

Abstract
Although the universal genetic code exhibits only minor variations in nature, Francis Crick proposed in 1955 that “the adaptor hypothesis allows one to construct, in theory, codes of bewildering variety.” The existing code has been expanded to enable incorporation of a variety of unnatural amino acids at one or two nonadjacent sites within a protein by using nonsense or frameshift suppressor aminoacyl-tRNAs (aa-tRNAs) as adaptors. However, the suppressor strategy is inherently limited by compatibility with only a small subset of codons, by the ways such codons can be combined, and by variation in the efficiency of incorporation. Here, by preventing competing reactions with aa-tRNA synthetases, aa-tRNAs, and release factors during translation and by using nonsuppressor aa-tRNA substrates, we realize a potentially generalizable approach for template-encoded polymer synthesis that unmasks the substantially broader versatility of the core translation apparatus as a catalyst. We show that several adjacent, arbitrarily chosen sense codons can be completely reassigned to various unnatural amino acids according to de novo genetic codes by translating mRNAs into specific peptide analog polymers (peptidomimetics). Unnatural aa-tRNA substrates do not uniformly function as well as natural substrates, revealing important recognition elements for the translation apparatus. Genetic programming of peptidomimetic synthesis should facilitate mechanistic studies of translation and may ultimately enable the directed evolution of small molecules with desirable catalytic or pharmacological properties.
The extraordinary synthetic capability of the translation apparatus, with its wide substrate diversity, capacity to synthesize long polymers, and genetic encodability using adaptors (F. Crick, quoted in ref. 1), has long made it an attractive target for biosynthetic engineering. Nevertheless, rewriting the central dogma in biology to enable information flow from nucleic acid templates to polymers of unnatural amino acids in a controllable and generalizable manner has not been realized, and decades of research have not established its feasibility. Thus, attempting this goal has value, apart from its potential applications, in testing our understanding of translation.
Early work, in which the amino acid moiety of natural aminoacyl-tRNAs (aa-tRNAs) was chemically modified after charging, showed that sense codons can be partially reassigned to either a different standard amino acid (2) or an α-hydroxy acid (3), ultimately leading to synthesis of polymers of indeterminate length containing a random mixture of ester and amide linkages (4). Although these studies suggested the potential of harnessing translation for synthesis of encoded unnatural polymers, the approach is restricted to chemically accessible derivatives of natural aa-tRNAs and also suffers from substantial competition with amino acids in the translation extract (3).
Nonsense-suppressing aa-tRNAs (5–8), synthesized completely in vitro chemoenzymatically (6, 9–11) and engineered for resistance to proofreading and recharging by the aa-tRNA synthetases (RSs), have found wide utility for the specific incorporation of a large variety of unnatural amino acids at a single site per protein (12) but are restricted to use at a maximum of three stop codons. The suppression approach has been extended to rarely used sense codons by combining frameshifting aa-tRNAs with mRNAs containing extra downstream stop codons to terminate nonframe-shifted products (13). With frameshifting aa-tRNAs, two nonadjacent codons have been reassigned in one mRNA (14–16), but extension to more than two codons will be restricted to the most rarely used of the 61 sense codons because of competition with natural aa-tRNAs and will require complicated overlapping reading-frame designs. In addition, extension of frameshift suppression for use at adjacent sites, necessitating the positioning of adjacent unnatural anticodons of more than three bases each on the ribosome, is likely to be problematic. Moreover, the engineering of new tRNA anticodons must circumvent inadvertent recognition by the RSs, because the anticodon is a major recognition element (17).
Efficiencies of successful single nonsense or frameshift suppressions with unnatural amino acids are frequently below 50% (12–18), theoretically incompatible with appreciable product synthesis if several incorporations are required, and many unnatural amino acids fail to incorporate at all (12, 18). Although the inefficiencies may be explained in part by competition with endogenous release factors (19, 20), RSs and aatRNAs (15), or by the use of a suboptimal suppressor tRNA (21), additional explanations are required for the dramatic differences observed between different unnatural amino acids carried by the same suppressor tRNA (12) or between different tRNA bodies carrying the same amino acid (21). Presumably, such differences alter recognition by elongation factor (EF)-Tu and/or the ribosome (see Discussion).
It was hypothesized that synthetic limitations with unnatural amino acids might be largely overcome by excluding the factors and activities leading to competition in translation (22). Indeed, translations performed according to these principles incorporated biotinylated lysine from a native tRNALys adaptor (22). However, in combining such a purified system (22, 23) with chemoenzymatically synthesized substrates to facilitate switching of amino acid identity and codon specificity, a concern is the potential deleterious effect of using tRNAs without native nucleoside modifications. Information on such effects is limited because chemoenzymatically synthesized substrates have been typically used in crude charging and/or translation systems known to contain tRNA modification activities (24). If unnatural substrate features are rejected by the translational machinery, efficiencies may not be improved simply by provision of longer times for incorporation. Here, we combine a purified translation system free of RSs with chemoenzymatically synthesized nonsuppressor aa-tRNA substrates to explore the versatility of translation in a potentially generalizable manner.
