mitochondrial respiratory chain complexes leading to a heterogeneous mitochondrial population with an overall decrease of complex I and complex V activities in liver of newborn PEX5-deficient mice.
Deficiency of the Respiratory Chain Enzymes Does Not Affect ATP Levels
Because the respiratory chain is responsible for the production of ATP by oxidative phosphorylation and the regeneration of NAD+, functional defects of respiratory chain enzymes (mostly complexes I and IV) are often accompanied by low ATP levels and increased ratios of lactate/pyruvate and β-hydroxybutyrate/acetoacetate. 58 Unfortunately, because of starvation of the newborn PEX5−/− mice, all redox markers were strongly depleted, interfering with the proper assessment of the redox status. Nevertheless, steady-state ATP levels were not altered in PEX5−/− mice as compared to control littermates (Table 2) ▶ , suggesting that despite the reduction of the activities of complexes I and V, the overall mitochondrial respiratory function of the whole hepatocyte population is still sufficient. These results are consistent with the so-called “threshold effect” 59 in patients with complex I deficiency, who may have normal ATP production despite severe reduction of complex I activity. 47,57
Indications Suggestive of Increased Oxidative Stress in PEX5−/− Knockout Mice
Mitochondrial electron transport has long been recognized as a major source of superoxide (.O2−) and H2O2. 60 The superoxide anion radical is generated by direct transfer of electrons from complex I or complex III to oxygen. Under physiological conditions, 2 to 3% of the consumed oxygen is converted to superoxide. To protect the cell from toxic injury, .O2− is converted to the less toxic H2O2 by MnSOD present in the mitochondrial matrix. This H2O2 may either diffuse out of mitochondria into the cytoplasm or may remain in the mitochondrial matrix and is reduced to H2O by glutathione peroxidases of the appropriate cellular compartment. If the balance between the generation and scavenging of these ROS is disturbed, increased oxidative stress may develop in mitochondria, 58,61 leading to oxidation of lipids in membranes, of thiol-containing proteins, and of mitochondrial DNA.
The ultrastructural alterations in the mitochondrial populations of different organs of PEX5−/− mice are suggestive of oxidative stress. These alterations, which have also been reported in mitochondrial respiratory chain disorders, include: 1) proliferation of pleomorphic mitochondria, 2) abnormal cristae (eg, parallel stacks, curvilinear forms), 3) fluffy matrix, and 4) swollen mitochondria. 62 Similar ultrastructural alterations have also been described in the liver in many other disease states associated with oxidative stress (eg, Reye’s syndrome, acute fatty liver of pregnancy) and intoxication with drugs (eg, peroxisome proliferators, chloramphenicol, nonsteroidal anti-inflammatory drugs) or toxins (ethanol, CCl4, antimycin A, and rotenone). 58,63,64 In Wilson’s (copper intoxication), Parkinson’s, or Huntington’s diseases and Friedreich’s ataxia similar mitochondrial alterations have also been described. 58,65 The early stage alterations observed in Wilson’s disease are similar to the mitochondrial changes in E18.5 PEX5−/− mice (except for crystalline inclusions). Interestingly, analogous mitochondrial alterations have been noted in iron overload, which is associated with substantial depletion of endogenous antioxidants and moderate lipid peroxidative damage. 66 Similar pathological alterations could play a role in the pathogenesis of the secondary mitochondrial changes in Zellweger syndrome, because iron overload and deposition have also been reported in several organs of Zellweger patients. 48,52,67
Ultrastructural alterations of mitochondria with circular cristae and dilation of intracristal spaces similar to those reported here were recently described in syncytiotrophoblastic cells of human placenta and it was suggested that they represented an adaptive response to oxidative stress because of elevation of oxygen tension at the end of the first trimester. 68 The further intensification of degenerative mitochondrial alterations in newborn (P0.5) PEX5−/− mice in comparison to 18.5-day embryos would also support the notion that oxidative stress, because of elevation of oxygen tension after the birth, is responsible for the worsening of the mitochondrial changes in PEX5 knockouts.
