

Abstract
Latency-associated nuclear antigen (LANA) of KSHV is expressed in all forms of Kaposi's sarcoma-associated herpesvirus (KSHV)-mediated tumors and is important for TR-mediated replication and persistence of the virus. LANA does not exhibit any enzymatic activity by itself but is critical for replication and maintenance of the viral genome. To identify LANA binding proteins, we used a LANA binding sequence 1 DNA affinity column and determined the identities of a number of proteins associated with LANA. One of the identified proteins was uracil DNA glycosylase 2 (UNG2). UNG2 is important for removing uracil residues yielded after either misincorporation of dUTP during replication or deamination of cytosine. The specificity of the ′LANA-UNG2 interaction was confirmed by using a scrambled DNA sequence affinity column. Interaction of LANA and UNG2 was further confirmed by in vitro binding and coimmunoprecipitation assays. Colocalization of these proteins was also detected in primary effusion lymphoma (PEL) cells, as well as in a cotransfected KSHV-negative cell line. UNG2 binds to the carboxyl terminus of LANA and retains its enzymatic activity in the complex. However, no major effect on TR-mediated DNA replication was observed when a UNG2-deficient (UNG−/−) cell line was used. Infection of UNG−/− and wild-type mouse embryonic fibroblasts with KSHV did not reveal any difference; however, UNG−/− cells produced a significantly reduced number of virion particles after induction. Interestingly, depletion of UNG2 in PEL cells with short hairpin RNA reduced the number of viral genome copies and produced infection-deficient virus.
Kaposi's sarcoma-associated herpesvirus (KSHV), also designated human herpes virus 8, is the biologic agent of Kaposi's sarcoma, primary effusion lymphoma (PEL), and multicentric Castleman's disease (12-14, 19, 65, 73). KSHV persists indefinitely in infected cells as an episome with the expression of a limited number of genes (20, 35, 39, 66, 75). Among these genes, the latency-associated nuclear antigen (LANA) is expressed from a polycistronic mRNA and is detected in all forms of KSHV tumors (18, 36). Cells infected with KSHV show anchorage-independent growth and increased telomerase activity, suggesting that KSHV induces cell immortalization (22, 37, 70). Also, KSHV infection induces chromosomal instability, an important event during tumorigenesis (52). These functions were found to be regulated by the major latent protein LANA (37, 64, 70). Additionally, LANA can also down regulate apoptotic pathways, p53, and pRb and induces cell immortalization in combination with Hras (23, 60). LANA also modulates the distribution of GSK-3β, a negative regulator of the Wnt signaling pathway, in a cell cycle-dependent manner and induces cells to enter S phase (10, 24).
LANA, which was initially detected by serum from a KSHV-infected patient in an immunofluorescence assay, is a large nuclear protein and is typically detected in a punctate nuclear pattern in KSHV-infected cells (34, 46, 61). The KSHV genome was detected at the sites of LANA on chromosome spreads of KSHV-infected cells, suggesting a role for LANA in KSHV genome tethering (4, 16). Later studies mapped the domains of LANA important for tethering to host chromosomes (5, 6, 17). LANA associates with human chromatin through the amino-terminal domain and remains attached during all of the phases of the cell cycle (7, 54, 63). LANA tethers the KSHV genome to host chromosomes through binding at the terminal repeats (TRs) of the KSHV genome (62). The TRs are 801 bp long, high-GC regions of the KSHV genome and are present as multicopy tandem repeats (44, 62). Each TR copy contains two LANA binding sequences (LBS1 and -2, high and low affinity, respectively) separated by a 22-bp DNA sequence (25). LANA binds to LBS through amino acids 936 to 1139 of the carboxyl-terminal domain (17, 38). Deletion mutant forms of this region showed that amino acids 1007 to 1021 are likely to be the DNA-contacting residues of LANA (38).
TR also supports replication of a plasmid in a LANA-dependent manner (28, 32). A single copy of the TR element is able to support replication, but the mechanism of replication is not fully understood (25, 28, 71). Sequence analysis and deletion mutation of TR mapped the minimal sequence essential for replication to a 29- to 32-bp-long GC-rich sequence upstream of LBS1 and LBS2 (33). LANA is critical for replication of TR-containing plasmids, but it does not have any enzymatic activity required for replication, thus suggesting the recruitment of necessary cellular proteins for replication at the TR. The mechanism of replication mediated by the TR is now beginning to be resolved, and the involvement of a number of cellular proteins is being investigated (67, 71). Among the proteins identified so far are human origin recognition complexes (ORCs), which interact with LANA at the TR (47, 71). EBNA1, a functional homolog of LANA, also interacts with ORCs and is essential for replication of the Epstein-Barr virus genome (reviewed in reference 8).
Besides replication, LANA is critical for the maintenance of KSHV episomal DNA. LANA-depleted cells, by using either recombinant virus (Bac36ΔLANA) or short hairpin RNA (shRNA) for LANA, failed to maintain KSHV episomes for the long term, suggesting its role in persistence and segregation (26, 74). Therefore, to understand the role of LANA in the process of replication and segregation, we wanted to identify LANA-interacting proteins when LANA is bound to its cognate sequence. Analysis of LANA-interacting proteins revealed a number of cellular proteins important for episomal persistence. Uracil DNA glycosylase 2 (UNG2) was identified as one of the LANA-interacting proteins.
UNG2 is encoded by the UDG gene, which spans approximately 13.8 kb and is located at chromosome 12q24.1 (42). This gene encodes mitochondrial UNG1 and nuclear UNG2, and both have the same catalytic domain but differ in their N-terminal amino acids (49). The major role of UNG2 is removal of dUTPs from nuclear DNA yielded either after misincorporation during the replication process or because of deamination of cytosine residues (43). UNG2 is located in the nucleoplasm and at replication foci. The mechanism of its translocation and association with replication foci is not well understood (41). However, it has been proposed that UNG2 is transported along with proliferating-cell nuclear antigen (PCNA), similar to DNA ligase I and DNA polymerase iota, postreplicative repair enzymes (41). UNG2 also associates with PCNA through its PCNA-interacting motif (Q4xxLxxFF11) (41). The N-terminal domain of UNG2 also interacts with replication protein A at the replication foci (51). Interaction of UNG2 with PCNA and replication protein A is important for base excision repair (BER) at the replication foci (43, 51).
In this report, we demonstrate that LANA binds to UNG2 through its carboxyl-terminal domain adjacent to the DNA binding domain. We also show the interaction of these two proteins in an immunofluorescence assay. The LANA-UNG2 complex possessed glycosylase activity, suggesting the involvement of UNG2 in the removal of any uracil residues misincorporated during the replication process. KSHV-infected cells with UNG2 levels depleted by shRNA showed a reduced viral genome copy number, suggesting that UNG2 is an important protein for persistence of the viral genome.
MATERIALS AND METHODS
Cells, plasmids, and antibodies.
BC-3 and BCBL-1, KSHV-positive primary effusion lymphoma (PEL) cell lines, and BJAB and DG75, KSHV-negative cell lines, were cultured in RPMI supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and penicillin-streptomycin (5 U/ml and 5 μg/ml, respectively). Human embryonic kidney 293 (HEK293) and HEK293T cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and penicillin-streptomycin (5 U/ml and 5 μg/ml, respectively). Primary UNG−/− and UNG+/+ mouse embryo fibroblast (MEF) cells (F11.1) were a gift from Hans E. Krokan (Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, Trondheim, Norway) and were established from transformed clones arising spontaneously after repeated passage in culture (2). MEFs were cultured in Dulbecco's modified Eagle's medium high in glucose with glutamine-Ham's F12 with sodium pyruvate (1:1), 10% fetal bovine serum, 1× nonessential amino acid solution, and penicillin-streptomycin. pBSpuroTR containing an entire (801-bp) TR unit was generated by cloning TR at the NotI site of pBS SKII+ (Stratagene, Inc., La Jolla, CA) as described previously (71). Expression vectors for full-length and truncated mutant LANA tagged with a myc epitope at its carboxyl terminus were described previously (70). pDsRed-LANA was described previously (64). UNG2 fused to green fluorescent protein, pUNG2EGFP, and rabbit UNG2-specific antibody (PU101) was a kind gift from Hans E. Krokan (Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, Trondheim, Norway). cDNA encoding the UNG2 open reading frame was PCR amplified with pUNG2EGFP as the template and cloned into pGEX2TK and pCDNA3.1HA to make a glutathione S-transferase (GST) fusion protein and a hemagglutinin (HA)-tagged protein, respectively. Rabbit anti-LANA polyclonal antibody was a kind gift from Bala Chandran (Rosalind Franklin University of Medicine and Science, North Chicago, Illinois). RTA was detected with a mouse monoclonal antibody as described previously (45).
DNA affinity column.
