

Abstract
PMR1 is an endonuclease that is activated by estrogen to degrade Xenopus albumin mRNA. A previous report showed that the functional unit of endonuclease-mediated mRNA decay is a ~680-kDa polysome-bound complex that contains both PMR1 and substrate mRNA. PMR1 contains two domains involved in endonuclease targeting to polysomes, an N-terminal domain that lies between residues 200 and 250, and a C-terminal domain that lies within the last 100 residues. Loss of either domain inactivated PMR1 targeting to polysomes and stabilized albumin mRNA. The current study identified a phosphorylated tyrosine residue within the C-terminal polysome-targeting domain and showed that this modification is required for PMR1-mediated mRNA decay. Changing this tyrosine to phenylalanine inactivated the targeting of PMR1 to polysomes, blocked binding of PMR1 to the functional complex containing its substrate mRNA, prevented the targeting of a green fluorescent protein fusion protein to this complex, and stabilized albumin mRNA to degradation by PMR1 in vivo. A general tyrosine kinase inhibitor inhibited the phosphorylation of PMR1, which in turn inhibited PMR1-catalyzed degradation of albumin mRNA. These results indicate that one or more tyrosine kinases functions as a regulator of endonuclease-mediated mRNA decay.
mRNA decay plays key role in regulating eukaryotic gene expression, and cells possess a number of distinct enzymatic pathways to both initiate mRNA decay and catalyze the degradation of the mRNA body. For many unstable mRNAs decay begins with shortening of the poly(A) tail, usually to <30 nucleotides (1). This disrupts the structure of the translating mRNP,1 and mRNA targeted for degradation becomes associated with a different set of proteins than those of the actively translating complex (2). In both yeast and mammalian cells, proteins involved in decapping and 5′-3′ mRNA degradation are concentrated in discrete cytoplasmic foci, termed P-bodies (3), or GW bodies (4) for the presence of a protein rich in glycine and tryptophan residues. Deadenylated yeast mRNAs relocate from actively translating polysomes to these foci before degradation of the mRNA body. The same biochemical process has yet to be shown for mammalian cells, but the similarities between the constituents of these complexes suggest this will also be the case. Exosome-catalyzed 3′-5′decay is the other principal pathway seen in yeast (5). Although earlier work suggested that this process was secondary to 5′-3′decay, the finding that a subset of mRNAs are up-regulated in cells deleted for Xrn1 or Dcp1 raises the possibility that exosome-catalyzed 3′-5′decay may play a greater role in yeast mRNA decay than previously appreciated (6). It is generally thought that most vertebrate mRNAs are degraded by the exosome in a 3′-5′manner; however, data supporting this come primarily from in vitro experiments (7) and a convincing in vivo demonstration yet to be presented.
In addition to exonuclease-mediated mRNA decay pathways there are numerous examples of endonuclease-mediated mRNA decay in vertebrate cells. Examples include avian apo-very low density lipoprotein II mRNA (8), vitellogenin (9), transferrin receptor mRNA (10), β-globin mRNA (11, 12), insulin-like growth factor II mRNA (13), and Xenopus serum albumin mRNA (14). On a functional level endonuclease cleavage within the mRNA body has the same effect as deadenylation in that it disrupts the physical relationship between the cap and the poly(A) tail. The best characterized mRNA endonuclease is polysomal ribonuclease 1, or PMR1. PMR1 was identified in Xenopus liver as a polysome-associated endonuclease whose activation after estrogen stimulation resulted in the selective degradation of serum protein mRNAs (15, 16). Unexpectedly, this protein is a member of the peroxidase gene family (17). Previous work showed that liver PMR1 is associated with polysomes and was released by EDTA as part of a ~680-kDa complex that could be recovered on oligo(dT) cellulose (18). In this tissue albumin mRNA decay results from estrogen stimulating a 20-fold increase in unit activity of the polysome-bound enzyme.
These data indicated that two critical events are involved in estrogen regulation of endonuclease-mediated mRNA decay. The first is the targeting of the endonuclease to polysomes, and the second is the activation of the polysome-bound enzyme. To characterize the targeting of PMR1 to polysomes we developed a form of the full-length protein that does not degrade substrate mRNA but shows the same sedimentation pattern in transfected Cos-1 cells as seen for PMR1 in Xenopus liver (19). Treating transfected cells with puromycin or adding EDTA to cell extracts released PMR1 in a ~680-kDa complex, and deletion mapping experiments identified 2 domains that mediate its targeting both to polysomes and to the ~680-kDa complex. The ~680-kDa complex also contains albumin mRNA, and deletions that disrupted the targeting of PMR1 to polysomes or to this complex interfered with the recovery of albumin mRNA by PMR1 bearing a C-terminal tandem affinity (TAP) tag. Intriguingly, PMR1-TAP selectively recovered its substrate albumin mRNA on IgG-Sepharose but did not recover luciferase mRNA. Experiments with the catalytically active form of the enzyme showed that PMR1 selectively degraded albumin but not luciferase mRNA. Albumin mRNA was stabilized if its translation was blocked by inserting a stable stem-loop into the 5′-untranslated region, indicating that it must be actively translated to be targeted for degradation by PMR1.
PMR1 has 2 domains that function in its targeting to polysomes (19). One domain lies between the first 200 and 250 amino acids, and a second domain lies somewhere between 50 and 100 amino acids upstream from the C terminus. Identifying the precise location of the C-terminal targeting domain proved problematic. Loss of the C-terminal 50 amino acids (Δ50C) produced a protein that could no longer bind to poly-somes or form an mRNP complex with albumin mRNA. However, this sequence was not sufficient to target a GFP fusion protein to either polysomes or the mRNP complex. This required an additional upstream 50 amino acids, a result suggesting that the 50-amino acid deletion disrupted the C-terminal targeting domain. A scan of sequence motifs identified a possible tyrosine phosphorylation site at position 650, 19 amino acids upstream of the Δ50C deletion site. Data presented here show that PMR1 is tyrosine-phosphorylated both in Xenopus liver and in transfected Cos-1 cells. Furthermore, tyrosine phosphorylation of PMR1 is required for its targeting to polysomes, for formation of the ~680-kDa polysome-bound complex with substrate mRNA, and for endonuclease-mediated mRNA decay.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
The preparation of plasmids expressing catalytically active PMR1, catalytically inactive PMR1, and C-terminal deletions with or without a TAP tag on the C terminus were described previously (19). The nomenclature used both there and in the present paper reflects the size of the mature PMR1 peptide, with PMR60 corresponding to the full-length, catalytically active form of the protein, and PMR60o corresponding to a catalytically inactive form of the protein having histidine residues at 393 and 479 changed to alanine. Deletions that remove 50, 100, or 150 amino acids from the C terminus are indicated as Δ50C, Δ100C, and Δ150C, respectively. All of the constructs have an Myc epitope tag on the N terminus and were detected using a monoclonal antibody to this tag. The mutations Y649F, Y650F, and the double mutant Y649,650F were generated by site-directed mutagenesis of the tyrosines at positions 649 or 650 using the GeneEditor mutagenesis kit (Promega). The preparation of Y649F PMR1 used the primer 5′-GACCGGTTTTTCTATGAGCAG, the preparation of Y650F PMR1 used the primer 5′-CGGTTTTACTTTGAG-CAGCCT, and the preparation of Y649,650F PMR1 used the primer 5′-GGAGACCGGTTTTTCTTTGAGCAGCCTTCAG. The Y650D mutation was generated by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene) using the forward primer 5′-GACCGGTTTTACGATGAGCAGCC and the reverse primer 5′-GGCTGCTCATCGTAAAACCGGTC. To prepare a tetracycline-regulated plasmid expressing PMR60, the sequence encoding the full-length protein was PCR-amplified from the pcDNA3-containing plasmid using the primers 5′-TGACAAGCTTCCGCCATTACAGGACAGTGC and 5′-GATCGCGGCCGCTTAAGCCACTTTCCAAGGAT, which contain restriction sites for NotI and HindIII, respectively. The amplified product was then digested with HindIII and NotI and ligated into the corresponding sites in pTRE-myc (Clontech). The sequences of all plasmids were confirmed before use in transfection experiments.