Materials and Methods
Substrates. Synthetic genes were cloned to enable in vitro synthesis of tRNAminusCA species (from FokI-cut templates) for ligation to an aa-pdCpA or synthesis of full-length tRNAs (from BstNI-cut templates). The tRNA sequences contained substitutions at their 5′ and 3′ termini to maintain the secondary structure of the aminoacyl stems while enabling efficient transcription initiation at the first nucleotide with GMP by T7 RNA polymerase. Nvoc-aa-pdCpA derivatives of eU (25), mS (26), bK (27), and yU§ (Fig. 3b) were prepared and ligated to tRNAminusCA species by using general methods (11). The concentrations of Nvoc-aa-tRNA ligation products were estimated by urea polyacrylamide gel electrophoresis at pH 5 (ligation of certain aa-pdCAs required a concentration of T4 RNA ligase (New England Biolabs) severalfold higher than that recommended (11); only efficient ligations were used). Natural aatRNAs were prepared from pure isoacceptors (Subriden RNA, Rolling Bay, WA) as described (22) or with pure recombinant RSs (20). The specific activities of 3H-labeled amino acids were 21,400 (Fig. 2b), 14,600 (Fig. 2c), and 16,900 (Fig. 3) dpm/pmol.
Fig. 3.

Translations incorporating three adjacent different unnatural amino acids by reassigning arbitrarily chosen codons. (a) Reassignment of NAAC,TACC, and VGUU codons of the universal genetic code to encode unnatural amino acids (U1–3) of our choosing. Two additional adaptors, termed 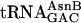 and
and 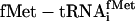 , were constructed to give the group of three chemoenzymatically synthesized tRNAs shown, differing only in their anticodon sequences (blue). The template (black), potentially encoding the natural translation product fMNTVE (green), was synthesized to test for the adjacent incorporation of three different unnatural amino acids by using these synthetic adaptors. (b) Rudimentary new genetic codes. Translation of the five-codon mRNA illustrated in a according to the blue code (Upper) would give the product fM-yU-mS-eU-E, whereas the purple code (Lower) would give fM-yU-eU-mS-E. (c) Dependence on each unnatural aa-tRNA for synthesis of fM-yU-mS-eU-E and fM-yU-eU-mS-E. All translations contained purified ribosomes and factors,
, were constructed to give the group of three chemoenzymatically synthesized tRNAs shown, differing only in their anticodon sequences (blue). The template (black), potentially encoding the natural translation product fMNTVE (green), was synthesized to test for the adjacent incorporation of three different unnatural amino acids by using these synthetic adaptors. (b) Rudimentary new genetic codes. Translation of the five-codon mRNA illustrated in a according to the blue code (Upper) would give the product fM-yU-mS-eU-E, whereas the purple code (Lower) would give fM-yU-eU-mS-E. (c) Dependence on each unnatural aa-tRNA for synthesis of fM-yU-mS-eU-E and fM-yU-eU-mS-E. All translations contained purified ribosomes and factors, 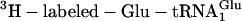 , and
, and 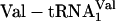 . The positive control translation (data not shown) was supplemented with mRNA MVE (22) and unlabeled
. The positive control translation (data not shown) was supplemented with mRNA MVE (22) and unlabeled 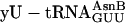 , and yielded 8,600 dpm of product. The fM-yU-mS-eU-E translation (blue open triangle) was supplemented instead with mRNA MNTVE (a) and substrates
, and yielded 8,600 dpm of product. The fM-yU-mS-eU-E translation (blue open triangle) was supplemented instead with mRNA MNTVE (a) and substrates 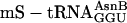 ,
, 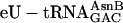 , and
, and 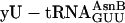 (charged according to the blue genetic code), each at 1 μM, whereas control translations (red open triangles) omitted the individual unnatural aa-tRNAs listed below the x axis. The fM-yU-eU-mS-E translation (purple filled triangle) was also supplemented with mRNA MNTVE and
(charged according to the blue genetic code), each at 1 μM, whereas control translations (red open triangles) omitted the individual unnatural aa-tRNAs listed below the x axis. The fM-yU-eU-mS-E translation (purple filled triangle) was also supplemented with mRNA MNTVE and 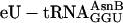 but differed in containing
but differed in containing 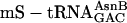 and
and 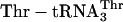 (see purple genetic code), whereas control translations (red filled triangles) omitted the aa-tRNAs listed. Background dpm obtained in a translation without mRNA was subtracted. (d) HPLC analysis of a replicate of the complete translation performed in c by using the blue code. Radiolabeled translation reaction was treated with alkali, mixed with authentic unlabeled marker peptide (fM-yU-mS-eU-E), and analyzed by reversed-phase HPLC on a C18 column using a 4–32% acetonitrile/water gradient (large plot), or mixed with two closely related markers of identical amino acid composition and analyzed by a shallower 9–14% gradient to maximize resolution (Inset). The elution positions of the marker peptides are indicated above the chromatograms. (e) HPLC analysis (as in d Inset) of a replicate of the complete translation performed in c by using the purple code, demonstrating synthesis of fM-yU-eU-mS-E.