Preparation of the DNA affinity column used was described previously (71). Briefly, an oligonucleotide containing the LBS (17 bp, in italics and underlined) and an additional three nucleotides at the 5′ end and six nucleotides at the 3′ end (italics) (GATCCGCCTCCCGCCCGGGCATGGGGCCGCGG) with overlapping BamHI sites (bold) was synthesized and annealed with its complementary strand. Multimerized double-stranded DNA was ligated to cyanogen bromide-activated Sepharose beads and packed onto a Bio-Rad column. Nuclear extracts (NE) from BC-3 (KSHV-positive) and BJAB (KSHV-negative) cells were prepared as described earlier and bound to the LBS affinity column as described previously (71). The eluted fractions were aliquoted and frozen at −80°C.
A control DNA column was prepared by synthesizing a similar-length DNA sequence with a scrambled (SCR) LBS (5′-GATCCGCCATCGTAGATCTGTACTGTACGCGG-3′). Similar amounts of total NE were bound to the DNA columns with the multimerized LBS and the SCR LBS. Proteins eluted after extensive washing of the column were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to either Western blotting or sequencing of the band after gel excision.
Matrix-assisted laser desorption ionization-time of flight and liquid chromatography quadrupole (LCQ) mass spectrometry (MS) analysis.
Distinct protein bands from KSHV-positive BC-3 cell NE were excised from a Coomassie G-250-stained gel (see Fig. 1A). These bands were subjected to matrix-assisted laser desorption ionization-time of flight and LCQ mass spectrometry proteomic analysis at the Proteomics Core Facility at the University of Pennsylvania School of Medicine. Proteins for each band with LCQ scores greater than 20 were reported.
FIG. 1.

LANA bound to its cognate sequence in an affinity column interacts with host cell proteins. (A) Proteins eluted with buffer containing 500 mM NaCl from an LBS affinity column were resolved by 10% SDS-PAGE and stained with Coomassie brilliant blue. Bands specific to BC-3 cells (indicated as LAPs) were excised and sequenced. LAP12 was identified as UNG2. The values on the right are molecular sizes in kilodaltons. (B) UNG2 elutes from the affinity column containing LBS DNA but not from the SCR LBS DNA column. Equal amounts of BC-3 NE were incubated with either an LBS or an SCR LBS DNA affinity column, followed by thorough washing to remove any loosely bound protein. Protein eluted at 500 mM NaCl-containing buffer was Western blotted for detection of UNG2. PCNA shows that equal amounts of NE were present in the binding reaction mixture. (C) In vitro-translated LANA specifically binds to GST-UNG2. [35S]methionine-labeled LANA was incubated with either GST or GST-UNG2, and the bound fractions were quantified. The control protein (luciferase) did not bind to GST-UNG2. (D) Lysates from HEK293T cells expressing LANA-myc were incubated with either GST or GST-UNG2, and the bound fraction was analyzed by Western blotting with anti-myc antibody. LANA was precipitated with GST-UNG2 but not in the GST lane. PC, precleared with glutathione-Sepharose beads. (E) LANA coimmunoprecipitated with UNG2 from BCBL-1 cells. The pA3MUNG2 or pA3M vector only was transfected into BCBL-1 cells and immunoprecipitated (IP) with myc ascites. Western immunoblot assay detection with anti-LANA antibody showed coimmunoprecipitation of LANA from pA3MUNG2-transfected cells but not from cells transfected with the vector only. PC, precleared with protein A and protein G beads.
Western blot analysis.
An aliquot of the fraction eluted with 500 mM NaCl-containing elution buffer was resolved by SDS-PAGE, followed by detection with a UNG2-specific antibody (rabbit polyclonal antibody PU101). The signals were detected with Alexa Fluor 680 (Molecular Probes, Carlsbad, CA)- and 800 (Rockland, Gilbertsville, PA)-conjugated antibodies by Odyssey infrared scanning technology (LiCor, Lincoln, NE). myc- and HA-tagged proteins were detected with 9E10 and 12CA5 hybridomas, respectively.
In vitro binding of UNG2 to LANA.
Full-length LANA (amino acids 1 to 1162) and different truncation mutant forms of LANA were in vitro translated by a coupled in vitro transcription-translation system (TNT; Promega, Inc., Madison, WI) according to the manufacturer's instructions in the presence of [35S]methionine-cysteine (Perkin-Elmer, Inc., Boston, MA). pGEX-UNG2 expressing GST-fused UNG2 protein was expressed in Escherichia coli as described previously (71). Approximately 10 μg of GST-UNG2 protein was used for binding with in vitro-translated LANA and its derivatives. In vitro-translated products were precleared with glutathione-Sepharose beads in binding buffer (1× phosphate-buffered saline [PBS], 0.1% NP-40, 0.5 mM dithiothreitol, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 μg of aprotinin per ml) for 30 min. Precleared LANA was then incubated with either GST or GST-UNG2 fusion protein in binding buffer. Binding was performed overnight at 4°C with constant rotation, followed by collection of the beads through centrifugation. The beads were washed three times with 1 ml of binding buffer and then resuspended in SDS lysis buffer and resolved by SDS-PAGE. The bound fraction was analyzed after the gel was dried and exposed to a PhosphorImager plate (Molecular Dynamics, Inc.).
myc-tagged LANA expressed in HEK293T cells was also tested for binding to GST-UNG2. HEK293T cells expressing myc-tagged LANA were lysed in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, pH 8.0) with protease inhibitors (1 mM PMSF, 10 μg/ml pepstatin, 10 μg/ml leupeptin, and 10 μg/ml aprotinin) and centrifuged at 15,000 rpm and 4°C to remove cell debris. GST-UNG2 fusion protein was added to the cell lysate and subjected to overnight binding in RIPA buffer. The bound complex was washed five times to remove loosely bound proteins and resolved by SDS-PAGE. LANA was detected with anti-myc antibody by Western blotting.
Immunolocalization of LANA and UNG2.
HEK293 cells were cotransfected with pA3M LANA and pUNG2EGFP. BC-3 cells were only transfected with pUNG2EGFP, and endogenous LANA was detected with rabbit anti-LANA serum. At 24 h posttransfection, cells were spread on glass slides and followed by fixation in acetone-methanol (1:1) for 10 min at −20°C. Slides were air dried and incubated with 20% normal goat serum in 1× PBS to block the nonspecific binding sites. LANA was detected with rabbit anti-LANA polyclonal serum at room temperature in a humidified chamber, followed by washing three times for 5 min in PBS. The LANA signal was detected with Alexa Fluor 594 (Molecular Probes, Carlsbad, CA). UNG2 was visualized by fluorescence from the enhanced green fluorescent protein fusion. Slides were then washed four times with 1× PBS, mounted with Paramount G, and visualized with an Olympus confocal laser scanning microscope.
Detection of UNG activity.
To detect the activity of UNG2 when it is bound to LANA, we used a previously described (11) PCR-based assay (see Fig. 4A). The use of dUTP instead of dTTP for PCR amplification results in an amplicon which contains uracil residues. Incubation of the amplicon with UNG2 results in the excision of uracil residues from dU-containing PCR products. Heat treatment and alkaline pH conditions result in degradation of the abasic polynucleotide of the dU-containing PCR product, which is further blocked from subsequent PCR reamplification. Any DNA fragment can be used, regardless of its origin, for the UNG assay; the only important criterion is that it should be amplifiable by PCR. In this assay, we used the KSHV K1 gene as template DNA and amplified it with primers S-K1 (5′-ATGTTCCTGTATGTTGTCTGCAG-3′) and AS-K1 (5′-TCAGTACCAATCCACTGGTTGC-3′) with either dUTP or dTTP in the mixtures of deoxynucleoside triphosphates, leading to a PCR product of 870 bp, designated the dU DNA template or the dT DNA template, respectively. Subsequent PCR amplification with the dU DNA or dT DNA template treated with UNG2-LANA was then performed for up to 30 amplification cycles under identical conditions. Briefly, 1 μl of either dU or dT template DNA was assayed in a final volume of 50 μl in the presence of 1.5 mM MgCl2, 200 nM each amplification primer, 200 nM each deoxynucleoside triphosphate, and 1.5 U of Taq polymerase. PCR products were resolved on a 1% agarose gel.
FIG. 4.

UNG2 retains its glycosylase activity when complexed with LANA. (A) Schematic for detection of UNG2 activity in a PCR assay. Amplification of any gene with a specific set of primers in the presence of dU incorporates uracil into the DNA. Treatment of this amplicon with UNG2 cleaves the glycosidic bond and generates AP sites, which blocks the next round of amplification. The dT-containing amplicon remains unaffected by UNG2 and therefore serves as the template for the next round of amplification. (B) Coimmunoprecipitation of UNG2 with LANA by anti-myc antibody. UNG2 coimmunoprecipitated with LANA (lane 4) was detected in an anti-HA Western blot (WB) assay. HA-UNG2 cotransfected with pA3M (vector only) was not detected in the myc immunoprecipitation (IP) lane, suggesting that the coimmunoprecipitation was specific (lane 6). (C) Immunoprecipitated UNG2 blocked reamplification of target DNA. The KSHV K1 gene, amplified with specific primers in the presence of dT or dU and treated with UNG2, blocked the reamplification of a dU-containing amplicon (lane 4). Lane 5 shows specific amplification of the K1 gene with purified DNA as the template. (D) Immunoprecipitated LANA did not block reamplification of dU-containing templates after UNG assay (lane 2). UNG2 coimmunoprecipitated with LANA blocked reamplification of dU-containing templates (lane 4). Lanes 5 and 6 contain the reamplification products obtained with dT- and dU-containing DNA as the template after a UNG assay with the complex coimmunoprecipitated from the pA3M vector and HA-UNG2-transfected cells. Lanes 7 and 8 contain the reamplication products from equal amounts of template DNA containing dT and dU used in the UNG assay. The reduced amounts of reamplification in dU-containing templates were most probably due to the instability of DNA containing the RNA base uracil.