Cell Culture and Transient Transfection
Cos-1 cells were cultured in Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum and 2 mM glutamine. For most experiments 2.5 × 106 cells in log phase growth were transfected with 10 μg (total) of plasmid DNA by using LipofectAMINE (Invitrogen) following the manufacturer’s protocol. Unless otherwise indicated cells were collected 40 h after transfection. In experiments using puromycin to disrupt ribosomes, 200 μg/ml puromycin (Sigma) was added to the medium for 30 min before harvest. Cos-1 (tTA) cells were prepared by stable transfection of Cos-1 cells with pCMVtetRVP16-hygro (kindly provided by Jose Garcia-Sanz) and selection in hygromycin-containing medium. For the experiment in Fig. 8A, 1.6 ×106 cells were first plated in a 100-mm dish in medium containing 1 μg/ml doxycycline. Twenty-four hours later they were transfected in doxycycline-containing medium with 10 μg of pTRE-myc-PMR60-TAP and kept for an additional 15 h in doxycycline-containing medium. Expression of myc-PMR60-TAP was induced by removing doxycycline from the medium and washing the cells twice with phosphate-buffered saline. AG9 or AG18 (100 μM) were added at the time of induction, and cells were harvested 3 and 6 h later for analysis of tyrosine-phosphorylated PMR60 recovered on IgG-Sepharose. The experiment in Fig. 7B used the same approach except that cells were transfected with 8 μg of pTRE-myc-PMR60 or empty vector plus 1 μg each of the albumin and luciferase expression plasmids. Doxycycline was removed from the medium 15 h later, and the cells were washed twice with phosphate-buffered saline and then incubated for 6 h in fresh medium containing either no added drug or 100 μM AG9 or AG18. Total RNA recovered from each culture was analyzed as described below by ribonuclease protection assay for albumin and luciferase mRNA.
Fig. 8. Tyrosine kinase inhibitor stabilizes albumin mRNA to degradation by PMR1.
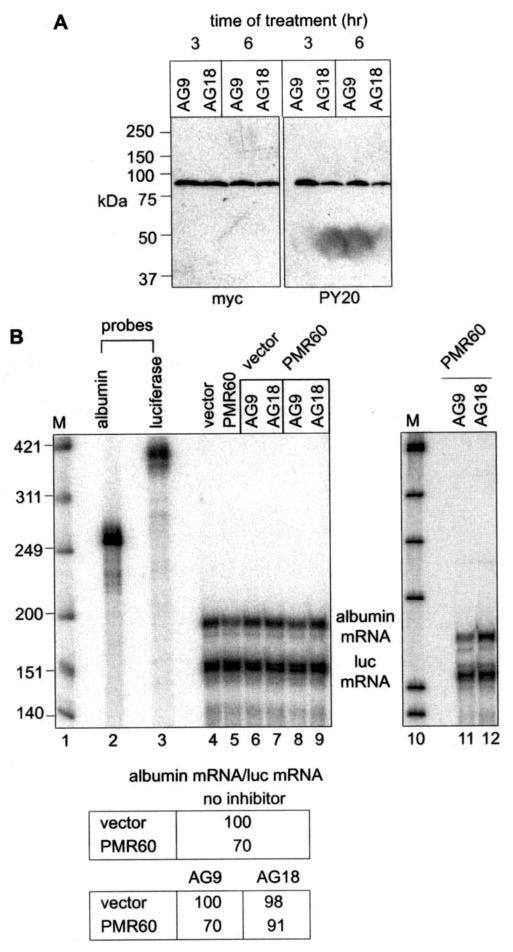
A, PMR60o-TAP was cloned into the tetracy-cline-regulated plasmid pTRE-myc and transfected into Cos-1 (tTA) cells in medium containing doxycycline. Fifteen hours later doxycycline was removed, and AG9 or AG18 were added to the medium. Cytoplasmic extracts prepared after 3 and 6 h were bound to IgG-Sepharose, eluted with SDS sample buffer, and analyzed by Western blot with antibody to the Myc tag or PY20. B, Cos-1 (tTA) cells were transfected in doxycycline-containing medium with plasmids expressing albumin and luciferase mRNA and either pTRE-myc (vector) or pTRE-PMR60. Doxycycline was removed 15 h later, and cells were incubated for 6 h with no additions (lanes 4 and 5), with AG9 (lanes 6, 8, and 11), or with AG18 (lanes 7, 9, and 12). The recovered RNA was analyzed by RNase protection assay, and albumin mRNA normalized to luciferase mRNA is presented in the table. The right panel (lanes 11 and 12) is an independent repeat of the data in lanes 8 and 9. M, Mr standards.
Fig. 7. The Y650F mutation stabilizes albumin mRNA to degradation by PMR1.
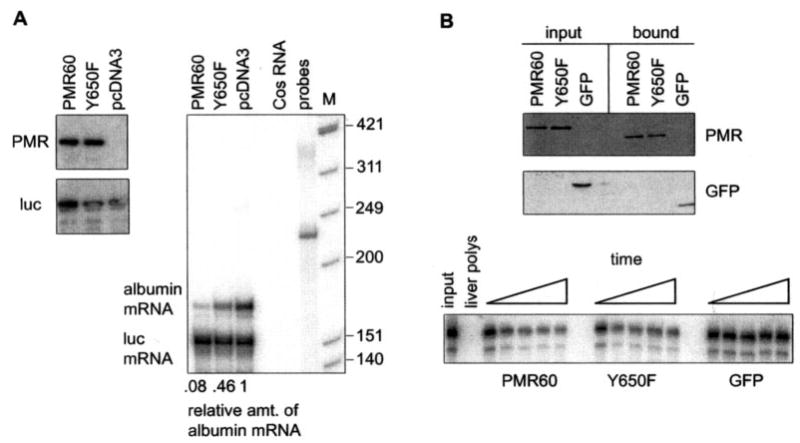
A, Cos-1 cells were transfected with pcDNA3, pcDNA expressing catalytically active PMR60, or pcDNA3 expressing PMR60 with the Y650F mutation together with plasmids expressing albumin and luciferase mRNA. The relative expression each protein was determined by Western blot (left panel), and their impact on steady-state levels of albumin and luciferase mRNA was determined by RNase protection (right panel). The relative amount of albumin mRNA normalized to luciferase mRNA determined by phosphorimaging analysis is shown beneath the autoradiogram. B, the catalytic activity of PMR60 and PMR60 with the Y650F mutation was determined by assaying the degradation of 5′32P-labeled albumin mRNA by PMR60-TAP, PMR60-TAP with the Y650F mutation, or GFP-TAP that were expressed in transfected cells and recovered on IgG-Sepharose. The upper panel is a Western blot of input protein and protein recovered from IgG-Sepharose Tev protease cleavage, and the lower panel is a time course of 0, 5, 10, 20, and 30 min of incubation with substrate RNA. M, Mr standards.
Preparation of Cell Extracts, Polysome Profile Analysis, and Glycerol Gradients
The preparation and analysis of cell extracts followed protocols used in our previous study (19). Cells were washed twice with ice-cold phosphate-buffered saline, then suspended in cell lysis buffer (10 mM HEPES-KOH, pH 7.5, 10 mM KCl, 5 mM MgCl2, 50 mM NaF, 0.5% Nonidet P-40 (v/v), 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 25 μl/ml protease inhibitor mixture (Sigma), 10 μl/ml phosphatase inhibitor (Sigma), and 10 μl/ml RNaseOUT (Invitrogen)). After incubation for 15 min on ice, the cells were homogenized with 30 strokes of a Dounce homogenizer (A pestle), and the homogenate was centrifuged for 10 min at 15,000 μg. To disassociate polysomes, post-mitochondrial supernatant was adjusted to 50 mM EDTA. Linear gradients of 10–40% (w/v) sucrose were prepared in 10 mM HEPES-KOH, pH 7.5, 2 mM dithiothreitol containing either 5 mM MgCl2 or 10 mM EDTA. Post-mitochondrial extracts were gently layered on top of the gradient followed by centrifugation for 3.5 h at 228,000 μg in a Sorvall TH641 rotor. 0.5-ml fractions were collected from the bottom of the tube. Linear gradients of 10–40% (v/v) glycerol were prepared in buffer containing 10 mM HEPES-KOH, pH 7.5, 5 mM MgCl2, and 2 mM dithiothreitol. Post-mitochondrial extracts were separated by centrifugation for 20 h at 83,000 μg in a Sorvall TH641 rotor. Molecular size markers containing a mixture of thyroglobulin (Mr 669,000), ferritin (Mr 440,000), catalase (Mr 232,000), lactate dehydrogenase (Mr 140,000),and bovine serum albumin (Mr 67,000) were fractionated on a parallel gradient. Fractions containing GFP and fusion proteins were transferred to 96-well plate and measured with a Tecan GENios fluorescence plate reader at 470 nm.