(see purple genetic code), whereas control translations (red filled triangles) omitted the aa-tRNAs listed. Background dpm obtained in a translation without mRNA was subtracted. (d) HPLC analysis of a replicate of the complete translation performed in c by using the blue code. Radiolabeled translation reaction was treated with alkali, mixed with authentic unlabeled marker peptide (fM-yU-mS-eU-E), and analyzed by reversed-phase HPLC on a C18 column using a 4–32% acetonitrile/water gradient (large plot), or mixed with two closely related markers of identical amino acid composition and analyzed by a shallower 9–14% gradient to maximize resolution (Inset). The elution positions of the marker peptides are indicated above the chromatograms. (e) HPLC analysis (as in d Inset) of a replicate of the complete translation performed in c by using the purple code, demonstrating synthesis of fM-yU-eU-mS-E.
Fig. 2.

Translations incorporating five adjacent unnatural amino acids site-specifically. (a) mRNA sequence, encoded natural translation product without Glu-tRNAGlu (green), and encoded unnatural translation product when 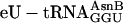 is replaced with
is replaced with 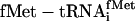 (blue). (b) Incorporation of five adjacent eU amino acids. Positive control translations (green) contained the purified ribosomes and factors, mRNA MT5V,
(blue). (b) Incorporation of five adjacent eU amino acids. Positive control translations (green) contained the purified ribosomes and factors, mRNA MT5V, 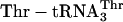 , ≈3 μM
, ≈3 μM 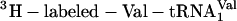 , and
, and  . In other translations, natural
. In other translations, natural 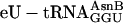 was omitted (negative controls in red) or replaced with ≈3 μM
was omitted (negative controls in red) or replaced with ≈3 μM 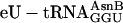 (blue). Product values (dpm after subtraction of background dpm obtained in control translations lacking mRNA) represent three experiments performed on three different occasions with three different preparations of
(blue). Product values (dpm after subtraction of background dpm obtained in control translations lacking mRNA) represent three experiments performed on three different occasions with three different preparations of 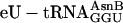 . Bars indicate standard deviations. X, amino acid variable. (c) HPLC analysis of a replicate of the translations performed with
. Bars indicate standard deviations. X, amino acid variable. (c) HPLC analysis of a replicate of the translations performed with 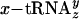 in b. Radiolabeled translation reaction was treated with alkali, mixed with authentic unlabeled marker peptide [fM(eU)5V dissolved in 88% formic acid], and analyzed by reversed-phase HPLC on a C18 column. The chromatogram shows a 27–71% acetonitrile/water linear gradient in the presence of 0.1% trifluoroacetic acid. The elution position of the marker peptide is indicated above the chromatogram. Peptide products were not detectable on a 2–32% acetonitrile/water gradient used for resolving less hydrophobic peptides such as fMT5V (ref. 22; data not shown).
in b. Radiolabeled translation reaction was treated with alkali, mixed with authentic unlabeled marker peptide [fM(eU)5V dissolved in 88% formic acid], and analyzed by reversed-phase HPLC on a C18 column. The chromatogram shows a 27–71% acetonitrile/water linear gradient in the presence of 0.1% trifluoroacetic acid. The elution position of the marker peptide is indicated above the chromatogram. Peptide products were not detectable on a 2–32% acetonitrile/water gradient used for resolving less hydrophobic peptides such as fMT5V (ref. 22; data not shown).
Translations. mRNAs and translation mixes were prepared as described (22), except that translation components differed by omission of polyethylene glycol, addition of His-tagged EF-Ts (28), further purification of initiation factor (IF)2 by gelfiltration chromatography, and additional washing of ribosomes. Ribosome salt washes were as described (22), except that an additional high-speed spin of 1 min preceded the final pelleting of the four-times-washed ribosomes to remove residual insoluble material. Ribosomes and factors were not contaminated with RSs or proteases, as measured by charging of total tRNA (Sigma) with 15 14C-labeled amino acids (New England Nuclear) and by stability of peptides. Macromolecular concentrations in translations were adjusted slightly to give 0.5 μM each of IF1, IF2, IF3, EF-G, and EF-Ts, 2.5 μM EF-Tu, four-times-washed ribosomes at 0.029 A260 unit/μl [27 nM estimated to be active (22)], 1 μM mRNA, 0.2 μM 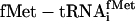 , and 0.5 μM (unless otherwise indicated) each elongator aa-tRNA, and translations were typically performed at 37°C for 30 min without preincubation. Translations analyzed by cation-exchange (treatment with alkali, acidification, then minichromatography to separate anionic formylated peptides from unformylated amino acids) were all performed on a 1-pmol scale with respect to limiting input
, and 0.5 μM (unless otherwise indicated) each elongator aa-tRNA, and translations were typically performed at 37°C for 30 min without preincubation. Translations analyzed by cation-exchange (treatment with alkali, acidification, then minichromatography to separate anionic formylated peptides from unformylated amino acids) were all performed on a 1-pmol scale with respect to limiting input  , whereas the scales varied for translations analyzed by HPLC. Peptide markers were synthesized on an Advanced Chemtech peptide synthesizer from commercial reagents.