UNG2 was coimmunoprecipitated with LANA from HEK293T cells as the source of UNG activity. LANA by itself was also used to test for any UNG activity after immunoprecipitation.
Coimmunoprecipitation of LANA and UNG2.
myc-tagged LANA and HA-tagged UNG2 were transfected either separately or together into HEK293T cells. Cells were harvested 36 h posttransfection and lysed in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, pH 8.0) with protease inhibitors (1 mM PMSF, 10 μg/ml pepstatin, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). Lysates were centrifuged to remove cell debris and precleared. Lysates were then incubated with anti-myc antibody (myc ascites) overnight at 4°C with rotation, followed by incubation with protein A+G-Sepharose beads at 4°C for 1 h. The resulting immunoprecipitates were collected by centrifugation at 2,000 × g for 2 min at 4°C, and the pellets were washed four times with 1 ml of ice-cold RIPA butter. Half of the immune complex was resuspended in 30 μl of 2× SDS protein sample buffer (62.5 mM Tris, pH 6.8, 40 mM dithiothreitol, 2% SDS, 0.025% bromophenol blue, 10% glycerol) for Western blotting. UNG2 was detected with anti-HA antibody, and then the membrane was reprobed with anti-myc antibody for LANA detection.
Short-term replication assay.
pBSpuroA3 (three copies of TR) was cotransfected into either UNG−/− or UNG+/+ MEFs with a LANA expression vector. At 96 h posttransfection, DNA was extracted by a modified form of Hirt's procedure as described previously (31). Extracted DNA was digested with EcoRI (to linearize it) or EcoRI-DpnI with sufficient enzyme overnight. Digested DNA was resolved on a 0.8% agarose gel and transferred to a nylon membrane. DpnI-resistant copies of TR plasmid were detected by hybridization with a 32P-labeled TR probe with PhosphorImager plates (Molecular Dynamics, Inc.).
BrdU labeling and immunoprecipitation of replicated DNA.
BrdU (5-bromo-2-deoxyuridine, a thymidine analog) was administered at 72 h posttransfection to MEFs and pulsed overnight to label the replicating DNA. DNA was extracted as described previously and digested with DpnI overnight, followed by immunoprecipitation of BrdU-labeled DNA from 90% of the digested product with an anti-BrdU mouse monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) (29). The remaining 10% of the digested DNA was used as input. One microliter of the above-described immunoprecipitated DNA was used for detection of incorporated BrdU and DpnI-resistant copies of pBSpuroA3 after replication with primers described previously (71).
Purification of KSHV virions.
Virions were purified and virion proteins were fractioned and detected as described previously (45, 71). UNG2 was detected with rabbit anti-UNG2 antibody in proteins from pelleted and gradient-purified virions.
Infection of MEF cells with KSHV and induction for virus production.
UNG−/− and UNG+/+ MEF cells grown to 50 to 70% confluence in 100-mm-diameter tissue culture dishes were infected with the same amount of concentrated virus from an identical number of BCBL-1 cells. Virus obtained from approximately 20 million BCBL-1 cells were used to infect a single 100-mm culture dish in the presence of Polybrene (Sigma, St. Louis, MO). Two days after infection, cells were passaged and replated after thorough washing to remove any virion particles adhering to the cell surface. Efficiency of infection of these MEFs by BCBL-1-derived virus was determined by immunofluorescence assay with rabbit anti-LANA antibody.
Equal numbers of UNG−/− and UNG+/+ MEFs (infected with BCBL-1 cell-derived virus) were plated in a 100-mm culture dish for induction to produce virus. Cells were induced with 20 ng of 12-O-tetradecanoylphorbol-13-acetate (TPA) per ml and 1.5 mM sodium butyrate (Sigma, St. Louis, MO) for 5 days, followed by supernatant collection and clearing by centrifugation at 2,000 rpm for 15 min to remove cells and cell debris. The supernatant was then filtered through 0.45-μm-pore-size filters, and virions were pelleted at 20,000 rpm for 2 h. Virion DNA was isolated by disrupting the virus at 60°C for 2 h in lysis buffer (10 mM Tris, pH 8.5, 1 mM EDTA, pH 8.0, 1% Sarcosyl, 0.1 mg/ml proteinase K). Lysates were phenol extracted and then CHCl3-indole-3-acetic acid extracted. The relative number of virions produced was quantified by real-time quantitative PCR (qPCR) with K1 gene amplification as described previously (72).
shRNA treatment and quantitation of KSHV episomal copies.
The UNG2-specific shRNA used was described previously (56), and the sequence is 5′-AUCGGCCAGAAGACGCUCUdTdT-3′ (sense, corresponding to the first exon [nucleotides 713 to 731] of the UNG genomic region). Firefly luciferase gene shRNA was used as a control shRNA (BD Clontech, Mountain View, CA). shRNA for UNG2 (5′-GATCCGATCGGCCAGAAGACGCTCTTTCAAGAGAAGAGCGTCTTCTGGCCGATCTTTTTTGATATCG-3′) was cloned into pSIRENRetroP (BD Clontech) after annealing with its complementary sequence. Fifteen micrograms of either pSIRENRetroP UNG2 shRNA or pSIRENRetroP Luc shRNA was transfected into 10 million BC-3 and BCBL-1 cells. Transfected cells were selected with puromycin (3 μg/ml) for 3 weeks. Puromycin-resistant colonies were assayed for UNG2 protein in a Western blot assay with anti-UNG2-specific antibodies.
Total DNA was extracted from these selected colonies by lysing of cells in 5 mM EDTA-1% sarcosyl, followed by proteinase K digestion (1). Relative numbers of KSHV episomal copies were calculated by real-time qPCR amplification of the K1 gene.
RESULTS
Host cellular protein binds to multimerized LBS affinity column.
Multiple studies have demonstrated a role for LANA in KSHV genome replication (28, 32, 71). However, LANA does not have any enzymatic activity associated with DNA replication, including DNA unwinding or ATPase activity, which is required for DNA replication. LANA is also important for persistence of the KSHV genome and replication of TR-containing plasmids (4, 16). Recombinant KSHV with LANA deleted as well as LANA transcript depletion by shRNA reduced the number of KSHV genomic copies, demonstrating that tethering is important for long-term persistence (26, 74). LANA has been shown to bind to cellular proteins to support tethering and replication of the KSHV genome (7, 16, 40, 63, 71). In order to identify cellular proteins associated with LANA when LANA is bound to its cognate sequence, we performed an LBS affinity binding assay (71). Fractions from BC-3 and BJAB cells (KSHV positive and negative, respectively) eluted at 500 mM NaCl were resolved by 10% SDS-PAGE. Coomassie staining of the gel detected unique bands in eluates from the BC-3 LBS affinity column (Fig. 1A). There were also a number of unique bands in BJAB NE; however, we decided to identify the bands from BC-3 NE as the immediate focus was to identify the LANA-associated proteins. These unique bands from BC-3 NE were designated LAP1 to -11 (LANA-associating proteins 1 to 11).
Identification of LAPs.
Coomassie-stained bands were excised from the gel and submitted for identification by liquid chromatography-tandem MS analysis at the Proteomics Core Facility of the University of Pennsylvania. The Mascot software, which uses a statistical scoring algorithm for identification of protein bands, was used (53). The MS spectra were also manually checked to ensure that each identification was of high confidence. Matching of the theoretical molecular weights of the identified bands also supported the MS results and the level of confidence. The identities of these bands are listed in Table 1. LAP11 was identified as a UNG (accession number NP_550433). UNG2 has been shown to remove uracil residues from replicating DNA through a BER pathway (41).
TABLE 1.
Identification of protein bands from NE of BC-3 cells detected in an LBS affinity column
| Band | Protein identity | Accession no. |
|---|---|---|
| LAP1 | DNA-PKcs | P78527 |
| LAP2 | Spectrin, beta, nonerythrocytic 1 isoform 1 | NP_003119 |
| LAP3 | Clathrin heavy chain 1 | NP_004850 |
| LAP4 | DNA topoisomerase 2-beta | Q02880 |
| LAP5 | Cyclin-dependent kinase-like protein 5 | NP_003150 |
| LAP6 | Splicing factor 3b, subunit 1b | NP_036565 |
| LAP7 | EBNA2 coactivator (p100) | NP_055205 |
| LAP8 | ATP-dependent DNA helicase II (Ku80) | NP_066964 |
| LAP9 | Heat shock protein hsp70 | NP_068814 |
| LAP10 | Actin binding protein ABP620 | BAA83821 |
| LAP11 | UNG2 | NP_550433 |
UNG2 preferentially binds to a DNA affinity column conjugated with the LBS.