Antibodies
Antibodies to the c-Myc epitope tag (9E10), GFP (B2), and ribosomal protein S6 (E13) were purchased from Santa Cruz Biotechnology. The antibodies to PMR1 were described previously (14, 17). Horseradish peroxidase-coupled rabbit anti-mouse IgG and mouse anti-rabbit IgG were purchased from Santa Cruz. The phosphotyrosine monoclonal antibody 4G10 was purchased from Upstate Biotechnology, and PY20 and RC20:biotin were purchased from BD Biosciences. Western blots were quantified by scanning densitometry of x-ray films using a calibrated scanner and ImageQuant™ software (Amersham Biosciences).
Immunoprecipitation of Xenopus Liver PMR1
Male Xenopus were injected 9 h before death with 1 mg of 17β-estradiol. Polysomes isolated from the excised livers were extracted with 0.4 M KCl to obtain a crude preparation of PMR1-containing protein (20). One mg of dialyzed extract was incubated with protein A-Sepharose to which was bound a polyclonal antibody to Xenopus PMR1 (14) or 4G10. After washing with radioimmune precipitation assay buffer, bound protein was eluted with SDS sample buffer and separated by SDS-PAGE. Protein recovered with PMR1 antibody was analyzed by Western blot using the phosphotyrosine monoclonal antibodies PY20 or RC20 or the epitope-specific PMR1 antibody 1277 (17). Protein recovered using 4G10 was analyzed by Western blot using antibody 1277. Horseradish peroxidase-coupled rabbit anti-mouse IgG was used as the secondary antibody for PY20, horseradish peroxidase-coupled mouse anti-rabbit IgG was used as the secondary antibody for PMR1 antibody 1277, and horseradish peroxidase-coupled streptavidin was used to identify the biotin tag on RC20.
Immunoprecipitation of Tyrosine-phosphorylated PMR1
20 μg of cytoplasmic extract prepared from cells transfected with PMR60o-TAP was incubated with gentle rocking for 2 h at 4 °C with 5 μl of RC20: biotin (BD Biosciences) or 5 μl of non-immune IgG. This was added to 20 μl of drained SoftLink avidin resin (Promega) that was blocked by overnight incubation at 4 °C with 200 μl of Cos-1 cytoplasmic extract and washed 3 times with washing buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Nonidet P-40). Each mixture was incubated for 2 h at 4 °C, the resin was washed with washing buffer, and bound proteins were eluted with SDS sample buffer. Input, unbound, and bound fractions were separated by SDS-PAGE and analyzed by Western blot with PY20 or antibody to the Myc epitope. The resulting film was quantified by scanning densitometry and analyzed using ImageQuant™ software (Amersham Biosciences).
IgG-Sepharose Selection of PMR1-TAP Complexes
Cells were transiently transfected with 6 μg of vector (pcDNA3), plasmid myc-PMR60o-TAP, or myc-PMR60o-TAP with the Y650F mutation plus 2 μg each of plasmids expressing full-length albumin (21) and luciferase mRNA. 250 μl of post-mitochondrial extract in lysis buffer containing 10 mM EDTA was mixed with 50 μl of drained IgG-SepharoseTM 6 beads (Amersham Biosciences) that was pre-blocked with lysate from non-transfected cells. This was incubated by end-over-end rotation for 1 h at 4 °C. The beads were washed 3 times with 0.5 ml of washing buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Nonidet P-40) and once with Tev buffer (10 mM Tris- HCl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.1%Non-idet P-40, and 1 mM dithiothreitol) at 4 °C. Bound complexes were eluted by cleavage with 50 units of Tev protease (Invitrogen) in 100 μl of Tev buffer at 4 °C for 2 h. Protein and RNA released from the resin were extracted using Trizol reagent, PMR1 was analyzed by Western blot using the monoclonal antibody against the Myc epitope tag or PY20, and RNA samples were analyzed by semi-quantitative RT-PCR. For the assay of endonuclease activity, protein recovered from IgG-Sepharose by Tev cleavage was incubated with 250 fmol of a 5′32P-labeled 160-nucleotide transcript derived from the portion of albumin mRNA bearing the mapped PMR1 cleavage sites. The reaction mixtures were separated on a denaturing 6% polyacrylamide-urea gel and analyzed by phosphorimaging.
RT-PCR Analysis of Albumin and Luciferase mRNA
Albumin and luciferase mRNA recovered by IgG-Sepharose beads were analyzed by RT-PCR as described previously (19). Reverse transcription was performed with oligo(dT)15 as primer and Superscript II reverse transcriptase (Invitrogen). Albumin mRNA was amplified using 5′-CAAAG-ACCAGCCTTCAAACTC-3′ (sense) and 5′-CACAGTTGAATGCTCTAA-GCA-3′ (antisense). Luciferase mRNA was amplified using 5′-AGATC-CACAACCTTCGCTTC-3′ (sense) and 5′-CTGAGGAGCCTTCAGGAT-TAC-3′ (antisense). In each set the sense primer was 5′32P-labeled. PCR amplification was performed for 18 cycles of 95 °C for 45 s, 61 °C for 45 s, and 72 °C for 60 s, conditions that were empirically determined to lie in the linear range. Serial dilutions of albumin and luciferase mRNA were analyzed in parallel, the products from each reaction were separated on a denaturing 6% polyacrylamide-urea gel, and the results were quantified by phosphorimaging analysis.
Ribonuclease Protection Assay
2 × 106 cells were transfected as above with plasmids expressing albumin and luciferase mRNA and co-transfected with plasmids expressing catalytically active myc-PMR60 (12) or protein bearing the Y650F mutation. Total RNA was isolated with Trizol reagent. The antisense albumin riboprobe was synthesized by in vitro transcription (Ambion) using T7 promoter from a pcRII-Topo plasmid containing exons 14 and 15 of albumin cDNA. The antisense firefly luciferase riboprobe was synthesized with T3 promoter from a pBluescript(SK) plasmid containing the first 153 nucleotides of firefly luciferase cDNA. Ribonuclease protection assay was carried out with 5 μg of total RNA hybridized to 40 pg of each riboprobe using the ribonuclease protection assay III kit (Am-bion), and protected probe was separated on a denaturing 6% polyacrylamide-urea gel and quantified by phosphorimaging analysis.
RESULTS
PMR1 Is Tyrosine-phosphorylated in Xenopus Liver and Transfected Cos-1 Cells
Signature motifs within the sequence of PMR1 are shown schematically in Fig. 1A. PMR1 is translated as an 80-kDa precursor that is processed to the catalytically active ~60-kDa form (17). The large open box shows the region of similarity to the peroxidases, with the polysometargeting domains bracketed beneath. Notable in this analysis were three proline-rich domains identified as potential SH3 ligands and a single predicted site for tyrosine phosphorylation (black rectangle). The C-terminal targeting domain lies within the last 100 amino acids of PMR1, and previous results indicated this domain was interrupted by a deletion that removed the terminal 50 amino acids (Δ50C (19)). The location of the putative tyrosine phosphorylation site at position 650 was, therefore, consistent with a possible role in the C-terminal targeting domain of PMR1.
Fig. 1. Mapping the tyrosine phosphorylation site on PMR1.
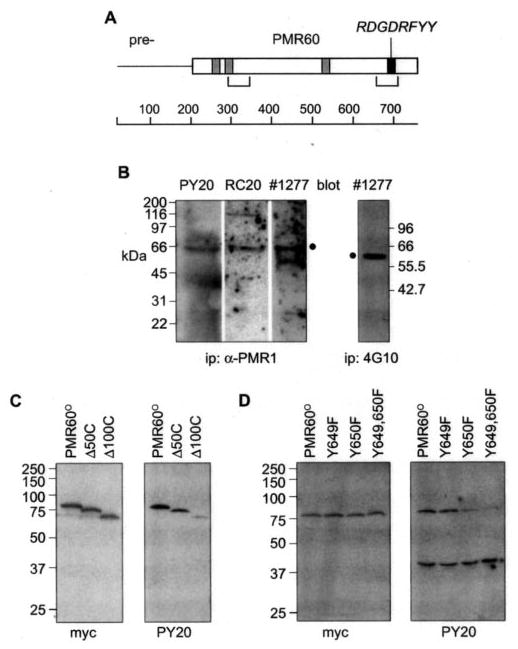
A, a schematic of the 80-kDa unprocessed PMR1 is shown with the mature 60-kDa form (PMR60) indicated, sequences with potential for binding to SH3 domains shown in gray boxes, a potential tyrosine kinase phospho-rylation site in the black box, and the locations of the two polysome targeting domains in brackets. B, a 0.4 M KCl extract of Xenopus liver polysomes was immunoprecipitated (ip) with a rabbit polyclonal antibody to PMR1 (three left panels) or with the phosphotyrosine mono-clonal antibody 4G10 (right panel), and recovered protein was separated by SDS-PAGE. Individual lanes of protein recovered with the PMR1 antibody were probed with the phosphotyrosine monoclonal antibodies PY20 or RC20:biotin or with a peptide-specific antibody to PMR1, 1277. The position of PMR1 is indicated with a filled circle (•). The lower band seen with the latter is IgG heavy chain. The blot of protein recovered with 4G10 was probed with the PMR1 antibody 1277. C, to map the general location of the tyrosine phosphorylation site Cos-1 cells were transfected with plasmids expressing TAP-tagged forms of the full-length protein (PMR60o) or deletions lacking 50 (~50C) or 100 (~100C) amino acids from the C terminus. Protein recovered on IgG-Sepharose was extracted with SDS sample buffer and analyzed by Western blot with antibody to the N-terminal Myc tag on PMR60oor PY20. D, the tyrosine residues at positions 649 (Y649F), 650 (Y650F), or both sites (Y649,650F) in PMR60o-TAP were mutated to phenylalanine, and the phosphorylation state of each protein expressed in transfected cells was determined as in C.