, whereas the scales varied for translations analyzed by HPLC. Peptide markers were synthesized on an Advanced Chemtech peptide synthesizer from commercial reagents.
Results
A Purified RS-Free Translation System with Modular tRNA Adaptors. Our system (Fig. 1a) was constructed from ribosomes purified exhaustively to remove measurable contaminating RS charging activities (see Materials and Methods), recombinant translation factors (22), in vitro-synthesized mRNAs, in vitro-charged native tRNA isoacceptors, and chemoenzymatically synthesized aatRNAs. In pilot studies, a tRNAAsn-based chemoenzymatically synthesized elongator tRNA that was arbitrarily chosen (termed tRNAGGU AsnB, where the subscript refers to the anticodon; Fig. 1b) was nonenzymatically charged with eU (structure shown in Fig. 3b) and assayed in the purified translation system for single incorporation directed by mRNA MTV (see Materials and Methods). It was comparable in specificity and efficiency to the natural 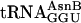 , even at 0.5 μM (a concentration 20 to 40-fold lower than typically needed in crude translation systems; refs.11 and 31), alleviating concerns that altering the tRNA body (Fig. 1b) or charging with the unnatural amino acid eU might be problematic. Control translations with unacylated full-length
, even at 0.5 μM (a concentration 20 to 40-fold lower than typically needed in crude translation systems; refs.11 and 31), alleviating concerns that altering the tRNA body (Fig. 1b) or charging with the unnatural amino acid eU might be problematic. Control translations with unacylated full-length 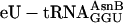 in place of
in place of 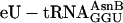 did not synthesize any full-length peptide, confirming that eU was indeed incorporated into products (data not shown).
did not synthesize any full-length peptide, confirming that eU was indeed incorporated into products (data not shown).
Fig. 1.

Our purified substrate-based translation system lacking RS activities. (a) The core translation machinery (blue) is depicted incorporating multiple unnatural amino acids (U1,U2,...) into peptidomimetic product. Escherichia coli served as the source for our natural components. IF1, IF2, IF3, His-tagged initiation factors; EF-Tu, EF-Ts, EF-G, His-tagged elongation factors. An mRNA template containing a Shine and Dalgarno ribosome binding site (SD) is colored purple, substrates are green, and products are red. Regeneration of GTP from GDP is catalyzed by pyruvate kinase using phosphoenolpyruvate substrate (data not shown). After translation, peptide products are released from the peptidyl-tRNAs by base-catalyzed hydrolysis [termination factors were omitted from the system for simplicity and because rapid product release would be undesirable for ribosome display experiments (29)]. (b) Natural E. coli tRNAAsn (ref. 30; black; the anticodon is purple) and our synthetic ligated derivative, tRNAAsnB (differences from the natural tRNA in blue).
Translating mRNA into a Specific Peptidomimetic Polymer. We then used the chemically aminoacylated 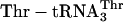 adaptor (Fig. 1b) to translate mRNA MT5V (Fig. 2a) to test for site-specific incorporation of several adjacent unnatural amino acids. This combination of adaptor and template allowed us to compare the efficiency of synthesis of the unnatural fM(eU)5V product to that of the natural fMT5V product, synthesized by translation of the same template with the native
adaptor (Fig. 1b) to translate mRNA MT5V (Fig. 2a) to test for site-specific incorporation of several adjacent unnatural amino acids. This combination of adaptor and template allowed us to compare the efficiency of synthesis of the unnatural fM(eU)5V product to that of the natural fMT5V product, synthesized by translation of the same template with the native 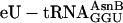 (Fig. 2b, green). When
(Fig. 2b, green). When 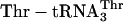 was substituted for
was substituted for 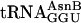 (Fig. 2b, blue), five adjacent eUs were incorporated into fM(eU)5V product at ≈30% overall yield in comparison with fMT5V, assuming similar recovery of fMT5V and fM(eU)5V during analysis. Control translations (Fig. 2b, red) without a cognate aa-tRNA for the T codon did not incorporate [3H]valine, confirming the specificity of decoding. HPLC analysis of the fM(eU)5V translation revealed comigration of the product with authentic fM(eU)5V marker prepared by chemical synthesis (Fig. 2c).