Another sign of oxidative stress in liver mitochondria of PEX5−/− mice was the significant up-regulation of MnSOD (Figure 6) ▶ , which is an important enzyme for the protection against the development of oxidative stress. 69,70 An increase in MnSOD was also reported in some patients with complex I deficiency. 47
As mentioned above, the sites of radical production in the respiratory chain are complexes I and III. A clear correlation between the degree of complex I inhibition, radical production, and cell injury by lipid peroxidation was shown in different experimental models. 71 Because in PEX5−/− mice, complex I activity was twofold reduced in liver homogenates and complexes I and III showed a lower immunostaining intensity in liver sections, it is plausible that increased mitochondrial radical production occurs in the peroxisome-deficient mouse livers. Interestingly, it has been demonstrated that radicals produced in mitochondria are themselves effective inhibitors of different respiratory chain enzymes and that the pattern of inactivation is different for distinct oxygen radicals. 72 Thus, a vicious cycle could be initiated whereby the reduced activities of complex I and possibly complex III would release ROS that in turn could further inhibit the activities of the respiratory chain enzymes.
Origin and Consequences of Mitochondrial Changes
The fact that the absence of functioning peroxisomes is associated with an altered mitochondrial structure and function in man as well as in mice indicates that a conserved mechanism underlies these subcellular changes. In some Zellweger patients no mitochondrial alterations were observed, but this might be related to the genetic heterogeneity of the syndrome that can indeed be caused by defects in at least 10 different genes. 17 In PEX2-deficient mice, another model of Zellweger syndrome, 73 even though no clear-cut mitochondrial alterations in hepatocytes were reported, a few morphologically abnormal mitochondria can be identified in the published electron micrographs (eg, Figure 6B ▶ in Faust and Hatten 73 ).
In speculating on a link between the absence of peroxisomes and the mitochondrial alterations two possibilities must be taken into consideration which are not necessarily mutually exclusive: 1) defective rescue function of peroxisomes against ROS and 2) defective detoxification function of peroxisomes.
A physiological function of peroxisomes in defense against oxidative stress has been well established in plant cells. 74 Moreover, the localization in mammalian liver peroxisomes of various enzymes involved in generation and detoxification of ROS such as xanthine oxidase, 75 superoxide dismutases, 76, 77 and glutathione peroxidase 78 suggest strongly that they must be involved in regulation of cellular redox processes. 3 Indeed, Reddy and Lalwani 79 were the first to draw attention to the generation of ROS in hepatic peroxisomes of rodents treated with peroxisome proliferators and its possible implications for hepatocarcinogenesis. The same group described recently the importance of peroxisome proliferation in regulation of H2O2 levels in the liver of acyl-CoA oxidase I knockout mice. 80 Moreover, the complex interaction between the peroxisomal and mitochondrial lipid metabolism in the pathogenesis of fatty liver was shown in mice nullizygous for both peroxisomal acyl-CoA oxidase and PPAR-α. 81 In oxidative stress induced by ischemia and reperfusion of kidney, degenerative changes of regular peroxisomes in proximal tubules was noted by Singh 3 and Gulati and colleagues 82 that is followed by a compensatory regeneration of smaller particles. Interestingly, in the same experimental model there is evidence of severe reduction of complex I activity in renal mitochondria, 83 suggesting an association between peroxisome degeneration and mitochondrial complex I inactivation, similar to PEX5−/− mice. Furthermore, fibroblasts from patients with peroxisome biogenesis disorders have been shown to be much more sensitive than controls to ROS generated by UV irradiation, 84,85 which has been suggested to be because of reduced plasmalogen levels, which might have an important anti-oxidative function in biomembranes. 86 Interestingly, HepG2 cells with a prominent peroxisome compartment 87 respond to UV irradiation by tubulation and proliferation of peroxisomes. 5 Finally, H2O2 was recently shown to induce several PEX genes that are involved in biogenesis of peroxisomes. 4 Taken together there is substantial evidence that peroxisomes have an important function in cellular rescue against ROS. How the cellular redox state is affected when both ROS-generating and -detoxifying enzymes are mislocalized to the cytoplasm such as in our PEX5 knockout mice, however requires further study. Nevertheless, it seems highly likely that the absence of peroxisomes leads to increased ROS levels in cells with prominent peroxisomal metabolism that in turn could impair the mitochondrial respiratory chain enzymes triggering a vicious cycle of mitochondrial degeneration as mentioned above.