Before embarking on a study of PMR1 phosphorylation in transfected Cos-1 cells we first asked whether the endogenous protein in Xenopus liver is tyrosine-phosphorylated. In the experiment in Fig. 1BXenopus liver polysomes were extracted with 0.4 m KCl to solubilize polysome-bound proteins (14). In the three left panels the dialyzed extract recovered by immunoprecipitation with a polyclonal antibody to PMR1 was analyzed by Western blot with two phosphotyrosine-specific antibodies (PY20, RC20:biotin) or an epitope-specific antibody to PMR1 (1277 (17)). Each of these identified the 62-kDa PMR1 peptide. The same polysome extract was immunoprecipitated using the phosphotyrosine monoclonal antibody 4G10, and the Western blot of recovered proteins was probed with the PMR1 epitope-specific antibody 1277 (right panel). This too identified a single 62-kDa protein, indicating that PMR1 in Xenopus liver is tyrosine-phosphorylated. The sample evaluated here came from frogs that received estrogen 9 h before death, a time that just precedes the major drop in albumin mRNA (22). Similar results were seen with frogs that did not receive estrogen, suggesting that the hormone does not induce major changes in tyrosine phosphorylation of PMR1.
Previous work used sequential deletions of a catalytically inactive form of PMR1 (PMR60o) bearing a an N-terminal Myc epitope tag to map the general locations of N- and C-terminal domains involved in endonuclease targeting to polysomes (19). The use of proteins bearing a C-terminal tandem affinity (TAP) tag facilitated the recovery of PMR1-containing complexes on IgG-Sepharose. To test whether PMR1 expressed in Cos-1 cells is tyrosine-phosphorylated within the C-terminal 100 amino acids, Cos-1 cells were transfected with TAP fusions of the full-length protein (PMR60o) or deletions that removed 50 (Δ50C) or 100 (Δ100C) amino acids from the C terminus. Polysome-bound complexes were dissociated by treating cells with puromycin for 30 min before harvest, and the PMR1-TAP fusion proteins were recovered on IgG-Sepharose. Western blot with antibody to the Myc tag showed that all three forms of PMR1 were recovered with equal efficiency (Fig. 1C, left panel). In contrast, only full-length PMR60o and the Δ50C deletion showed appreciable tyrosine phosphorylation (Fig. 1C, right panel). Therefore, PMR1 is tyrosine-phosphorylated in Xenopus liver and in transfected Cos-1 cells, and as predicted, the tyrosine phosphorylation site lies within the last 100 amino acids.
The sequence RDGDRFYY located between amino acids 643 and 650 matches a consensus tyrosine phosphorylation sequence (R/K)XX(D/G)XXXY. The presence of adjacent ty-rosines within this site raised the possibility that PMR1 might be phosphorylated on either or both of these residues. To pinpoint the residue that was phosphorylated, each tyro-sine was changed to phenylalanine (Y649F, Y650F) or both were changed together (Y649,650F). TAP fusions of these proteins were transfected together with GFP-TAP into Cos-1 cells, and protein recovered on IgG-Sepharose was analyzed as in Fig. 1C. All four forms of PMR60o were expressed and recovered with equal efficiency (Fig. 1D, left panel) but only those forms that retained tyrosine at position 650 (Tyr-650) were phosphorylated (Fig. 1D, right panel). Interestingly, GFP was also tyrosine-phosphorylated, and the similar recovery of this in each sample served as an internal control for protein binding to IgG-Sepharose. These data map the phosphorylation site to the tyrosine at position 650 within the C-terminal targeting domain.
Most of the PMR1 Expressed in Cos-1 Cells Is Tyrosine-phosphorylated
To estimate the extent of PMR1 tyrosine phosphorylation, we compared the efficiency of recovery of total PMR1 versus tyrosine-phosphorylated PMR1 by immunoprecipitation with the phosphotyrosine-specific antibody RC20: biotin. In the experiment in Fig. 2 Cos-1 cells were transfected with PMR60o -TAP. Forty-eight hours later puromycin was added to dissociate polysome-bound complexes, and cytoplasmic extracts were incubated with RC20:biotin or nonimmune IgG before binding onto immobilized avidin. Bound proteins were eluted with SDS sample buffer, and input, unbound, and bound fractions were separated by SDS-PAGE. The blot in the left panel was probed with PY20 to determine the efficiency of recovering tyrosine-phosphorylated PMR60o -TAP with RC20: biotin. Tyrosine-phosphorylated proteins were only recovered from extract that received RC20:biotin, and PMR60o -TAP was the only o 75-kDa tyrosine-phosphorylated protein expressed in these cells (indicated with a diamond). Scanning densitometry of the PY20 blot showed that 67% of input tyrosine-phosphorylated PMR60o -TAP was recovered with RC20:biotin on the avidin resin. A blot of the same fractions probed with antibody to the Myc tag showed that 40% of the input PMR60o-TAP was recovered in the bound fraction. Normalizing this percent recovery to the 67% binding efficiency, one obtains a value of 60% for tyrosine-phosphorylated PMR60o -TAP. This probably represents a minimum value because some degree of dephosphorylation likely occurred in the course of this experiment.
Fig. 2. Estimating the population of PMR1 that is tyrosine-phosphorylated.
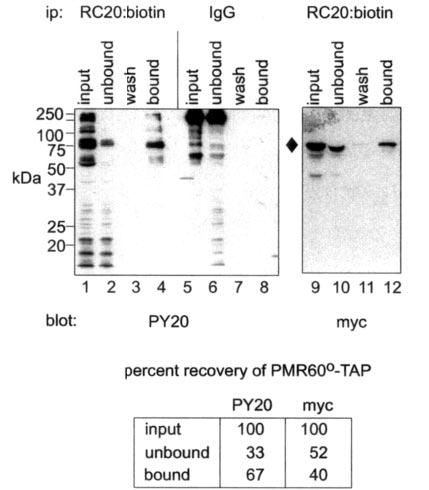
Cytoplasmic extracts from cells transfected with PMR60o -TAP were incubated with RC20:biotin or nonimmune IgG before binding to immobilized avidin. Input, unbound, wash and bound fractions eluted with SDS sample buffer were separated by SDS-PAGE and analyzed by Western blot with PY20 or antibody to the Myc tag on PMR60o -TAP. The blot in the left panel probed with PY20 (lanes 1–8) determined the specificity and efficiency for recovering tyrosine-phosphorylated protein, and the blot in the right panel probed with Myc antibody (lanes 9–12) determined the efficiency of recovering PMR60o -TAP. The data quantified by scanning densitometry are shown beneath the figure. The recovery of Myc staining protein on avidin resin was normalized to the efficiency of recovery for tyrosine-phosphorylated PMR60o -TAP determined with PY20. ip, immunoprecipitate.
Tyrosine-phosphorylated PMR1 Sediments with Polysomes and mRNP Complexes on Sucrose Gradients
When cytoplasmic extracts are fractionated on sucrose density gradients, PMR1 is localized to two complexes (18, 19). One is a polysome-bound complex that contains PMR1 together with its translating substrate mRNA (19). The second, more slowly sedimenting complex is similar in size to mRNPs. To examine the phosphorylation state of PMR1 in each of these complexes, Cos-1 cells were transfected with PMR60o -TAP, and cytoplasmic extracts were separated on a 10–40% sucrose gradient. Because Western blotting with PY20 would identify PMR1 and all other tyrosine-phosphorylated proteins, we instead used binding to IgG-Sepharose to selectively recover PMR60o -TAP in each fraction. Bound protein was eluted with SDS sample buffer and analyzed by Western blot using antibody to the Myc tag and PY20 (Fig. 3A). Both antibodies revealed the same distribution of protein across the gradient, with one population bound to polysomes and another sedimenting with mRNPs. This distribution matched that of PMR60o (19) and supports results in Fig. 2 showing that the majority of PMR1 expressed in Cos-1 cells is tyrosine-phosphorylated.