(Fig. 2b, blue), five adjacent eUs were incorporated into fM(eU)5V product at ≈30% overall yield in comparison with fMT5V, assuming similar recovery of fMT5V and fM(eU)5V during analysis. Control translations (Fig. 2b, red) without a cognate aa-tRNA for the T codon did not incorporate [3H]valine, confirming the specificity of decoding. HPLC analysis of the fM(eU)5V translation revealed comigration of the product with authentic fM(eU)5V marker prepared by chemical synthesis (Fig. 2c).
Creation of Unnatural Genetic Codes. To test the feasibility of creating rudimentary genetic codes, we constructed an mRNA containing three adjacent different test codons (AAC, ACC, and GUU) and also constructed the necessary two additional tRNA adaptors by using the tRNAAsnB body (Fig. 3a). Importantly, the test codons had been chosen arbitrarily and are present in three different codon boxes (standard groupings) of the universal genetic code, whereas the adaptors for their translation had been created rationally, differing only by anticodon mutations guided by the rules of codon–anticodon base pairing. We charged each adaptor chemically with an unnatural amino acid according to our blue genetic code (Fig. 3b Upper) and used these substrates for translation of the five-codon template (Fig. 3a). The encoded fM-yU-mS-eU-E product is indeed synthesized, based on the incorporation of radioactivity into product (Fig. 3c, blue open triangle), omission experiments (see below), and the comigration on HPLC with authentic synthetic peptide (Fig. 3d). We further examined the modularity and specificity of the approach by translating the same mRNA (Fig. 3a) by using the purple genetic code (Fig. 3b Lower) to synthesize the closely related sequence fM-yU-eU-mS-E. This translation required the preparation of two new combinations of unnatural amino acids and tRNAAsnB bodies and yielded the expected fM-yU-eU-mS-E product, as judged by incorporation of radioactivity into product (Fig. 3c, purple filled triangle) and comigration with authentic marker peptide on HPLC (Fig. 3e). The lack of read-through by noncognate aa-tRNAs in more stringent experiments, in which each chemically aminoacylated tRNA was individually omitted, further demonstrates the specificity of decoding in these translation reactions (Fig. 3c, red triangles).
The yield of fM-yU-mS-eU-E is ≈55% when compared with the amount of product translated from mRNA MVE with natural aa-tRNA substrates (Fig. 3c legend), and the amount of fM-yU-eU-mS-E is lower (Fig. 3c). When the 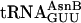 adaptor is chemically aminoacylated with the bulky biotinyllysine derivative (Fig. 3b Right) and tested for a single incorporation, the yield is only 20% [measured by binding to avidin (22); data not shown]. In practice, therefore, it is advisable first to test each charged tRNA individually, as is currently done with crude systems. It is also apparent that even our tRNAs charged with smaller unnatural amino acids give somewhat lower yields of products in translations requiring three to five adjacent unnatural amino acid incorporations in comparison with control translations containing only natural aa-tRNAs.
adaptor is chemically aminoacylated with the bulky biotinyllysine derivative (Fig. 3b Right) and tested for a single incorporation, the yield is only 20% [measured by binding to avidin (22); data not shown]. In practice, therefore, it is advisable first to test each charged tRNA individually, as is currently done with crude systems. It is also apparent that even our tRNAs charged with smaller unnatural amino acids give somewhat lower yields of products in translations requiring three to five adjacent unnatural amino acid incorporations in comparison with control translations containing only natural aa-tRNAs.
Discussion
Prior engineering of translation has been limited to the specific reassignment of the amino acid identity of only a small subset of the 64 codons at two nonadjacent codon positions within an mRNA because of the inherent restrictions of suppressor tRNAs and crude translation systems. Here, our combination of a purified translation system free of RSs with chemoenzymatically synthesized nonsuppressor aa-tRNA substrates enabled several, adjacent, arbitrarily chosen codons to be completely reassigned to unnatural amino acids in a potentially generalizable manner. The plasticity of translation illustrated by our studies supports the notion that the universality of the genetic code is primarily due to intrinsic constraints imposed not by the core translation apparatus but rather by RSs and the rest of the proteome. This idea is consistent with the known greater divergence of mitochondrial genetic codes, which encode very few proteins (32).