In order to confirm the specificity of LANA and UNG2 binding to the LBS affinity column, we incubated BC-3 NE with LBS and SCR LBS DNA columns under similar condition. After thorough washing of the columns, proteins were eluted from both of the columns and resolved by 10% SDS-PAGE. Western blot analysis for detection of UNG2 in eluates from these two columns revealed that UNG2 predominantly associated with the LBS column rather than the SCR LBS column, although a faint signal was seen in the SCR eluate (Fig. 1B). LANA was not detected in the SCR LBS column, confirming the inability of LANA to bind to the SCR sequence (data not shown).
UNG2 forms complexes with LANA in vitro and in human cells expressing LANA.
We confirmed the above-described findings on UNG2 binding to LANA by in vitro binding assays with GST-UNG2 as bait. In vitro-translated LANA incubated with GST-UNG2 fusion protein showed precipitation of LANA (Fig. 1C, lane 3), suggesting that these proteins can interact in vitro. GST alone showed no obvious or visible precipitation of LANA (Fig. 1C, lane 2), and GST-UNG2 did not show binding to the control protein, luciferase (Fig. 1C, lane 6), suggesting that the interaction of GST-UNG2 with LANA is likely to be specific in the cells.
We also used LANA heterologously expressed in HEK293T cells to test its ability to bind GST-UNG2. HEK293T cell lysate containing LANA was precleared with glutathione-Sepharose beads and incubated with either GST-UNG2 or GST (Fig. 1D). Bound LANA was detected by anti-myc antibody in the GST-UNG2 lane (Fig. 1D, lane 3) but not in the GST lane (Fig. 1D, lane 6), corroborating the above-described results showing binding of these two proteins.
KSHV-infected cells express LANA during latent infection; therefore, we wanted to determine whether endogenous LANA expressed from the KSHV genome is also capable of interacting with UNG2. PEL (BCBL-1) cells transfected with either pA3M (myc vector) or pA3MUNG2 (myc-UNG2) were lysed and immunoprecipitated for UNG2 with myc antibody. Detection of LANA in pA3MUNG2-transfected BCBL-1 cells (Fig. 1E, lane 3) was due to coimmunoprecipitation of LANA with UNG2, supporting the association of these two proteins in KSHV-positive cells. The absence of LANA in the myc immunoprecipitation and pA3M transfection lane supports the specificity of the association in this assay (Fig. 1E, lane 6).
UNG2 colocalizes with LANA in transiently transfected and KSHV-infected cells.
HEK293 cells were transfected with pDSRed-LANA and pEGFPUNG2 and plated on glass slides. At 12 h posttransfection, cells were stained with 4′,6′-diamidino-2-phenylindole (DAPI) and analyzed by confocal laser microscopy. Detection of LANA and UNG2 showed a punctate nuclear pattern and that most of the UNG2 signal was colocalized with red fluorescent protein-LANA, again corroborating the above-described finding that these two proteins interact in vivo, as they were in same nuclear compartments (Fig. 2). We further detected colocalization of LANA and UNG2 in the KSHV-infected PEL cell line BC-3 (Fig. 2). We transfected pEGFPUNG2 into BC-3 cells and detected LANA with rabbit anti-LANA antibody, followed by staining with goat anti-rabbit Alexa Fluor 594 (red). The merged image of these two proteins showed distinct colocalization of LANA and UNG2 in BC-3 cells.
FIG. 2.

UNG2 colocalizes with LANA. HEK293 cells transfected with pDSRed-LANA (red) and pEGFPUNG2 (green) showed colocalization (yellow) of these two proteins detected in cotransfected cells (merge panel). Cells stained with DAPI show nuclear staining (blue). UNG2 colocalizes with LANA in BC-3 cells. BC-3 cells transfected with pEGFPUNG2 were stained with anti-LANA antibody, and then detection with Alexa Fluor 594 (red) showed nuclear punctate staining of LANA. Detection of UNG2 as a green fluorescent protein fusion showed the colocalization of these two proteins.
UNG2 binds to the C terminus of LANA.
Different domains of LANA have been shown to be involved in various functions, including genome tethering and transcriptional modulation (73). Therefore, we wanted to determine the domain of LANA important for the recruitment of UNG2. Mutant forms of LANA with specific domains deleted were in vitro translated and assayed for binding to GST-UNG2 with 10% of the total translated product as the input (Fig. 3). As indicated above, GST did not show any significant binding to the deletion mutant forms of LANA. Additionally, mutant forms of LANA lacking the C terminus did not show any binding to GST-UNG2 (Fig. 3, LANA amino acids 1 to 435, 1 to 756, and 1 to 950). LANA deletion mutant forms which contained the extreme C terminus (amino acids 946 to 1162) showed significant binding to UNG2, indicating that UNG2 can bind to LANA in close proximity to the DNA binding domain of LANA (amino acids 936 to 1139) (38).
FIG. 3.

UNG2 binds to the carboxyl terminus of LANA. The indicated LANA deletion mutant forms were in vitro translated with a transcription-translation kit in the presence of [35S]methionine and analyzed for binding with either GST or GST-UNG2. LANA deletion mutant forms containing the carboxyl terminus (amino acids 762 to 1162, 1 to 327, 929 to 1172, and 946 to 1162) bound to GST-UNG2. Bound fractions were quantified, and the relative binding with each LANA deletion mutant form, calculated on the basis of three independent experiments, is shown as a bar graph.
Cellular UNG2 retains its activity when complexed with LANA.
We wanted to determine whether UNG2, when complexed with LANA, retains its enzymatic activity. We used an indirect PCR based assay (11) to detect UNG2 activity as described in Materials and Methods and the schematic in Fig. 4A. If the template DNA containing dU is incubated with purified UNG2 and heated at alkaline pH, UNG2 excises the uracil residues from the template and thus generates nicks throughout its entire length. This prevents reamplification of the template DNA. In contrast to this, DNA containing dT remains intact and can be reamplified. We used purified UNG2 expressed in E. coli (data not shown) and immunoprecipitated UNG2 expressed in HEK293 cells for the UNG assay. Results of the UNG2 assay of dT- and dU-containing K1 gene amplicons are shown in Fig. 4C. Immunoprecipitates from HEK293 cells transfected with the vector-alone control did not affect reamplification of the K1 gene in both the dT- and dU-containing templates. As expected, immunoprecipitates from HA-UNG2-expressing cells blocked the reamplification of dU-containing templates but not dT-containing templates, demonstrating the presence of UNG activity in the complex.
In order to assay the activity of UNG2 in a complex with LANA, we used UNG2 coimmunoprecipitated with LANA by anti-myc antibody from LANA-myc- and HA-UNG2-transfected HEK293 cells as the source of UNG2. We confirmed the coimmunoprecipitation of UNG2 by anti-HA Western blotting (Fig. 4B, lanes 3 and 4). Immunoprecipitates from cells transfected with LANA-myc and HA-UNG2 separately were also analyzed by Western blotting with anti-myc and anti-HA antibodies (Fig. 4B, lanes 1 and 2 and lanes 5 and 6, respectively). For the UNG assay, dT- and dU-containing K1 amplicons were incubated with the complex immunoprecipitated from LANA-myc-, LANA-myc-HA-UNG2-, and HA-UNG2-transfected cells. Templates from these combinations were subjected to reamplification, and the results are shown in Fig. 4D. Immunoprecipitates from LANA-myc-transfected cells did not block reamplification of the target DNA, suggesting that LANA by its own does not have any UNG activity (Fig. 4D, lanes 1 and 2). Incubation of dU residue-containing template DNA with the immunoprecipitate from LANA-myc-HA-UNG2-transfected cells blocked reamplification, suggesting the presence of UNG activity in the immunoprecipitated complex (Fig. 4D, lanes 3 and 4). Immunoprecipitates obtained with anti-myc antibody from HA-UNG2-transfected cells did not block reamplification of K1 template DNA, ruling out the possibility of nonspecific precipitation of UNG2 with anti-myc antibody (Fig. 4D, lanes 5 and 6). This suggested that the UNG2 activity associated with the LANA-UNG2 complex is mostly due to the UNG2 bound to LANA. Importantly, this assay is not a quantitative assay but rather a qualitative assay and detects the presence or absence of DNA glycosylase activity. The reduction in reamplification of dU-containing templates treated with anti-myc immunoprecipitates from LANA-myc and HA-UNG2 could be due to the unstable nature of DNA containing the RNA base uracil. Additionally, endogenous UNG2 immunoprecipitated with LANA-myc may also have contributed to the UNG2 activity detected.