An alternative explanation for the damage to the mitochondrial compartment might be the toxic effects of accumulation of compounds that are normally degraded by peroxisomal β-oxidation eg, very long-chain fatty acids, dicarboxylic acids, bile acid intermediates, and branched chain fatty acids. Notably, it was recently found that bile duct ligation in rats causes impairment of the mitochondrial respiratory chain, possibly by altering the lipid composition of the inner mitochondrial membrane. 88 Furthermore, it is well known that free fatty acids act as potent detergents that can damage cellular membranes. 89 In this respect it is important to mention that the mitochondrial changes in the PEX5 knockout mice are most prominent in tissues with an active lipid metabolism and a high content of peroxisomes (eg, liver, proximal renal tubules, and adrenals) and that the mitochondrial damage described in this report seems to be a cell autonomous phenomenon. The latter was exemplified in liver where liver parenchymal cells displayed severe mitochondrial changes whereas neighboring endothelial cells, Kupffer cells, and connective tissue cells did not show any abnormalities.
Interestingly, Mathis and colleagues 90 have noted that the mitochondrial changes in the liver of Zellweger patients were already present before the development of major hepatic disease. Indeed, in our newborn peroxisome-deficient mice, also no severe liver pathology was observed, suggesting that the mitochondrial alterations could play a role in the pathogenesis of the liver dysfunctions at later stages of the disease. In conclusion, the severely altered mitochondria in various organs of peroxisome-deficient mice that may be induced by oxidative stress could play a major role in the pathogenesis of the multiple organ dysfunctions observed in this devastating disorder.
Acknowledgments
We thank Heike Steininger, Gabie Krämer, Richard Morlang, Benno Das, and Fred Thoné for excellent technical assistance.
Footnotes
Address reprint requests to Prof. Dr. med. H. Dariush Fahimi, Institut für Anatomie und Zellbiologie, Abteilung Medizinische Zellbiologie, Universität Heidelberg, Im Neuenheimer Feld 307, D-69120 Heidelberg, Deutschland. E-mail: h.dariush.fahimi@urz.uni-heidelberg.de.
Supported by the BioMed II Program of European Community (grant BMH4-98-3569); the Deutsche Forschungsgemeinschaft, Bonn, Germany (grant Ba 1155/1-4 and SFB 601-B1), and the Vlaamse Gemeenschap, Belgium (grant GOA/99/09).
E. B. and I. V. contributed equally to this article.
Present address of E. B.: Department of Biological Chemistry, Johns Hopkins University SOM/725 N. Wolfe/409 Physiology, Baltimore, MD 21205.
References
- 1.Baes M, Gressens P, Baumgart E, Carmeliet P, Casteels M, Fransen M, Evrard P, Fahimi D, Declercq PE, Collen D, van Veldhoven PP, Mannaerts GP: A mouse model for Zellweger syndrome. Nat Genet 1997, 17:49-57 [DOI] [PubMed] [Google Scholar]
- 2.De Duve C, Baudhuin P: Peroxisomes (microbodies and related particles). Physiol Rev 1966, 46:323-357 [DOI] [PubMed] [Google Scholar]
- 3.Singh I: Mammalian peroxisomes: metabolism of oxygen and reactive oxygen species. Ann NY Acad Sci 1996, 804:612-627 [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Huertas E, Charlton WL, Johnson B, Graham IA, Baker A: Stress induces peroxisome biogenesis genes. EMBO J 2000, 19:6770-6777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schrader M, Wodopia R, Fahimi HD: Induction of tubular peroxisomes by UV irradiation and reactive oxygen species in HepG2 cells. J Histochem Cytochem 1999, 47:1141-1148 [DOI] [PubMed] [Google Scholar]