Fig. 3. Mutating Y650 blocks the targeting PMR1 to polysomes.
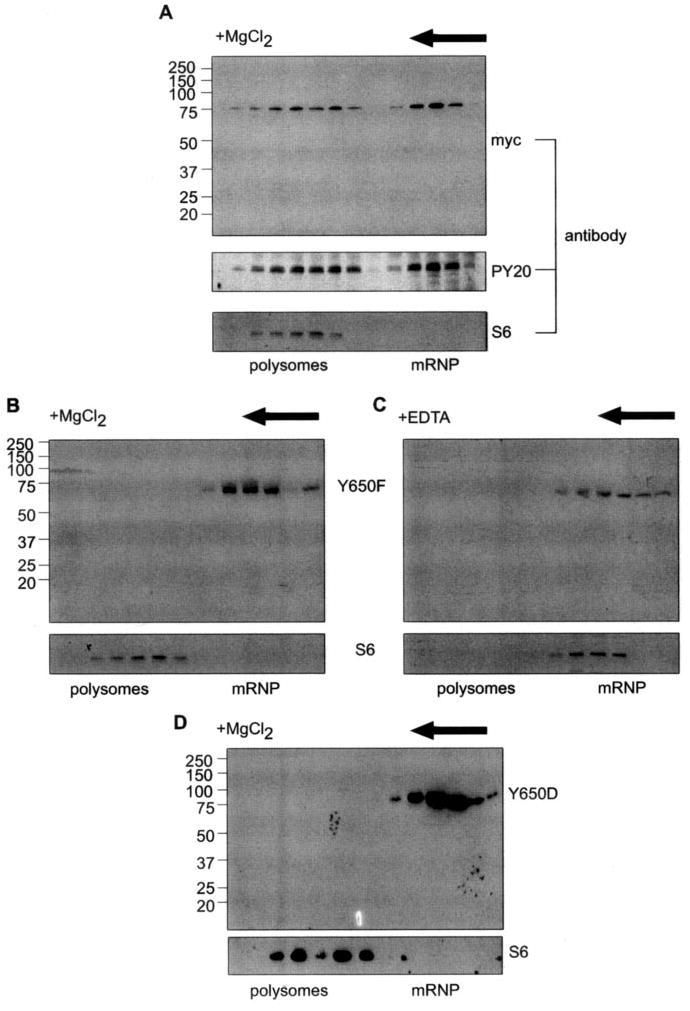
A, Cos-1 cells were transfected with PMR60o -TAP, and cytoplasmic extracts were separated on a 10–40% sucrose gradient that contained MgCl2 to maintain ribosome integrity. Even-numbered fractions were treated with EDTA to dissociate polysome-bound complexes, applied to IgG-Sepharose, and eluted with SDS sample buffer. The recovered proteins were analyzed by Western blot with antibody to the Myc tag or with PY20. The sedimentation of polysomes was determined by Western blot of unselected gradient fractions with a monoclonal antibody to ribosomal protein S6. B, Cos-1 cells with a plasmid expressing PMR60o with the Y650F mutation and fractionating cytoplasmic extract on a 10–40% sucrose gradient. Even-numbered fractions were analyzed by Western blot with antibody to the Myc tag or with antibody to S6 to map the sedimentation of polysomes on the gradient. C, the sample analyzed in B was sedimented on a gradient containing 5 mM EDTA, and even-numbered fractions were analyzed as in B with antibodies to the Myc tag or S6. D, cells were trans-fected with PMR60o, in which Tyr-650 was changed to aspartic acid (Y650D), and the sedimentation of PMR1 was analyzed on a gradient containing MgCl2 as in B.
Phosphorylation at Tyrosine 650 Is Required for Targeting PMR1 to Polysomes
The results in Fig. 3A suggest either that tyrosine phosphorylation is not required for targeting PMR1 to polysomes or that the smaller PMR1-containing complex is a precursor to the polysome-bound complex. If the first option is true, then inactivating the tyrosine phosphorylation site with the Y650F mutation should have no effect on the gradient distribution of PMR1. Alternatively, if tyrosine phosphorylation is required for targeting PMR1 to polysomes, protein with the Y650F mutation should sediment only in the smaller complex. The gradient profile in Fig. 3B shows that PMR60o with the Y650F mutation is restricted to the slowly sedimenting complex. Adding EDTA to dissociate polysomes had no impact on the sedimentation of (Fig. 3C), and the sedimentation profiles in Figs. 3, B and C, are identical to that of PMR1 released by EDTA dissociation of polysomes (19).
Because most of the PMR1 in the cell is tyrosine-phosphorylated, it was formally possible that the Y650F mutation disrupted polysome targeting by replacing a negatively charged residue with a hydrophobic residue. This was examined by substituting aspartic acid for tyrosine at position 650 (Y650D, Fig. 3D). As with the Y650F mutation, PMR60o with the Y650D mutation did not target to polysomes and was instead restricted to the mRNP complex like PMR60o with the Y650F mutation. Therefore, tyrosine phosphorylation, and not a negative charge at this position, is required for targeting PMR1 to polysomes.
The Y650F Mutation Blocks Formation of the Complex of PMR1 with Substrate mRNA
A central observation of our previous study was that PMR1 forms a selective complex with its translating, polysome-bound mRNA, and it is in this context that endonuclease cleavage initiates mRNA decay (19). A key piece of data supporting this was the observation that albumin mRNA could only be recovered on IgG-Sepharose by TAP-tagged forms of the protein that retained the ability to associate with polysomes on sucrose density gradients. The similarity between the results in Fig. 3 and those obtained with N- or C-terminal targeting domain deletions raised the possibility that tyrosine phosphorylation is required to form the complex of PMR1 with polysome-bound substrate mRNA. The first test of this hypothesis compared the recovery of albumin mRNA on IgG-Sepharose by PMR60o-TAP with PMR60o-TAP bearing the Y650F mutation. In the experiment in Fig. 4, Cos-1 cells were transfected with vectors expressing albumin mRNA, luciferase mRNA, and PMR60o-TAP, the Y650F mutant form of the protein, or PMR1 with the C-terminal targeting domain deleted (Δ150C-TAP). The Δ150C deletion was shown previously to inactivate PMR1 targeting to polysomes and block the recovery of albumin mRNA by PMR1 (19). Polysomes were dissociated by treating cells with puromycin for 30 min before harvest, and PMR1-containing complexes recovered on IgG-Sepharose were analyzed by Western blot for recovered protein and by RT-PCR for recovered mRNA. Somewhat more of the Y650F mutation and the Δ150C deletion forms of PMR60o were recovered from IgG-Sepharose than the full-length protein (left panel). Equal amounts of albumin and luciferase mRNA were expressed in each of the transfectants; however, albumin mRNA was only recovered with full-length PMR60o-TAP (right panel). The selective nature of this process was confirmed by the absence of luciferase mRNA recovered with any form of the protein. Together with the results in Fig. 3, these data indicate that the mutation that blocks tyrosine phosphorylation of PMR1 prevents incorporation of this protein into a complex with its substrate mRNA.
Fig. 4. The Y650F mutation prevents recovery of substrate mRNA by PMR1.
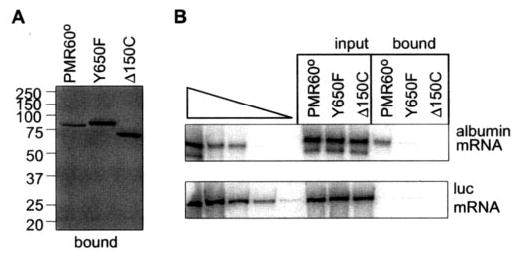
Full-length PMR60o -TAP, PMR60o-TAP with the Y650F mutation (Y650F), or PMR60o -TAP deleted of the C-terminal 150 amino acids (Δ150C) were transfected into Cos-1 cells together with plasmids expressing albumin and luciferase mRNA. Extracts from puromycin-treated cells were bound to IgG-Sepharose and eluted by cleavage with Tev protease. Protein recovered in the bound complexes was analyzed by Western blot with antibody to the Myc tag (left panel), and RNA extracted from the bound complexes was analyzed by quantitative RT-PCR and phosphorimaging analysis (19) (right panel).