The study of translation using custom-designed substrates with our purified system (a purified “polypeptide polymerase”) has advantages over crude systems. Although inefficiencies with unnatural aa-tRNAs in crude systems are at least partly due to competing activities, this cannot be the case in our system because competitors have been deliberately excluded. Thus, the interesting observation here that unnatural substrates are still less efficient than natural ones directly reveals substrate determinants necessary for efficient translation (Figs. 1b and 3a) and suggests the existence of other competing reactions [one possibility is peptidyl-tRNA drop-off from the ribosome (23, 33)]. Nevertheless, the findings that our incorporations were more efficient than generally observed in crude systems, despite using lower concentrations of unnatural aa-tRNAs and the omission of all termination factors (33), and that adjacent unnatural amino acids can be incorporated broadens the range of experimental possibilities. For example, appropriately chosen pairs of adjacent unnatural amino acids might be used to probe the chemical mechanism of ribosomal peptide bond formation.
What recognition elements have been altered by the introduction of unnatural features into our substrates? Given that the affinities of EF-Tu for native aa-tRNAs are similar, whereas its affinities for mismatched combinations of amino acids and tRNA bodies are very different, one hypothesis is that efficient delivery by EF-Tu to the ribosome of each amino acid may require matching with a tRNA body of appropriate compensatory affinity for EF-Tu (34). Alternatively, tRNA nucleoside modifications can be important for efficient translation in vivo (35), whereas interpretation of in vitro studies with unmodified tRNAs by using crude charging or translation systems is complicated by the presence of endogenous modification activities (24). The loss of anticodon loop nucleoside modifications in many tRNAs is hypothesized to decrease anticodon–codon stability on the ribosome (36), which could lead to increased dissociation of cognate aa-tRNAs from the ribosome at both the initial selection and proofreading steps (37). Perhaps unnatural amino acid incorporation from our substrates could be improved by altering EF-Tu or the ribosome.
Our system should facilitate unambiguous definition of substrate elements that affect translational activity, including the enigmatic nucleoside modifications. Synthetic aa-tRNAs could be constructed to more closely resemble readily available natural aa-tRNAs for comparative studies, e.g., by using tRNA sequences other than that of tRNAAsn and unmutated tRNA sequences created by cleavage of in vitro synthesized precursors (38, 39). These findings may also be helpful in extending the synthetic scope of our initial system and existing suppressor systems.
Finally, we propose a potential application of our system for ligand discovery. Although the scalability of our system is not designed to provide an alternative to solid-phase peptide synthesis for preparation of individual peptides or to suppressor technology for preparation of proteins containing one or two nonadjacent unnatural amino acids, it is designed to produce the largest screenable libraries of small peptidomimetics. Theoretically, translation with 20 different aa-tRNAs of mRNA templates containing 10 random codons gives a library of 1013 different peptidomimetics (we synthesized 1012 peptidomimetics in a 70-μl translation, a fraction of which was analyzed in the experiment depicted in Fig. 2c) when ≈0.01 nmol of each aa-tRNA is incorporated (the typical research scale for aa-tRNA preparation is 1 nmol). Recycling of aa-tRNAs is also plausible by extrapolation of generalizable methods for engineering new synthetase specificities (8, 40, 41). Our approach therefore provides another route to create libraries for the discovery of small-molecule ligands or stereospecific catalysts (42), complementing other potential approaches for generating genetically encoded degradation-resistant lead compounds, such as mirror-image ligand display (43, 44) and nonribosomal peptide synthesis (45). The potential attraction of such approaches is that directed Darwinian evolution in vitro (29) is much faster than the many person-years of random chemical syntheses typically needed in industry for lead optimization, and their unrivalled library sizes may produce ligands with higher affinities. It is even conceivable that such selections could yield drug candidates directly (compare the orally available, 11-residue cyclic peptide cyclosporin A) when building blocks such as N-methyl amino acids are chosen to encode pharmacologically desirable properties such as protease resistance and membrane permeability.
Acknowledgments
We thank Dr. H. Weissbach for advice, Drs. S. Altman, A. R. Benson, M. Ehrenberg, M. Larvie, and C. T. Walsh for comments on the manuscript, Drs. Y.-W. Hwang and D. L. Miller for the EF-Tu and EF-Ts clones, and Dr. T. Ueda for the RS clones. This work was supported by grants from the National Institutes of Health (to A.C.F. and S.C.B.), an Army Idea Award from the Department of Defense (DAMD17-00-1-0163 to S.C.B.), and a National Science Foundation CAREER Award (to V.W.C.). S.C.B. is a Pew Scholar in the biomedical sciences and an Established Investigator of the American Heart Association. V.W.C. is the recipient of a Beckman Young Investigator Award, a Burroughs Wellcome Fund New Investigator Award in the Toxicological Sciences, and a Camille and Henry Dreyfus New Faculty Award.
Abbreviations: EF, elongation factor; IF, initiation factor; aa-tRNA, aminoacyl-tRNA; RS, aa-tRNA synthetase; tRNAminusCA, tRNA from which the 3′-terminal CA has been deleted; 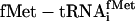 , x = charged amino acid, y = amino acid specificity of either the natural isoacceptor or the natural isoacceptor on which the chemoenzymatic sequence is based, and z = either the natural isoacceptor designation or the anticodon sequence (5′ to 3′) of chemoenzymatic tRNA sequence.