Replication of TR-containing plasmids was similar in UNG−/− and UNG+/+ (F11.1) MEFs.
Since UNG2 binds to the carboxyl terminus of LANA, which is important for replication, we wanted to determine whether UNG2 is capable of modulating TR-mediated replication. We performed a DpnI sensitivity assay to determine the replication of TR-containing plasmids in UNG-deficient MEFs. This DpnI assay relies on the fact that DpnI digests DNA only if the adenine (A) of its recognition site (GATC) is methylated. Since eukaryotic cells do not possess dam methylase, plasmid DNA replicated in these cells lacks methylation at adenine and thus becomes resistant to DpnI digestion. We used TR-containing plasmid DNA prepared in E. coli (dam+) for the replication assay by transfecting it into MEFs and determining the number of DpnI-resistant copies by Southern blotting and by PCR. pBSpuroA3 (a three-TR-containing plasmid) was cotransfected with a LANA-expressing vector into UNG-deficient (UNG−/−) and wild-type (UNG+/+) MEFs. Episomal DNA extracted by a modified form of Hirt's procedure was digested with either EcoRI (to linearize) or EcoRI and DpnI overnight and transferred to a GeneScreen membrane, followed by detection with a 32P-labeled TR probe. DpnI-resistant bands were quantified, and the relative density is plotted with normalization of the input as 10%. Relative densities of DpnI-resistant bands in UNG−/− and UNG+/+ cells did not show much difference, suggesting that UNG2 does not directly affect DNA replication (Fig. 5A). Western blotting shows the absence of UNG2 in UNG−/− MEFs (Fig. 5B).
FIG. 5.

TR-containing plasmid pBSpuroA3 replicates in UNG−/− MEFs with an efficiency similar to that seen in wild-type (UNG+/+) MEFs. (A) pBSpuroA3 containing three copies of TRs was cotransfected with pA3MLANA into UNG−/− and F11.1 (UNG+/+) cells. Plasmids extracted after 96 h posttransfection were digested with either EcoRI or EcoRI and DpnI, followed by Southern detection of DpnI-resistant copies, which indicated replication of the plasmids. Densitometric analysis of DpnI-resistant bands from three experiments indicates the presence of similar numbers of replicated copies in these two MEFs. (B) Western blot (WB) assay detection of UNG2 in UNG−/− and UNG+/+ MEFs. A β-actin assay was included to show equal protein loading. (C) Quantitation of replicated copies of TR-containing plasmids by BrdU labeling. pBSpuroA3- and pA3MLANA-cotransfected UNG−/− and UNG+/+ cells were pulsed with BrdU, followed by plasmid extraction. Ninety percent of the DpnI-digested Hirt DNA was immunoprecipitated (IP) by anti-BrdU antibody, followed by purification of bound DNA. Equal volumes of the purified DNAs from UNG−/− and UNG+/+ cells were used as templates for amplification of the vector backbone. Results of densitometric analysis of amplified bands from three independent experiments are shown as a bar diagram.
We further supported the above-described results by BrdU incorporation in newly replicated DNA. UNG−/− and UNG+/+ cells cotransfected with pBSpuroA3 and pA3MLANA were pulsed with BrdU for 12 h, followed by extraction of episomal DNA. Ninety percent of the DpnI-digested Hirt DNA was immunoprecipitated by anti-BrdU antibody. A region of the vector backbone from the input, as well as immunoprecipitated DNA, was amplified, and the relative densities of the amplicons were determined with 10% as the input. Relative densities of the amplicons from UNG−/− and UNG+/+ cells did not show much difference, and thus supported the above-described result (Fig. 5C).
UNG2 is packaged into virion particles.
We and others have previously shown that virions carry cellular, as well as viral, proteins, most probably for rapid establishment of latent infection following a short burst of lytic reactivation (39, 45). Therefore, we wanted to determine whether cellular UNG2 is packaged into virion particles. We determined the presence of UNG2 in pelleted and gradient-purified virions in a Western blot assay. A protein preparation used previously for detection of RTA and LANA ORCs was used for detection of UNG2. Specifically, Western blot analysis showed the presence of UNG2 predominantly in the core of KSHV virion particles, although there was some signal associated with the tegument (Fig. 6).
FIG. 6.
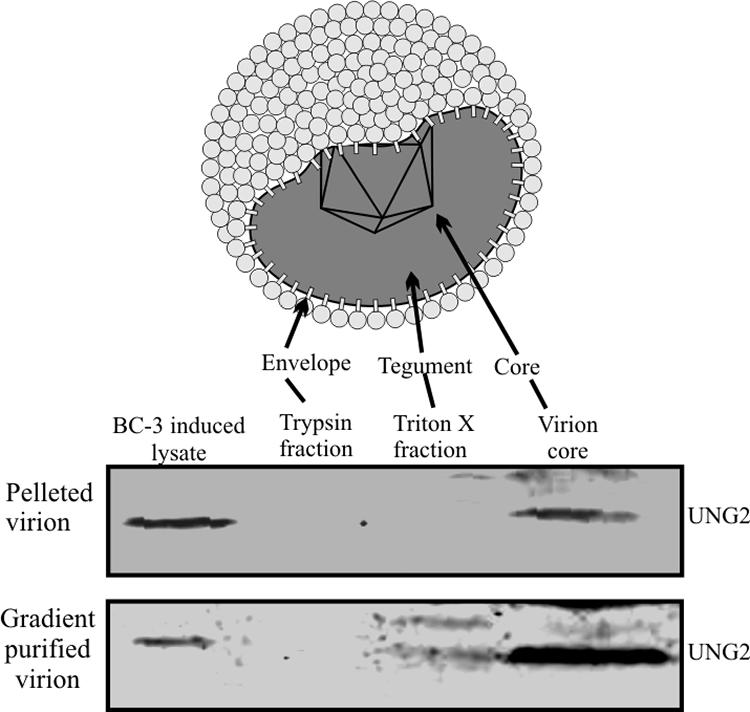
KSHV virion packages cellular UNG2. Virions obtained from 500 million BC-3 cells were pelleted and purified by sucrose gradient centrifugation. Different fractions of the virions were extracted as described previously (71), followed by Western blot assay detection of UNG2.
KSHV virions obtained from BCBL-1 cells are capable of infecting UNG−/− MEF cells.
KSHV virions produced after TPA and sodium butyrate induction were concentrated and purified for infection assays. Equal amounts of virion-containing suspension were added to UNG−/− and UNG+/+ MEFs. Infected cells were stained with anti-LANA antibody after one passage to detect infection and establishment of latent infection. LANA staining was indistinguishable in UNG−/− and UNG+/+ cells, and representative staining is shown in Fig. 7A. Since UNG2 is a base excision repair enzyme responsible for removal of any misincorporated uracil residues, it may not have reflected a difference in initial infection. Therefore, we passaged MEFs (UNG−/− and UNG+/+) infected with KSHV for several rounds in order to accumulate mutations due to misincorporation of uracil and deamination of cytosine in the genome. If the inserted mutation occurs in any regulatory gene, that would most likely affect the production of virions after induction. In fact, our virion quantitation data showed an approximately threefold reduction in the number of virions obtained from UNG−/− cells versus the number of virions obtained from UNG+/+ cells (Fig. 7B).
FIG. 7.

KSHV virions infect UNG2-deficient (UNG−/−) MEFs with an efficiency similar to that with which they infect wild-type (UNG+/+) MEFs. (A) Immunolocalization of LANA in KSHV-infected MEFs. (B) Quantitation of KSHV virions obtained from infected UNG−/− and UNG+/+ MEFs showed an approximately 30% reduction in the number of virions obtained from equal numbers of infected MEFs. (C) Detection of RTA in KSHV-infected UNG−/− and UNG+/+ cells after induction with TPA and sodium butyrate. (D) LANA immunostaining in HEK293 cells infected with KSHV virions obtained from UNG−/− and UNG+/+ cells. Infection of HEK293 cells with the viruses obtained from MEFs was greatly reduced, with almost no LANA detection in HEK293 cells infected with virus from UNG−/− cells, and only a few UNG+/+ cells produced virus.
We further wanted to determine the levels of RTA in these MEFs, as RTA is essential in activating the expression of KSHV lytic genes (69). As expected, we found reduced levels of RTA (Fig. 7C) in UNG−/− MEFs, explaining in part the reduction in the number of virions produced. We next examined the infectivity of the virions produced by and purified from these UNG−/− and UNG+/+ MEFs. We used equivalent amounts of virion particles (quantified with virion DNA by qPCR) from UNG−/− and UNG+/+ cells for infection of HEK293 cells. Infectivity was detected by immunostaining for LANA in these cells. LANA staining suggested that the infectivity of the virus progeny obtained from the MEFs was highly retarded, with very little or no infectivity of virions from UNG−/− cells. However, few cells infected with virions from UNG+/+ cells were detected by LANA staining (Fig. 7D).
PEL cells treated with UNG2 shRNA show a reduced number of episomal copies.