- 6.Mannaerts GP, Van Veldhoven PP: Functions and organization of peroxisomal β-oxidation. Ann NY Acad Sci 1996, 804:99-115 [DOI] [PubMed] [Google Scholar]
- 7.Sumida C, Graber R, Nunez E: Role of fatty acids in signal transduction: modulators and messengers. Prostaglandins Leukot Essent Fatty Acids 1993, 48:117-122 [DOI] [PubMed] [Google Scholar]
- 8.Masters CJ: Cellular signalling: the role of the peroxisome. Cell Signal 1996, 8:197-208 [DOI] [PubMed] [Google Scholar]
- 9.Price PT, Nelson CM, Clarke SD: Omega-3 polyunsaturated fatty acid regulation of gene expression. Curr Opin Lipidol 2000, 11:3-7 [DOI] [PubMed] [Google Scholar]
- 10.Krisans S: Cell compartmentalization of cholesterol biosynthesis. Ann NY Acad Sci 1996, 804:142-164 [DOI] [PubMed] [Google Scholar]
- 11.Hajra AK, Das AK: Lipid biosynthesis in peroxisomes. Ann NY Acad Sci 1996, 804:129-141 [DOI] [PubMed] [Google Scholar]
- 12.Goldfischer S, Moore CL, Johnson AB, Spiro AJ, Valsamis MP, Wisniewski HK, Ritch RH, Norton WT, Rapin I, Gartner LM: Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science 1973, 182:62-64 [DOI] [PubMed] [Google Scholar]
- 13.Goldfischer S, Collins J, Rapin I, Coltoff-Schiller B, Chang CH, Nigro M, Black VH, Javitt NB, Moser HW, Lazarow PB: Peroxisomal defects in neonatal-onset and X-linked adrenoleukodystrophies. Science 1985, 227:67-70 [DOI] [PubMed] [Google Scholar]
- 14.Moser HW: Molecular genetics of peroxisomal disorders. Front Biosci 2000, 5:298-306 [DOI] [PubMed] [Google Scholar]
- 15.Lazarow PB, Moser HW: Disorders of peroxisome biogenesis. ed 7 Scriver CR Beaudet AL Sly WS Valle D eds. The Metabolic And Molecular Basis of Inherited Disease, 1995, vol 12:pp 2287-2349 McGraw-Hill, New York [Google Scholar]
- 16.Dammai V, Subramani S: The human peroxisomal targeting receptor, Pex 5p, is translocated into the peroxisomal matrix and recycled to the cytosol. Cell 2001, 105:187-196 [DOI] [PubMed] [Google Scholar]
- 17.Gould SJ, Vallee D: Peroxisome biogenesis disorders. Trends Genet 2000, 16:340-345 [DOI] [PubMed] [Google Scholar]
- 18.Gressens P, Baes M, Leroux P, Lombet A, Van Veldhoven P, Janssen A, Vamecq J, Marret S, Evrard P: Neuronal migration disorder in Zellweger mice is secondary to glutamate receptor dysfunction. Ann Neurol 2000, 48:336-343 [PubMed] [Google Scholar]
- 19.Janssen A, Baes M, Gressens P, Mannaerts GP, Declercq P, Van Veldhoven PP: Docosahexaenoic acid deficit is not a major pathogenic factor in peroxisome-deficient mice. Lab Invest 2000, 80:31-35 [DOI] [PubMed] [Google Scholar]
- 20.Mooi WJ, Dingemans KP, Van den Bergh Weerman MA, Jöbsis AC, Heymans HAS, Barth PG: Ultrastructure of the liver in the cerebrohepatorenal syndrome of Zellweger. Ultrastruct Pathol 1983, 5:135-144 [DOI] [PubMed] [Google Scholar]
- 21.Kelley RI: The cerebrohepatorenal syndrome of Zellweger, morphologic and metabolic aspects. Am J Med Genet 1983, 16:503-517 [DOI] [PubMed] [Google Scholar]
- 22.Hughes JL, Poulos A, Robertson E, Chow CW, Sheffield LJ, Christodoulou J, Carter RF: Pathology of hepatic peroxisomes and mitochondria in patients with peroxisomal disorders. Virchows Arch A Pathol Anat 1990, 416:255-264 [DOI] [PubMed] [Google Scholar]
- 23.Trijbels JMF, Berden JA, Monnens LAH, Willems JL, Janssen AJM, Schutgens RBH, Van den Broek-Van Essen M: Biochemical studies in the liver and muscle of patients with Zellweger syndrome. Pediatr Res 1983, 17:514-517 [DOI] [PubMed] [Google Scholar]