, x = charged amino acid, y = amino acid specificity of either the natural isoacceptor or the natural isoacceptor on which the chemoenzymatic sequence is based, and z = either the natural isoacceptor designation or the anticodon sequence (5′ to 3′) of chemoenzymatic tRNA sequence.
Footnotes
In our nomenclature for individual nonstandard amino acids, amino acids other than the standard 20 are represented by an uppercase letter and a lowercase letter prefix. The uppercase letter refers to the standard abbreviation of the related natural amino acid side chain and the prefix refers to the nonstandard functional group. When an unnatural amino acid side chain is unrelated to a natural one, we designate the uppercase letter part as U for unrelated unnatural. Thus, formylmethionine is fM, biotinyllysine is bK, O-methylserine is mS, and 2-amino-4-pentenoic acid and 2-amino-4-pentynoic acid are eU and yU, respectively (eU is also known as allylglycine; structures are shown in Fig. 3b). Nvoc indicates the amino-protecting group 6-nitroveratryloxycarbonyl.
References
- 1.Judson, H. F. (1979) The Eighth Day of Creation: Makers of the Revolution in Biology (Simon & Schuster, New York), p. 293.
- 2.Chapeville, F., Lipmann, F., von Ehrenstein, G., Weisblum, B., Ray, W. J. & Benzer, S. (1962) Proc. Natl. Acad. Sci. USA 48, 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fahnestock, S. & Rich, A. (1971) Nature New Biol. 229, 8–10. [DOI] [PubMed] [Google Scholar]
- 4.Fahnestock, S. & Rich, A. (1971) Science 173, 340–343. [DOI] [PubMed] [Google Scholar]
- 5.Noren, C. J., Anthony-Cahill, S. J., Griffith, M. C. & Schultz, P. G. (1989) Science 244, 182–188. [DOI] [PubMed] [Google Scholar]
- 6.Bain, J. D., Glabe, C. G., Dix, T. A., Chamberlin, A. R. & Diala, E. S. (1989) J. Am. Chem. Soc. 111, 8013–8014. [Google Scholar]
- 7.Nowak, M. W., Kearney, P. C., Sampson, J. R., Saks, M. E., Labarca, C. G., Silverman, S. K., Zhong, W., Thorson, J., Abelson, J. N., Davidson, N., et al. (1995) Science 268, 439–442. [DOI] [PubMed] [Google Scholar]
- 8.Wang, L., Brock, A., Herberich, B. & Schultz, P. G. (2001) Science 292, 498–500. [DOI] [PubMed] [Google Scholar]
- 9.Hecht, S. M., Alford, B. L., Kuroda, Y. & Kitano, S. (1978) J. Biol. Chem. 253, 4517–4520. [PubMed] [Google Scholar]
- 10.Baldini, G., Martoglio, B., Schachenmann, A., Zugliani, C. & Brunner, J. (1988) Biochemistry 27, 7951–7959. [DOI] [PubMed] [Google Scholar]
- 11.Ellman, J., Mendel, D., Anthony-Cahill, S., Noren, C. J. & Schultz, P. G. (1991) Methods Enzymol. 202, 301–336. [DOI] [PubMed] [Google Scholar]
- 12.Cornish, V. W., Mendel, D. & Schultz, P. G. (1995) Angew. Chem. Int. Ed. Engl. 34, 621–633. [Google Scholar]
- 13.Ma, C., Kudlicki, W., Odom, O. W., Kramer, G. & Hardesty, B. (1993) Biochemistry 32, 7939–7945. [DOI] [PubMed] [Google Scholar]
- 14.Hohsaka, T., Ashizuka, Y., Sasaki, H., Murakami, H. & Sisido, M. (1999) J. Am. Chem. Soc. 121, 12194–12195. [Google Scholar]
- 15.Hohsaka, T., Ashizuka, Y., Taira, H., Murakami, H. & Sisido, M. (2001) Biochemistry 40, 11060–11064. [DOI] [PubMed] [Google Scholar]
- 16.Anderson, R. D., Zhou, J. & Hecht, S. M. (2002) J. Am. Chem. Soc. 124, 9674–9675. [DOI] [PubMed] [Google Scholar]
- 17.Schimmel, P. & Söll, D. (1997) Proc. Natl. Acad. Sci. USA 94, 10007–10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornish, V. W., Benson, D. R., Altenbach, C. A., Hideg, K., Hubbell, W. L. & Schultz, P. G. (1994) Proc. Natl. Acad. Sci. USA 91, 2910–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Short, G. F., Golovine, S. Y. & Hecht, S. M. (1999) Biochemistry 38, 8808–8819. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu, Y., Inoue, A., Tomari, Y., Suzuki, T., Yokogawa, T., Nishikawa, K. & Ueda, T. (2001) Nat. Biotechnol. 19, 751–755. [DOI] [PubMed] [Google Scholar]
- 21.Cload, S. T., Liu, D. R., Froland, W. A. & Schultz, P. G. (1996) Chem. Biol. 3, 1033–1038. [DOI] [PubMed] [Google Scholar]
- 22.Forster, A. C., Weissbach, H. & Blacklow, S. C. (2001) Anal. Biochem. 297, 60–70. [DOI] [PubMed] [Google Scholar]
- 23.Cenatiempo, Y., Robakis, N., Reid, B. R., Weissbach, H. & Brot, N. (1982) Arch. Biochem. Biophys. 218, 572–578. [DOI] [PubMed] [Google Scholar]
- 24.Samuelsson, T., Boren, T., Johansen, T.-I. & Lustig, F. (1988) J. Biol. Chem. 263, 13692–13699. [PubMed] [Google Scholar]
- 25.Cornish, V. W. (1996) Ph.D. thesis (Univ. of California, Berkeley).