BC-3 and BCBL-1 cells selected with UNG2 shRNA for 3 weeks were assayed for UNG2 expression levels. UNG2 shRNA-treated PEL cells, BC-3 cells, and BCBL-1 cells showed efficient depletion of UNG2, i.e., close to an 85% reduction compared to control luciferase shRNA-treated cells (Fig. 8A). We further determined the number of latently persisting copies of episomal DNA in these shRNA-treated cells by real-time qPCR assay. The relative number of KSHV episomal copies was significantly reduced in UNG2 shRNA-treated PEL cells (Fig. 8B). Upon induction of these cells with TPA and sodium butyrate, the number of virions produced was further reduced, which may be a consequence of increased accumulation of mutations in the KSHV genome (Fig. 8C). This result corroborates the data obtained from UNG2-deficient MEFs. In order to detect the infectivity of virions from UNG2 shRNA-treated PEL cells, we infected HEK293 cells with equivalent numbers of virions as quantified by qPCR and detected LANA expression, which is a hallmark of latent infection. LANA staining suggested that virions from UNG2 shRNA-treated cells were also able to infect HEK293 cells but with significantly reduced efficiency compared to that of control shRNA-treated cells (Fig. 8D). The bar diagrams at the bottom of Fig. 8D represent the number of LANA-positive cells in a total of approximately 100 cells in the optical field. Representations of LANA-staining cells are shown in Fig. 8D.
FIG. 8.

UNG2 shRNA-treated PEL cells showed reduced numbers of episomal copies and produced fewer virions after reactivation. (A) Detection of UNG2 in shRNA-treated BCBL-1 and BC-3 cells showed a significant knockdown of UNG2 levels because of UNG2 shRNA-treated cells but not with control luciferase shRNA. β-Actin blot assays show equal protein loading. (B) UNG2 shRNA-treated BCBL-1 and BC-3 cells showed 40 to 70% decreases in viral genome copy numbers during latency. Viral copies were calculated by real-time qPCR with the K1 gene as the target for amplification. (C) Relative numbers of virions produced after TPA and sodium butyrate treatment of BCBL-1 and BC-3 cells selected with UNG2 and Luc shRNAs. Virions were quantified by isolating DNA from purified virions and quantification by K1 gene amplification in a real-time qPCR. (D) Immunostaining of LANA in HEK293 cells infected with virions obtained from UNG2 and Luc shRNA-treated cells. Equal numbers of virions from UNG2 and Luc shRNA-selected cells were used for infection of HEK293 cells. The average number of LANA-positive cells per optical field was determined, and the results are presented as a bar graph. On the basis of LANA staining, virions obtained from UNG2 shRNA-treated PEL cells showed three- to fourfold reduced infectivity.
DISCUSSION
LANA of KSHV is critical for tethering of viral episomes and replication (4, 16). The mechanism of LANA-mediated replication is not fully understood, but the evidence so far suggests that LANA can recruit components of the cellular replication machinery at the latent replication origin, the TR element (47, 67, 71). Our LBS affinity column data strengthen this hypothesis that host cellular replication machinery is involved in the replication of KSHV episomes (71). Most of the proteins which were identified by the affinity column contribute to DNA replication. These proteins include the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), DNA topoisomerase 2β, ATP-dependent DNA helicase II (Ku80), and UNG2. The DNA-PKcs of ∼460 kDa is a serine/threonine kinase and belongs to the phosphatidylinositol 3-kinase (p110) family (30). Ku80 is an ATP-dependent DNA helicase which forms a dimeric regulatory component with K70 proteins (21, 27). DNA-PKcs, Ku80, and Ku70 form a heterotrimeric enzyme (DNA-PK) which was previously shown to be involved in phosphorylation of DNA-bound proteins and transcription factors, including Sp1, p53, and the carboxy-terminal domain of RNA polymerase II; DNA damage; and the DNA-activated protein kinase (3). This suggests that DNA-PKcs may play a role in regulating transcription, replication, and recombination, as well as DNA repair (50). DNA topoisomerase 2β plays a critical role in controlling DNA topology, which is important for transcription, replication, and recombination (reviewed in reference 9). Interestingly, LANA has previously been shown to be involved in replication and KSHV episome segregation during cell division. However, the mechanism of segregation is not understood. Association of these proteins with LANA provides clues as to how LANA may function in replication and genome segregation.
The role of UNG2, which specifically removes the RNA base uracil from DNA, in the base excision repair pathway has been well studied (41, 43). Incorporation of uracil can occur in DNA either by misincorporation of dUTP during the replication process or by deamination of cytosine residues. Deamination of cytosine generates G · U mispairing, which leads to an A · T transition unless it is repaired before the next round of replication (43). In the genomic DNA, the frequency of cytosine deamination is on the order of 50 to 600 cytosines per genome per day (41). Uracil misincorporation into the genome is due to the direct incorporation of dUTP during DNA replication. Incorporation of dUTP into DNA during replication is prevented by a specific enzyme, dUTPase, which converts dUTP into dUMP, thus lowering the ratio of dUTP to dTTP and therefore reduces the chance of dUTP incorporation (reviewed in references 15 and 41). Damaged bases may be miscoding, cytotoxic, or both (43). To counteract these effects, cells have several defense mechanisms either to eliminate the defective cells or repair the damage. This damage then causes cell cycle arrest, allowing repair to take place before cells proceed to the next round of replication, thus maintaining the integrity of the cellular genome (41, 43).
Large-DNA human herpesvirus encodes dUTPase and UNG (reviewed in references 15 and 41). However, it is unclear why this virus produces the genes that encode these repair enzymes if the host cells contain the genes for them (reviewed in references 15 and 41). It has been postulated that the virus might need these genes to prevent incorporation and/or retention of uracil in viral DNA under certain physiological conditions, including viral infection and terminal differentiation, which reduce UNG2 and dUTPase activities (55, 58, 68). Viruses deficient for dUTPase and UNG2 have been reported to have decreased replication and virulence (58). Vaccinia virus deficient for UNG2 showed impaired replication and viability (48, 68). UNG2 encoded by herpes simplex virus is dispensable for viral replication in immortalized cells but has been shown to be important for efficient viral reactivation and latency in the murine nervous system (57, 59). Recombinant HSV-1 lacking UNG2 has been shown to accumulate mutations over time in culture (59). Cytomegalovirus also encodes dUTPase and UNG2, and mutant forms lacking these enzymes have been shown to have defects in DNA replication (55). These studies have suggested that UNG2 is an important enzyme for maintaining the integrity of the viral genome.
KSHV, like other herpesviruses, encodes a dUTPase enzyme, but expression of the gene for this enzyme was only detected during the lytic cycle of the virus (data not shown). Therefore, dUTPase encoded by KSHV most likely helps in reducing the ratio of dUTP to dTTP in reactivated cells and thus prevents misincorporation of uracil into newly synthesized viral DNA before it has been packaged into virion particles.
Most uracil lesions of DNA are repaired by a mechanism called BER, which is carried out by the DNA glycosylases (41). Uracil DNA glucosylase hydrolyzes the glycosydic bond between the target base, uracil, and deoxyribose, generating an apurinic-apyrimidinic (AP) site in the DNA and releasing a free base (41, 43). These AP sites are repaired by the AP endonuclease deoxyribophosphodiesterase and filled in by DNA polymerase β, followed by ligation with DNA ligase III (41). Additionally, human UNG2 has been shown to interact with replication protein A, which is important for short patch repair pathways in association with DNA polymerase and PCNA (41).
Detection of human UNG2 as a LAP led to the hypothesis that LANA may also have a role in maintaining the integrity of KSHV episomal DNA during latent infection by recruiting UNG2 in close proximity to replication foci. It is well established that LANA is critical for TR-mediated plasmid replication (28, 32). It is not entirely clear how LANA facilitates TR plasmid replication, but growing evidence suggests that LANA binds to the ORCs at the TR (71). Binding of MCMs to TRs was also confirmed by indirect binding assays (chromatin immunoprecipitation assays) (71). Since TR-containing plasmids cannot replicate without LANA, LANA either recruits cellular replication machinery to the TR or stabilizes the association of these proteins at the replicator element of the TR. Understanding the role of LANA in KSHV replication is an important area of research and is ongoing in our laboratory.
Interaction of UNG2 with the carboxyl terminus of LANA, which is also the DNA binding domain (16, 38), suggests that UNG2 remains in close proximity to the TR DNA (Fig. 9). Thus, it maybe involved in the removal of any incorporated uracil residues postreplication. In vitro assays have previously shown that uracil is rapidly removed from replicatively incorporated dUTP residues in isolated nuclei (51). That UNG2 retained its glycosylase activity when complexed with LANA (demonstrated by PCR-based assay) indicates that LANA may recruit UNG2 for the repair of any misincorporated uracil residues in TR DNA postreplication.
FIG. 9.