- 24.Vamecq J, Draye JP, van Hoof F, Misson JP, Evrard P, Verellen G, Eyssen HJ, Eldere J, Schutgens RB, Wanders RJ, Roels F, Goldfischer SL: Multiple peroxisomal enzymatic deficiency disorders. A comparative biochemical morphologic study of Zellweger cerebrohepatorenal syndrome and neonatal adrenoleukodystrophy. Am J Pathol 1986, 125:524-535 [PMC free article] [PubMed] [Google Scholar]
- 25.Beier K, Völkl A, Hashimoto T, Fahimi HD: Selective induction of peroxisomal enzymes by the hypolipidemic drug bezafibrate. Detection of modulations by automatic image analysis in conjunction with immunoelectron microscopy and immunoblotting. Eur J Cell Biol 1988, 46:383-393 [PubMed] [Google Scholar]
- 26.Munim A, Asayama K, Dobashi K, Suzuki K, Kawaoi A, Kato K: Immunohistochemical localization of superoxide dismutases in fetal and neonatal rat tissues. J Histochem Cytochem 1992, 40:1705-1713 [DOI] [PubMed] [Google Scholar]
- 27.Grabenbauer M, Fahimi HD, Baumgart E: Detection of peroxisomal proteins and their mRNAs in fetal and newborn mice. J Histochem Cytochem 2001, 49:155-164 [DOI] [PubMed] [Google Scholar]
- 28.Speel EJ, Hopman AH, Komminoth P: Amplification methods to increase the sensitivity of in situ hybridization: play card(s). J Histochem Cytochem 1999, 47:281-288 [DOI] [PubMed] [Google Scholar]
- 29.Schad A, Fahimi HD, Völkl A, Baumgart E: Nonradioactive in situ hybridization for detection of mRNAs encoding for peroxisomal proteins: heterogeneous hepatic lobular distribution after treatment with a single dose of bezafibrate. J Histochem Cytochem 1996, 44:825-834 [DOI] [PubMed] [Google Scholar]
- 30.Karnovsky MJ: Use of ferrocyanide reduced osmium tetroxide in electron microscopy. J Cell Biol 1971, 51:146A [Google Scholar]
- 31.Fahimi HD: Cytochemical localization of catalase in rat hepatic microbodies (peroxisomes). J Cell Biol 1969, 43:275-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angermüller S, Fahimi HD: Selective cytochemical localization of peroxidase, cytochrome oxidase and catalase in rat with 3,3′-diaminobenzidine. Histochemistry 1981, 71:33-44 [DOI] [PubMed] [Google Scholar]
- 33.Seligman AM, Karnovsky MJ, Wasserkrug HL, Hanker JS: Nondroplet ultrastructural demonstration of cytochrome oxidase activity with a polymerizing osmiophilic reagent, diaminobenzidine (DAB). J Cell Biol 1968, 38:1-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson JM, Karnovsky MJ: Ultrastructural localization of several phosphatases with cerium. J Histochem Cytochem 1983, 31:1197-1208 [DOI] [PubMed] [Google Scholar]
- 35.Baumgart E, Völkl A, Hashimoto T, Fahimi HD: Biogenesis of peroxisomes: immunocytochemical investigation of peroxisomal membrane proteins in proliferating rat liver peroxisomes and in catalase-negative membrane loops. J Cell Biol 1989, 108:2221-2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newman GR, Jasani B, Williams ED: A simple post-embedding system for the rapid demonstration of tissue antigens under the electron microscope. Histochem J 1983, 15:543-555 [DOI] [PubMed] [Google Scholar]
- 37.Fahimi HD, Reich D, Völkl A, Baumgart E: Contributions of the immunogold technique to investigation of the biology of peroxisomes. Histochem Cell Biol 1996, 106:105-114 [DOI] [PubMed] [Google Scholar]
- 38.Goping G, Kuijpers GA, Vinet R, Pollard HB: Comparison of LR White and Unicryl as embedding media for light and electron immunomicroscopy of chromaffin cells. J Histochem Cytochem 1996, 44:289-295 [DOI] [PubMed] [Google Scholar]