- 26.Mendel, D., Ellman, J. A., Chang, Z., Veenstra, D. L., Kollman, P. A. & Schultz, P. G. (1992) Science 256, 1798–1802. [DOI] [PubMed] [Google Scholar]
- 27.Gallivan, J. P., Lester, H. A. & Dougherty, D. A. (1997) Chem. Biol. 4, 739–749. [DOI] [PubMed] [Google Scholar]
- 28.Hwang, Y.-W., Sanchez, A., Hwang, M.-C. C. & Miller, D. L. (1997) Arch. Biochem. Biophys. 348, 157–162. [DOI] [PubMed] [Google Scholar]
- 29.Dower, W. J. & Mattheakis, L. C. (2002) Curr. Opin. Chem. Biol. 6, 390–398. [DOI] [PubMed] [Google Scholar]
- 30.Ohashi, K., Harada, F., Ohashi, Z., Nishimura, S., Stewart, T. S., Vögeli, G., McCutchan, T. & Söll, D. (1976) Nucleic Acids Res. 3, 3369–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steward, L. E. & Chamberlin, A. R. (1998) Methods Mol. Biol. 77, 325–354. [DOI] [PubMed] [Google Scholar]
- 32.Knight, R. D., Landweber, L. F. & Yarus, M. (2001) J. Mol. Evol. 53, 299–313. [DOI] [PubMed] [Google Scholar]
- 33.Karimi, R., Pavlov, M. Y., Heurgué-Hamard, V., Buckingham, R. H. & Ehrenberg, M. (1998) J. Mol. Biol. 281, 241–252. [DOI] [PubMed] [Google Scholar]
- 34.Asahara, H. & Uhlenbeck, O. C. (2002) Proc. Natl. Acad. Sci. USA 99, 3499–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curran, J. F. (1998) in Modification and Editing of RNA, eds. Grosjean, H. & Benne, R. (Am. Soc. Microbiol., Washington, DC), pp. 493–516.
- 36.Grosjean, H., Houssier, C., Romby, P. & Marquet, R. (1998) in Modification and Editing of RNA, eds. Grosjean, H. & Benne, R. (Am. Soc. Microbiol., Washington, DC), pp. 113–133.
- 37.Yarus, M. & Smith, D. (1995) in tRNA: Structure, Biosynthesis, and Function, eds. Söll, D. & RajBhandary, U. (Am. Soc. Microbiol., Washington, DC), pp. 443–469.
- 38.Forster, A. C. & Altman, S. (1990) Science 249, 783–786. [DOI] [PubMed] [Google Scholar]
- 39.Forster, A. C., Davies, C., Sheldon, C. C., Jeffries, A. C. & Symons, R. H. (1988) Nature 334, 265–267. [DOI] [PubMed] [Google Scholar]
- 40.Bessho, Y., Hodgson, D. R. W. & Suga, H. (2002) Nat. Biotechnol. 20, 723–728. [DOI] [PubMed] [Google Scholar]
- 41.Santoro, S. W., Wang, L., Herberich, B., King, D. S. & Schultz, P. G. (2002) Nat. Biotechnol. 20, 1044–1048. [DOI] [PubMed] [Google Scholar]
- 42.Gani, D. (2001) Nature 414, 703–705. [DOI] [PubMed] [Google Scholar]
- 43.Schumacher, T. N. M., Mayr, L. M., Minor, D. L., Jr., Milhollen, M. A., Burgess, M. W. & Kim, P. S. (1996) Science 271, 1854–1857. [DOI] [PubMed] [Google Scholar]
- 44.Forster, A. C. (1996) M.D. thesis (Harvard University, Boston).
- 45.Gewolb, J. (2002) Science 295, 2205–2207. [DOI] [PubMed] [Google Scholar]