Proposed model of LANA-UNG2 interaction at KSHV TRs. LANA is a large (1,162-amino-acid) nuclear protein important for tethering of the viral genome to the host chromosome (4, 16). The three-dimensional structure of LANA, predicted with Robetta, a free protein structure prediction server (http://robetta.bakerlab.org/), shows distinct amino- and carboxyl-terminal domains. The amino-terminal domain of LANA shows a distinct chromosome binding sequence (CBS, amino acids 5 to 22). The DNA binding domain (DBD) of LANA, which lies between amino acids 996 and 1139 (38), is marked in blue (LANA-C). The interaction of LANA with UNG2 was mapped to the carboxyl terminus (amino acids 945 to 1162). The interaction of LANA and UNG2 is most likely important for bringing UNG2 into close proximity to the TR DNA, which is most likely important for the removal of any uracil residues incorporated during the replication process. Incorporation of uracil, if left unrepaired, may lead to the production of defective virus. PEL cells depleted of UNG2 with shRNA showed reduced KSHV genomic copies, possibly because of the accumulation of mutations caused by unrepaired incorporation of uracil residues during successive rounds of replication.
UNG2 is not required for latent DNA replication, as UNG2-deficient (UNG−/−) MEFs did not show any significant differences in the replication ability of TR-containing plasmids compared with wild-type MEFs. LANA was not detected in the virion tegument (71), but we detected UNG2 in purified virions, suggesting that it may be important during infection or for maintaining viral genome integrity. In order to determine its role, we infected UNG−/− and UNG+/+ cells with virions from BCBL-1 cells which showed comparable levels of infection. However, the numbers of virions obtained from UNG2-deficient MEFs after induction were significantly reduced, most likely because of the defect in the uracil repair pathway and generation of defective copies of latently persisting episomal DNA. Detection of lower levels of RTA in UNG−/− MEFs partly explains the lower number of virions obtained from these KSHV-infected MEFs. Of note, a reduction in the number of latently persisting KSHV episomal copies in UNG−/− cells may occur just because of the lack of UNG2 and not primarily because of the interaction with LANA. Therefore, strategies to disrupt the interaction of UNG2 with LANA are under development in our laboratory.
The KSHV-positive PEL cell lines BCBL-1 and BC-3, with reduced levels of UNG2 targeted by shRNA, showed reduced copies of the episomally persisting KSHV genome. Induction of these UNG2 shRNA-expressing cells produced three- to fourfold lower numbers of KSHV virions compared to cells expressing a control shRNA. Importantly, the cells selected with the shRNA specific for UNG2 were unable to grow for extended periods in culture. This may be due to loss of UNG2 function, growth retardation because of a lower number of KSHV episomal copies, or a combination of these two effects. Infection of HEK293 cells with virions obtained from shRNA-treated cells was less effective, as determined on the basis of LANA staining in HEK293 cells infected with equal numbers of virions obtained from either UNG2 or luciferase (control) shRNA-treated PEL cells. In conclusion, the data presented here suggest that LANA most likely brings UNG2 in close proximity to its episomal DNA through interaction with its DNA binding domain (C terminus) and therefore helps in removing any misincorporated uracils during postreplication repair. Cells lacking UNG2 fails to remove these misincorporated uracils, and the successive accumulation of these mutations leads to the generation of defective episomal copies and thus production of defective virions (Fig. 9). Therefore, targeting of UNG2 function could potentially be a useful strategy for blocking KSHV persistence in infected cells.
Acknowledgments
This work was supported by Public Health Service grants CA072510 and CA091792 from NCI and DE01436 and DE017335 from NIDCR (E.S.R.). E.S.R. is a scholar of the Leukemia and Lymphoma Society of America.
We thank Hans E. Krokan and Geir Slupphaug, Norwegian University of Science and Technology, Trondheim, Norway, for providing reagents and helpful suggestions. We also thank Chao-Xing Yuan and the Proteomics Core Facility at the University of Pennsylvania School of Medicine for the identification of protein bands.
Footnotes
Published ahead of print on 23 August 2006.
REFERENCES
- 1.Aiyar, A., C. Tyree, and B. Sugden. 1998. The plasmid replicon of EBV consists of multiple cis-acting elements that facilitate DNA synthesis by the cell and a viral maintenance element. EMBO J. 17:6394-6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersen, S., T. Heine, R. Sneve, I. Konig, H. E. Krokan, B. Epe, and H. Nilsen. 2005. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis 26:547-555. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, C. W. 1993. DNA damage and the DNA-activated protein kinase. Trends Biochem. Sci. 18:433-437. [DOI] [PubMed] [Google Scholar]
- 4.Ballestas, M. E., P. A. Chatis, and K. M. Kaye. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641-644. [DOI] [PubMed] [Google Scholar]
- 5.Ballestas, M. E., and K. M. Kaye. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 75:3250-3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbera, A. J., M. E. Ballestas, and K. M. Kaye. 2004. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 N terminus is essential for chromosome association, DNA replication, and episome persistence. J. Virol. 78:294-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbera, A. J., J. V. Chodaparambil, B. Kelley-Clarke, V. Joukov, J. C. Walter, K. Luger, and K. M. Kaye. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856-861. [DOI] [PubMed] [Google Scholar]
- 8.Bell, S. P., and A. Dutta. 2002. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71:333-374. [DOI] [PubMed] [Google Scholar]
- 9.Berger, J. M., and J. C. Wang. 1996. Recent developments in DNA topoisomerase II structure and mechanism. Curr. Opin. Struct. Biol. 6:84-90. [DOI] [PubMed] [Google Scholar]
- 10.Boshoff, C. 2003. Kaposi virus scores cancer coup. Nat. Med. 9:261-262. [DOI] [PubMed] [Google Scholar]
- 11.Bouhamdan, M., S. Benichou, F. Rey, J. M. Navarro, I. Agostini, B. Spire, J. Camonis, G. Slupphaug, R. Vigne, R. Benarous, and J. Sire. 1996. Human immunodeficiency virus type 1 Vpr protein binds to the uracil DNA glycosylase DNA repair enzyme. J. Virol. 70:697-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 13.Chang, Y. 1997. Kaposi's sarcoma and Kaposi's sarcoma associated herpesvirus (human herpesvirus 8): where are we now? J. Natl. Cancer Inst. 89:1829-1831. [DOI] [PubMed] [Google Scholar]
- 14.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 15.Chen, R., H. Wang, and L. M. Mansky. 2002. Roles of uracil-DNA glycosylase and dUTPase in virus replication. J. Gen. Virol. 83:2339-2345. [DOI] [PubMed] [Google Scholar]
- 16.Cotter, M. A., II, and E. S. Robertson. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254-264. [DOI] [PubMed] [Google Scholar]
- 17.Cotter, M. A., II, C. Subramanian, and E. S. Robertson. 2001. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy terminus. Virology 291:241-259. [DOI] [PubMed] [Google Scholar]
- 18.Dittmer, D., M. Lagunoff, R. Renne, K. Staskus, A. Haase, and D. Ganem. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dupin, N., T. L. Diss, P. Kellam, M. Tulliez, M. Q. Du, D. Sicard, R. A. Weiss, P. G. Isaacson, and C. Boshoff. 2000. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 95:1406-1412. [PubMed] [Google Scholar]
- 20.Dupin, N., C. Fisher, P. Kellam, S. Ariad, M. Tulliez, N. Franck, E. van Marck, D. Salmon, I. Gorin, J. P. Escande, R. A. Weiss, K. Alitalo, and C. Boshoff. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 96:4546-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dvir, A., S. R. Peterson, M. W. Knuth, H. Lu, and W. S. Dynan. 1992. Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc. Natl. Acad. Sci. USA 89:11920-11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flore, O., S. Rafii, S. Ely, J. J. O'Leary, E. M. Hyjek, and E. Cesarman. 1998. Transformation of primary human endothelial cells by Kaposi's sarcoma-associated herpesvirus. Nature 394:588-592. [DOI] [PubMed] [Google Scholar]
- 23.Friborg, J., Jr., W. Kong, M. O. Hottiger, and G. J. Nabel. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889-894. [DOI] [PubMed] [Google Scholar]
- 24.Fujimuro, M., F. Y. Wu, C. ApRhys, H. Kajumbula, D. B. Young, G. S. Hayward, and S. D. Hayward. 2003. A novel viral mechanism for dysregulation of beta-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat. Med. 9:300-306. [DOI] [PubMed] [Google Scholar]
- 25.Garber, A. C., J. Hu, and R. Renne. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 277:27401-27411. [DOI] [PubMed] [Google Scholar]
- 26.Godfrey, A., J. Anderson, A. Papanastasiou, Y. Takeuchi, and C. Boshoff. 2005. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 105:2510-2518. [DOI] [PubMed] [Google Scholar]
- 27.Gottlieb, T. M., and S. P. Jackson. 1993. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 72:131-142. [DOI] [PubMed] [Google Scholar]
- 28.Grundhoff, A., and D. Ganem. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 77:2779-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansen, R. S., T. K. Canfield, M. M. Lamb, S. M. Gartler, and C. D. Laird. 1993. Association of fragile X syndrome with delayed replication of the FMR1 gene. Cell 73:1403-1409. [DOI] [PubMed] [Google Scholar]