Tyrosine Phosphorylation Is Required for PMR1 to Join the Functional ~680-kDa Polysome Complex with Its Substrate mRNA
The functional unit of endonuclease-mediated mRNA decay is a ~680-kDa RNP complex containing PMR1 and its polysome-bound substrate mRNA, and deletions that inactivate PMR1 targeting to polysomes also prevent its binding to this complex, block the recovery of albumin mRNA with PMR1, and stabilize albumin mRNA to degradation by PMR1 in vivo (19). Therefore, if tyrosine phosphorylation is required for PMR1-mediated mRNA decay, the Y650F mutation should block formation of the ~680-kDa complex. In the experiment in Fig. 5, A and B, PMR60o-TAP or PMR60o-TAP with the Y650F mutation were co-transfected with plasmids expressing albumin and luciferase mRNA, and cytoplasmic extracts prepared from puromycin-treated cells were separated on glycerol gradients. Western blots using antibody to the Myc tag showed that PMR60o-TAP with the Y650F mutation was restricted to the complex that sediments at ≤140 kDa, whereas wild-type PMR60o-TAP was present both in this and the ~680-kDa complex (Fig. 5A). For simplicity we term this complex I and the ≤140 kDa form complex II.
Fig. 5. The Y650F mutation blocks targeting of PMR1 to the functional ~680-kDa complex.
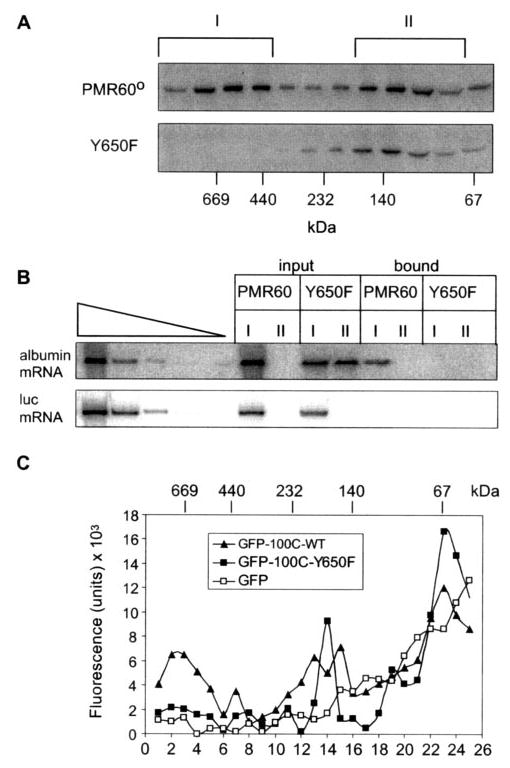
A, Cos-1 cells transfected with PMR60o or PMR60o with the Y650F mutation plus plasmids expressing albumin and luciferase mRNA were treated with puromycin before harvest, and cytoplasmic extracts were separated on 10–40% glycerol gradients. Even-numbered fractions were analyzed by Western blot with antibody to the Myc tag. B, the fractions identified with brackets in A were pooled and identified as complex I and complex II. Half of each pooled complex was bound to IgG-Sepharose and eluted by cleavage with Tev protease, and RNA extracted from the bound and unbound complexes was analyzed by quantitative RT-PCR as in Fig. 4. C, the C-terminal 100 amino acids of PMR1 bearing the native sequence or the Y650F mutation were fused to the C terminus of GFP. Cytoplasmic extracts from Cos-1 cells transfected with these or GFP alone were fractionated on a 10–40% glycerol gradient, and individual fractions were assayed for fluorescence. WT, wild type.
Gradient fractions corresponding to complex I and complex II were pooled as indicated in Fig. 5A and divided into two portions. RNA was extracted from one portion to determine the distribution of albumin mRNA and luciferase mRNA in the two complexes (Fig. 5B, input). The other portion was used to determine which of these complexes contained both PMR1 and albumin mRNA. This was accomplished by binding the pooled fractions to IgG-Sepharose and extracting RNA from material recovered by cleavage with Tev protease (Fig. 5B, bound). Albumin mRNA and luciferase mRNA in the input and bound fractions were then assayed by semiquantitative PCR. The input samples showed that albumin mRNA and luciferase mRNA were both present in complex I. For unknown reasons albumin mRNA was also in the smaller complex II in cells expressing PMR60o-TAP with the Y650F mutation but not in cells expressing wild-type PMR60o-TAP. A different picture emerged for RNA recovered on IgG-Sepharose. Here albumin mRNA present in complex I was only recovered with wild-type PMR60o-TAP, and no mRNA was recovered with the Y650F mutation. These data indicate that the inability of PMR60o-TAP with the Y650F mutation to target to polysomes in Fig. 3 or recover albumin mRNA in Fig. 4 resulted from a block in the formation of complex I containing PMR1 and its substrate mRNA.
The results described above point to tyrosine phosphorylation at position 650 as a critical modification required for targeting PMR1 to its polysome-bound substrate mRNA. However, PMR1 has two independent polysome targeting domains, and the above experiments were all performed with the full-length protein. Previous work showed that the C-terminal 100 amino acids of PMR1 are sufficient to target a GFP fusion protein both to polysomes and to the ~680-kDa complex I (19). The role of a phosphotyrosine at position 650 in this process was examined on glycerol gradients, which compared the sedimentation of GFP with a that of a fusion protein with the wild-type C-terminal 100-amino acids (GFP-100C-WT) or a fusion protein with the Y650F mutation (GFP-100C-Y650F). The results in Fig. 5C show that the Y650F mutation inactivated the targeting of the GFP fusion protein to complex I. These data point to phosphorylation of Tyr-650 as a critical determinant for the function of the C-terminal targeting domain in binding PMR1 to complex I.
Total and Tyrosine-phosphorylated PMR1 Have Overlapping but Not Identical Glycerol Gradient Sedimentation Profiles
A portion of the PMR1 expressed in transfected Cos-1 cells is not tyrosine-phosphorylated (Fig. 2), and results with the Y650F mutation suggest that unphosphorylated protein should behave similarly to that of protein that cannot be phosphorylated. To examine this possibility we compared the glycerol gradient distribution of total PMR60o versus tyro-sine-phosphorylated PMR60o. In the experiment in Fig. 6 cells expressing PMR60o-TAP were treated with puromycin before harvest to dissociate polysome-bound mRNPs, and PMR1-containing complexes recovered from IgG-Sepharose by cleavage with Tev were fractionated on a glycerol gradient. The total distribution of PMR60o was determined by Western blot with antibody to the Myc tag (upper panel, Myc), and the distribution of tyrosine-phosphorylated PMR1 was determined by Western blot with PY20 (lower panel). To facilitate this comparison the data were quantified by densi-tometry, and the percent distribution of Myc- and PY20-staining protein was plotted. Although there was good agreement between the signals for total and tyrosine-phosphorylated PMR60o in complex I, the lighter complex II showed an overlapping pattern, with the peak of tyrosine-phosphorylated PMR60o sedimenting slightly faster than the peak of total PMR1 protein. The trailing edge of Myc-staining protein in complex II is close in size to PMR60o with the calmodulin-binding protein tag that is left after Tev protease cleavage, raising the possibility that this represents monomeric protein.
Fig. 6. Total and tyrosine-phosphorylated PMR1 have overlapping but not identical sedimentation profiles.
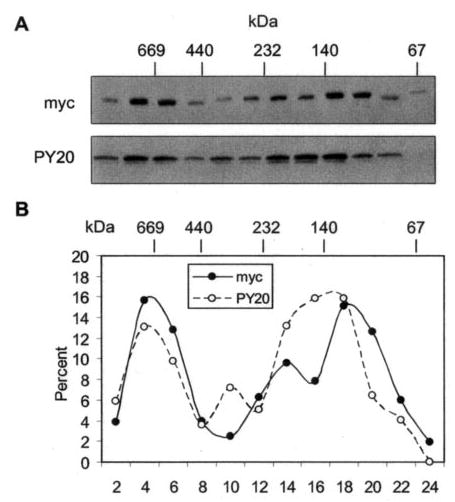
A, cytoplasmic extracts prepared from cells with PMR60o-TAP were bound to IgG-Sepharose, and complexes recovered by Tev protease cleavage were fractionated on a 10–40% glycerol gradient. Gradient fractions were analyzed by Western blot with antibody to the Myc tag to detect the total distribution of PMR1 and PY20 to detect the distribution of tyrosine-phosphorylated PMR1. B, the relative amount of protein determined with each antibody was determined by scanning densitometry and plotted as the percent present in each fraction of the gradient.