- 39.Baumgart E: Morphology of peroxisomes in light- and electron microscopy. Latruffe N Bugaut M eds. Peroxisomes. 1994, :pp 37-57 Springer-Verlag, Heidelberg [Google Scholar]
- 40.Slot JW, Geuze HJ: Sizing of protein A-colloidal gold probes for immunoelectron microscopy. J Cell Biol 1981, 90:533-536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rahman S, Blok RB, Dahl HM, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR: Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996, 39:343-351 [DOI] [PubMed] [Google Scholar]
- 42.Jaworek D, Welsch J: Adenosine 5′-triphosphate. UV-method with phosphoglycerate kinase. ed 3 Bergmeyer HU eds. Methods of Enzymatic Analysis, 1985, :pp 340-346 Verlag Chemie, Weinheim [Google Scholar]
- 43.Noll F: L-(+)-lactate. ed 3 Bergmeyer HU eds. Methods in Enzymatic Analysis, 1985, :pp 582-588 Verlag Chemie, Weinheim [Google Scholar]
- 44.Lamprecht W, Heinz F: Pyruvate. ed 3 Bergmeyer HU eds. Methods in Enzymatic Analysis, 1985, :pp 570-577 Verlag Chemie, Weinheim [Google Scholar]
- 45.Vassault A, Bonnefort JP, Specola N, Saudubray JM: Techniques in Diagnostic Human Biochemical Genetics. Homme FA eds. A Laboratory Manual. 1991, :pp 285-308 Wiley-Liss, New York [Google Scholar]
- 46.Salto R, Giron MD, del Mar Sola M, Vargas AM: Evolution of pyruvate carboxylase and other biotin containing enzymes in developing rat liver and kidney. Mol Cell Biochem 1999, 200:111-117 [DOI] [PubMed] [Google Scholar]
- 47.Pitkänen S, Robinson BH: Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J Clin Invest 1996, 98:345-351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Versmold HT, Bremer HJ, Herzog V, Siegel G, von Bassewitz DB, Irle U, von Voss H, Lombeck I, Brauser B: A metabolic disorder similar to Zellweger syndrome with hepatic acatalasia and absence of peroxisomes, altered content and redox state of cytochromes, and infantile cirrhosis with hemosiderosis. Eur J Pediatr 1972, 124:261-275 [DOI] [PubMed] [Google Scholar]
- 49.Lazarow PB, Black V, Shio H, Fujiki Y, Hajra A, Datta NS, Bangaru BS, Dancis J: Zellweger syndrome: biochemical and morphological studies on two patients treated with clofibrate. Pediatr Res 1985, 19:1356-1364 [DOI] [PubMed] [Google Scholar]
- 50.Aubourg P, Robain O, Rocchiccioli F, Dancea S, Scotto J: The cerebro-hepato-renal (Zellweger) syndrome: lamellar lipid profiles in adrenocortical, hepatic mesenchymal, astrocyte cells and increased levels of very long chain fatty acids and phytanic acid in the plasma. J Neurol Sci 1985, 69:9-25 [DOI] [PubMed] [Google Scholar]
- 51.Sarnat HB, Machin G, Darwish HZ, Rubin S: Mitochondrial myopathy of cerebro-hepato-renal (Zellweger) syndrome. Can J Neurol Sci 1983, 10:170-177 [DOI] [PubMed] [Google Scholar]
- 52.Müller-Höcker J, Walther JU, Bise K, Pongratz D, Hübner G: Mitochondrial myopathy with loosely coupled oxidative phosphorylation in a case of Zellweger syndrome. Virchows Arch Cell Pathol 1984, 45:125-138 [DOI] [PubMed] [Google Scholar]
- 53.Dimmick JE: Pathology of peroxisomal disorders. Applegarth DA Dimmick JE Hall JG eds. Organelle Diseases. 1997, :pp 211-232 Chapman and Hall Medical, London [Google Scholar]
- 54.Powers JM, Moser HW: Peroxisomal disorders: genotype, phenotype, major neuropathological lesions, and pathogenesis. Brain Pathol 1998, 8:101-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Powers JM: The pathology of peroxisomal disorders with pathogenetic considerations. J Neuropathol Exp Neurol 1995, 54:710-719 [PubMed] [Google Scholar]