- 30.Hartley, K. O., D. Gell, G. C. Smith, H. Zhang, N. Divecha, M. A. Connelly, A. Admon, S. P. Lees-Miller, C. W. Anderson, and S. P. Jackson. 1995. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 82:849-856. [DOI] [PubMed] [Google Scholar]
- 31.Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. [DOI] [PubMed] [Google Scholar]
- 32.Hu, J., A. C. Garber, and R. Renne. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 76:11677-11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu, J., and R. Renne. 2005. Characterization of the minimal replicator of Kaposi's sarcoma-associated herpesvirus latent origin. J. Virol. 79:2637-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kedes, D. H., M. Lagunoff, R. Renne, and D. Ganem. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi's sarcoma-associated herpesvirus. J. Clin. Investig. 100:2606-2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kedes, D. H., E. Operskalski, M. Busch, R. Kohn, J. Flood, and D. Ganem. 1996. The seroepidemiology of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 2:918-924. [DOI] [PubMed] [Google Scholar]
- 36.Kellam, P., C. Boshoff, D. Whitby, S. Matthews, R. A. Weiss, and S. J. Talbot. 1997. Identification of a major latent nuclear antigen, LNA-1, in the human herpesvirus 8 genome. J. Hum. Virol. 1:19-29. [PubMed] [Google Scholar]
- 37.Knight, J. S., M. A. Cotter II, and E. S. Robertson. 2001. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus transactivates the telomerase reverse transcriptase promoter. J. Biol. Chem. 276:22971-22978. [DOI] [PubMed] [Google Scholar]
- 38.Komatsu, T., M. E. Ballestas, A. J. Barbera, B. Kelley-Clarke, and K. M. Kaye. 2004. KSHV LANA1 binds DNA as an oligomer and residues N-terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 319:225-236. [DOI] [PubMed] [Google Scholar]
- 39.Krishnan, H. H., P. P. Naranatt, M. S. Smith, L. Zeng, C. Bloomer, and B. Chandran. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krithivas, A., M. Fujimuro, M. Weidner, D. B. Young, and S. D. Hayward. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76:11596-11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krokan, H. E., F. Drablos, and G. Slupphaug. 2002. Uracil in DNA—occurrence, consequences and repair. Oncogene 21:8935-8948. [DOI] [PubMed] [Google Scholar]
- 42.Krokan, H. E., M. Otterlei, H. Nilsen, B. Kavli, F. Skorpen, S. Andersen, C. Skjelbred, M. Akbari, P. A. Aas, and G. Slupphaug. 2001. Properties and functions of human uracil-DNA glycosylase from the UNG gene. Prog. Nucleic Acid Res. Mol. Biol. 68:365-386. [DOI] [PubMed] [Google Scholar]
- 43.Krokan, H. E., R. Standal, and G. Slupphaug. 1997. DNA glycosylases in the base excision repair of DNA. Biochem. J. 325(Pt. 1):1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lagunoff, M., and D. Ganem. 1997. The structure and coding organization of the genomic termini of Kaposi's sarcoma-associated herpesvirus. Virology 236:147-154. [DOI] [PubMed] [Google Scholar]
- 45.Lan, K., D. A. Kuppers, S. C. Verma, N. Sharma, M. Murakami, and E. S. Robertson. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J. Virol. 79:7453-7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lennette, E. T., D. J. Blackbourn, and J. A. Levy. 1996. Antibodies to human herpesvirus type 8 in the general population and in Kaposi's sarcoma patients. Lancet 348:858-861. [DOI] [PubMed] [Google Scholar]
- 47.Lim, C., H. Sohn, D. Lee, Y. Gwack, and J. Choe. 2002. Functional dissection of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J. Virol. 76:10320-10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Millns, A. K., M. S. Carpenter, and A. M. DeLange. 1994. The vaccinia virus-encoded uracil DNA glycosylase has an essential role in viral DNA replication. Virology 198:504-513. [DOI] [PubMed] [Google Scholar]
- 49.Nilsen, H., M. Otterlei, T. Haug, K. Solum, T. A. Nagelhus, F. Skorpen, and H. E. Krokan. 1997. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 25:750-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Odegaard, E., C. R. Yang, and D. A. Boothman. 1998. DNA-dependent protein kinase does not play a role in adaptive survival responses to ionizing radiation. Environ. Health Perspect. 106(Suppl. 1):301-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Otterlei, M., E. Warbrick, T. A. Nagelhus, T. Haug, G. Slupphaug, M. Akbari, P. A. Aas, K. Steinsbekk, O. Bakke, and H. E. Krokan. 1999. Post-replicative base excision repair in replication foci. EMBO J. 18:3834-3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan, H., F. Zhou, and S. J. Gao. 2004. Kaposi's sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 64:4064-4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perkins, D. N., D. J. Pappin, D. M. Creasy, and J. S. Cottrell. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551-3567. [DOI] [PubMed] [Google Scholar]
- 54.Piolot, T., M. Tramier, M. Coppey, J. C. Nicolas, and V. Marechal. 2001. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J. Virol. 75:3948-3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prichard, M. N., G. M. Duke, and E. S. Mocarski. 1996. Human cytomegalovirus uracil DNA glycosylase is required for the normal temporal regulation of both DNA synthesis and viral replication. J. Virol. 70:3018-3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Priet, S., N. Gros, J. M. Navarro, J. Boretto, B. Canard, G. Querat, and J. Sire. 2005. HIV-1-associated uracil DNA glycosylase activity controls dUTP misincorporation in viral DNA and is essential to the HIV-1 life cycle. Mol. Cell 17:479-490. [DOI] [PubMed] [Google Scholar]
- 57.Pyles, R. B., N. M. Sawtell, and R. L. Thompson. 1992. Herpes simplex virus type 1 dUTPase mutants are attenuated for neurovirulence, neuroinvasiveness, and reactivation from latency. J. Virol. 66:6706-6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pyles, R. B., and R. L. Thompson. 1994. Evidence that the herpes simplex virus type 1 uracil DNA glycosylase is required for efficient viral replication and latency in the murine nervous system. J. Virol. 68:4963-4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pyles, R. B., and R. L. Thompson. 1994. Mutations in accessory DNA replicating functions alter the relative mutation frequency of herpes simplex virus type 1 strains in cultured murine cells. J. Virol. 68:4514-4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Radkov, S. A., P. Kellam, and C. Boshoff. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6:1121-1127. [DOI] [PubMed] [Google Scholar]
- 61.Rainbow, L., G. M. Platt, G. R. Simpson, R. Sarid, S. J. Gao, H. Stoiber, C. S. Herrington, P. S. Moore, and T. F. Schulz. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J. Virol. 71:5915-5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Russo, J. J., R. A. Bohenzky, M. C. Chien, J. Chen, M. Yan, D. Maddalena, J. P. Parry, D. Peruzzi, I. S. Edelman, Y. Chang, and P. S. Moore. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 93:14862-14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shinohara, H., M. Fukushi, M. Higuchi, M. Oie, O. Hoshi, T. Ushiki, J. Hayashi, and M. Fujii. 2002. Chromosome binding site of latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus is essential for persistent episome maintenance and is functionally replaced by histone H1. J. Virol. 76:12917-12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Si, H., and E. S. Robertson. 2006. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 80:697-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soulier, J., L. Grollet, E. Oksenhendler, P. Cacoub, D. Cazals-Hatem, P. Babinet, M. F. d'Agay, J. P. Clauvel, M. Raphael, L. Degos, and F. Sigaux. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276-1280. [PubMed] [Google Scholar]
- 66.Staskus, K. A., W. Zhong, K. Gebhard, B. Herndier, H. Wang, R. Renne, J. Beneke, J. Pudney, D. J. Anderson, D. Ganem, and A. T. Haase. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stedman, W., Z. Deng, F. Lu, and P. M. Lieberman. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J. Virol. 78:12566-12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stuart, D. T., C. Upton, M. A. Higman, E. G. Niles, and G. McFadden. 1993. A poxvirus-encoded uracil DNA glycosylase is essential for virus viability. J. Virol. 67:2503-2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Verma, S. C., S. Borah, and E. S. Robertson. 2004. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 78:10348-10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verma, S. C., T. Choudhuri, R. Kaul, and E. S. Robertson. 2006. Latency-associated nuclear antigen (LANA) of Kaposi's sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 80:2243-2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verma, S. C., K. Lan, T. Choudhuri, and E. S. Robertson. 2006. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen modulates K1 expression through its cis-acting elements within the terminal repeats. J. Virol. 80:3445-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verma, S. C., and E. S. Robertson. 2003. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol. Lett. 222:155-163. [DOI] [PubMed] [Google Scholar]
- 74.Ye, F. C., F. C. Zhou, S. M. Yoo, J. P. Xie, P. J. Browning, and S. J. Gao. 2004. Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J. Virol. 78:11121-11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhong, W., H. Wang, B. Herndier, and D. Ganem. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 93:6641-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]