The Y650F Mutation Stabilizes Albumin mRNA to PMR1-mediated Degradation
A deletion that disrupted the C-terminal polysome targeting domain of PMR1 stabilized albumin mRNA to degradation by PMR1 (19). Because tyrosine phos-phorylation is required for targeting PMR1 to polysome-bound substrate mRNA, the Y650F mutation should have the same stabilizing effect as that deletion. To test this Cos-1 cells were co-transfected with empty vector (pcDNA3) or catalytically active forms of PMR60 or PMR60 with the Y650F mutation and plasmids expressing albumin and luciferase mRNA. The Western blot in Fig. 7A, left panel, shows that PMR60 and the Y650F mutant were equally expressed. Compared with vector control, PMR60 catalyzed a 12-fold decrease in the steady-state level of albumin mRNA without affecting the level of luciferase mRNA (Fig. 7A, right panel). The Y650F mutation stabilized albumin mRNA such that its level returned to ~50% of that seen with the vector control. This degree of stabilization is indistinguishable from that obtained when the C-terminal targeting domain was disrupted by deleting the last 50 amino acids (19).
To ensure that the Y650F mutation did not alter the catalytic activity of PMR1, we examined the ability of wild-type and mutant protein to degrade albumin mRNA in vitro. In the experiment in Fig. 7B cells were transfected with TAP fusions of PMR60, PMR60 with the Y650F mutation, or GFP. Cytoplasmic extracts from puromycin-treated cells were applied to IgG-Sepharose, and bound proteins were recovered by Tev protease cleavage. The Western blot in the upper panel of Fig. 7B shows that both forms of PMR60 were recovered with equal efficiency. A 5′32P-labeled albumin mRNA substrate transcript was degraded equally well by both proteins, whereas no ribonuclease activity was observed with GFP (Fig. 7B, lower panel). Thus, the Y650F mutation stabilized albumin mRNA by preventing tyrosine phosphorylation of PMR60 and consequent targeting of this endonuclease to its polysome-bound substrate mRNA.
Inhibiting Tyrosine Kinases Reduces the Phosphorylation of PMR1 and Stabilizes Albumin mRNA to Degradation by PMR1
The preceding results suggest that endonuclease-mediated mRNA decay may be regulated by alterations in the activity of one or more tyrosine kinases. To determine the feasibility of this, we examined the impact of tyrphostin AG18, a global tyrosine kinase inhibitor, on both tyrosine phosphorylation of PMR60 and the degradation of albumin mRNA. Initial experiments showed levels of the drug that exceeded those used here were toxic and inhibited the expression of PMR60. Therefore, for these experiments PMR60 was placed under tetracy-cline repressor control so that it could be induced when AG18 was added to the medium, and cells were treated for the shortest time that gave a reproducible response. To determine the impact of AG18 on phosphorylation of PMR1 Cos-1 (tTA) cells were transfected with a plasmid expressing PMR60-TAP in medium containing doxycycline. Fifteen hours later doxycy-cline was removed, and AG18 or the inactive congener AG9 was added to the medium. Cytoplasmic extracts isolated after 3 and 6 h of treatment were bound to IgG-Sepharose, and protein eluted with SDS sample buffer was analyzed by Western blot with antibody to the Myc tag or PY20 (Fig. 8A). Under these conditions AG18 had little effect on the overall expression of PMR60-TAP, but it reduced tyrosine phosphorylation of PMR60-TAP by 50%.
We next examined the impact of AG18 on PMR60 degradation of albumin mRNA. In this experiment cells were co-trans-fected with the cytomegalovirus-promoter-controlled plasmids expressing albumin and luciferase mRNA and either empty vector (pTRE-myc) or pTRE expressing PMR60. Cells were kept in doxycycline-containing medium for 15 h, during which time albumin and luciferase mRNA accumulated before induction of PMR60. PMR60 expression was induced for 6 h by removing doxycycline from the medium, and RNA from control, AG9, or AG18-treated cells was analyzed by RNase protection using the same probes as in Fig. 7A. We reasoned that newly induced PMR60 that targeted to polysomes would degrade a portion of the accumulated albumin mRNA, but the overall level of degradation would be less than that seen in Fig. 7A. Inhibiting tyrosine kinases with AG18 should inhibit the PMR60-catalyzed decrease in albumin mRNA but have no effect on albumin mRNA in cells transfected with empty vector.
In cells expressing PMR60 the steady-state level of albumin mRNA dropped to 70% that of the vector control vector (Fig. 8B, lanes 4 and 5), confirming our expectation that only a portion of the accumulated albumin mRNA would be degraded by the newly expressed endonuclease. Neither AG9 nor AG18 had any effect on the steady-state level of albumin mRNA in cells that were transfected with empty vector (lanes 6 and 7). In PMR60-expressing cells treated with AG9, the level of albumin mRNA again dropped to 70% of the vector control (compare lane 8 with lane 5); however, in cells treated with AG18 the level of albumin mRNA was 91% that of control (lane 9). A somewhat greater effect of AG18 versus AG9 was observed in an independent repeat of this experiment (lanes 10–12), where the level of albumin mRNA in AG18-treated cells was twice that in cells treated with AG9. These results are consistent with those in Fig. 7 using the Y650F mutant form of PMR1 that cannot be tyrosine-phosphorylated and demonstrate that changes in tyrosine phosphorylation modulate the effectiveness of endonuclease-mediated mRNA decay.
DISCUSSION
Endonuclease-mediated mRNA decay occurs in an ~680-kDa polysome-bound complex containing PMR1 and its translating substrate mRNA, and mRNA is stabilized by insertion of a secondary structure in the 5′-untranslated region that blocks translation of substrate mRNA or by deletions that inactivate targeting of PMR1 to this complex (19). Sequential 50-amino acid deletions of the N and C termini of PMR1 identified two targeting domains, and results presented here show that phosphorylation of Tyr-650 in the C-terminal targeting domain is required for endonuclease-mediated mRNA decay. Several lines of evidence support this conclusion. First, the targeting of PMR1 to polysomes was inactivated by mutations that changed tyrosine at position 650 to phenylalanine (Y650F). This is the result one might expect if tyrosine phosphorylation is required for targeting PMR1 to polysomes. Next, aspartic acid at position 650 (Y650D) did not substitute for tyrosine, so it is unlikely that a negative charge at this location was the primary determinant for polysome binding. Changing Tyr-650 to phenylala-nine inactivated the recovery of albumin mRNA by the TAP-labeled protein as effectively as deletion of the C-terminal targeting domain (Fig. 4). Because the formation of a complex containing PMR1 and substrate mRNA lies at the core of en-donuclease-mediated mRNA decay, the inability of PMR60o with the Y650F mutation to recover albumin mRNA indicates that this single amino acid alteration prevented the formation of this complex.
Glycerol gradient analysis played a key role in identifying the functional unit for endonuclease-mediated mRNA decay (18, 19). We term this ~680-kDa polysome-bound RNP complex I. Results in Fig. 5A showed that Y650F mutation restricted PMR60o to a complex (termed complex II) that sedimented at ≤140 kDa. In this experiment both wild-type PMR60o and protein with the Y650F mutation had a C-terminal TAP tag to facilitate the recovery of RNP complexes. Albumin mRNA was present in complex I, but it was not recovered on IgG-Sepha-rose because the Y650F mutation restricted PMR1 to the lighter complex II (Fig. 5B). These data provide the biochemical explanation for data in Fig. 4. The behavior of the parent protein extended to a GFP fusion bearing the C-terminal 100 amino acids of PMR1 (Fig. 5C). Whereas the wild-type sequence targeted GFP to complex I, protein bearing the Y650F mutation was restricted to the slowly sedimenting complexes.
Based on these results we hypothesized that the targeting of PMR1 to polysomes involves a stepwise process beginning with tyrosine phosphorylation that leads first to the formation of complex II (which sediments with the mRNP fraction on sucrose gradients) and then to the formation of polysome-bound complex I with its substrate mRNA. Support for this is seen in Fig. 6. In this experiment cells transfected with PMR60o-TAP were treated with puromycin to dissociate polysome-bound complexes. Cytoplasmic extracts were then bound to IgG-Sepharose, and material recovered by cleavage with Tev pro-tease was fractionated on a glycerol gradient. The recovered fractions were then analyzed by Western blot with antibody to the Myc tag to determine the distribution of total PMR60o protein, and PY20, to determine the distribution of tyrosine-phosphorylated PMR60o. This was similar for protein present in complex I but not for the more slowly sedimenting material. The graphical representation in Fig. 6B shows that a portion of PMR60o that is not tyrosine-phosphorylated trails that of the tyrosine-phosphorylated protein in complex II. Based on the results obtained with the Y650F mutation on sucrose and glycerol gradients, we interpret this to indicate that PMR1 joins complex II either at the time of tyrosine phosphorylation or thereafter, and this complex serves as the precursor for forming the ~680-kDa polysome-bound complex I.