- 56.Poderoso JJ, Lisdero C, Schöpfer F, Riobo N, Carreras MC, Cadenas E, Boveris A: The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. J Biol Chem 1999, 274:37709-37716 [DOI] [PubMed] [Google Scholar]
- 57.Pitkänen S, Feigenbaum A, Laframboise R, Robinson BH: NADH-coenzyme Q reductase (complex I) deficiency: heterogeneity in phenotype and biochemical findings. J Inher Metab Dis 1996, 19:675-686 [DOI] [PubMed] [Google Scholar]
- 58.Treem WR, Sokol RJ: Disorders of the mitochondria. Semin Liver Dis 1998, 18:237-253 [DOI] [PubMed] [Google Scholar]
- 59.Rossignol R, Malgat M, Mazat J-P, Letellier T: Threshold effect and tissue specificity. Implications for mitochondrial cytophathies. J Biol Chem 1999, 274:33426-33432 [DOI] [PubMed] [Google Scholar]
- 60.Chance B, Sies H, Boveris A: Hydroperoxide metabolism in mammalian organs. Physiol Rev 1979, 59:527-605 [DOI] [PubMed] [Google Scholar]
- 61.Pessayre D, Mansouri A, Haouzi D, Fromenty B: Hepatotoxicity due to mitochondrial dysfunction. Cell Biol Toxicol 1999, 15:367-373 [DOI] [PubMed] [Google Scholar]
- 62.Bove KE: Mitochondriopathy. Appelgarth DA Dimmick JE Hall JG eds. Organelle Diseases. 1997, :pp 365-379 Chapman and Hall Medical London [Google Scholar]
- 63.Ghadially FN: Mitochondria. Ultrastructural Pathology of the Cell. A Text and Atlas of Physiological and Pathological Alterations in Cell Fine Structure, chapt 3. 1975, :pp 101-185 Butterworths, London [Google Scholar]
- 64.Phillips MJ, Poucell S, Patterson J, Valencia P: The Liver. An atlas and text of ultrastructural pathology. 1987, :pp 1-585 Raven Press, New York [Google Scholar]
- 65.Schapira AH, Gu M, Taanman JW, Tabrizi SJ, Seaton T, Cleeter M, Cooper JM: Mitochondria in the etiology and pathogenesis of Parkinson’s disease. Ann Neurol 1998, 44(Suppl 1):S89-S98 [DOI] [PubMed] [Google Scholar]
- 66.Dabbagh AJ, Mannion T, Lynch SM, Frei B: The effect of iron overload on rat plasma and liver oxidant status in vivo. Biochem J 1994, 300:799-803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Powers JM, Moser HW, Moser AB, Upshur JK, Bradford BF, Pai SG, Kohn PH, Frias J, Tiffany C: Fetal cerebrohepatorenal (Zellweger syndrome): dysmorphic, radiologic, biochemical, and pathologic findings in four affected fetuses. Hum Pathol 1985, 16:610-620 [DOI] [PubMed] [Google Scholar]
- 68.Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ: Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol 2000, 157:2111-2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robinson BH: The role of manganese superoxide dismutase in health and disease, review. J Inherit Metab Dis 1998, 21:598-603 [DOI] [PubMed] [Google Scholar]
- 70.Huang T-T, Carlson EJ, Raineri I, Gillespie AM, Kozy H, Epstein CJ: The use of transgenic mice to study oxygen free radical metabolism. Ann NY Acad Sci 1999, 893:95-112 [DOI] [PubMed] [Google Scholar]
- 71.Barrientos A, Moraes CT: Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem 1999, 274:16188-16197 [DOI] [PubMed] [Google Scholar]
- 72.Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ: The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem 1990, 265:16330-16336 [PubMed] [Google Scholar]
- 73.Faust PL, Hatten ME: Targeted deletion of the PEX2 peroxisome assembly gene in mice provides a model for Zellweger syndrome, a human neuronal migration disorder. J Cell Biol 1997, 139:1293-1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Corpas FJ, Barroso JB, del Rio LA: Peroxisomes as a source of reactive oxygen species and nitric oxide signal molecules in plant cells. Trends Plant Sci 2001, 6:145-150 [DOI] [PubMed] [Google Scholar]