The above data indicate that tyrosine phosphorylation activates the C-terminal polysome targeting domain, thus licensing the endonuclease to join the complex containing its translating substrate mRNA. We showed previously that expression of catalytically active PMR1 in transfected cells reduced the steady-state level of albumin mRNA 11-fold compared with cells transfected with vector alone (19). This was reversed when polysome targeting was inactivated by deleting the C-terminal 50 amino acids. This deletion had no effect on catalytic activity, yet in these cells the steady-state level of albumin mRNA returned to 50% that of the vector control. Because the targeting of PMR1 to polysomes is required for endonuclease-mediated mRNA decay, the Y650F mutation should stabilize albumin mRNA as effectively as the Δ50C deletion. The results in Fig. 7 show this to be the case. As in our earlier study, PMR60 catalyzed a 12-fold decrease in albumin mRNA without affecting the steady-state level of luciferase mRNA (Fig. 7A). The Y650F mutation had no impact on the catalytic activity of PMR1 (Fig. 7B) but nonetheless stabilized albumin mRNA to endonuclease-mediated degradation (Fig. 7A). In agreement with all of the preceding data, the degree of stabilization observed here was similar to that of the Δ50C mutation (19). The tyrosine at position 650 is both present and phosphorylated in the Δ50C deletion of PMR1 (Fig. 1C) but the Δ50C deletion removed additional downstream sequence that is needed for targeting to polysomes.2
The computer analysis that identified the tyrosine phosphorylation site on PMR1 suggested c-abl as a potential kinase for this sequence. However, RNA-mediated interference of c-abl by >90% had no effect on the tyrosine phosphorylation of PMR1 (data not shown). Therefore, the responsible kinase(s) remains to be identified. Although much of the PMR1 expressed in this model system is tyrosine-phosphorylated (Fig. 2), such may not be the case for all PMR1-expressing cells, and changes in PMR1 phosphorylation may constitute a key step in regulating mRNA decay. Support for this was seen in Fig. 8, where inhibiting tyrosine kinases with AG18 reduced the tyrosine phosphoryl-ation of PMR60 and stabilized albumin mRNA. In addition to confirming that tyrosine phosphorylation is the key step in licensing PMR1 to target and degrade its substrate mRNA, these data provide direct evidence linking the activity of one or more tyrosine kinases to endonuclease-mediated mRNA decay. Changes in the phosphorylation of RNA-binding proteins modulate the turnover of numerous unstable mRNAs (for example see Refs. 23–25), but to the best of our knowledge this is the first demonstration of tyrosine phosphorylation regulating the function of an effector nuclease involved in the catalysis of mRNA decay. They also suggest that inhibitors of tyrosine kinases that are under development for cancer and other diseases might have a previously unappreciated impact on the turnover of mRNAs that are targeted by endonuclease-mediated mRNA decay.
Acknowledgments
We thank Kris Cunningham, Mark Hanson, and Arnold DuBell for assistance with experiments on tyrosine phosphoryl-ation of PMR1 in Xenopus liver, Jose Garci-Sanz for the tetracycline repressor expression plasmid used to prepare Cos-1 (tTA) cells, and members of the Schoenberg lab for helpful comments.
Footnotes
This work was supported by National Institutes of Health Grant GM38277 (to D. R. S.). Support for core facilities was provided by National Cancer Institute Grant P30 CA16058 to the Ohio State University Comprehensive Cancer Center.
The abbreviations used are: mRNP, messenger ribonucleoprotein particle; PMR1, polysomal ribonuclease 1; PMR60, catalytically active 60-kDa processed form of PMR1; PMR60o, catalytically inactive form of PMR60; TAP, tandem affinity tag; GFP, green fluorescent protein; RT, reverse transcription.
F. Yang, unpublished information.
References
- 1.Wilusz CJ, Wormington M, Peltz SW. Nat Rev Mol Cell Biol. 2001;2:237–246. doi: 10.1038/35067025. [DOI] [PubMed] [Google Scholar]
- 2.Tharun S, Parker R. Mol Cell. 2001;8:1075–1083. doi: 10.1016/s1097-2765(01)00395-1. [DOI] [PubMed] [Google Scholar]
- 3.Sheth U, Parker R. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eystathioy T, Jakyymiw A, Chan EKL, Seraphin B, Cougot N, Fritzler MJ. RNA (N Y) 2003;9:1171–1173. doi: 10.1261/rna.5810203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Hoof A, Staples RR, Baker RE, Parker R. Mol Cell Biol. 2000;20:8230–8243. doi: 10.1128/mcb.20.21.8230-8243.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He F, Li X, Spatrick P, Casillo R, Dong S, Jacobson A. Mol Cell. 2003;12:1439–1452. doi: 10.1016/s1097-2765(03)00446-5. [DOI] [PubMed] [Google Scholar]
- 7.Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M. Cell. 2001;107:451–464. doi: 10.1016/s0092-8674(01)00578-5. [DOI] [PubMed] [Google Scholar]
- 8.Binder R, Hwang SP, Ratnasabapathy R, Williams DL. J Biol Chem. 1989;264:16910–16918. [PubMed] [Google Scholar]
- 9.Cunningham KS, Dodson RE, Nagel MA, Shapiro DJ, Schoenberg DR. Proc Natl Acad Sci U S A. 2000;97:12498–12502. doi: 10.1073/pnas.220425497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Binder R, Horowitz JA, Basilion JP, Koeller DM, Klausner RD, Harford JB. EMBO J. 1994;13:1969–1980. doi: 10.1002/j.1460-2075.1994.tb06466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevens A, Wang Y, Bremer K, Zhang J, Hoepfner R, Antoniou M, Schoenberg DR, Maquat LE. Proc Natl Acad Sci U S A. 2002;99:12741–12746. doi: 10.1073/pnas.192442399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bremer KA, Stevens A, Schoenberg DR. RNA (N Y) 2003;9:1157–1167. doi: 10.1261/rna.5720303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Dijk EL, Sussenbach JS, Holthuizen PE. J Mol Biol. 2000;300:449–467. doi: 10.1006/jmbi.2000.3856. [DOI] [PubMed] [Google Scholar]
- 14.Dompenciel RE, Garnepudi VR, Schoenberg DR. J Biol Chem. 1995;270:6108–6118. doi: 10.1074/jbc.270.11.6108. [DOI] [PubMed] [Google Scholar]
- 15.Pastori RL, Moskaitis JE, Schoenberg DR. Biochemistry. 1991;30:10490–10498. doi: 10.1021/bi00107a018. [DOI] [PubMed] [Google Scholar]
- 16.Pastori RL, Moskaitis JE, Buzek SW, Schoenberg DR. Mol Endocrinol. 1991;5:461–468. doi: 10.1210/mend-5-4-461. [DOI] [PubMed] [Google Scholar]
- 17.Chernokalskaya E, DuBell AN, Cunningham KS, Hanson MN, Dompenciel RE, Schoenberg DR. RNA (N Y) 1998;4:1537–1548. doi: 10.1017/s1355838298980451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cunningham KS, Hanson MN, Schoenberg DR. Nucleic Acids Res. 2001;29:1156–1162. doi: 10.1093/nar/29.5.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang F, Schoenberg DR. Mol Cell. 2004;14:435–445. doi: 10.1016/j.molcel.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Schoenberg DR, Cunningham KS. Methods. 1999;17:60–73. doi: 10.1006/meth.1998.0708. [DOI] [PubMed] [Google Scholar]
- 21.Das Gupta J, Gu H, Chernokalskaya E, Gao X, Schoenberg DR. RNA (N Y) 1998;4:766–776. doi: 10.1017/s1355838298971837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schoenberg DR, Moskaitis JE, Smith JH, Jr, Pastori RL. Mol Endocrinol. 1989;3:805–814. doi: 10.1210/mend-3-5-805. [DOI] [PubMed] [Google Scholar]
- 23.Chen CY, Del Gatto-Konczak F, Wu Z, Karin M. Science. 1998;280:1945–1949. doi: 10.1126/science.280.5371.1945. [DOI] [PubMed] [Google Scholar]
- 24.Mahtani KR, Brook M, Dean JLE, Sully G, Saklatvala J, Clark AR. Mol Cell Biol. 2001;21:6461–6469. doi: 10.1128/MCB.21.9.6461-6469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ming XF, Kaiser M, Moroni C. EMBO J. 1998;17:6039–6048. doi: 10.1093/emboj/17.20.6039. [DOI] [PMC free article] [PubMed] [Google Scholar]