- 75.Angermüller S, Bruder G, Völkl A, Wesch H, Fahimi HD: Localization of xanthine oxidase in crystalline cores of peroxisomes. A cytochemical and biochemical study. Eur J Cell Biol 1987, 45:137-144 [PubMed] [Google Scholar]
- 76.Keller GA, Warner TG, Steimer KS, Hallewell RA: Cu, Zn superoxide dismutase is a peroxisomal enzyme in human fibroblasts and hepatoma cells. Proc Natl Acad Sci USA 1991, 88:7381-7385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Singh AK, Dobashi K, Gupta MP, Asayama K, Singh I, Orak JK: Manganese superoxide dismutase in rat liver peroxisomes: biochemical and immunochemical evidence. Mol Cell Biochem 1999, 197:7-12 [DOI] [PubMed] [Google Scholar]
- 78.Singh AK, Dhaunsi GS, Gupta MP, Orak JK, Asayama K, Singh I: Demonstration of glutathione peroxidase in rat liver peroxisomes and its intraorganellar distribution. Arch Biochem Biophys 1994, 315:331-338 [DOI] [PubMed] [Google Scholar]
- 79.Reddy JK, Lalwani ND: Carcinogenesis by hepatic peroxisome proliferators: evaluation of the risk of hypolipidemic drugs and industrial plasticizers to humans. CRC Critic Rev Toxicol 1984, 12:1-58 [DOI] [PubMed] [Google Scholar]
- 80.Fan CY, Pan J, Chu R, Lee D, Kluckman KD, Usuda N, Singh I, Yeldandi AV, Rao MS, Maeda N, Reddy JK: Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fatty acyl-coenzyme A oxidase gene. J Biol Chem 1996, 271:24698-24710 [DOI] [PubMed] [Google Scholar]
- 81.Hashimoto T, Fujita T, Usada N, Cook W, Qi C, Peters JM, Gonzalez FJ, Yelandi AV, Rao MS, Reddy JK: Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty acyl-CoA oxidase. Genotype correlation with fatty liver phenotype. J Biol Chem 1999, 274:19228-19236 [DOI] [PubMed] [Google Scholar]
- 82.Gulati S, Singh AK, Irazu C, Orak J, Rajagopalan PR, Fitts CT, Singh I: Ischemia-reperfusion injury: biochemical alterations in peroxisomes of rat kidney. Arch Biochem Biophys 1992, 295:90-100 [DOI] [PubMed] [Google Scholar]
- 83.Weinberg JM, Venkatachalam MA, Roeser NF, Nissim I: Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci USA 2000, 97:2826-2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hoefler G, Paschke E, Hoefler S, Moser AB, Moser HW: Photosensitized killing of cultured fibroblasts from patients with peroxisomal disorders due to pyrene fatty acid-mediated ultraviolet damage. J Clin Invest 1991, 88:1873-1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spisni E, Cavazzoni M, Griffoni C, Caluolari E, Tomasi V: Evidence that photodynamic stress kills Zellweger fibroblasts by a nonapoptotic mechanism. Biochim Biophys Acta 1998, 1402:61-69 [DOI] [PubMed] [Google Scholar]
- 86.Paltauf F: Ether lipids in biomembranes. Chem Phys Lipids 1994, 74:101-139 [DOI] [PubMed] [Google Scholar]
- 87.Schrader M, Baumgart E, Volkl A, Fahimi HD: Heterogeneity of peroxisomes in human hepatoblastoma cell line HepG2. Evidence of distinct subpopulations. Eur J Cell Biol 1994, 64:281-294 [PubMed] [Google Scholar]
- 88.Krahenbuhl S, Talos C, Lauterburg BH, Reichen J: Reduced antioxidative capacity in liver mitochondria from bile duct ligated rats. Hepatology 1995, 22:607-612 [DOI] [PubMed] [Google Scholar]
- 89.Ho JK, Moser H, Kishimoto Y, Hamilton JA: Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest 1995, 96:1455-1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mathis RK, Watkins JB, Szczepanik-Van Leeuween P, Lott I: Liver in the cerebro-hepato-renal syndrome: defective bile acid synthesis and abnormal mitochondria. Gastroenterology 1980, 79:1311-1317 [PubMed] [Google Scholar]